

Abstract
IPX066 (extended‐release carbidopa‐levodopa [ER CD‐LD]) is an oral extended‐release capsule formulation of carbidopa and levodopa. The single‐dose pharmacokinetics of ER CD‐LD (as 2 capsules; total dose, 97.5 mg‐390 mg CD‐LD) versus immediate‐release (IR) CD‐LD (25 mg‐100 mg), sustained‐release (CR) CD‐LD (25 mg‐100 mg), and CD‐LD‐entacapone (25 mg‐100 mg‐200 mg) was evaluated in healthy subjects. Following IR dosing, LD reached peak concentrations (Cmax) at 1 hour; LD concentrations then decreased rapidly and were less than 10% of peak by 5 hours. With CR CD‐LD and CD‐LD‐entacapone, LD Cmax occurred at 1.5 hours, and concentrations were less than 10% of peak by 6.3 and 7.5 hours, respectively. The initial increase in LD concentration was similar between ER CD‐LD and IR CD‐LD and faster than for CR CD‐LD and CD‐LD‐entacapone. LD concentrations from ER CD‐LD were sustained for approximately 5 hours and did not decrease to 10% of peak until 10.1 hours. Dose‐normalized LD Cmax values for ER CD‐LD were significantly lower (P< .05) than for the other CD‐LD products. Bioavailability of LD from ER CD‐LD was 83.5%, 78.3%, and 58.8% relative to IR CD‐LD, CR CD‐LD, and CD‐LD‐entacapone, respectively.
Keywords: IPX066, levodopa, extended release, pharmacokinetics, Parkinson's disease
Parkinson's disease is an age‐related progressive neurodegenerative disease that typically presents with motor symptoms of bradykinesia, rigidity, postural instability, and resting tremor. Levodopa (LD) is the most effective and widely used therapeutic agent in the treatment of Parkinson's disease.1, 2, 3, 4 Levodopa is actively absorbed, mainly in the proximal small intestine and is rapidly metabolized by aromatic L‐amino acid decarboxylase (AADC). Therefore, LD products are formulated with an AADC inhibitor, commonly carbidopa (CD), to prevent peripheral metabolism and increase the fraction of levodopa transported to the brain for conversion to dopamine. When combined with CD, immediate‐release (IR) LD has a half‐life of approximately 1.5 hours. Levodopa in combination with a decarboxylase inhibitor is a mainstay in the treatment of Parkinson's disease.
During the early stages of Parkinson's disease, dosing 3 times a day with IR CD‐LD is generally adequate to provide clinical effect. However, because of the short half‐life of LD, as disease progresses, the response duration from a single dose of IR CD‐LD progressively decreases and the “on” states (ie, period in which the medication is providing benefit in regard to motor function) may last less than 1 hour.5
Motor complications, namely, dyskinesia and motor “on/off” fluctuations, develop in about 50% of patients within 5 years of treatment.6 These deleterious effects have been attributed, at least in part, to fluctuations in plasma concentrations of LD.7, 8, 9, 10 Data from intravenous11, 12 and intraduodenal13, 14 LD infusion studies indicate that motor complications may be less likely to develop with more physiologic, continuous dopaminergic stimulation. A goal of Parkinson's disease pharmacotherapy has been the development of an oral formulation with the advantages of continuous infusion, a rapid onset of effect followed by a sustained therapeutic plasma concentration. The only sustained‐release product currently approved in the United States is Sinemet CR, which is absorbed over 4 to 6 hours.15 However, initial LD absorption is delayed, often requiring coadministration of IR CD‐LD, particularly for the first morning dose.16, 17 Formulations that add entacapone, a catechol‐O‐methyl transferase (COMT) inhibitor, to CD‐LD increase the exposure to LD by about 33% to 46%,18, 19, 20 and reduce “off” time by about 1 hour21 compared with IR CD‐LD. However, CD‐LD products with entacapone have been associated with a shorter time to onset of dyskinesia and increased frequency of dyskinesia compared with CD‐LD products.22
IPX066 (RYTARYTM; carbidopa and levodopa extended‐release capsules) is a multiparticulate extended‐release (ER) oral capsule formulation of CD‐LD for the treatment of Parkinson's disease, postencephalitic parkinsonism, and symptomatic parkinsonism that may follow carbon monoxide intoxication or manganese intoxication. The ER CD‐LD capsule formulation comprises different types of beads including immediate‐release and extended‐release beads combined in a specific ratio to provide the desired LD plasma profile. The immediate‐release component provides the initial rapid increase in LD concentrations with the extended‐release beads providing the subsequent delayed and extended‐release of LD. Inactive ingredients in the formulation include microcrystalline cellulose, mannitol, tartaric acid, ethylcellulose, hypromellose, sodium starch glycolate, sodium lauryl sulfate, povidone, talc, methacrylic acid copolymers, triethyl citrate, croscarmellose sodium, and magnesium stearate. The CD:LD ratio in ER CD‐LD is 1:4, which is similar to marketed formulations of CD‐LD. This study was designed to characterize the pharmacokinetics of ER CD‐LD in comparison with 3 commonly used CD‐LD combination products in healthy volunteers.
