

Abstract
Purpose
Previously, patients with RHO mutations and a class A phenotype were found to have severe early-onset loss of rod function, whereas patients with a class B phenotype retained rod function at least in certain retinal regions. Here class B patients were studied at different disease stages to understand the topographic details of the phenotype in preparation for therapies of this regionalized retinopathy.
Methods
A cohort of patients with RHO mutations and class B phenotype (n = 28; ages 10–80 years) were studied with rod and cone perimetry and optical coherence tomography (OCT).
Results
At least three components of the phenotype were identified in these cross-sectional studies. Patients could have hemifield dysfunction, pericentral loss of function, or a diffuse rod sensitivity loss across the visual field. Combinations of these different patterns were also found. Colocalized photoreceptor layer thicknesses were in agreement with the psychophysical results.
Conclusions
These disorders with regional retinal variation of severity require pre-evaluations before enrollment into clinical trials to seek answers to questions about where in the retina would be appropriate to deliver focal treatments, and, for retina-wide treatment strategies, where in the retina should be monitored for therapeutic efficacy (or safety).
Keywords: cone, optical coherence tomography, perimetry, rod
The patterns of visual field loss in forms of retinitis pigmentosa (RP) were a topic of study before as well as after the current era of known genotypes.1,2 The midperipheral ring scotoma is the pattern most commonly associated with RP.2 When mutations in the rhodopsin gene (RHO) were found to cause autosomal dominant (ad) RP, evidence emerged for different disease patterns.3,4 Certain RHO mutations were found to be associated with severe early-onset loss of rod function but protracted cone degeneration; these phenotypic subtypes were named diffuse or type 1 or class A. The other phenotype was termed regional or type 2 or class B.3,4 The common visual field pattern for class B phenotype was an altitudinal (superior hemifield) loss of rod and cone function with relatively preserved inferior visual field function.5–11 The early publications about the phenotype in class B were followed by many other studies indicating that the altitudinal pattern was a recognizable feature.
As we move from visual field patterns to an era of potential therapies, we have to know more details of phenotype and answers to specific questions about how to measure outcome of therapy. For example, is the altitudinal loss a pattern that simply increases in size, moving from peripheral to central retina? Would a subretinal injection of therapy be best at a certain eccentricity—is it just a matter of determining a transition zone from healthier to unhealthier retina and using that as a marker of where to introduce a treatment? Is something like the in–out boundary at ellipsoid zone (EZ) line edge a potentially useful outcome for any trial?12,13 When a therapy is not localized, what retinal regions should be monitored? Looking at the visual fields shown in various class B patients with RHO mutations, there seems to be an extension from the superior field around and under the central field. Other than some general concept of progressive disease, why is this retinal regional pattern appearing?
We studied a cohort of class B patients at different disease stages to try to understand more details of the phenotypic patterns in preparation for focal and retina-wide therapies for this regionalized retinopathy. At least three components of the phenotype were found in these cross-sectional studies.
Methods
Subjects
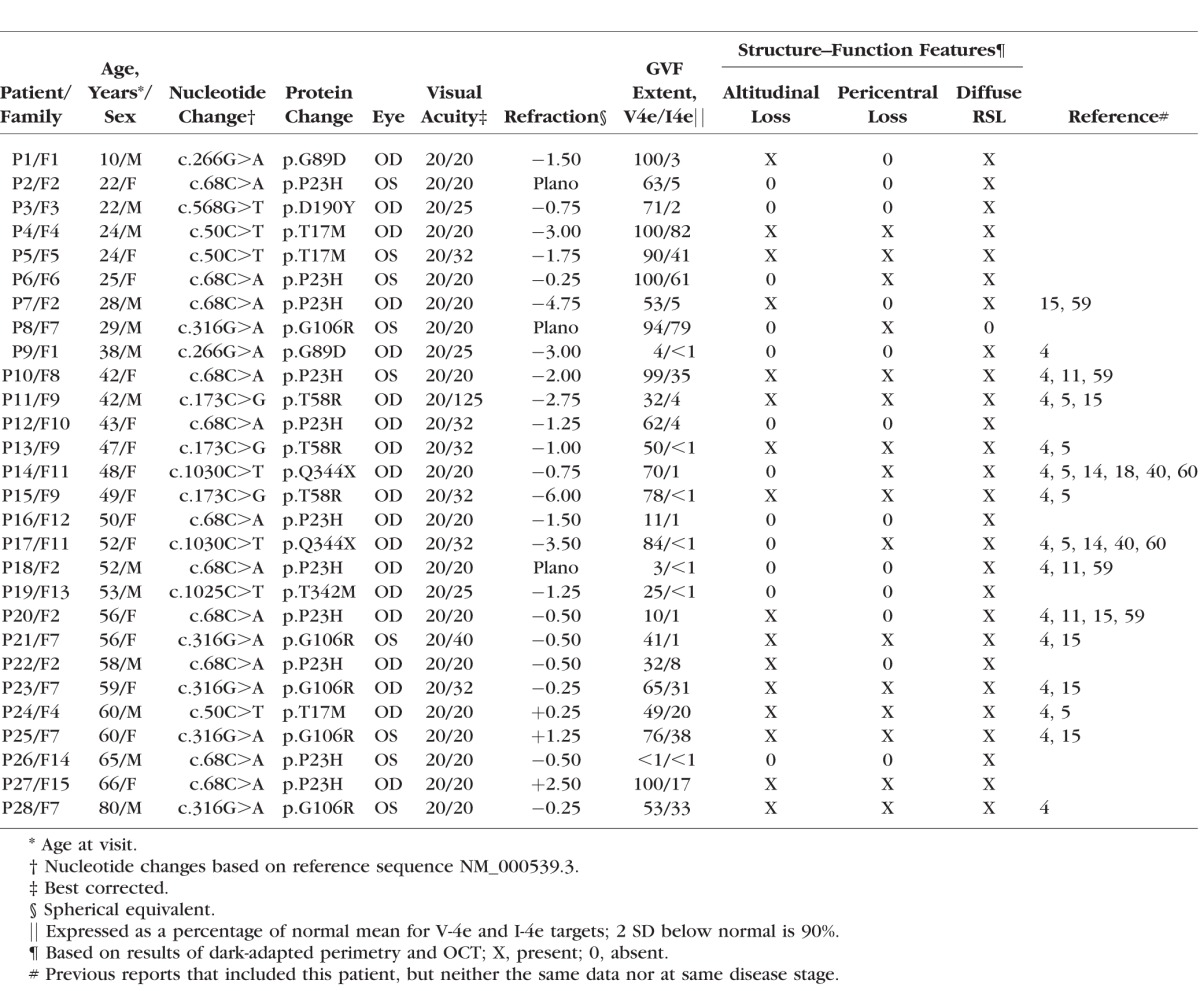
Patients with RHO gene mutations were included in this study (n = 28, representing 15 families; Table). Molecular testing of the patients and their families has been previously reported.4,5,11,14,15 Procedures followed the Declaration of Helsinki and the study was approved by the Institutional Review Board (IRB). Informed consent, assent, and parental permission were obtained, and the work was Health Insurance Portability and Accountability Act compliant. Patients underwent a complete eye examination including best-corrected visual acuity and Goldmann kinetic visual fields (using V-4e and I-4e test targets).
