

Abstract
High-throughput sequencing of messenger RNA (RNA-seq) has become the standard method for measuring and comparing the levels of gene expression in a wide variety of species and conditions. RNA-seq experiments generate very large, complex data sets that demand fast, accurate, and flexible software to reduce the raw read data to comprehensible results. HISAT, StringTie, and Ballgown are free, open-source software tools for comprehensive analysis of RNA-seq experiments. Together, they allow scientists to align reads to a genome, assemble transcripts including novel splice variants, compute the abundance of these transcripts in each sample, and compare experiments to identify differentially expressed genes and transcripts. This protocol describes all the steps necessary to process a large set of raw sequencing reads and create lists of gene transcripts, expression levels, and differentially expressed genes and transcripts. The protocol’s execution time depends on the computing resources, but typically takes under 45 minutes of computer time.
Keywords: RNA-seq, transcriptome, gene expression, transcript assembly, splicing, sequence alignment
Editor Summary
Pertea et al. describe a protocol to analyze RNA-seq data using HISAT, StringTie, and Ballgown (the “new Tuxedo” package). The protocol can be used for assembly of transcripts, quantification of gene expression levels and differential expression analysis.
Introduction
RNA sequencing (RNA-seq) experiments capture the total messenger RNA from a collection of cells and then sequence that RNA in order to determine which genes were active, or expressed, in those cells. Using high-throughput sequencing machines, a single experiment can capture the expression levels of thousands of genes at once with high accuracy1–3. These experiments generate enormous numbers of raw sequencing reads, typically numbering in the tens of millions even for a modest-sized assay. The number of reads produced from each gene can be used as a measure of gene abundance, and with proper design RNA-seq can detect which genes are expressed at significantly different levels in two or more conditions. RNA-seq data can easily detect genes and gene variants that are not included in standard annotation, including noncoding RNA genes. With the appropriate software, RNA-seq can also be used to discover conditions in which distinct isoforms of a single gene are differentially regulated and expressed.
RNA-seq experiments must be analyzed with accurate, efficient software that is carefully designed to handle the very large sequencing volumes generated by most experiments. The analysis pipeline can be conceptually divided into four main tasks: (i) alignment of the reads to the genome; (ii) assembly of the alignments into full-length transcripts, (iii) quantification of the expression levels of each gene and transcript, and (iv) calculation of the differences in expression for all genes among the different experimental conditions. Some of us previously developed two software tools, TopHat25 and Cufflinks4, that together could accomplish all four of these tasks, as described in an earlier protocol6. Recently we have developed three new software tools that accomplish the same tasks while running much faster, using substantially less memory, and providing more accurate overall results. HISAT7 aligns RNA-seq reads to a genome and discovers transcript splice sites, running far faster than TopHat2 and requiring much less computer memory than other methods. StringTie8 assembles the alignments into full and partial transcripts, creating multiple isoforms as necessary, and estimates the expression levels of all genes and transcripts. Ballgown9 takes the transcripts and expression levels from StringTie and applies rigorous statistical methods to determine which transcripts are differentially expressed between two or more experiments. We describe here a protocol to use these tools for RNA-seq data analysis; Figure 1 shows the software used in this protocol and highlights the main steps performed by each tool. All the tools are fully documented on the web and actively maintained by the developers.
Figure 1.
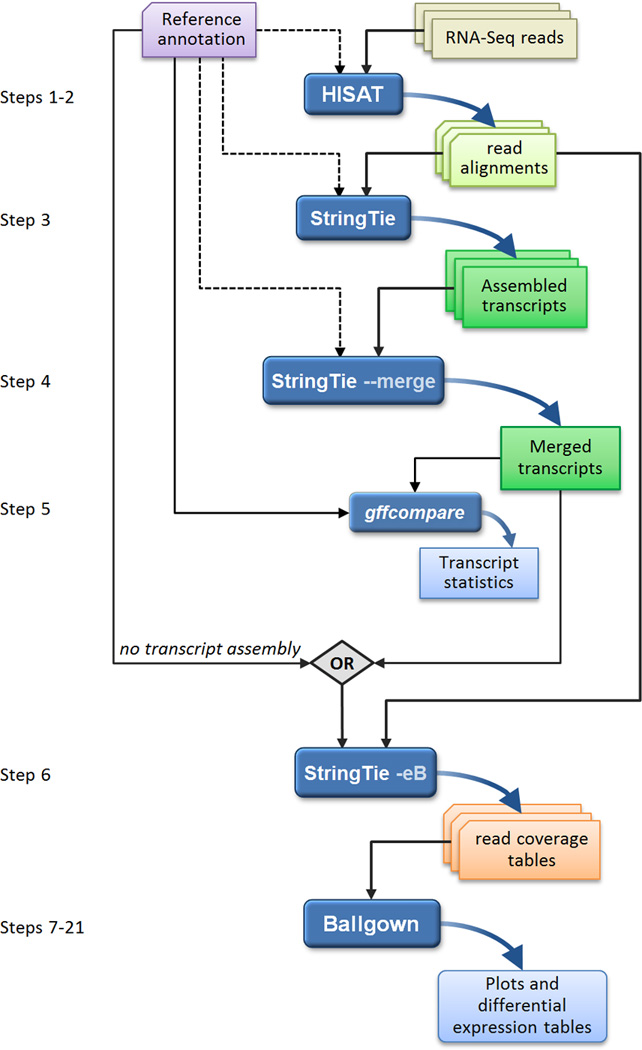
An overview of the "new Tuxedo" protocol. In an experiment involving multiple RNA-seq data sets, reads are first mapped to the genome using HISAT (Steps 1–2). Annotation of reference genes and transcripts can be provided as input, but this is optional, as indicated by the dotted line. The alignments are then passed to StringTie (Step 3), which assembles and quantifies the transcripts in each sample. (In the alternative protocol, the alignments from Step 2 are passed directly to Step 6, skipping all assembly steps. Step 6 will then estimate abundance only for known, annotated transcripts.) After initial assembly, the assembled transcripts are merged together (Step 4) by a special StringTie module, which creates a uniform set of transcripts for all samples. StringTie can use annotation in both of these steps, as shown by the dotted lines. The gffcompare program then compares the genes and transcripts to the annotation and reports statistics on this comparison (Step 5). In Step 6, StringTie processes the read alignments and either the merged transcripts or the reference annotation (through the diamond labeled "OR"). Using this input, StringTie re-estimates abundances where necessary and creates new transcript tables for input to Ballgown. Ballgown then compares all transcripts across conditions and produces tables and plots of differentially expressed genes and transcripts (Steps 7–21). Black and curved blue lines in the figure represent input to and output from the programs, respectively. Optional inputs are represented by dotted lines.
Overview of the protocol
RNA-seq experiments can be used to measure many phenomena. For simplicity, the protocol described here is intended to resemble experiments that will correspond closely to many users’ designs. We consider an experiment that compares two biological conditions, such as case versus control, wild type vs. mutant, or disease vs. normal comparisons. For each condition we include 6 replicates, noting that 3 is the minimum number of replicates for valid statistical results. The software will support many other designs, including time-course experiments and comparisons among more than two conditions.
The data used in this protocol comprises human RNA-seq samples, although the protocol will work for any species with a sequenced genome, including mouse, rat, Drosophila, Arabidopsis, yeast, and many others. (Some program parameters may require adjustment to optimize the results for genomes with smaller intron sizes.) The data files are very large, as is often the case for high-throughput RNA-seq experiments; thus to make the protocol faster and simpler for novice users, we have extracted a subset of the reads mapping to human chromosome X, a relatively gene-rich chromosome that spans 151 megabases (Mb), about 5% of the genome. The protocol describes the end-to-end analysis of these reads, but it will work equally well with the full data set, for which it will require significantly more computing time.
This protocol begins with raw RNA-seq reads collected from all samples, and produces several useful outputs, including lists of genes, transcripts, and expression levels for each sample, tables showing differentially expressed genes between the two conditions, and accompanying statistical measures of significance. First, reads from each sample are mapped to the reference genome with HISAT (Figure 1). The user can provide a file of annotated gene positions as an option, and HISAT will use that file but will also detect splice sites missing from the annotation. Next, the alignments are passed to StringTie for transcript assembly. StringTie assembles the genes for each data set separately, estimating the expression levels of each gene and each isoform as it assembles them. After assembling each sample, the full set of assemblies is passed to StringTie’s merge function, which merges together all the gene structures found in any of the samples. This step is required because transcripts in some of the samples might only be partially covered by reads, and as a consequence only partial versions of them will be assembled in the initial StringTie run. The merge step creates a set of transcripts that is consistent across all samples, so that the transcripts can be compared in subsequent steps. The merged transcripts are then fed back to StringTie one more time so that it can re-estimate the transcript abundances using the merged structures. The re-estimation uses the same algorithm as the original assembly, but reads may need to be re-allocated for transcripts whose structures were altered by the merging step. StringTie also provides additional read-count data for each transcript that are required by Ballgown. Finally, Ballgown takes all the transcripts and abundances from StringTie, groups them by experimental condition, and determines which genes and transcripts are differentially expressed between conditions. Ballgown includes plotting tools as part of the R/Bioconductor package that help to visualize the results.
This protocol does not require programming expertise, but it does assume familiarity with the Unix command line interface and the ability to run basic R scripts. Users should be comfortable running programs from the command line and editing text files in the Unix environment.
Alternative analysis packages
HISAT, StringTie, and Ballgown provide a complete analysis package (the "new Tuxedo" package) that begins with raw read data and produces gene lists and expression levels for each RNA-seq sample as well as lists of differentially expressed genes for an overall experiment. Other RNA-seq analysis packages have been developed that can be used instead of or in combination with these tools, most notably the TopHat2 and Cufflinks systems (the original "Tuxedo" package). The alignment step requires a spliced alignment algorithm that allows reads to span introns and that does not require annotation, for which several alternative tools are available5, 11, 12. Alignments from these other tools could be used as input to StringTie. Alternative methods have been developed for the transcriptome assembly and quantification steps as well4, 13, 14. Several methods can reconstruct transcripts de novo, without the use of a reference genome15–17, a different problem from the one considered in this protocol. Other RNA-seq analysis programs can quantitate transcripts by using annotated, known genes10, 18, 19; by skipping the transcript assembly step these programs offer substantial speed advantages. We note that even for very well-studied organisms such as human, mouse, and fruit fly, the annotation of protein-coding genes, splice variants, and noncoding RNA genes is far from complete, and these alternative programs cannot report any novel transcripts or splice variants.
After all samples have been assembled, our protocol uses StringTie to merge the transcripts, but the cuffmerge program, part of the Cufflinks package, could be used instead. Finally, multiple tools for computing differential expression have been developed20–22, and these could be used as alternatives to Ballgown. The input requirements for some of these alternative programs vary, and may require data format conversion steps before they can be used with the programs included here. Such customization is beyond the scope of this protocol, and we recommend that users follow the protocol described below.