Materials and Methods
An institutional review board (St. Charles Community Institutional Review Board, St. Charles, Missouri) approved the study protocol, and each subject gave written informed consent before any protocol‐specified procedures or evaluations were performed. The study was conducted at a single center in the United States in accordance with the ethical principles embodied in the Declaration of Helsinki, Good Clinical Practice guidelines, and the International Conference on Harmonisation guidelines.
Subjects
Healthy male and female subjects (aged 18–45 years, inclusive) with a body mass index (BMI) between 18 and 29.5 kg/m2 and a supine blood pressure within the range of 90 to 139 mm Hg systolic and 50 to 89 mm Hg diastolic, were eligible to participate in this study. Subjects were excluded if they had any clinically relevant abnormalities as determined by medical history, physical examination, blood chemistry, complete blood count, urinalysis, and electrocardiogram (ECG) or if they had a positive urine drug screen or alcohol breath test. Subjects were also excluded if they had a history of glaucoma or peptic ulcer disease or had used any prescription or over‐the‐counter medications (excluding contraceptives, acetaminophen, or multivitamins) within 14 days (21 days for monoamine oxidase inhibitors) before study start and throughout the study period. All subjects were required to use a medically accepted method of contraception throughout the study period and for 1 month after study completion. Alcohol and grapefruit consumption were not permitted beginning 72 hours before each dose through 12 hours after dosing.
Study Design
This was a randomized, single‐dose, open‐label, 4‐sequence, 4‐treatment crossover study to assess the pharmacokinetics of ER CD‐LD versus 3 commercially available formulations of CD‐LD.
During each treatment period, subjects received a single oral dose of 1 of 4 treatments (according to the randomization sequence) under fasted conditions: 2 ER CD‐LD capsules (48.75 mg CD–195 mg LD; total dose, 97.5 mg CD–390 mg LD), 1 IR CD‐LD tablet (Sinemet® 25–100; 25 mg CD–100 mg LD), 1 sustained‐release (CR) CD‐LD tablet (Sinemet® CR 25–100; 25 mg CD–100 mg LD), or 1 CD‐LD‐entacapone tablet (Stalevo® 100; 25 mg CD, 100 mg LD, and 200 mg entacapone). There was a washout period of at least 6 days between treatments. The LD dose (100 mg) selected for the commercial products was a clinically relevant dose. The LD dose for ER CD‐LD (390 mg) was chosen to approximately match the expected peak LD concentrations for the commercial products.
Plasma Sample Analysis
During each treatment period blood was collected from each subject into tubes containing K2‐EDTA prior to dosing (within 1 hour of dosing) and 0.25, 0.5, 1, 1.5, 2, 2.5, 3,3.5, 4, 4.5, 5, 6, 7, 8, 10, and 12 hours after dosing. Blood samples were centrifuged and plasma samples were transferred to prechilled polypropylene tubes containing hydrazine dihydrochloride and sodium metabisulfite and were stored at −70°C until analysis.
A validated high‐performance liquid chromatography (HPLC)/tandem mass spectrometry method using multiple reaction monitoring was developed to measure plasma concentrations of LD and CD. Plasma samples were mixed with internal standard (levodopa‐d3 and carbidopa‐d3) and extracted using solid‐phase extraction. Following processing, samples were injected into an HPLC coupled with an API 4000 mass analyzer. Sample response was monitored using peak area ratio. The calibration curves were linear over the range of 10 to 2000 ng/mL for levodopa and 2 to 400 ng/mL for carbidopa (r ≥ 0.99). Both analytes were stable in plasma through 4 freeze‐thaw cycles. The interassay precision, as measured by the percent coefficient of variation (%CV) for quality control samples in the study ranged from 2.3% to 7.3% for LD and 2.0% to 7.1% for CD. The interassay accuracy measured as the percent difference ranged from −2.2% to 1.2% for LD and −1.3% to 0.7% for CD.