Table.
Clinical and Molecular Characteristics of the RHO Mutation Patients
Visual Function
Dark- and light-adapted static threshold perimetry (200-ms duration, 1.7°-diameter target; 500- and 650-nm stimuli in the dark-adapted state; and white light on a white background) was performed. Sensitivity was measured at 2° intervals along the horizontal and vertical meridians spanning 60° and with a full-field test of 72 loci on a 12° grid.16 Special attention was paid to the known dark adaptation abnormalities in class B patients with RHO mutations4,5,11; dark-adapted testing preceded all light-adapted testing and all imaging or clinical examinations performed in the light. The difference between dark-adapted sensitivities to 500- and 650-nm stimuli was used to determine if rods or cones mediated vision of the 500-nm stimulus. Separate aggregate metrics for the superior and inferior hemifields were obtained by averaging rod sensitivity losses (RSL) at locations on the 12° grid with vertical eccentricities above 4° (n = 27) and below 4° (n = 34), respectively. Rod sensitivity losses were obtained by subtracting from location-specific mean normal sensitivity (500-nm stimulus) when perception was rod mediated. For locations where rod sensitivity was not measurable (target either not seen or cone mediated), the upper limit of possible rod sensitivities was used in the subtraction. Other details of the techniques, data analyses, and normal results have been described previously.16,17
Pericentral dysfunction was defined by perimetry, mainly dark-adapted two-color static perimetry, as described above. Rod sensitivity loss within an annular region (between 3° and 40°18) surrounding the central island of function in the patients was considered pericentral, whether in a complete or incomplete annulus. Most often, there were correlative retinal optical coherence tomography (OCT) structural abnormalities in the regions of the RSL.
Optical Coherence Tomography
Optical coherence tomography was performed with a spectral-domain (SD) system (mainly with RTVue-100 but, in a limited number of subjects, using RTVue XR Avanti; Optovue, Inc., Fremont, CA, USA). For analysis of outer nuclear layer (ONL) and rod outer segment (ROS) thickness profiles colocalized to visual profiles, overlapping OCT scans (9 mm in length; 1019 longitudinal reflectivity profiles [LRPs], each averaging 17–32) were used to cover the horizontal and vertical meridians up to 9-mm eccentricity from the fovea. Postacquisition processing of the data was performed with custom programs (MATLAB 7.5; MathWorks, Natick, MA, USA). Longitudinal reflectivity profiles making up the scans were aligned by straightening the major RPE reflection. Outer nuclear layer and ROS layer thicknesses in patients were quantified (details previously reported13,19), plotted as a function of eccentricity, and compared with the normal ranges (mean ± 2 SD; n = 15; age range, 8–62 years). Ellipsoid zone (EZ, also called IS/OS) line extent was determined as previously published.13 For topographic analysis of ONL thickness, overlapping raster scans were used (size 6 × 6 mm containing 101 lines of 513 LRPs for RTVue-100 or 12 × 9 mm containing 320 lines of 320 LRPs for Avanti). Precise location and orientation of each scan relative to fundus features (blood vessels, intraretinal pigment, and optic nerve head) were determined by integrating backscatter intensity of each raster scan and visualizing such features on the generated images. Individual LRPs forming all registered raster scans were allotted to regularly spaced bins (1° × 1°) in a rectangular coordinate system centered at the fovea; LRPs in each bin were aligned and averaged. Boundaries corresponding to ONL layers were segmented semiautomatically with manual override using both intensity and slope information of backscatter signal along average LRP. Missing data were interpolated bilinearly; thicknesses were mapped to a pseudocolor scale; and the locations of blood vessels and optic nerve head were overlaid for reference.20–22
Results
Hemifield Dysfunction and Complexities of This Pattern
The most described phenotypic feature in class B patients with RHO mutations is superior hemifield loss but preserved inferior field function. Details of function and structure in four representative patients, all with 20/20 to 20/32 visual acuity, are provided in the next paragraphs. First (patient [P] 25, family [F] 7), a very clear altitudinal defect by kinetic perimetry shows both rod and cone dysfunction and photoreceptor laminar defects by OCT colocalized to the psychophysical results. A second patient (P24, F4) illustrates not only an altitudinal loss but also an extension of dysfunction and structural abnormality inferior to the central island. The extension is in a pericentral distribution. A third patient (P23, F7) similarly shows an altitudinal loss and a pericentral extension of abnormality, but the extent of the pericentral defect is greater. A fourth patient (P27, F15) shows focal loss of function and structure in the superior field, possibly representing the initial defect of an altitudinal process. A mild diffuse rod-mediated sensitivity loss with photoreceptor structural correlates is also present.
Patient 25, Family 7.
A 60-year-old woman with a G106R RHO mutation has an altitudinal absolute scotoma with kinetic perimetry (Fig. 1A). There is retained detection of the V-4e target in the far peripheral superior field and across the entire inferior field; and with the I-4e target, there is a residual abnormally reduced inferior island. A sensitivity profile along the horizontal 60° indicates that cone (light adapted) function is within normal limits; vertical profile, however, shows the loss of cone sensitivity in the superior field (marked A for altitudinal defect). Dark-adapted sensitivities along the horizontal meridian are at the lower limit of normal; in the superior field, there is loss of rod sensitivity, and inferiorly, the sensitivities are again at the lower limit of normal. Structure–function relationships are shown using cross-sectional OCT images colocalized to the visual profiles. In the horizontal and vertical meridians there is a central peak of normal ONL thickness. Beyond the central few degrees (horizontally) ONL is subnormal, as is ROS thickness. Superior field–inferior retina ONL and ROS thicknesses eccentric to the center are reduced to no detectable structure. Interestingly, ONL in the inferior field–superior retina is subnormal while ROS falls within normal limits. The EZ line is detectable across the horizontal meridian but cannot be detected inferior to the central few degrees in the vertical meridian.
Figure 1.