We also note that although the protocol presented here detects differentially expressed transcripts and genes, it is not designed to detect differential exon usage alone, a task that several other programs (DEXseq23, rMATS24, and MISO25) have been developed specifically to address.
Limitations of the protocol and software
HISAT, StringTie, and Ballgown are not the only methods for gene and transcript expression analysis from RNA-seq data, and they will not handle all RNA-seq experiments. For example, some RNA-seq experiments may require pre-processing of the raw RNA-seq data to remove contaminants, adapters, low-quality sequences, and other artifacts. This protocol does not include such pre-processing, but tools such as the FASTX toolkit (http://hannonlab.cshl.edu/fastx_toolkit) and FastQC (http://www.bioinformatics.babraham.ac.uk/projects/fastqc) can help ensure that high-quality data is provided to the initial spliced alignment step. We also assume that RNA-seq data has been generated on an Illumina sequencing machine, but the much longer, high-error-rate reads from third-generation sequences such as those from Pacific Biosciences or Oxford Nanopore may require different software, especially for the alignment step.
Ballgown can be used to load data from several assemblers including StringTie, Cufflinks4 and RSEM10; however, other transcriptome assembly methods produce output that is not yet compatible with Ballgown. The methods are also primarily designed for use at the transcript level, although the Bioconductor object allows for the application of any Biconductor package for gene- or exon-level analysis. Note that the default parameters for Ballgown were created with the assumption that sample sizes would be modest, ranging from as few as 3 up to a few hundred. However, the parameters can be tuned to a wide range of specific cases should users seek to apply the software in ways other than that described in this protocol.
Experimental Design
Read alignment with HISAT
RNA-seq analysis begins by mapping reads against a reference genome to identify their genomic positions. This mapping information allows us to collect subsets of the reads corresponding to each gene, and then to assemble and quantify transcripts represented by those reads. Several very fast algorithms for aligning reads to a genome have been developed, of which Bowtie26, 27 and BWA28 are among the most widely used. Both of these methods use a data structure called the Burrows-Wheeler transform (BWT), which stores the reference genome in a highly compressed form. Through the use of a special indexing scheme called the Ferragina-Manzini (FM) index29, these programs can search a genome extremely rapidly, yielding alignment speeds measured in millions of reads per hour.
RNA-seq mappers need to solve an additional problem not encountered in DNA-only alignment: many RNA-seq reads will span introns. The RNA sequencing processing captures and sequences mature messenger RNA molecules, from which introns have been removed by splicing. Thus a single short read might align to two locations that are separated by 10,000 bp or more (the average human intron length is >6000 bp, and some introns are > 1 Mbp in length). For a typical human RNA-seq data set using 100-bp reads, more than 35% of the reads will span multiple exons. Aligning these multi-exon spanning reads is a much more difficult task than aligning reads contained within one exon.
HISAT7 (Hierarchical Indexing for Spliced Alignment of Transcripts) employs two types of indexes for alignment: a global, whole-genome index and tens of thousands of small local indexes. Both indexes are constructed using the same BWT/FM index as Bowtie2, and the HISAT system even uses some of the Bowtie2 code. Thanks to its use of these efficient data structures and algorithms, HISAT generates spliced alignments several times faster than Bowtie and BWA while using only about twice as much memory. HISAT was designed as a successor to TopHat and TopHat2 (which were developed by some of the authors of this protocol) and has compatible outputs, but runs approximately 50 times faster. For alignment to the human genome, the index requires less than 8 gigabytes (GB) of memory, allowing it to run on a conventional desktop, and 20 human RNA-seq samples, with 100M reads per sample, should take in less than a day to process on one computer. For species with smaller genomes, memory and processing requirements will be correspondingly reduced. In this protocol, we use HISAT2, which incorporates algorithmic improvements that yield slightly higher accuracy than the original HISAT system.
Alignments of RNA-seq data can be used to detect novel splice sites, transcription initiation sites, and transcription termination sites. Like TopHat, HISAT will detect all of these events if they are present in the data. Users can also provide gene annotations, which specify the location of known genes including all of their exon and intron boundaries. The use of high-quality annotation may improve HISAT’s alignments in the vicinity of known genes, but this input is optional.
Transcript assembly and quantification with StringTie
Analysis of RNA-seq experiments requires accurate reconstructions of all the isoforms expressed from each gene as well as estimates of the relative abundance of those isoforms. Because annotation remains incomplete, even for the most intensively studied species such as humans, we cannot provide our programs with a complete list of all genes and all isoforms. Nonetheless, accurate quantification benefits from knowledge of precisely which reads originated from each isoform, which cannot be computed perfectly because reads are much shorter than transcripts. StringTie8 assembles transcripts from RNA-seq reads that have been aligned to the genome, first grouping the reads into distinct gene loci and then assembling each locus into as many isoforms as are needed to explain the data. StringTie uses a network flow algorithm that begins with the most highly-expressed transcript, which it assembles and quantitates simultaneously. It then removes the reads associated with that transcript and repeats the process, assembling more isoforms until all the reads are used, or else until the number of reads remaining is below the (user-adjustable) level of transcriptional noise.
StringTie’s network flow algorithm can re-construct transcripts more accurately than some previous methods because it computes abundance and exon-intron structure at the same time. The algorithm also has computational run-time efficiencies that allow it to run many times faster and to use much less memory than its predecessor, Cufflinks. Most RNA-seq runs can be processed by StringTie on a conventional desktop computer with as little as 8 GB of RAM, and even large data sets run in under an hour.
Users have the option of providing StringTie with a file containing reference gene models; this annotation file contains a specification of the exon-intron structure for "known" genes along with names for those genes. If the user provides this annotation file, StringTie will use it as a guide to assembly, and it will also tag the genes it assembles with names from that file. Annotation can be helpful in the reconstruction of low-abundance genes, where the number of reads is too low to allow accurate assembly. Note that the annotation file is optional, and StringTie will assemble additional, unannotated genes as necessary to explain the data. After assembling transcripts with StringTie, a user can use the gffcompare utility (see Box 1 and Table 1) to determine how many assembled transcripts match annotated genes either fully or partially, and to compute how many are entirely novel. Alternatively, the users can skip the assembly of novel genes and transcripts, and use StringTie simply to quantify all the transcripts provided in an annotation file (see Figure 1).
Box 1: Comparing a set of transcripts to a known gene list.
Comparing lists of genes and transcripts requires first that the lists are in the same format. Many bioinformatics programs utilize GFF (gene finding format) to represent genes and transcripts. GFF is a simple tab-delimited text format that describes the locations and the attributes of genes, transcripts, exons, and introns on a genome. StringTie uses Gene Transfer Format (GTF), a special version of GFF that contains some additional data types (see http://www.ensembl.org/info/website/upload/gff.html for details).
We have developed the gffcompare utility (available from http://ccb.jhu.edu/software/stringtie/gff.shtml or http://github.com/gpertea/gffcompare) to compare one or more GTF files produced by StringTie to a reference annotation file in either GFF or GTF. Assuming that StringTie’s output is in transcripts.gtf and the reference annotation file is chrX.gtf, the gffcompare program can be run using the following command:
$ gffcompare –G –r chrX.gtf transcripts.gtf
The –r option is followed by the annotation file to use as reference, and the –G option tells gffcompare to compare all transcripts in the input transcripts.gtf file, even those that might be redundant.
The gffcompare program is based on the CuffCompare utility, which is part of the Cufflinks/Tuxedo suite4, and many of the usage options and outputs documented in the CuffCompare manual (http://cole-trapnell-lab.github.io/cufflinks/cuffcompare) apply to the gffcompare program as well. All files generated by gffcompare will have the prefix gffcmp unless the user chooses a different prefix with the –o option. When used as shown above gffcompare produces an output file, called gffcmp.annotated.gtf, which adds to each transcript a "class code" (described in Table 1) and the name of the transcript from the reference annotation file. This allows the user to quickly check how the predicted transcripts relate to an annotation file. The gffcompare command shown here will also compute sensitivity and precision statistics for different gene features (e.g., exons, introns, transcripts, and genes) in the gffcmp.stats output file. Sensitivity is defined as the proportion of genes from the annotation that are correctly reconstructed, while precision (also known as positive predictive value) captures the proportion of the output that overlaps the annotation. Thus both these measures are relative to the input annotation and might not fully capture the true accuracy of the assembled genes and transcripts. The gffcmp.stats file also contains several other measures such as the total number of novel exons, introns, or genes contained in the StringTie output.
Table 1.
Class codes used to describe how assembled transcripts compare to reference annotation.
| Class code | Description |
|---|---|
| = | Predicted transcript has exactly the same introns as the reference transcript. |
| c | Predicted transcript is contained within the reference transcript. |
| j | Predicted transcript is a potential novel isoform that shares at least one splice junction with a reference transcript. |
| e | Single exon predicted transcript overlaps a reference exon plus at least 10 bp of a reference intron, indicating a possible pre-mRNA fragment. |
| i | Predicted transcript falls entirely within a reference intron. |
| o | Exon of predicted transcript overlaps a reference transcript. |
| p | Predicted transcript lies within 2 Kb of a reference transcript (possible polymerase run-on fragment). |
| r | Predicted transcript has more than 50% of its bases overlapping a soft-masked (repetitive) reference sequence. |
| u | Predicted transcript is intergenic in comparison to known reference transcripts. |
| x | Exon of predicted transcript overlaps reference but lies on the opposite strand. |
| s | Intron of predicted transcript overlaps a reference intron on the opposite strand. |
In most experimental designs, including the protocol here, the experiment includes multiple RNA-seq samples. The genes and isoforms present in one sample are rarely identical to those present in all other samples, but they need to be assembled in a consistent manner so that they can be compared. We address this problem by merging all assemblies together using StringTie’s merge function, which merges all the genes found in any of the samples. Thus a transcript that was missing an exon in one sample due to lack of coverage might be restored to its full length; see Figure 2 for an illustration of how merging can improve assemblies.
Figure 2.
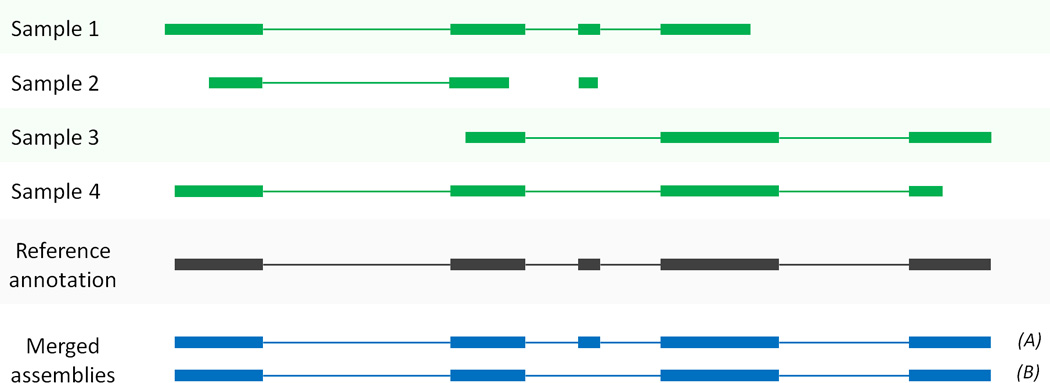
Merging transcript assemblies using StringTie’s merge function. In this example, four partial assemblies from four different samples are merged into two transcripts A and B. Samples 1 and 2 are both consistent with the reference annotation, which is used here to merge and extend them to create transcript A. Samples 3 and 4 are consistent with each other but not with the annotation, and these are merged to create transcript B.