Pharmacokinetic and Statistical Analyses
Pharmacokinetic parameters for LD and CD were estimated using noncompartmental pharmacokinetic methods (Phoenix WinNonlin, version 6.2). The maximum plasma concentration (Cmax), and time to peak concentration (Tmax) were observed values. The apparent elimination half‐life (t1/2) was calculated as −(ln2)/k, where k is the slope of the log‐linear regression of the terminal phase of the concentration‐versus‐time curve. The area under the plasma concentration‐versus‐time curve from hour 0 to the last quantifiable concentration at time t (AUCt) was determined by the linear trapezoidal method. The AUC extrapolated to infinity (AUCinf) was calculated as AUCinf = AUCt + (Ct)/k, where Ct is the last measurable concentration. In addition, the duration that LD concentrations were above 50% of Cmax, time for LD concentrations to decrease to less than 10% of Cmax, and the ratio of Cmax/concentration at 6 hours (C6 h) was estimated to characterize the LD profiles of the various formulations. To compare the sustained nature of the LD profile for the various treatments, the time course of Cmax/Ct and the percent deviation from the mean LD concentration (Cavg) were also estimated. In addition, the expected deviation from Cavg at steady state was predicted based on superposition of the single‐dose pharmacokinetics. In this prediction, ER CD‐LD was dosed every 6 hours and the other CD‐LD products were dosed every 3 hours.
Statistical analyses were conducted to evaluate the Cmax and AUC of LD absorption from ER CD‐LD relative to the 3 reference formulations. A mixed‐effect analysis of variance model, which included treatment, period, and sequence as fixed factors and subject‐within‐sequence as a random effect, was used for the analysis of log‐transformed (dose‐normalized) AUC and Cmax. The least‐squares estimate of the mean for the ratio and a 90% confidence interval (CI) are presented. Statistical analyses were conducted using SAS, version 9.1.3 (Cary, North Carolina). Subjects who completed all treatments were included in the statistical analyses.
Assuming a ratio of unity for the mean AUC values for a test and reference formulation and a standard deviation of 0.4 (SD in logarithmic scale), a sample size of 20 subjects would assure with 90% confidence that the ratio of the AUC means is between 0.81 and 1.23. Twenty‐four subjects were enrolled to account for any dropouts.
Safety Assessments
Adverse events were monitored throughout the study. Vital signs (heart rate, blood pressure, and respiratory rate) were measured periodically following administration of study drugs after the subject had been resting supine for 5 minutes and again after the subject had been standing for 2 minutes. Safety assessments performed prestudy and poststudy included physical examinations, electrocardiograms (12‐lead), and clinical laboratory tests (chemistry, hematology, and urinalysis).
Results
A total of 24 healthy subjects (13 men and 11 women) were enrolled and treated; 22 subjects completed the study. Two subjects did not receive all treatments: 1 subject withdrew consent, and 1 subject was withdrawn for a protocol violation of a positive drug screen. Of the 24 subjects enrolled, 9 were white and 15 were African American, and the mean age was 24.6 years (range, 18–42 years). The mean body weight was 68.6 kg (range, 51.0–95.7 kg), and mean BMI was 23.6 kg/m2 (range, 19.0–28.8 kg/m2).
Pharmacokinetics of Levodopa and Carbidopa
The mean plasma concentration–time profiles of levodopa and carbidopa following a single dose of ER CD‐LD, IR CD‐LD, CR CD‐LD, and CD‐LD‐entacapone are displayed in Figure 1. The estimated LD and CD pharmacokinetic parameters for the 4 treatments are summarized in Table 1. With IR CD‐LD, LD was rapidly absorbed with median time to peak plasma concentrations at 1 hour. Levodopa plasma concentrations then decreased rapidly and were less than 10% of peak by 5 hours. With both CR CD‐LD and CD‐LD‐entacapone, median time to peak LD plasma concentrations was 1.5 hours, and concentrations were less than 10% of peak by 6.3 and 7.5 hours, respectively. With ER CD‐LD, LD plasma concentrations increased rapidly, reaching an initial plateau at approximately 1 hour, with median Tmax noted at approximately 4.5 hours. Plasma concentrations were less than 10% of peak at 10.1 hours.
Figure 1.