Function and structure in RHO mutation patients with class B phenotype. (A) P25, with a G106R mutation. Kinetic visual field has a superior hemifield scotoma that does not cross the horizontal meridian into the inferior field. Sensitivity profiles for light- and dark-adapted conditions are shown across the horizontal and vertical meridians (icons to right upper of each part of figure indicate location of data). Colocalized OCT cross-sectional images without and with highlighted laminae ONL (blue), EZ line (orange), and ROS (light blue). ONL thickness, EZ line visibility, and ROS thickness are plotted. Gray: normal limits for comparison with patient data. Hatched area is the location of the optic nerve head. (B) P24, with a T17M mutation. Kinetic field (V-4e) has a superior hemifield scotoma that does cross the horizontal meridian, and there is an extension of the scotoma around the central field, inferiorly (i.e., a pericentral defect). Sensitivity profiles across the horizontal and vertical meridians with colocalized and quantified OCTs (below). Mediation of dark-adapted sensitivities (based on sensitivity difference with two-color perimetry) is shown above data: R, rod; M, mixed rod and cone; C, cone; A, altitudinal; Pc, pericentral.
Patient 24, Family 4.
A 60-year-old man with a T17M RHO mutation shows a different version of the altitudinal kinetic field abnormality (Fig. 1B). There is no detection of V-4e in the superior field but both target sizes are seen inferiorly. The absolute scotoma not only is present superiorly but also extends below the horizontal meridian and inferior to the central island. Sensitivity profiles along horizontal and vertical meridians indicate both cone and rod function. The central cone island has an adjacent scotoma between 18° and 26° in the nasal field, and then function returns at further eccentricities (marked Pc for pericentral defect); there is no detection of this stimulus on the temporal side of the physiological blind spot. Dark-adapted sensitivity centrally is not as wide nasally as the cone island; the adjacent scotoma extends from 10° to 24° and there is detectable rod function at further eccentricities. Vertical profiles show the central cone and rod islands and the superior field altitudinal scotoma. A notch of cone and RSL is also present in the inferior field (most pronounced at 12°) but there is a return to normal cone function and near-normal rod function farther inferiorly.
Cross-sectional OCT images colocalized to the visual profiles in the horizontal and vertical meridians show only a central island of normal ONL thickness extending for a few degrees to either side; EZ line is also detectable, as is ROS thickness. There is no detectable outer retinal structure in the superior field–inferior retina beyond the preserved central island. The inferior field–superior retina has a notch of structural loss (ONL, EZ line, and ROS) that corresponds to the region of cone and rod dysfunction at 12° to 15°.
Patient 23, Family 7.
A further example of a patient with a superior field scotoma that extends inferiorly around a central island is shown (Fig. 2A). This 59-year-old woman with a G106R RHO mutation retains detectable function superior to the altitudinal scotoma (similar to P24) and there is a large region of function in the inferior field (with both large and small targets by kinetic perimetry). Light-adapted sensitivities are normal centrally; along the horizontal meridian, there is reduced sensitivity for cones between 10° and 20° in the nasal field and limited or no function temporal to the physiological blind spot. Under dark-adapted conditions the same general pattern is present, but the spatial extent of retained rod function along this meridian is less than for cones. The vertical sensitivity profile captures the superior field scotoma for cones and rods and the less extensive notch of dysfunction between ∼10° and 20° in the inferior field.
Figure 2.

Function and structure in RHO mutation patients with class B phenotype. (A) P23, with a G106R mutation. Kinetic field has an altitudinal scotoma and an incomplete pericentral extension into the inferior field. Sensitivity profiles across horizontal and vertical meridians and colocalized OCT data. (B) P27, with a P23H mutation. Kinetic field shows an absolute scotoma in superior temporal field and a relative scotoma in the superior field in the inferior pericentral region. Sensitivity profiles as well as colocalized OCT data show a pronounced pericentral loss of function and structure in the superior field–inferior retina.
Central ONL thickness is preserved (with complicating macular edema), but integrity of this lamina is lost after a few degrees eccentricity, both nasally and temporally. Outer nuclear layer thickness is evident at ∼28° in the temporal retina. Ellipsoid zone line extent and ROS thickness follow a similar pattern horizontally. There is ONL loss in the inferior retina paralleling the dysfunction. The notch of ONL loss in the superior retina, EZ line disappearance, and ROS thickness loss are also consistent with the dysfunction.
Patient 27, Family 15.
A 66-year-old woman with a P23H RHO mutation has a localized superior–temporal field absolute scotoma on kinetic perimetry, and relative scotomas in the superior field and below fixation (Fig. 2B). Light-adapted sensitivity is at the lower limit of normal across the horizontal meridian, but rod function is abnormal across this region. A notch of cone dysfunction is present superiorly in the field between 10° and 20° in the vertical meridian; when tested under dark-adapted conditions, the region shows rod dysfunction. Optical coherence tomography indicates normal central ONL but a slight decrease in ONL thickness across the horizontal meridian; EZ line is visible across this meridian, and ROS thickness is reduced but measurable. The notch of dysfunction in the vertical meridian has anatomic correlates with ONL reduction, EZ line disruption, and ROS abnormalities at ∼10° to 20° in the inferior retina.
Pericentral Notches of Dysfunction Without Hemifield Loss
Phenotypes of two patients illustrate that pericentral structure–function abnormalities can also be detected apart from profound superior hemifield defects (Fig. 3). A notch of rod dysfunction can be detected in the region between 10° and 24° eccentric to the fovea, either inferior (Fig. 3A) or superior (Fig. 3B) in the retina.
Figure 3.

Function and structure in RHO mutation patients with milder class B phenotypes. (A) P4, with a T17M mutation. Kinetic field is normal. Sensitivity profiles reveal a pericentral defect in the superior field–inferior retina, and colocalized OCT indicates a structural correlation. (B) P6, with a P23H mutation. Kinetic field has a full extent with V-4e but some limitation with I-4e. Only the dark-adapted sensitivity profile reveals a pericentral defect, and this is in the inferior field–superior retina. A structural correlate at this pericentral location is noted on OCT, although there is also a more generalized reduction in ONL and ROS thickness in this patient, except for the central few degrees.
Patient 4, Family 4.
A 24-year-old man with a T17M RHO mutation has a normal kinetic field (Fig. 3A). Light- and dark-adapted sensitivities across 60° of horizontal meridian are also within normal limits. A notch of cone and rod dysfunction in the superior field–inferior retina is evident in the vertical profiles. Colocalized outer retinal structure along the horizontal meridian indicates normal central ONL but subnormal ONL and ROS thicknesses outside the central few degrees. A vertical scan indicates abnormalities in the ONL, ROS, and EZ line in a location corresponding to the notch of rod dysfunction noted above.
Patient 6, Family 6.
A 25-year-old woman with a P23H RHO mutation has a normal kinetic field with V-4e but generalized constriction using I-4e (Fig. 3B). Along the horizontal meridian, light-adapted sensitivities are within normal limits but rod sensitivity is subnormal. Vertical profiles are normal in the light-adapted state, but a notch of rod dysfunction is present in the inferior field–superior retina, superimposed on the subnormal sensitivity. Outer retinal structure is normal in the central few degrees and then is reduced at greater eccentricities. Outer nuclear layer and ROS thicknesses are abnormal, but EZ line visibility is present across the horizontal meridian. Vertically, there is reduced ONL, disruption of EZ line, and ROS loss at approximately 20° in the superior retina, corresponding to the notch of rod dysfunction.