After the merging step, StringTie is then run one more time to re-estimate the abundances of the merged transcripts. The re-estimation step uses the same abundance estimation algorithm as in the initial steps.
Many if not all users of RNA-seq are interested in discovering differentially expressed genes, and many are focused on well-studied genes. Thus it might be tempting to use a method that relies entirely on annotation and cannot discover new genes and new isoforms, especially for well-studied species such as humans, mice, and fruit flies. However, the standard annotation files available for these species are likely missing thousands of isoforms as well as many genes. For example, a recent study30 used ribosome footprint profiling to validate 7,273 previously unannotated human genes, most of which were discovered when they ran StringTie on a very large set (2.8 billion reads) of RNA-seq data. StringTie automatically detects new genes and new isoforms if they are present in your experimental data, regardless of whether or not they appear in standard annotation files, and the protocol described here can discover differential expression affecting these genes.
Differential expression analysis with Ballgown
Exploratory analysis, visualization, and statistical modeling are the next steps after assembling and quantifying transcripts. The R programming language and the Bioconductor software suite host a comprehensive set of tools ranging from plotting raw data, to normalization, to downstream statistical modeling. Our pipeline uses the Ballgown package, which is designed to be a bridge between upstream command line software like HISAT and StringTie and the downstream functionality of the Bioconductor community.
StringTie is designed to produce a linked set of tables that can be directly read into R using functions in the Ballgown package. The resulting data in R includes information in three tables: (1) phenotype data – information about the samples being collected; (2) expression data – normalized and un-normalized measures of the amount each exon, junction, transcript, and gene expressed in each sample; and (3) genomic information – coordinates giving the location of the exons, introns, transcripts, and genes as well as annotation including information such as gene names.
Once the information has been read into the R session, analysis proceeds interactively. In other words, any package or function available in R and Bioconductor is available for use. Most differential expression analyses proceed along this path: (1) data visualization and inspection, (2) statistical tests for differential expression, (3) multiple test correction, and (4) downstream inspection and summarization of results. Ballgown provides functions for each of these steps that can be combined with other R commands when there are specific issues with data sets, as typically arise in RNA-seq analysis.
The Ballgown portion of the protocol begins with loading the data into R. This requires loading the abundance data produced by StringTie and the phenotype information describing the samples. A critical step in the process is to ensure that the identifiers (IDs) from the genomic samples match the IDs from the phenotype data. Ballgown will produce an error if the samples do not appear to match. After loading the data, the next step is to inspect the distribution of abundance estimates for the transcripts. The abundance estimates (expressed as FPKM values, Fragments Per Kilobase of transcript per Million mapped reads) have already been library-size normalized, so any extreme differences in the overall distribution between samples should lead to concern that there has been a problem with that sample, the alignment, or the abundance estimation.
Differential expression analysis proceeds using a linear model. The FPKM values attached to transcripts are typically highly skewed; thus to stabilize the variance, Ballgown’s built-in functions apply a log transformation and then fit standard linear models that can be used to test for differential expression. Ballgown permits the analysis of both timecourse and fixed condition differential expression analyses. One common problem that arises in RNA-seq analysis is failure to account for confounders – variables such as batch effects – that are not of interest for the analysis but may affect the expression levels of genes. Using the Ballgown stattest function you can directly specify any known confounders. Ballgown allows you to perform your analysis at the gene, transcript, exon, or junction level. The result is a table with information on the feature tested for differential expression, the fold change between conditions if appropriate, the p-value, and the q-value for differential expression.
These results can then be used for further exploration with downstream gene set analysis software, visual inspection of significant results, or exporting of results to tables that can be shared and manually inspected using programs such as Excel.
MATERIALS
EQUIPMENT
Data (example RNA-seq reads, indexes, and gene annotations for use in this protocol are available at ftp://ftp.ccb.jhu.edu/pub/RNAseq_protocol, see EQUIPMENT SETUP for details)
HISAT2 software (http://ccb.jhu.edu/software/hisat2) or http://github.com/infphilo/hisat2 (version 2.0.1 or later)
StringTie software (http://ccb.jhu.edu/software/stringtie) or https://github.com/gpertea/stringtie (version 1.2.2 or later)
SAM tools (http://samtools.sourceforge.net) (version 0.1.19 or later)
R (https://www.r-project.org) (version 3.2.2 or later)
Hardware (64-bit computer running either Linux or Mac OS X (10.7 Lion or later); 4 GB of RAM (8 GB preferred); see EQUIPMENT SETUP)
EQUIPMENT SETUP
CRITICAL The commands used in the protocol should all be run from the Unix shell prompt within a terminal window up to the differential expression analysis, which is performed in the R environment. We encourage users to create a single directory (e.g., "my_rnaseq_exp") in which to store all example data and files created by the analysis. All commands are described under the assumption that the user is working in this directory. The protocol also includes small sections of code to be run in the R statistical computing environment. Commands meant to be executed from the Unix shell (e.g., bash or csh) are prefixed with a ‘$’ character. Commands meant to be run from either an R script or at the R interactive shell are prefixed with a ‘>’ character.
Required data
The protocol is illustrated with an example experiment from chromosome X of Homo sapiens that you can analyze to familiarize yourself with the full suite of software. All of the data you will need is available in the file chrX_data.tar.gz, described below. To use HISAT2 and StringTie for gene expression analysis, you must be working with an organism whose genome has been sequenced. Both tools can also use an annotation file of genes and transcripts, although this is optional. HISAT2 requires an index for the genome, which is provided in the data download file. Pre-built indexes for multiple other species are available at the HISAT2 website. Box 2 provides instructions on how to create a HISAT2 index if one is not already available.
Box 2: Creating a HISAT2 index.
Pre-built HISAT2 indices for the human genome and many model organisms can be downloaded from the HISAT website. If the required index is not available there, a user may have to build a custom index. Here we describe how to build this index for human chromosome X; to build it for another genome, replace the genome file chrX.fa and the annotation file chrX.gtf with files corresponding to that genome and its annotation.
First, using the python scripts included in the HISAT2 package, extract splice site and exon information from the gene annotation file:
$ extract_splice_sites.py chrX_data/genes/chrX.gtf > chrX.ss $ extract_exons.py chrX_data/genes/chrX.gtf > chrX.exon
Second, build a HISAT2 index:
$ hisat2-build --ss chrX.ss --exon chrX.exon chrX_data/genome/chrX.fachrX_tran
The --ss and --exon options can be omitted in the command above if annotation is not available.
Note that the index-building step requires a larger amount of memory than the alignment step, and might not be possible on a desktop computer. For example, indexing requires 9 GB of RAM for chromosome X, and 160 GB for the whole human genome. The amount of memory is much smaller if one omits annotation information. Indexing chromosome X using 1 CPU core takes less than 10 minutes. It should take ~2 hours to build an index for the whole human genome using 8 CPU cores.
Downloading and organizing required data
Unpack the data (ftp://ftp.ccb.jhu.edu/pub/RNAseq_protocol/chrX_data.tar.gz) and inspect the contents.
$ tar xvzf chrX_data.tar.gz
Assuming we stored the data at my_rnaseq_exp/, the package expands to contain a folder chrX_data/, which has the following structure: samples/ indexes/ genome/ genes/ (i.e., four directories).
The samples directory contains paired-end RNA-seq reads for 12 samples, with 6 samples from each of two populations, GBR (British from England) and YRI (Yoruba from Ibadan, Nigeria). For each population there are 3 samples each from male and female subjects (see Table 2 for the population and sex associated with each sample). All sequence is in compressed "fastq" format, which stores each read on 4 lines. Each sample in turn is contained in two files, one for read 1 and another for read 2 of each pair. Thus for example, sample ERR188245 is contained in files ERR188245_chrX_1.fastq.gz and ERR188245_chrX_2.fastq.gz, where each file contains 855,983 reads. Note that the original data files contain reads mapping to the entire genome rather than just chromosome X, and these files are approximately 25 times larger. The README file at ftp://ftp.ccb.jhu.edu/pub/RNAseq_protocol contains instructions on how to download the full data sets for those who are interested. Note that HISAT2 also provides an option, called --sra-acc, to directly work with SRA data31 over the internet. This eliminates the need to manually download SRA reads and convert them into fasta/fastq format, without significantly affecting the run time. For example, the following HISAT2 command will download and align one of the 12 samples used in this protocol (ERR188245):
$ hisat2 -p 8 --dta -x chrX_data/indexes/chrX_tran --sra-acc ERR188245 –S ERR188245_chrX.sam
Table 2.
Population and sex associated with each sample used in the protocol.
| Sample ID | Sex | Population |
|---|---|---|
| ERR188245 | female | GBR |
| ERR188428 | female | GBR |
| ERR188337 | female | GBR |
| ERR188401 | male | GBR |
| ERR188257 | male | GBR |
| ERR188383 | male | GBR |
| ERR204916 | female | YRI |
| ERR188234 | female | YRI |
| ERR188273 | female | YRI |
| ERR188454 | male | YRI |
| ERR188104 | male | YRI |
| ERR188044 | male | YRI |
The indexes directory contains the HISAT2 indexes for chromosome X, including chrX_tran.1.ht2, chrX_tran.2.ht2, chrX_tran.3.ht2, chrX_tran.4.ht2, chrX_tran.5.ht2, chrX_tran.6.ht2, chrX_tran.7.ht2, and chrX_tran.8.ht2.
The genome directory contains one file, chrX.fa, the sequence of human chromosome X (GRCh38 build 81). Note that if you were using the full genome, this directory would contain a single file containing all the chromosomes. The genes directory contains one file, chrX.gtf, containing human gene annotations for GRCh38 from the RefSeq database.
The chrX_data directory also contains two more files: mergelist.txt and geuvadis_phenodata.csv. These files are text files that you need to run the protocol, as explained below. Normally you would create these yourself in a text editor, but we provide them here for use as examples.
Downloading and installing software
Create a directory to store all of the executable programs used in this protocol (if none already exists):
$ mkdir $HOME/bin
Add the above directory to your PATH environment variable:
$ export PATH=$HOME/bin:$PATH
To install the SAM tools, download them from http://samtools.sourceforge.net, unpack the SAMtools tarfile, and cd to the SAM tools source directory (the protocol will work with all versions of SAMtools starting with 0.1.19):
$ tar jxvf samtools-0.1.19.tar.bz2 $ cd samtools-0.1.19 $ make $ cd ..
Copy the samtools binary to a directory in your PATH:
$ cp samtools-0.1.19/samtools $HOME/bin
To install HISAT2, download the latest binary package from http://ccb.jhu.edu/software/hisat2 or http://github.com/infphilo/hisat2, unpack the HISAT2 zip archive, and cd to the unpacked directory:
$ unzip hisat2-2.0.1-beta-OSX_x86_64.zip
Next, copy the HISAT2 executables to a directory in your PATH:
$ cp hisat2-2.0.1-beta/hisat2* hisat2-2.0.1-beta/*.py $HOME/bin
To install StringTie, download the latest binary package from http://ccb.jhu.edu/software/stringtie, unpack the StringTie tarfile, and cd to the unpacked directory:
$ tar xvzf stringtie-1.2.2.OSX_x86_64.tar.gz
(Note that this instruction uses the Mac OS X executable. If you are using Linux, get this executable instead: stringtie-1.2.2.Linux_x86_64.tar.gz.) Next, copy the StringTie package’s executable files to some directory in your PATH:
$ cp stringtie-1.2.2.OSX_x86_64/stringtie $HOME/bin
StringTie can also be obtained from https://github.com/gpertea/stringtie.
To install gffcompare download the latest binary package from http://github.com/gpertea/gffcompare, and follow the instructions provided in the README.md file.
To Install Ballgown, start an R session:
$ R R version 3.2.2 (2015-08–14) -- "Fire Safety" Copyright (C) 2015 The R Foundation for Statistical Computing Platform: x86_64-apple-darwin13.4.0 (64-bit)
The R introductory message will end with a ">" prompt. Install the Ballgown packages and other dependencies in R as follows:
> install.packages("devtools",repos="http://cran.us.r-project.org")
> source("http://www.bioconductor.org/biocLite.R")
> biocLite(c("alyssafrazee/RSkittleBrewer","ballgown",
"genefilter","dplyr","devtools"))
Bioconductor Version 3.0 or greater and R version 3.1 are required to run this protocol.
PROCEDURE
Align the RNA-seq reads to the genome (TIMING: <20 m)
CRITICAL Supplementary Software includes a shell script and other dependent files that can be used to run all the steps this protocol. Also included is a README file that explains how to run the script. These files are also available from ftp://ftp.ccb.jhu.edu/pub/RNAseq_protocol.
-
1.Map the reads for each sample to the reference genome:
$ hisat2 -p 8 --dta -x chrX_data/indexes/chrX_tran -1 chrX_data/samples/ERR188044_chrX_1.fastq.gz -2 chrX_data/samples/ERR188044_chrX_2.fastq.gz -S ERR188044_chrX.sam $ hisat2 -p 8 --dta -x chrX_data/indexes/chrX_tran -1 chrX_data/samples/ERR188104_chrX_1.fastq.gz -2 chrX_data/samples/ERR188104_chrX_2.fastq.gz -S ERR188104_chrX.sam $ hisat2 -p 8 --dta -x chrX_data/indexes/chrX_tran -1 chrX_data/samples/ERR188234_chrX_1.fastq.gz -2 chrX_data/samples/ERR188234_chrX_2.fastq.gz -S ERR188234_chrX.sam $ hisat2 -p 8 --dta -x chrX_data/indexes/chrX_tran -1 chrX_data/samples/ERR188245_chrX_1.fastq.gz -2 chrX_data/samples/ERR188245_chrX_2.fastq.gz -S ERR188245_chrX.sam $ hisat2 -p 8 --dta -x chrX_data/indexes/chrX_tran -1 chrX_data/samples/ERR188257_chrX_1.fastq.gz -2 chrX_data/samples/ERR188257_chrX_2.fastq.gz -S ERR188257_chrX.sam $ hisat2 -p 8 --dta -x chrX_data/indexes/chrX_tran -1 chrX_data/samples/ERR188273_chrX_1.fastq.gz -2 chrX_data/samples/ERR188273_chrX_2.fastq.gz -S ERR188273_chrX.sam $ hisat2 -p 8 --dta -x chrX_data/indexes/chrX_tran -1 chrX_data/samples/ERR188337_chrX_1.fastq.gz -2 chrX_data/samples/ERR188337_chrX_2.fastq.gz -S ERR188337_chrX.sam $ hisat2 -p 8 --dta -x chrX_data/indexes/chrX_tran -1 chrX_data/samples/ERR188383_chrX_1.fastq.gz -2 chrX_data/samples/ERR188383_chrX_2.fastq.gz -S ERR188383_chrX.sam $ hisat2 -p 8 --dta -x chrX_data/indexes/chrX_tran -1 chrX_data/samples/ERR188401_chrX_1.fastq.gz -2 chrX_data/samples/ERR188401_chrX_2.fastq.gz -S ERR188401_chrX.sam $ hisat2 -p 8 --dta -x chrX_data/indexes/chrX_tran -1 chrX_data/samples/ERR188428_chrX_1.fastq.gz -2 chrX_data/samples/ERR188428_chrX_2.fastq.gz -S ERR188428_chrX.sam $ hisat2 -p 8 --dta -x chrX_data/indexes/chrX_tran -1 chrX_data/samples/ERR188454_chrX_1.fastq.gz -2 chrX_data/samples/ERR188454_chrX_2.fastq.gz -S ERR188454_chrX.sam $ hisat2 -p 8 --dta -x chrX_data/indexes/chrX_tran -1 chrX_data/samples/ERR204916_chrX_1.fastq.gz -2 chrX_data/samples/ERR204916_chrX_2.fastq.gz -S ERR204916_chrX.sam
-
2.Sort and convert the SAM files to BAM:
$ samtools sort -@ 8 -o ERR188044_chrX.bam ERR188044_chrX.sam $ samtools sort -@ 8 -o ERR188104_chrX.bam ERR188104_chrX.sam $ samtools sort -@ 8 -o ERR188234_chrX.bam ERR188234_chrX.sam $ samtools sort -@ 8 -o ERR188245_chrX.bam ERR188245_chrX.sam $ samtools sort -@ 8 -o ERR188257_chrX.bam ERR188257_chrX.sam $ samtools sort -@ 8 -o ERR188273_chrX.bam ERR188273_chrX.sam $ samtools sort -@ 8 -o ERR188337_chrX.bam ERR188337_chrX.sam $ samtools sort -@ 8 -o ERR188383_chrX.bam ERR188383_chrX.sam $ samtools sort -@ 8 -o ERR188401_chrX.bam ERR188401_chrX.sam $ samtools sort -@ 8 -o ERR188428_chrX.bam ERR188428_chrX.sam $ samtools sort -@ 8 -o ERR188454_chrX.bam ERR188454_chrX.sam $ samtools sort -@ 8 -o ERR204916_chrX.bam ERR204916_chrX.sam
The above commands work with samtools version 1.3 or newer. For older versions of samtools the user should consult Box 3.
Box 3: Sorting and converting SAM files using samtools version 1.2.1 and older.
Recent versions of samtools can convert SAM to BAM in a single step. For those using older versions, two steps are required, as shown here.
Convert SAM files to binary BAM files:
$ samtools view -bS ERR188044_chrX.sam > ERR188044_chrX_unsorted.bam $ samtools view -bS ERR188104_chrX.sam > ERR188104_chrX_unsorted.bam $ samtools view -bS ERR188234_chrX.sam > ERR188234_chrX_unsorted.bam $ samtools view -bS ERR188245_chrX.sam > ERR188245_chrX_unsorted.bam $ samtools view -bS ERR188257_chrX.sam > ERR188257_chrX_unsorted.bam $ samtools view -bS ERR188273_chrX.sam > ERR188273_chrX_unsorted.bam $ samtools view -bS ERR188337_chrX.sam > ERR188337_chrX_unsorted.bam $ samtools view -bS ERR188383_chrX.sam > ERR188383_chrX_unsorted.bam $ samtools view -bS ERR188401_chrX.sam > ERR188401_chrX_unsorted.bam $ samtools view -bS ERR188428_chrX.sam > ERR188428_chrX_unsorted.bam $ samtools view -bS ERR188454_chrX.sam > ERR188454_chrX_unsorted.bam $ samtools view -bS ERR204916_chrX.sam > ERR204916_chrX_unsorted.bam
Sort the BAM files:
$ samtools sort -@ 8 ERR188044_chrX_unsorted.bam ERR188044_chrX $ samtools sort -@ 8 ERR188104_chrX_unsorted.bam ERR188104_chrX $ samtools sort -@ 8 ERR188234_chrX_unsorted.bam ERR188234_chrX $ samtools sort -@ 8 ERR188245_chrX_unsorted.bam ERR188245_chrX $ samtools sort -@ 8 ERR188257_chrX_unsorted.bam ERR188257_chrX $ samtools sort -@ 8 ERR188273_chrX_unsorted.bam ERR188273_chrX $ samtools sort -@ 8 ERR188337_chrX_unsorted.bam ERR188337_chrX $ samtools sort -@ 8 ERR188383_chrX_unsorted.bam ERR188383_chrX $ samtools sort -@ 8 ERR188401_chrX_unsorted.bam ERR188401_chrX $ samtools sort -@ 8 ERR188428_chrX_unsorted.bam ERR188428_chrX $ samtools sort -@ 8 ERR188454_chrX_unsorted.bam ERR188454_chrX $ samtools sort -@ 8 ERR204916_chrX_unsorted.bam ERR204916_chrX
Optionally, the user might want to delete temporary files:
$ rm *.sam *unsorted.bam
Assemble and quantify expressed genes and transcripts (TIMING: ~15 m)
-
3.Assemble transcripts for each sample:
$ stringtie -p 8 -G chrX_data/genes/chrX.gtf -o ERR188044_chrX.gtf –l ERR188044 ERR188044_chrX.bam $ stringtie -p 8 -G chrX_data/genes/chrX.gtf -o ERR188104_chrX.gtf –l ERR188104 ERR188104_chrX.bam $ stringtie -p 8 -G chrX_data/genes/chrX.gtf -o ERR188234_chrX.gtf –l ERR188234 ERR188234_chrX.bam $ stringtie -p 8 -G chrX_data/genes/chrX.gtf -o ERR188245_chrX.gtf –l ERR188245 ERR188245_chrX.bam $ stringtie -p 8 -G chrX_data/genes/chrX.gtf -o ERR188257_chrX.gtf –l ERR188257 ERR188257_chrX.bam $ stringtie -p 8 -G chrX_data/genes/chrX.gtf -o ERR188273_chrX.gtf –l ERR188273 ERR188273_chrX.bam $ stringtie -p 8 -G chrX_data/genes/chrX.gtf -o ERR188337_chrX.gtf –l ERR188337 ERR188337_chrX.bam $ stringtie -p 8 -G chrX_data/genes/chrX.gtf -o ERR188383_chrX.gtf –l ERR188383 ERR188383_chrX.bam $ stringtie -p 8 -G chrX_data/genes/chrX.gtf -o ERR188401_chrX.gtf –l ERR188401 ERR188401_chrX.bam $ stringtie -p 8 -G chrX_data/genes/chrX.gtf -o ERR188428_chrX.gtf –l ERR188428 ERR188428_chrX.bam $ stringtie -p 8 -G chrX_data/genes/chrX.gtf -o ERR188454_chrX.gtf –l ERR188454 ERR188454_chrX.bam $ stringtie -p 8 -G chrX_data/genes/chrX.gtf -o ERR204916_chrX.gtf –l ERR204916 ERR204916_chrX.bam
-
4.Merge transcripts from all samples:
$ stringtie --merge -p 8 -G chrX_data/genes/chrX.gtf -o stringtie_merged.gtf chrX_data/mergelist.txt
?TROUBLESHOOTING
Here mergelist.txt is a text file that has the names of the GTF files created in the previous step, with each file name on a single line (see ChrX_data for an example file). If you do not run StringTie in the same directory where all the GTF files are, then you also need to include the full path in each GTF file name in mergelist.txt. Use any text editor to create or edit the (plaintext) mergelist.txt file.