Photoreceptor Nuclear Layer Topography of the Pericentral Defects
The observations from horizontal and vertical profiles of function and colocalized structure in patients with RHO mutations and class B phenotype provided the impetus to examine further, with ONL thickness topography, the pericentral region in a subset of patients (Fig. 4). The normal ONL by OCT across the central retina is a composite thickness measurement of both cone and rod photoreceptor nuclei (Fig. 4A, left). For reference, normal rod and cone cell density topographies are shown (Fig. 4A, middle and right;23). Peak OCT ONL thickness at the fovea is due to the high density of cone photoreceptors; at the fovea, there is a rod-free zone. The rod:cone ratio is 1:1 at approximately 1.5° eccentricity and continues to increase to 30:1 to 40:1 into the midperiphery. Highest rod density (the rod hotspot) is at approximately 10° to 16° eccentric to the fovea.23
Figure 4.

ONL thickness topography maps in the central retina of RHO mutation patients. (A) Left: normal ONL thickness topography (average of data from five subjects with normal vision; ages 22–32 years). Middle: rod topography based on photoreceptor density map.23 Right: cone topography. Pseudocolor scales have been adjusted to allow comparison with the ONL thickness topography maps. (B–E) Examples of ONL thickness topography with different degrees of altitudinal and pericentral loss of photoreceptors in the central retina of patients with RHO mutations. (F–H) Examples of patients showing pericentral ONL loss without an altitudinal defect. All images are depicted as left eyes. Grayscale maps at the lower right corner of the ONL thickness map represent ONL as fraction of average normal ONL thickness. Each map has overlaid traces of major blood vessels for reference; location of optic nerve is shown as hatched circle. Pseudocolor scales are the same for all ONL thickness maps and shown at the right side of normal ONL in (A); value of the gray levels is shown in (E). Spatial scale for all pseudocolor images is shown below (F).
Our data for P24 and P23 (Figs. 1B, 2A) indicated that an altitudinal loss of function and structure can be accompanied by defects extending into the pericentral region around the residual central island of vision. Outer nuclear layer topographic maps (Figs. 4B–E) suggest a progression sequence that may occur in patients who eventually have the findings in P24 and P23. Patient 8 (Fig. 4B) shows normal ONL thickness superior to the foveal region and relatively mild ONL thickness reduction inferior to the central island. There is a crescent-shaped area of greater ONL reduction within the area of decreased ONL, suggesting that this may be an earlier or more vulnerable region for loss of structure. The grayscales (insets in Figs. 4B–H) complement the pseudocolor scale maps and show the relationship to average normal topography (n = 5; ages 22–32 years). There is half of a pericentral annulus of structural abnormality inferiorly with a region of greater ONL reduction within it. The inferior retinal ONL thickness deficits in P4 (Fig. 4C) and in P25 (Fig. 4D) are more extensive. There is a pericentral zone within the inferior area of ONL reduction in P4, but it is more extensive than in P8. The ONL topography in P25 appears to illustrate a more advanced stage. There is dense inferior ONL loss and a complete pericentral annulus contiguous with this inferior deficit. The pericentral losses are not the same in degree. Above the optic nerve head and temporal to the retained foveal structure are less severe regions, and directly superior to the preserved foveal island, ONL loss is not as pronounced. Patient 5 has a complete pericentral ring within which is a similar degree of ONL loss. There is also a more restricted central region of ONL (Fig. 4E).
Outer nuclear layer topography in other RHO patients suggested a pericentral loss without an altitudinal defect (Figs. 4F–H). Patient 6 has a pericentral ring that appears to be superimposed on a background of ONL loss that is less prominent—similar in degree to the inferior regional ONL thickness reduction in P8 (Fig. 4B). Patients 10 and 6 both retain a substantial central island of normal ONL thickness; but in contrast to P6, P10 has more reduction in ONL in the pericentral annulus. The surrounding retina (superior and inferior) is abnormally reduced in thickness, generally comparable to the losses surrounding the pericentral defect in P6. The disease effect in P15 is even more pronounced (Fig. 4H); the central island, although less affected than the pericentral region, is more reduced in thickness than in any of the other patients shown in this series.
Rod Sensitivity Losses Across the Visual Field and Hypothetical Disease Sequences
In addition to superior hemifield defects and pericentral notches of dysfunction, we also observed that patients could have reductions in rod-mediated function in other areas of the visual field. This was first noted in patients with superior hemifield dysfunction. Inferior field rod-mediated sensitivities, although assumed to be normal, were not. This prompted examination of our entire cohort of RHO mutation patients with class B phenotype for rod-mediated dysfunction (Fig. 5), specifically RSL in the visual field maps. As a first step, we looked at the inferior hemifields in the patients with superior hemifield defects. Average superior (black bars) and inferior (gray bars) hemifield rod sensitivity deficits were calculated for all patients except those with only cone-mediated central islands remaining (n = 24). The hemifield values were ordered left to right by inferior field loss; the accompanying superior hemifield losses were plotted next to those from each patient's inferior field (Fig. 5A). In half of the patients (12/24; 50%), the superior hemifield had more RSL (by ≥10 dB) than the inferior hemifield. There was no constant difference in the RSL of the two hemispheres. The superior hemifield was more severely affected, with 67% of patients having average RSLs > 35 dB, whereas for the inferior hemifield, 29% of the patients had RSL > 35 dB. The data indicate that there can be rod dysfunction across the visual field, to different degrees, in most patients with the class B phenotype.
Figure 5.

Rod sensitivity loss (RSL) across the visual field and hypothetical disease sequence in class B patients from RSL maps. (A) Average RSL for superior (black bars) and inferior (gray bars) hemifields in 24 patients with measurable rod function outside the central field. Patient results are ordered left to right by increasing inferior field losses. Test loci with vertical eccentricities less than 4° were excluded from either average. Inset: subsets of loci in each hemifield average. Dashed line: 2 SD limit for normal subjects. (B, C) RSL is mapped throughout the visual field in grayscale for one eye of each of the 28 patients. Arrows indicate a presumed progression from mild to more severe phenotypes, although this was not always possible. Fixation is marked with a red dot. Rod sensitivity may be nearly normal in early disease stages, and then RSL continues in a recognizable pattern with the development of superior hemifield defects and pericentral dysfunction. Most severe stages (lowest maps) show no evidence of rod function.