-
5.Examine how the transcripts compare to the reference annotation (optional):
$ gffcompare –r chrX_data/genes/chrX.gtf –G –o merged stringtie_merged.gtf
the –o option specifies the prefix to use for output files that gffcompare will create.The command above will generate multiple files explained in the gffcompare documentation; see Box 1 for more details.
-
6.Estimate transcript abundances and create table counts for Ballgown:
$ stringtie –e –B -p 8 -G stringtie_merged.gtf -o ballgown/ERR188044/ERR188044_chrX.gtf ERR188044_chrX.bam $ stringtie –e –B -p 8 -G stringtie_merged.gtf -o ballgown/ERR188104/ERR188104_chrX.gtf ERR188104_chrX.bam $ stringtie –e –B -p 8 -G stringtie_merged.gtf -o ballgown/ERR188234/ERR188234_chrX.gtf ERR188234_chrX.bam $ stringtie –e –B -p 8 -G stringtie_merged.gtf -o ballgown/ERR188245/ERR188245_chrX.gtf ERR188245_chrX.bam $ stringtie –e –B -p 8 -G stringtie_merged.gtf -o ballgown/ERR188257/ERR188257_chrX.gtf ERR188257_chrX.bam $ stringtie –e –B -p 8 -G stringtie_merged.gtf -o ballgown/ERR188273/ERR188273_chrX.gtf ERR188273_chrX.bam $ stringtie –e –B -p 8 -G stringtie_merged.gtf -o ballgown/ERR188337/ERR188337_chrX.gtf ERR188337_chrX.bam $ stringtie –e –B -p 8 -G stringtie_merged.gtf -o ballgown/ERR188383/ERR188383_chrX.gtf ERR188383_chrX.bam $ stringtie –e –B -p 8 -G stringtie_merged.gtf -o ballgown/ERR188401/ERR188401_chrX.gtf ERR188401_chrX.bam $ stringtie –e –B -p 8 -G stringtie_merged.gtf -o ballgown/ERR188428/ERR188428_chrX.gtf ERR188428_chrX.bam $ stringtie –e –B -p 8 -G stringtie_merged.gtf -o ballgown/ERR188454/ERR188454_chrX.gtf ERR188454_chrX.bam $ stringtie –e –B -p 8 -G stringtie_merged.gtf -o ballgown/ERR204916/ERR204916_chrX.gtf ERR204916_chrX.bam
Run the differential expression analysis protocol (TIMING: ~5 m)
-
7.Load relevant R packages. These include the Ballgown package that you will use for performing most of the analyses but a few other packages that help with these analyses, specifically: RSkittleBrewer (for setting up colors), genefilter (for fast calculation of means and variances), dplyr (for sorting and arranging results), and devtools (for reproducibility and installing packages):
$ R R version 3.2.2 (2015-08–14) -- "Fire Safety" Copyright (C) 2015 The R Foundation for Statistical Computing Platform: x86_64-apple-darwin13.4.0 (64-bit) > library(ballgown) > library(RSkittleBrewer) > library(genefilter) > library(dplyr) > library(devtools)
-
8.Load the phenotype data for the samples. An example file called geuvadis_phenodata.csv is included with the data files for this protocol (ChrX_data). In general, you will have to create this file yourself. It contains information about your RNA sequencing samples, formatted as illustrated in this csv (comma-separated values) file. Each sample should be described on one row of the file and each column should contain one variable. To read this file into R we use the command read.csv. In this file the values are separated by commas, but if the file were tabdelimited you could use the function read.table.
> pheno_data = read.csv("geuvadis_phenodata.csv")?TROUBLESHOOTING
-
9.Read in the expression data that was calculated by StringTie. Ballgown also currently supports reading of data from Cufflinks6 and RSEM10. To do this we use the ballgown command with the following three parameters: the directory where the data are stored (dataDir, which here is named simply “ballgown”), a pattern that appears in the sample names (samplePattern), and the phenotypic information we loaded in the previous step (pData). Note that once a Ballgown object is created any other Bioconductor32 package can be applied for data analysis or data visualization. Here we present a standardized pipeline that can be used to perform standard differential expression analysis.
> bg_chrX = ballgown(dataDir = "ballgown", samplePattern = "ERR", pData=pheno_data)
?TROUBLESHOOTING
-
10.Filter to remove low abundance genes. One common issue with RNA-seq data is that genes often have very few or zero counts. A common step is to filter out some of these. Another approach that has been used for gene expression analysis is to apply a variance filter. Here we remove all transcripts with a variance across samples less than one:
> bg_chrX_filt = subset(bg_chrX,"rowVars(texpr(bg_chrX)) > 1",genomesubset=TRUE)
-
11.
Identify transcripts that show statistically significant differences between groups. One thing we might want to do is make sure we account for variation in expression due to other variables. As an example, we will look for transcripts differentially expressed between sexes, while correcting for any differences in expression due to the population variable. We can do this using the stattest function from Ballgown. We set the getFC=TRUE parameter so we can look at the confounder-adjusted fold change between the two groups.
Note that Ballgown’s statistical test is a standard linear model based comparison. For small sample sizes (n < 4 per group) it is often better to perform regularization. This can be done using the limma33 package in Bioconductor. Other regularized methods such as DESeq23 and edgeR20 can be applied to gene or exon counts, but are not appropriate for direct application to FPKM abundance estimates. The statistical test uses a cumulative upper quartile normalization34.> results_transcripts = stattest(bg_chrX_filt, feature="transcript", covariate="sex", adjustvars = c("population"), getFC=TRUE, meas="FPKM") -
12.Identify genes that show statistically significant differences between groups. For this we can run the same function we used to identify differentially expressed transcripts, but here we set feature=“gene” in the stattest command:
> results_genes = stattest(bg_chrX_filt, feature="gene", covariate="sex", adjustvars = c("population"), getFC=TRUE, meas="FPKM") -
13.Add gene names and gene IDs to the results_transcripts data frame:
> results_transcripts = data.frame(geneNames=ballgown::geneNames(bg_chrX_filt), geneIDs=ballgown::geneIDs(bg_chrX_filt), results_transcripts)
-
14.Sort the results from smallest p-value to largest:
> results_transcripts = arrange(results_transcripts,pval) > results_genes = arrange(results_genes,pval)
-
15.Write the results to a CSV (comma-separated values) file that can be shared and distributed:
> write.csv(results_transcripts, "chrX_transcript_results.csv", row.names=FALSE) > write.csv(results_genes, "chrX_gene_results.csv", row.names=FALSE)
-
16.Identify transcripts and genes with a q-value of less than 0.05:
> subset(results_transcripts,results_transcripts$qval<0.05) > subset(results_genes,results_genes$qval<0.05)
The Ballgown output will appear on the screen for each of these commands; we have shown the results below in Table 3 (transcripts) and Table 4 (genes). As shown in the tables, chromosome X has 9 transcripts that are differentially expressed between the sexes (using a q-value threshold of 0.05), three of which corresponding to isoforms of known genes (XIST, TSIX, and PNPLA4). At the gene level (Table 4), chromosome X has 10 differentially expressed genes at the same q-value cutoff.
?TROUBLESHOOTING
Table 3.
Differentially expressed transcripts between sexes (q-val<5%). Fold change refers to the ratio between expression in females versus males; thus values below 1 mean that the transcript was expressed at a lower level in males.
| gene name |
gene id |
transcript name |
id | fold change | p-val | q-val |
|---|---|---|---|---|---|---|
| XIST | MSTRG.506 | NR_001564 | 1729 | 0.003255 | 7.0447e-10 | 1.6160e-06 |
| <none> | MSTRG.506 | MSTRG.506.1 | 1728 | 0.016396 | 1.2501e-08 | 1.4339e-05 |
| TSIX | MSTRG.505 | NR_003255 | 1726 | 0.083758 | 2.4939e-06 | 1.9070e-03 |
| <none> | MSTRG.506 | MSTRG.506.2 | 1727 | 0.047965 | 3.7175e-06 | 2.1319e-03 |
| <none> | MSTRG.585 | MSTRG.585.1 | 1919 | 7.318925 | 9.3945e-06 | 3.7715e-03 |
| PNPLA4 | MSTRG.56 | NM_004650 | 203 | 0.466647 | 9.8645e-06 | 3.7715e-03 |
| <none> | MSTRG.506 | MSTRG.506.5 | 1731 | 0.046993 | 2.1350e-05 | 6.9968e-03 |
| <none> | MSTRG.592 | MSTRG.592.1 | 1923 | 9.186257 | 3.5077e-05 | 1.0058e-02 |
| <none> | MSTRG.518 | MSTRG.518.1 | 1744 | 11.972859 | 4.4476e-05 | 1.1336e-02 |
Table 4.
Differentially expressed genes between sexes (q-val<5%). Fold change is defined as in Table 3.
| gene id | fold change | p-val | q-val |
|---|---|---|---|
| MSTRG.506 | 0.00267 | 6.8069e-11 | 6.7593e-08 |
| MSTRG.56 | 0.54688 | 3.6604e-06 | 1.8174e-03 |
| MSTRG.585 | 7.28272 | 6.9974e-06 | 2.3161e-03 |
| MSTRG.505 | 0.08930 | 1.1660e-05 | 2.8948e-03 |
| MSTRG.356 | 0.56441 | 1.7126e-05 | 3.4013e-03 |
| MSTRG.592 | 9.14996 | 3.2120e-05 | 5.3159e-03 |
| MSTRG.518 | 12.23489 | 4.3775e-05 | 6.2098e-03 |
| MSTRG.492 | 0.65092 | 1.9811e-04 | 2.4590e-02 |
| MSTRG.594 | 7.71803 | 2.2935e-04 | 2.5305e-02 |
| MSTRG.788 | 1.74428 | 3.5797e-04 | 3.5547e-02 |
Data visualization (TIMING: variable)
CRITICAL You can use Ballgown to visualize RNA-seq results in a variety of ways. The plots produced by Ballgown make it easier to view and compare expression data, and some commands can generate publication-ready figures.
-
17.Make the plots pretty. This step is optional, but if you do run it you will get the plots in the nice colors we used to generate our figures:
> tropical= c(‘darkorange’, ‘dodgerblue’, ‘hotpink’, ‘limegreen’, ‘yellow’) > palette(tropical)
-
18.Show the distribution of gene abundances (measured as FPKM values) across samples, colored by sex (Figure 3). Ballgown stores a variety of measurements that can be compared and visualized. For this protocol we will compare the FPKM measurements for the transcripts, but you can also obtain measurements for each splice junction, exon, and gene in the data set. The first command below accesses the FPKM data. The plots will be easier to visualize if you first transform the FPKM data; here we use a log2 transformation that adds one to all FPKM values because log2(0) is undefined (second command below). The third command actually creates the plot:
> fpkm = texpr(bg_chrX,meas="FPKM") > fpkm = log2(fpkm+1) > boxplot(fpkm,col=as.numeric(pheno_data$sex),las=2,ylab=‘log2(FPK M+1)’)
-
19.Make plots of individual transcripts across samples. For example, here we show how to create a plot for the 12th transcript in the data set (Figure 4). The first two commands below show the name of the transcript (NM_012227), and the name of the gene that contains it (GTPBP6):
> ballgown::transcriptNames(bg_chrX)[12] ## 12 ## "NM_012227" > ballgown::geneNames(bg_chrX)[12] ## 12 ## "GTPBP6" > plot(fpkm[12,] ~ pheno_data$sex, border=c(1,2), main=paste(ballgown::geneNames(bg_chrX)[12],’ : ‘, ballgown::transcriptNames(bg_chrX)[12]), pch=19, xlab="Sex", ylab=‘log2(FPKM+1)’) > points(fpkm[12,] ~ jitter(as.numeric(pheno_data$sex)), col=as.numeric(pheno_data$sex))
-
20.Plot the structure and expression levels in a sample of all transcripts that share the same gene locus. For example, in Figure 5 we show all the transcripts from the gene that contains the first transcript, NR_001564, in Table 3. This is the 1729th transcript in the ballgown object according to the id column in the table, and we can easily find the gene identifier of the transcript by using ballgown::geneIDs(bg_chrX)[1729]. This reveals that NR_001564 is an isoform of the gene XIST, which is known to be much more highly expressed in females than in males. We can see in Table 3 that other isoforms of this gene appear to be differentially expressed as well. We can plot their structure and expression levels by passing the gene name and the ballgown object to the plotTranscripts function. The plot shows one transcript on each row, colored by its FPKM level. By default the transcripts at that locus are colored by their expression levels in the first sample, but we can tell the plotTranscripts function what sample we are interested in – here we choose sample ERR188234:
> plotTranscripts(ballgown::geneIDs(bg_chrX)[1729], bg_chrX, main=c(‘Gene XIST in sample ERR188234’), sample=c(‘ERR188234’))
-
21.We can also plot the average expression levels for all transcripts of a gene within different groups using the plotMeans function. We need to specify which gene to plot, which Ballgown object to use, and which variable to group by. As an example, the following command plots the second gene in Table 4, MSTRG.56.
> plotMeans(‘MSTRG.56’, bg_chrX_filt,groupvar="sex",legend=FALSE)
Figure 3.
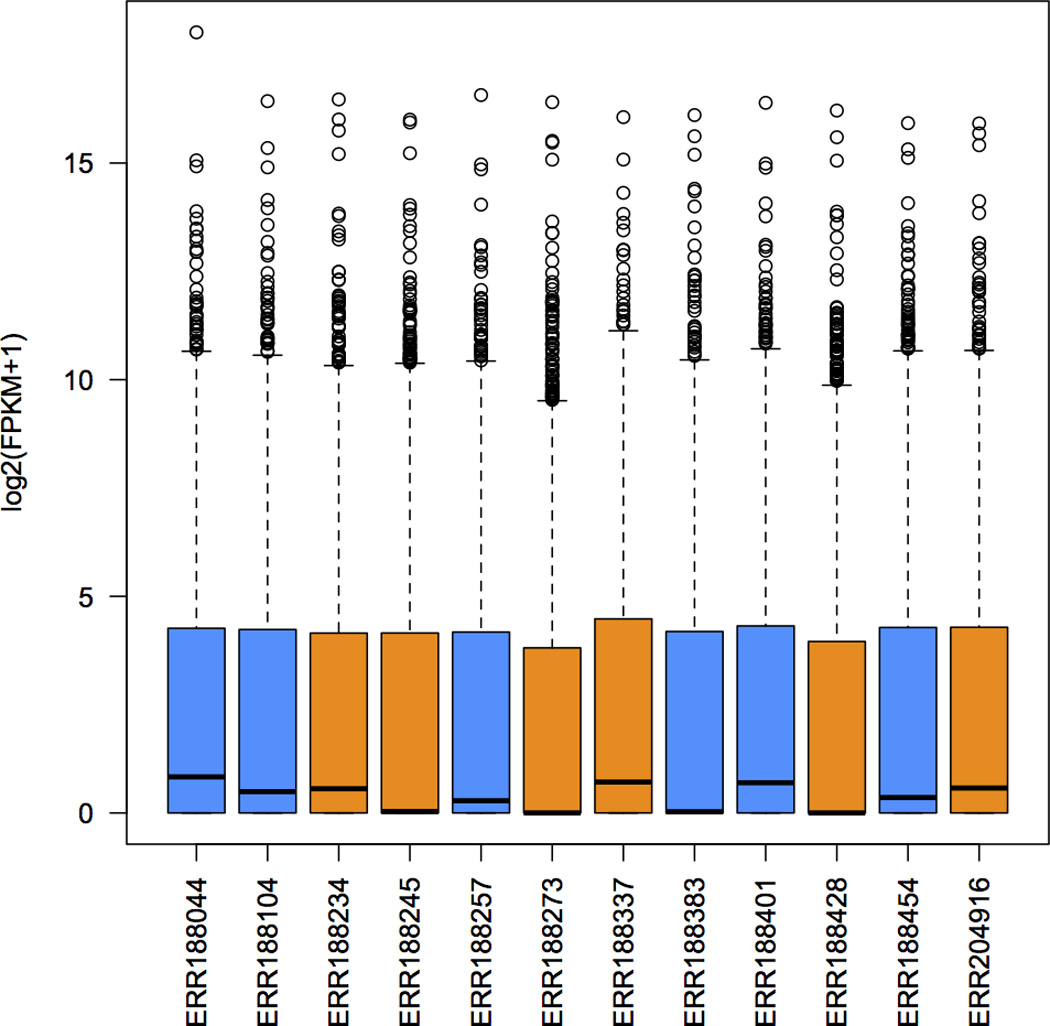
Distribution of FPKM values across the 12 samples. Samples from the same sex are shown in the same color: males in blue, and females in orange.
Figure 4.
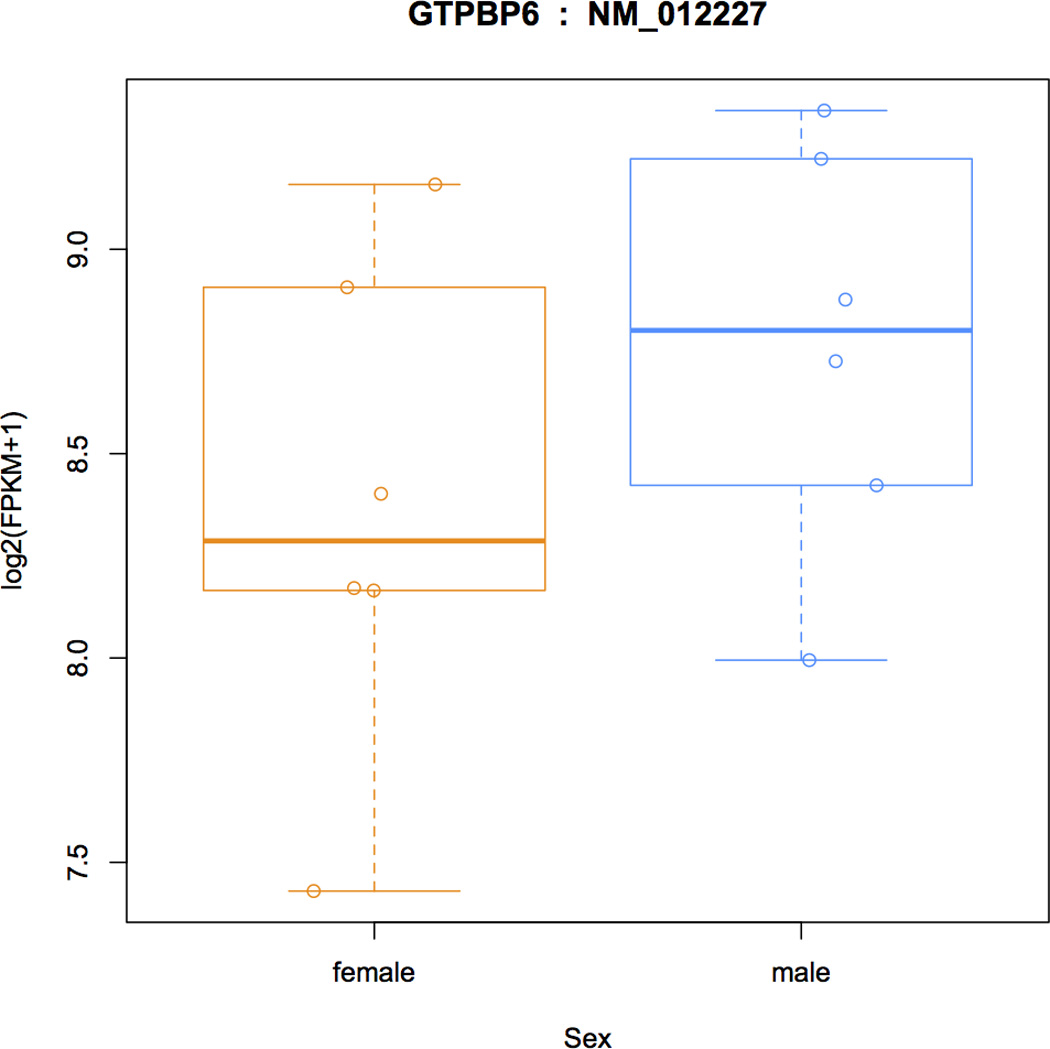
FPKM distributions in males and females for transcript NM_012227 from gene GTPBP6 (GTP Binding Protein 6), a gene that is known to be more highly expressed in males.
Figure 5.