Given the complexity of the patterns of RSL in class B patients, only longitudinal data can determine a disease sequence. The cross-sectional data in this study, however, allow us to propose a sequence. Recognition of the different patterns can also permit hypotheses about where (in the retina) various interventions could be directed or expected to have an effect. Rod sensitivity loss maps (in grayscale) were generated for each of the 28 patients (Figs. 5B, 5C). Maps were arranged according to their patterns of visual field loss and ordered from mildest to most severe RSL. Arrows between maps suggest a possible sequence. The spectrum of severity was wide; rod sensitivity could be nearly normal at an early stage (Fig. 5B; P8, upper right) and nondetectable at a later stage (P26, lowest left). There were different patterns in the intermediate phenotypes. Rod dysfunction could be severe in the superior visual field with a less affected inferior field (P25, P5, P10, P11, and P28). Patients 24, 13, and 21 could be considered as showing patterns that are the result of progression from a more exclusively altitudinal stage to a superior field defect that extends into the inferior field and encircles the fovea in a pericentral ring. Of interest, some patients can retain far superior field rod sensitivity (P23 and P15), and by the direction of the arrows we have assumed that this retained rod sensitivity is lost and a complete altitudinal defect occurs before loss of all rod sensitivity at later stages. From a pattern like that in P21, there could be more RSL in the inferior field (e.g., P1, P20). Residual peripheral islands and more of a typical midperipheral loss (e.g., P14, P19) may also be preludes to final loss of all rod sensitivity.
Another sequence that may represent the sectoral pattern often spoken of in the literature6,7,24 is also illustrated (Fig. 5C). From a relatively diffuse RSL (P6; not revealing the pericentral ONL reduction as in Fig. 4F), there are examples of superior–temporal field scotomas (P27) that may extend to include most of the superior field (P7 and P22) and leave only an inferior nasal field island of RSL that has more function than surrounding field. Other residual rod sensitivity around the central field (P12, P2, and P17) is shown as if in the sequence from inferior–nasal island to latest stages (P9 and connecting to the other group of maps, Fig. 5B), but this remains speculative and to be proven by longitudinal data.
Discussion
Therapeutic intervention in certain types of RP has become a more frequent topic to discuss since the molecular basis of disease has become better understood.25–29 Now that there is potential for therapy of these otherwise incurable forms of blindness, many practical clinical questions will be raised and in want of resolution. For example, in disorders with regional retinal variation of severity, where in the retina is it appropriate to deliver focal treatments? And, for retina-wide treatment strategies, where in the retina should we monitor for therapeutic efficacy (or toxicity)?
With such questions in mind, we reinvestigated the phenotype of a molecularly clarified disease group that was well studied phenotypically when the RHO gene was first discovered to be a cause of RP.30,31 In our previous analyses of this disease group,4 we divided the patients into mainly two subgroups and produced average phenotypes, based on rod and cone perimetry. Patients with class A phenotype had only cone function measurable at the earliest ages evaluated. Those with class B phenotype had measurable rod function, and the average pattern showed a hemifield (superior) rod scotoma with a gradient of lesser rod dysfunction within the inferior field.
A three-component pattern in class B patients has not been commented upon previously. In the present study, we confirmed that the altitudinal (inferior retina–superior field disease) predilection was present in more than half (n = 15; 54%) of the patients (Table). An unexpected finding, however, was that there was another disease predilection: pericentral visual loss. In rare patients (n = 4), pericentral disease was detected independent of an altitudinal defect, but it was mostly found in combination with the altitudinal defect (n = 11; 39%). In the pericentral retinal degeneration literature, there have been reports of Norwegian adRP families with RHO mutations and pericentral dysfunction detected by light-adapted kinetic perimetry.32 We previously noted a pericentral disease distribution in a patient with a RHO T58R mutation (Fig. 4C15). Among the genotypes we recently identified in patients with pericentral retinal degeneration, there was one patient with a RHO mutation (P1518). In the current study, that patient was restudied and a sibling was also found to have this pattern (Table; P14, P17).
The third phenotypic feature in most patients was a moderate reduction in rod sensitivity and loss of ONL thickness in the hemifield not expected to be abnormal (until the late stages): the superior retina–inferior field. In most patients this milder rod dysfunction was present when there was also a pronounced altitudinal defect, but in some patients it was a diffuse loss across the retina with or without a pericentral defect.
Is there human postmortem eye donor histopathology that lends support to our psychophysical and OCT results in class B patients?33 There are several reports of patients with P23H RHO mutations; most were at advanced stages when regional differences could not be discerned.34 A donor retina from a patient with a T17M RHO mutation showed correlation of the premortem altitudinal field loss with photoreceptor cell loss in the inferior retina.35 A P23H RHO patient had an altitudinal defect, and there were abnormalities in the superior retina but greater loss of rods in the inferior retina.36 The pericentral retina has not been a region of particular interest in donor retina studies, although a recent report that included a P23H RHO patient (and two class A patients) commented on very severe degeneration in “perifoveal” (along the horizontal meridian) and peripheral regions of retina.37 Considering the necessity of sampling in histopathologic studies, the late disease stage of most postmortem donors, and the specific interests of different investigators, it becomes difficult to relate findings in these rare donor retinas to the earlier stages of the in vivo observations described herein.
How does this further characterization of class B phenotype help in the design of future clinical trials? If the therapeutic strategy was focal and involved regional subretinal delivery of a gene therapy, for example, it would be important to determine where a “bleb” should be located. A RHO-specific gene therapy would be to prevent further degeneration of rod function and structure (and secondary extrafoveal cone loss) and would not be targeting foveal cones. At a screening visit for such a trial, patients would need measurements of extracentral retinal structure and rod function to understand the disease stage and to determine which or how many of the regional patterns are present. Given severe RSL and photoreceptor layer structural loss restricted to the inferior retina, a strategy could be to locate the bleb superior to the fovea and have it extend into a wide region of superior retina that would include a segment of a possible pericentral annulus. The latter, if treatment were efficacious, would theoretically prevent encircling of the central retina and decrease difficulties that patients experience with finding their place when reading printed material or looking for a mouse cursor on a computer monitor. Baseline and posttherapy monitoring could be reduced to a vertical profile of colocalized OCT structure and dark-adapted rod sensitivity extending from a region of the inferior retina through the fovea (to localize serial measurements) and into the superior retinal bleb. A more complete outcome would also include a light-adapted (cone-mediated) visual profile; alternatively, a cluster of loci could be tested with dark- and light-adapted perimetry and colocalized OCT that extended to either side of the vertical meridian.
With rod dysfunction and abnormal structure in the inferior retina and at a stage when there are already losses in the pericentral annulus, a superior retinal bleb approach would still be advisable (involving the presumably less affected superior retina but without encroachment into the fovea). The goal of this approach would again be to prevent further degeneration of rod function and structure—especially in the inferior field to permit continued patient mobility under dimly lit conditions. The degree of pericentral loss and extent of the central cone island at the time of therapy would determine whether the therapy would positively affect reading ability long-term (by keeping a segment of the annulus from progressive degeneration).