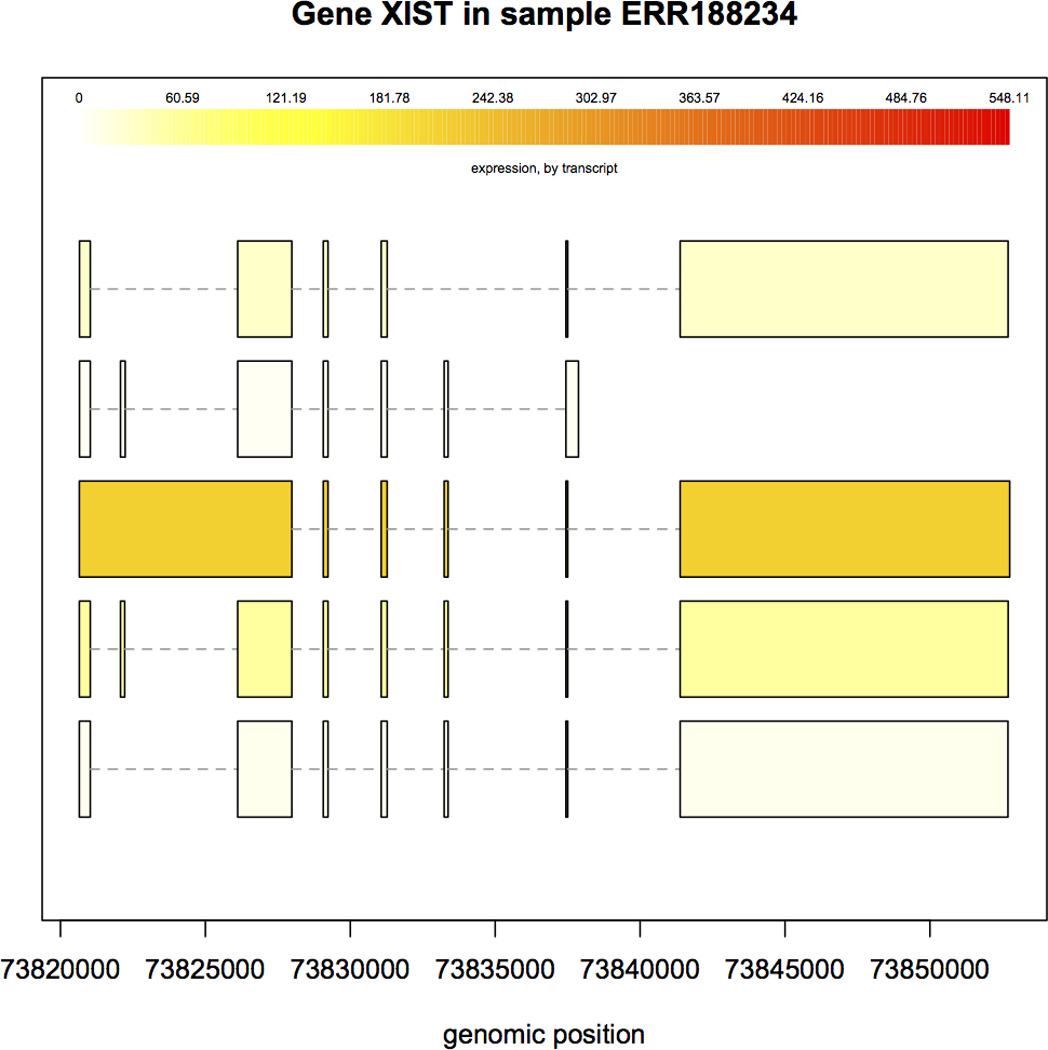
Structure and expression levels of five distinct isoforms of the XIST gene in sample ERR188234. Expression levels are shown in varying shades of yellow. The third isoform is expressed at a much higher level than the others, as indicated by the darker color.
TROUBLESHOOTING
Troubleshooting advice can be found in Table 5.
Table 5.
Troubleshooting advice.
| Step | Problem | Possible reason | Solution |
|---|---|---|---|
| 4 | StringTie reports that it cannot open the files in the input mergelist.txt file |
The GTF files specified in mergelist.txt do not exist, or you did not run the merge step in the same directory where the GTF files were created. |
Either run StringTie in the same directory as the input GTF files or edit the mergelist.txt file to include the full path to each GTF file name description. |
| 8 | You get an error message that the file geuvadis_phenodata.csv cannot be opened. |
The directory where you started your R session does not contain the phenotype data. |
Before proceeding, you need to tell R where all the files are. You can do this with the setwd command. For example if the directory containing the chrX_data is your home directory, then you would use the following R command: > setwd(“~/chrX_data”) You can then run the getwd() command to make sure that the directory has been properly set. |
| 9 | The Ballgown function results in an error that the first column of pData does not match the names of the folders containing the ballgown data. |
The sample names you have stored in your phenotype file do not match the file names of the samples you ran with StringTie. |
To make sure that the file names match the IDs in the phenotype file, run the following R command: > all(pheno_data$ids == list.files("ballgown")) This command should return TRUE. |
| 16 | The transcripts and genes you assembled (shown in Table 3 and Table 4 respectively) have different values for the ‘gene id’ and ‘transcript name’ than what you see in the protocol here. |
The transcript and gene identifiers assigned by StringTie (by default preceded by the string ‘MSTRG’) are generated in the order that the bundles of reads are processed. This order can differ from one run of StringTie to another when multiple threads are used (with the –p option). |
This is not actually an error. If a transcript matches a known, annotated transcript, then the ‘transcript name’ will be consistent from run to run. Otherwise this field can change when you re-run the protocol. |
TIMING
Running this protocol on the chrX data provided will take less than 1 h on a computer with eight processing cores and 4 GB of RAM. The most time-consuming steps are aligning the reads and assembling and quantifying the genes and transcripts. On the data provided, it takes Ballgown less than a minute to load the required files and generate the differentially expressed genes and transcripts. In contrast, it takes HISAT2 and StringTie about 13 minutes on an Intel i7–2600 desktop CPU at 3.4 GHz with 4 processing cores, and about 23 minutes on an AMD Opteron 6172 server at 2.1 GHz with 8 processing cores. Larger data sets will of course require more time and more memory. For instance, running this protocol on all 12 samples in the full human data set (see ftp://ftp.ccb.jhu.edu/pub/RNAseq_protocol/README for instructions on downloading the full set) on a machine with eight processor cores and at least 8 GB of RAM will require about 12.5 hours to align the reads, and less than 8 hours for transcript assembly and expression analyses.
ANTICIPATED RESULTS
RNA-seq alignments
Accurate transcript assembly and quantification is strongly correlated with the quality of the spliced read alignments. If a large percentage of the reads are not aligned, the transcriptome assembler will have a hard time reconstructing genes, especially those expressed at low levels. Thus the spliced aligner should have high sensitivity and precision. Mis-aligned reads also negatively influence the downstream assembly, especially those that incorrectly link together separate bundles of reads. These false positive alignments will disrupt the flow algorithm in StringTie, and skew the expression estimates of the assembled transcripts. To ensure a low rate of false positives, the alignment mode recommended in the HISAT2 step of this protocol requires longer "anchors" for the de novo discovery of splice sites. This leads to fewer alignments with short anchors, which helps StringTie (and other transcript assemblers) significantly improve its computational time and memory usage requirements.
Because we prepared the data for this protocol by aligning all reads in the initial data sets to the whole genome and then extracting only those reads that aligned to chromosome X and their mates, we expect a mapping rate close to 100% for the reads in our reduced data set. Table 6 shows the alignment rates for the full data set, which are comparable to what you might expect for typical RNA-seq experiments. Although in general more than 95% of the reads are aligned, a high percentage of them (>20%) are mapped in more than one place. Multi-mapped reads can severely affect the expression levels of genes, and the user should be very cautious when drawing conclusions about differentially expressed genes that contain a high percentage of multi-mapped reads35.
Table 6.
Read mapping statistics on the full human data set.
| Sample ID | Uniquely aligned reads (%) |
Multi-mapped reads (%) |
Overall alignment rate (%) |
|---|---|---|---|
| ERR188044 | 73.1 | 21.9 | 95.0 |
| ERR188104 | 75.8 | 21.2 | 96.9 |
| ERR188234 | 68.5 | 26.4 | 94.9 |
| ERR188245 | 63.8 | 33.2 | 97.0 |
| ERR188257 | 67.7 | 27.6 | 95.3 |
| ERR188273 | 71.4 | 20.9 | 92.3 |
| ERR188337 | 65.2 | 31.5 | 96.7 |
| ERR188383 | 67.9 | 29.3 | 97.2 |
| ERR188401 | 72.1 | 24.8 | 96.9 |
| ERR188428 | 39.7 | 57.7 | 97.4 |
| ERR188454 | 66.3 | 31.0 | 97.3 |
| ERR204916 | 69.5 | 26.8 | 96.3 |
Transcriptome assembly
The annotation reference file we provide in this protocol contains 2098 transcripts from 1086 genes on chromosome X. By running gffcompare (as shown in Step 6) on the GTF files produced by StringTie in Steps 3–4, we can collect summary statistics describing how the assembled transcripts compare to the annotation (Table 7). The number of annotated genes that are expressed varies across the samples, illustrating the tissue-specific nature of gene expression. After assembling all the samples, we merge the gene and transcript models using the StringTie merge operation (Step 4 of our protocol).
Table 7. Transcriptome assembly statistics.
Shown are the number of genes assembled from the RNA-seq reads for each of the 12 files, and the total number in the merged assembly with and without using the reference annotation. The total number of transcripts after merging with annotation (bottom row) includes only those transcripts that had non-zero expression levels. Also shown are the number of assembled genes and transcripts that are novel; i.e., that do not match any annotated genes.
| Sample ID | Number of assembled genes |
Novel genes | Transcripts matching annotation |
Novel transcripts |
|---|---|---|---|---|
| ERR188044 | 808 | 288 | 675 | 615 |
| ERR188104 | 793 | 294 | 651 | 630 |
| ERR188234 | 838 | 322 | 656 | 659 |
| ERR188245 | 677 | 196 | 579 | 470 |
| ERR188257 | 706 | 228 | 614 | 504 |
| ERR188273 | 631 | 177 | 536 | 406 |
| ERR188337 | 852 | 343 | 668 | 660 |
| ERR188383 | 707 | 225 | 608 | 497 |
| ERR188401 | 794 | 287 | 653 | 624 |
| ERR188428 | 631 | 173 | 588 | 414 |
| ERR188454 | 745 | 241 | 622 | 547 |
| ERR204916 | 761 | 265 | 643 | 564 |
| Merged without annotation | 908 | 382 | 661 | 1225 |
| Merged with annotation | 1258 | 414 | 1408 | 1257 |
When the annotation file is used during the merge step, the output will include all transcripts present in the annotation file, even those with expression levels of zero, as well as all novel transcripts. We recommend using the annotation file whenever it is available, because it helps StringTie assemble transcripts more accurately, especially those expressed at low levels. As shown in Table 7 (bottom rows), the number of transcripts matching the annotation increases substantially when annotation is provided. Table 7 also shows that in these data, there are nearly as many novel transcripts (isoforms) as known transcripts in each sample. Most of the transcriptome diversity is due to alternative splicing, and it is not unusual to observe that a large fraction of isoforms in an RNA-seq experiment are novel. Despite recent technological advances, the transcriptome landscape of many species, including human, is far from being completely known36, and it is quite plausible that most of these novel transcripts are genuine. In the absence of an experimental test for their validity, it is hard to determine which transcripts are real and which are assembly artifacts or transcriptional "noise."
Quantification and differential expression analysis
The assembly of RNA-seq reads reconstructs the exon-intron structure of genes and their isoforms, but we also need to estimate how much of each transcript was present in the original sample. Step 18 in the protocol shows how we can quickly inspect the distribution of the FPKM measurements of transcripts across samples, shown in Figure 3. For the samples used here, most of the log2(FPKM) values are less than 5, which suggest that only a small fraction of genes are expressed at very high levels. Genes expressed at lower levels are harder to assemble, which can lead to spurious genes and transcripts that show statistically significant differences between groups. Figure 6 shows the overall distribution of p-values that measure differential expression between males and females at both transcript and gene levels. We can see there is not a large difference between the sexes, but if there were, we would many p-values near zero. The figure also shows a spike near 0 in the distribution of gene-level p-values, suggesting (as expected) that we have better statistical power to detect differential expression between genes rather than transcripts. As shown in Table 3 and Table 4, for this data you should find in 9 transcripts and 10 genes whose differential expression has a q-value less than 0.05 (which is admittedly an arbitrary cutoff).
Figure 6.
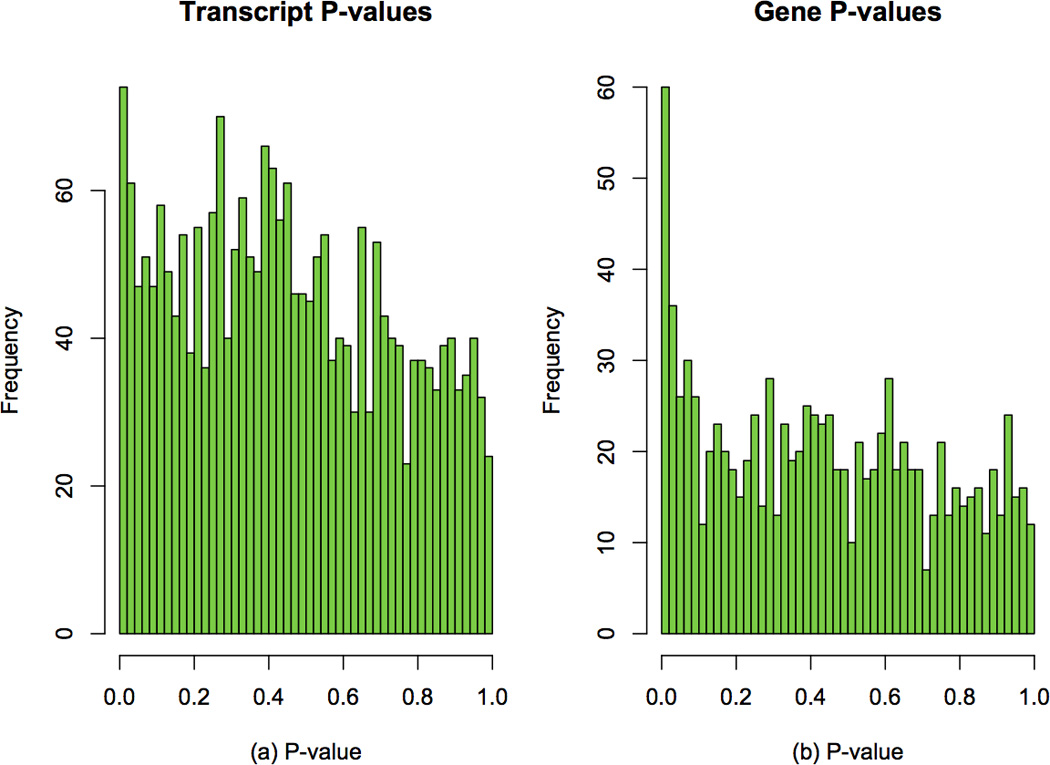
Overall distribution of differential expression p-values in females and males. Frequencies of p-values are shown for (a) transcripts and (b) genes.
Looking at the transcripts in Table 3 that correspond to known genes, all of them are known to be differentially expressed between males and females. In females the XIST gene is expressed exclusively from the inactive X chromosome, and is essential for the initiation and spread of X inactivation, an early developmental process that transcriptionally silences one of the pair of X chromosomes 37. TSIX is a non-coding RNA gene that binds XIST during X chromosome inactivation 38. PNPLA4 has been previously shown to have higher expression in females versus males 39.
Supplementary Material
Acknowledgments
This work was supported in part by the National Institutes of Health under grants R01-HG006677 (SLS), R01-GM083873 (SLS), R01-GM105705 (JTL), and by the National Science Foundation under grant DBI-1458178 (MP).
Footnotes
Author contributions
M.P. led the development of the protocol, with help from all the authors. D.K. is the main developer of HISAT, M.P. led the development of StringTie, and J.T.L. is the senior author of Ballgown. G.P. developed gffcompare and contributed to StringTie. All authors contributed to the writing of the manuscript. S.L.S. supervised the entire project.
Competing financial interests
The authors declare that they have no competing financial interests.
Supplementary Software: The supplement contains four files: (1) a Unix shell script that will run the entire protocol automatically; (2) a configuration file required by the shell script; (3) an R file containing the R commands that the script will run; and (4) a README file that describes how to download the required software packages, the chromosome X data, and how to configure and run the shell script.
REFERENCES
- 1.Lister R, et al. Highly integrated single-base resolution maps of the epigenome in Arabidopsis. Cell. 2008;133:523–536. doi: 10.1016/j.cell.2008.03.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Methods. 2008;5:621–628. doi: 10.1038/nmeth.1226. [DOI] [PubMed] [Google Scholar]
- 3.Cloonan N, et al. Stem cell transcriptome profiling via massive-scale mRNA sequencing. Nat Methods. 2008;5:613–669. doi: 10.1038/nmeth.1223. [DOI] [PubMed] [Google Scholar]
- 4.Trapnell C, et al. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nature Biotechnology. 2010;28:511–U174. doi: 10.1038/nbt.1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kim D, et al. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013;14:R36. doi: 10.1186/gb-2013-14-4-r36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Trapnell C, et al. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nature Protocols. 2012;7:562–578. doi: 10.1038/nprot.2012.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kim D, Langmead B, Salzberg SL. HISAT: a fast spliced aligner with low memory requirements. Nat Methods. 2015;12:357–360. doi: 10.1038/nmeth.3317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pertea M, et al. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat Biotechnol. 2015;33:290–295. doi: 10.1038/nbt.3122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Frazee AC, et al. Ballgown bridges the gap between transcriptome assembly and expression analysis. Nat Biotechnol. 2015;33:243–246. doi: 10.1038/nbt.3172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li B, Dewey CN. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics. 2011;12:323. doi: 10.1186/1471-2105-12-323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wu TD, Nacu S. Fast and SNP-tolerant detection of complex variants and splicing in short reads. Bioinformatics. 2010;26:873–881. doi: 10.1093/bioinformatics/btq057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dobin A, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Guttman M, et al. Ab initio reconstruction of cell type-specific transcriptomes in mouse reveals the conserved multi-exonic structure of lincRNAs. Nat Biotechnol. 2010;28:503–510. doi: 10.1038/nbt.1633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li W, Feng J, Jiang T. IsoLasso: a LASSO regression approach to RNA-Seq based transcriptome assembly. J Comput Biol. 2011;18:1693–1707. doi: 10.1089/cmb.2011.0171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Grabherr MG, et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nature Biotechnology. 2011;29:644–652. doi: 10.1038/nbt.1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schulz MH, Zerbino DR, Vingron M, Birney E. Oases: robust de novo RNA-seq assembly across the dynamic range of expression levels. Bioinformatics. 2012;28:1086–1092. doi: 10.1093/bioinformatics/bts094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Xie Y, et al. SOAPdenovo-Trans: de novo transcriptome assembly with short RNA-Seq reads. Bioinformatics. 2014;30:1660–1666. doi: 10.1093/bioinformatics/btu077. [DOI] [PubMed] [Google Scholar]
- 18.Roberts A, Pachter L. Streaming fragment assignment for real-time analysis of sequencing experiments. Nat Methods. 2013;10:71–73. doi: 10.1038/nmeth.2251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Patro R, Mount SM, Kingsford C. Sailfish enables alignment-free isoform quantification from RNA-seq reads using lightweight algorithms. Nat Biotechnol. 2014;32:462–464. doi: 10.1038/nbt.2862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26:139–140. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Trapnell C, et al. Differential analysis of gene regulation at transcript resolution with RNA-seq. Nat Biotechnol. 2013;31:46–53. doi: 10.1038/nbt.2450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Anders S, Reyes A, Huber W. Detecting differential usage of exons from RNA-seq data. Genome Res. 2012;22:2008–2017. doi: 10.1101/gr.133744.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shen S, et al. rMATS: robust and flexible detection of differential alternative splicing from replicate RNA-Seq data. Proc Natl Acad Sci U S A. 2014;111:E5593–E5601. doi: 10.1073/pnas.1419161111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Katz Y, Wang ET, Airoldi EM, Burge CB. Analysis and design of RNA sequencing experiments for identifying isoform regulation. Nat Methods. 2010;7:1009–1015. doi: 10.1038/nmeth.1528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ferragina P, Manzini G. Opportunistic data structures with applications. Proceedings 41st Annual Symposium on Foundations of Computer Science. 2000:390–398. [Google Scholar]
- 30.Raj A, et al. Thousands of novel translated open reading frames in humans inferred by ribosome footprint profiling. bioRxiv. 2015 doi: 10.7554/eLife.13328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kodama Y, Shumway M, Leinonen R. The Sequence Read Archive: explosive growth of sequencing data. Nucleic acids research. 2012;40:D54–D66. doi: 10.1093/nar/gkr854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Huber W, et al. Orchestrating high-throughput genomic analysis with Bioconductor. Nat Methods. 2015;12:115–121. doi: 10.1038/nmeth.3252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ritchie ME, et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43:e47. doi: 10.1093/nar/gkv007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Paulson JN, Stine OC, Bravo HC, Pop M. Differential abundance analysis for microbial marker-gene surveys. Nat Methods. 2013;10:1200–1202. doi: 10.1038/nmeth.2658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Robert C, Watson M. Errors in RNA-Seq quantification affect genes of relevance to human disease. Genome Biol. 2015;16:177. doi: 10.1186/s13059-015-0734-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pertea M. The human transcriptome: an unfinished story. Genes. 2012;3:344–360. doi: 10.3390/genes3030344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chow JC, et al. Inducible XIST-dependent X-chromosome inactivation in human somatic cells is reversible. Proc Natl Acad Sci U S A. 2007;104:10104–10109. doi: 10.1073/pnas.0610946104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lee JT, Davidow LS, Warshawsky D. Tsix, a gene antisense to Xist at the X-inactivation centre. Nat Genet. 1999;21:400–404. doi: 10.1038/7734. [DOI] [PubMed] [Google Scholar]
- 39.Talebizadeh Z, Simon SD, Butler MG. X chromosome gene expression in human tissues: male and female comparisons. Genomics. 2006;88:675–681. doi: 10.1016/j.ygeno.2006.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.