Sectoral patterns (such as illustrated in Fig. 5C) with mainly residual inferior–nasal field would invite placement of the bleb more eccentric, including the vertical midline but also more of the superior–temporal retina, and preferably avoiding the fovea. Monitoring outcome would again be a vertical profile (of structure and function) crossing the fovea from the inferior retina to the superior retina, but another vertical profile parallel to the midline or a diagonal profile crossing the treated region would be valuable. A more complete outcome would be a cluster of loci extending from the area of lost function and structure into the treated region with better-preserved retina.
A retina-wide therapy, whether administered intraocularly or systemically, with the purpose to preserve retinal regions with abnormal but not lost rod sensitivity and rod cells, will also require knowledge of the location of areas of residual function and structure. The most practical method for an initial assay would be with RSL maps (as in Fig. 5). Ideally, a full visual map would be used to monitor rod function before and after therapy. Alternatively and possibly more efficient with respect to time, visual and OCT profiles (vertical, horizontal, or customized), based on the findings in a map, can be used. A vertical profile through the fovea would suit many patterns, but also other transition zones can be assayed (more in some patients but less so in others).
There has been much discussion over decades about possible mechanisms underlying various regional disease patterns in inherited retinal degenerations.38,39 Specifically in RP from RHO mutations, many studies have explored photoreceptor cell death pathways in vitro and in RHO mutant animals.40–43 In general, results suggest there may not be a single mechanism but multiple mechanisms involved in causing rod degeneration, such as protein misfolding, mislocalization, release of toxic products, and aberrant signaling.42–44 From a more macroscopic and clinical viewpoint, all class B patients may have different amounts of retina-wide RSL, which may be due to a single mechanism or a mechanism specific to their mutation. Superimposed on this retina-wide rod disease could be, for example, a trigger for more rapid regional retinal degeneration, such as has been suggested as the basis for the altitudinal loss. This inferior retinal loss has been thought to be associated with increased vulnerability from light damage.4–6 The concept of light-exacerbated altitudinal defects in patients with RHO mutations has had support from experiments in animal models indicating that light accelerates rod cell death in various species with mutant rhodopsin.45–50
The pericentral distribution may also be a pattern superimposed on the retina-wide rod disease. This pattern corresponds to the retinal region in humans with the highest density of rod photoreceptors or rod ring23 (Fig. 4A). The theory of one-hit death kinetics of photoreceptor cell loss 51,52 would suggest that there would be no greater rod cell loss in the rod ring than elsewhere in the retina. A generalized reduction of ONL thickness, such as we have reported in maps in other retinal degenerations,20,21,53 would be expected. It has long been discussed that there can be cellular interactions between photoreceptors in forms of RP, attempting mainly to explain cone loss even in patients with primary rod cell diseases, such as RHO mutations.54 Natural variations of molecular and cellular environments across the retina55–57 are likely to drive the onset of disease at earlier ages at some regions but not others. Once apoptotic mechanisms are set in motion, noncell autonomous effects between rods could also be postulated to contribute to progression, such as diffusible factors from dying cells that may be toxic to neighboring photoreceptors.52 These combinations of effects could have more impact in the pericentral area of high rod density. In chimeric retinas, cell death in mutant rods can even precipitate death in neighboring wild-type rod cells.58 The underlying bases for the different regional patterns in patients with RHO mutations and class B phenotypes are outside the scope of this study, but recognition of their existence is key to prepare for therapy and how to monitor the effects of novel interventions.
Acknowledgments
The authors thank William C. Nyberg for his continuous support of all retinal imaging acquisition and analyses over the decades of his relationship with our Center for Hereditary Retinal Degenerations at Scheie Eye Institute. He set the foundations and standards for clinical and research work as exemplified in this article. His generosity of spirit, kindness to patients, and professional expertise will not be forgotten.
Supported by the Chatlos Foundation; National Eye Institute, National Institutes of Health (R24 EY 022012; P30 EY 001583); Foundation Fighting Blindness; Research to Prevent Blindness.
Disclosure: S.G. Jacobson, None; D.B. McGuigan III, None; A. Sumaroka, None; A.J. Roman, None; M.L. Gruzensky, None; R. Sheplock, None; J. Palma, None; S.B. Schwartz, None; T.S. Aleman, None; A.V. Cideciyan, None
References
- 1. Massof RW,, Finkelstein D. Two forms of autosomal dominant primary retinitis pigmentosa. Doc Ophthalmol. 1981; 51: 289–346. [DOI] [PubMed] [Google Scholar]
- 2. Grover S,, Fishman GA,, Brown J., Jr. Patterns of visual field progression in patients with retinitis pigmentosa. Ophthalmology. 1998; 105: 1069–1075. [DOI] [PubMed] [Google Scholar]
- 3. Gal A,, Apfelstedt-Sylla E,, Janecke AR,, Zrenner E. Rhodopsin mutations in inherited retinal dystrophies and dysfunctions. Prog Retin Eye Res. 1997; 16: 51–79. [Google Scholar]
- 4. Cideciyan AV,, Hood DC,, Huang Y,, et al. Disease sequence from mutant rhodopsin allele to rod and cone photoreceptor degeneration in man. Proc Natl Acad Sci U S A. 1998; 95: 7103–7108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Jacobson SG,, Kemp CM,, Sung CH,, Nathans J. Retinal function and rhodopsin levels in autosomal dominant retinitis pigmentosa with rhodopsin mutations. Am J Ophthalmol. 1991; 112: 256–271. [DOI] [PubMed] [Google Scholar]
- 6. Heckenlively JR,, Rodriguez JA,, Daiger SP. Autosomal dominant sectoral retinitis pigmentosa two families with transversion mutation in codon 23 of rhodopsin. Arch Ophthalmol. 1991; 109: 84–91. [DOI] [PubMed] [Google Scholar]
- 7. Stone EM,, Kimura AE,, Nichols BE,, Khadivi P,, Fishman GA,, Sheffield VC. Regional distribution of retinal degeneration in patients with proline to histidine mutation in codon 23 of the rhodopsin gene. Ophthalmology. 1991; 98: 1806–1813. [DOI] [PubMed] [Google Scholar]
- 8. Fishman GA,, Stone EM,, Gilbert LD,, Kenna P,, Sheffield VC. Ocular findings associated with rhodopsin gene codon 58 transversion mutation in autosomal dominant retinitis pigmentosa. Arch Ophthalmol. 1991; 109: 1387–1393. [DOI] [PubMed] [Google Scholar]
- 9. Fishman GA,, Stone EM,, Gilbert LD,, Sheffield VC. Ocular findings associated with rhodopsin gene codon 106 mutation glycine-to-arginine change in autosomal dominant retinitis pigmentosa. Arch Ophthalmol. 1992; 110: 646–653. [DOI] [PubMed] [Google Scholar]
- 10. Fishman GA,, Stone EM,, Sheffield VC,, Gilbert LD,, Kimura AE. Ocular findings associated with rhodopsin gene codon 17 and codon 182 transition mutations in dominant retinitis pigmentosa. Arch Ophthalmol. 1992; 110: 54–62. [DOI] [PubMed] [Google Scholar]
- 11. Kemp CM,, Jacobson SG,, Roman AJ,, Sung CH,, Nathans J. Abnormal rod dark adaptation in autosomal dominant retinitis pigmentosa with proline-23-histidine rhodopsin mutation. Am J Ophthalmol. 1992; 113: 165–174. [DOI] [PubMed] [Google Scholar]
- 12. Birch DG,, Locke KG,, Felius J,, et al. Rates of decline in regions of the visual field defined by frequency-domain optical coherence tomography in patients with RPGR-mediated x-linked retinitis pigmentosa. Ophthalmology. 2015; 122: 833–839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Sumaroka A,, Matsui R,, Cideciyan AV,, et al. Outer retinal changes including the ellipsoid zone (EZ) band in Usher syndrome 1B due to MYO7A mutations. Invest Ophthalmol Vis Sci. 2016; 57: OCT253–OCT261. [DOI] [PubMed] [Google Scholar]
- 14. Sung CH,, Davenport CM,, Hennessey JC,, et al. Rhodopsin mutations in autosomal dominant retinitis pigmentosa. Proc Natl Acad Sci U S A. 1991; 88: 6481–6485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Aleman TS,, Cideciyan AV,, Sumaroka A,, et al. Retinal laminar architecture in human retinitis pigmentosa caused by rhodopsin gene mutations. Invest Ophthalmol Vis Sci. 2008; 49: 1580–1590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Jacobson SG,, Voigt WJ,, Parel JM,, et al. Automated light- and dark-adapted perimetry for evaluating retinitis pigmentosa. Ophthalmology. 1986; 93: 1604–1611. [DOI] [PubMed] [Google Scholar]
- 17. Roman AJ,, Schwartz SB,, Aleman TS,, et al. Quantifying rod photoreceptor-mediated vision in retinal degenerations: dark-adapted thresholds as outcome measures. Exp Eye Res. 2005; 80: 259–272. [DOI] [PubMed] [Google Scholar]
- 18. Matsui R,, Cideciyan AV,, Schwartz SB,, et al. Molecular heterogeneity within the clinical diagnosis of pericentral retinal degeneration. Invest Ophthalmol Vis Sci. 2015; 56: 6007–6018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Cideciyan AV,, Hufnagel RB,, Carroll J,, et al. Human cone visual pigment deletions spare sufficient photoreceptors to warrant gene therapy. Hum Gene Ther. 2013; 1006: 1–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Jacobson SG,, Cideciyan AV,, Aleman TS,, et al. Photoreceptor layer topography in children with Leber congenital amaurosis caused by RPE65 mutations. Invest Ophthalmol Vis Sci. 2008; 49: 4573–4577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Jacobson SG,, Aleman TS,, Sumaroka A,, et al. Disease boundaries in the retina of patients with Usher syndrome caused by MYO7A gene mutations. Invest Ophthalmol Vis Sci. 2009; 50: 1886–1894. [DOI] [PubMed] [Google Scholar]
- 22. Beltran WA,, Cideciyan AV,, Iwabe S,, et al. Successful arrest of photoreceptor and vision loss expands the therapeutic window of retinal gene therapy to later stages of disease. Proc Natl Acad Sci U S A. 2015; 112: E5844–E5853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Curcio CA,, Sloan KR,, Kalina RE,, Hendrickson AE. Human photoreceptor topography. J Comp Neurol. 1990; 292: 497–523. [DOI] [PubMed] [Google Scholar]
- 24. Shah SP,, Wong F,, Sharp DM,, Vincent AL. A novel rhodopsin point mutation, proline-170-histidine, associated with sectoral retinitis pigmentosa. Ophthalmic Genet. 2014; 35: 241–247. [DOI] [PubMed] [Google Scholar]
- 25. Jacobson SG, Cideciyan AV,. . Treatment possibilities for retinitis pigmentosa. N Engl J Med. 2010; 363: 1669–1671. [DOI] [PubMed] [Google Scholar]
- 26. Sahni JN,, Angi M,, Irigoyen C,, Semeraro F,, Romano MR,, Parmeggiani F. Therapeutic challenges to retinitis pigmentosa: from neuroprotection to gene therapy. Curr Genomics. 2011; 12: 276–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Petrs-Silva H,, Linden R. Advances in gene technologies to treat retinitis pigmentosa. Clin Ophthalmol. 2014; 8: 127–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Guadagni V,, Novelli E,, Piano I,, Gargini C,, Strettoi E. Pharmacological approaches to retinitis pigmentosa: a laboratory perspective. Prog Retin Eye Res. 2015; 48: 62–81. [DOI] [PubMed] [Google Scholar]
- 29. Sacchetti M,, Mantelli F,, Merlo D,, Lambiase A,, et al. Systematic review of randomized clinical trials on safety and efficacy of pharmacological and nonpharmacological treatments for retinitis pigmentosa. J Ophthalmol. 2015; 2015: 737053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Dryja TP,, McGee TL,, Hahn LB,, et al. Mutations within the rhodopsin gene in patients with autosomal dominant retinitis pigmentosa. N Engl J Med. 1990; 323: 1302–1307. [DOI] [PubMed] [Google Scholar]
- 31. Farrar GJ,, McWilliam P,, Bradley DG,, et al. Autosomal dominant retinitis pigmentosa: linkage to rhodopsin and evidence for genetic heterogeneity. Genomics. 1990; 8: 35–40. [DOI] [PubMed] [Google Scholar]
- 32. Grøndahl J,, Riise R,, Heiberg A,, Leren T,, Christoffersen T,, Bragadottir R. Autosomal dominant retinitis pigmentosa in Norway: a 20-year clinical follow-up study with molecular genetic analysis. Two novel rhodopsin mutations: 1003delG and I179F. Acta Ophthalmol Scand. 2007; 85: 287–297. [DOI] [PubMed] [Google Scholar]
- 33. Milam AH,, Li Z-Y,, Fariss RN. Histopathology of the human retina in retinitis pigmentosa. Prog Retin Eye Res. 1998; 17: 175–205. [DOI] [PubMed] [Google Scholar]
- 34. Ben-Arie-Weintrob Y,, Berson EL,, Dryja TP. Histopathologic-genotypic correlations in retinitis pigmentosa and allied diseases. Ophthalmic Genet. 2005; 26: 91–100. [DOI] [PubMed] [Google Scholar]
- 35. Li ZY,, Jacobson SG,, Milam AH. Autosomal dominant retinitis pigmentosa caused by the threonine-17-methionine rhodopsin mutation: retinal histopathology and immunocytochemistry. Exp Eye Res. 1994; 58: 397–408. [DOI] [PubMed] [Google Scholar]
- 36. To K,, Adamian M,, Dryja TP,, Berson EL. Histologic study of variation in severity of retinitis pigmentosa due to the dominant rhodopsin mutation Pro23His. Am J Ophthalmol. 2002; 134: 290–293. [DOI] [PubMed] [Google Scholar]
- 37. Bonilha VL,, Rayborn ME,, Bell BA,, et al. Retinal histopathology in eyes from patients with autosomal dominant retinitis pigmentosa caused by rhodopsin mutations. Graefes Arch Clin Exp Ophthalmol. 2015; 253: 2161–2169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Bird AC. Retinitis pigmentosa–a review. Trans Ophthalmol Soc N Z. 1977; 29: 51–58. [PubMed] [Google Scholar]
- 39. Bird AC. Investigation of disease mechanisms in retinitis pigmentosa. Ophthalmic Paediatr Genet. 1992; 13: 57–66. [DOI] [PubMed] [Google Scholar]
- 40. Sung CH,, Schneider BG,, Agarwal N,, Papermaster DS,, Nathans J. Functional heterogeneity of mutant rhodopsins responsible for autosomal dominant retinitis pigmentosa. Proc Natl Acad Sci U S A. 1991; 88: 8840–8844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Sung C,, Davenport C,, Nathans J. Rhodopsin mutations responsible for autosomal dominant retinitis pigmentosa. Clustering of functional classes along the polypeptide chain. J Biol Chem. 1993; 268: 26645–26649. [PubMed] [Google Scholar]
- 42. Mendes HF,, van der Spuy J,, Chapple P,, Cheetham ME. Mechanisms of cell death in rhodopsin retinitis pigmentosa: implications for therapy. Trends Mol Med. 2005; 11: 177–185. [DOI] [PubMed] [Google Scholar]
- 43. Rakoczy EP,, Kiel C,, McKeone R,, Stricher F,, Serrano L. Analysis of disease-linked rhodopsin mutations based on structure function and protein stability calculations. J Mol Biol. 2011; 405: 584–606. [DOI] [PubMed] [Google Scholar]
- 44. Malanson KM,, Lem J. Rhodopsin-mediated retinitis pigmentosa. Prog Mol Biol Transl Sci. 2009; 88: 1–31. [DOI] [PubMed] [Google Scholar]
- 45. Naash ML,, Peachey NS,, Li ZY,, et al. Light-induced acceleration of photoreceptor degeneration in transgenic mice expressing mutant rhodopsin. Invest Ophthalmol Vis Sci. 1996; 37: 775–782. [PubMed] [Google Scholar]
- 46. Wang M, Lam TT, Tso MO, Naash MI,. . Expression of a mutant opsin gene increases the susceptibility of the retina to light damage. Vis Neurosci. 1997; 14: 55–62. [DOI] [PubMed] [Google Scholar]
- 47. Kijas JW,, Cideciyan AV,, Aleman TS,, et al. Naturally occurring rhodopsin mutation in the dog causes retinal dysfunction and degeneration mimicking human dominant retinitis pigmentosa. Proc Natl Acad Sci U S A. 2002; 99: 6328–6333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Vaughan DK,, Coulibaly SF,, Darrow RM,, Organisciak DT. A morphometric study of light-induced damage in transgenic rat models of retinitis pigmentosa. Invest Ophthalmol Vis Sci. 2003; 44: 848–855. [DOI] [PubMed] [Google Scholar]
- 49. Cideciyan AV,, Jacobson SG,, Aleman TS,, et al. In vivo dynamics of retinal injury and repair in the rhodopsin mutant dog model of human retinitis pigmentosa. Proc Natl Acad Sci U S A. 2005; 102: 5233–5238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. White DA,, Fritz JJ,, Hauswirth WW,, Kaushal S,, Lewin AS. Increased sensitivity to light-induced damage in a mouse model of autosomal dominant retinal disease. Invest Ophthalmol Vis Sci. 2007; 48: 1942–1951. [DOI] [PubMed] [Google Scholar]
- 51. Clarke G,, Collins RA,, Leavitt BR,, et al. A one-hit model of cell death in inherited neuronal degenerations. Nature. 2000; 406: 195–199. [DOI] [PubMed] [Google Scholar]
- 52. Burns J,, Clarke G,, Lumsden CJ. Photoreceptor death: spatiotemporal patterns arising from one-hit death kinetics and a diffusible cell death factor. Bull Math Biol. 2002; 64: 1117–1145. [DOI] [PubMed] [Google Scholar]
- 53. Herrera W,, Aleman TS,, Cideciyan AV,, et al. Retinal disease in Usher syndrome III caused by mutations in the clarin-1 gene. Invest Ophthalmol Vis Sci. 2008; 49: 2651–2660. [DOI] [PubMed] [Google Scholar]
- 54. Wong F. Investigating retinitis pigmentosa: a laboratory scientist's perspective. Prog Retin Eye Res. 1997; 16: 353–373. [Google Scholar]
- 55. Bernstein SL,, Wong P. Regional expression of disease-related genes in human and monkey retina. Mol Vis. 1998; 4: 24. [PubMed] [Google Scholar]
- 56. Hjelmeland LM,, Fujikawa A,, Oltjen SL,, Smit-McBride Z,, Braunschweig D. Quantification of retinal pigment epithelial phenotypic variation using laser scanning cytometry. Mol Vis. 2010; 16: 1108–1121. [PMC free article] [PubMed] [Google Scholar]
- 57. Li M,, Jia C,, Kazmierkiewicz KL,, et al. Comprehensive analysis of gene expression in human retina and supporting tissues. Hum Mol Genet. 2014; 23: 4001–4014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Huang PC,, Gaitan AE,, Hao Y,, Petters RM,, Wong F. Cellular interactions implicated in the mechanism of photoreceptor degeneration in transgenic mice expressing a mutant rhodopsin gene. Proc Natl Acad Sci U S A. 1993; 90: 8484–8488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Sakami S,, Maeda T,, Bereta G,, et al. Probing mechanisms of photoreceptor degeneration in a new mouse model of the common form of autosomal dominant retinitis pigmentosa due to P23H opsin mutations. J Biol Chem. 2011; 286: 10551–10567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Jacobson SG,, Kemp CM,, Cideciyan AV,, Macke JP,, Sung CH,, Nathans J. Phenotypes of stop codon and splice site rhodopsin mutations causing retinitis pigmentosa. Invest Ophthalmol Vis Sci. 1994; 35: 2521–2534. [PubMed] [Google Scholar]