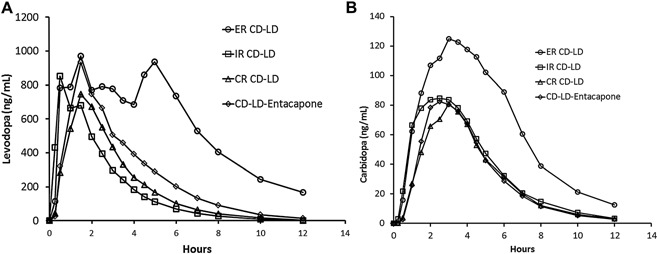
Mean levodopa (A) and carbidopa (B) plasma concentration–time profiles following a single dose of 2 capsules of ER CD‐LD (total dose, 97.5 mg CD‐390 mg LD), IR CD‐LD (25 mg‐100 mg), CR CD‐LD (25 mg‐100 mg), and CD‐LD‐entacapone (25 mg‐100 mg‐200 mg) under fasting conditions. ER, extended release, CR, sustained release; IR, immediate release; CD, carbidopa; LD, levodopa.
Table 1.
Single‐Dose Plasma Pharmacokinetic Parameters for Levodopa and Carbidopa, Fasting Conditions (n = 22)
| Pharmacokinetic Parameter a | Treatment | |||
|---|---|---|---|---|
| ER CD‐LD (97.5 mg‐390 mg) | IR CD‐LD (25 mg‐100 mg) | CR CD‐LD (25 mg‐100 mg) | CD‐LD‐Entacapone (25 mg‐100 mg‐200 mg) | |
| Levodopa | ||||
| Cmax (ng/mL) | 1326 ± 268 | 1094 ± 401 | 855 ± 299 | 1027 ± 284 |
| Cmax/D (ng/[mL · mg]) | 3.4 ± 0.7 | 10.9 ± 4.0 | 8.55 ± 3.0 | 10.3 ± 2.8 |
| Tmax (h) | 4.5 (0.5–8.0) | 1.0 (0.5–2.0) | 1.5 (1.0–2.0) | 1.5 (1.0–2.0) |
| t1/2 (h) | 1.9 ± 0.7 | 1.6 ± 0.2 | 1.6 ± 0.2 | 1.6 ± 0.2 |
| AUCinf (ng · h/mL) | 7244 ± 2553 | 2251 ± 664 | 2403 ± 680 | 3291 ± 1149 |
| AUCinf/D (ng · h/[mL · mg]) | 18.6 ± 6.5 | 22.5 ± 6.6 | 24.0 ± 6.8 | 32.9 ± 11 |
| Relative bioavailability (%) | — | 83.5 ± 21 | 78.3 ± 20 | 58.8 ± 18 |
| Cmax/C6 h | 2.4 ± 1.5 | 19.0 ± 13 | 9.4 ± 4.0 | 6.0 ± 2.4 |
| %CV in concentration (%) | 64.9 ± 12 | 121.4 ± 25 | 101.7 ± 12 | 92.4 ± 12 |
| Duration LD Cmax/Ct ≤ 2 | 4.0 ± 2.5 | 1.5 ± 0.7 | 2.1 ± 0.7 | 2.1 ± 1.0 |
| Duration LD concentrations above 50% Cmax (h) | 4.9 ± 2.4 | 1.5 ± 0.7 | 2.1 ± 1.0 | 2.1 ± 1.0 |
| Time LD concentrations decrease below 10% Cmax (h) | 10.1 ± 1.7 | 5.0 ± 1.1 | 6.3 ± 1.0 | 7.5 ± 1.1 |
| Carbidopa | ||||
| Cmax (ng/mL) | 148 ± 49 | 106 ± 43 | 86 ± 32 | 92 ± 29 |
| Cmax/D (ng/[mL · mg]) | 1.5 ± 0.5 | 4.2 ± 1.7 | 3.4 ± 1.3 | 3.7 ± 1.2 |
| Tmax (h) | 3.5 (1.5–6.0) | 2.5 (1.5–5.0) | 3.0 (2.0–4.5) | 2.5 (2.0–4.0) |
| t1/2 (h) | 2.5 ± 1.0 | 1.8 ± 0.2 | 2.0 ± 0.4 | 1.8 ± 0.3 |
| AUCinf (ng · h/mL) | 822 ± 276 | 448 ± 157 | 373 ± 117 | 381 ± 112 |
| AUCinf/D (ng · h/[mL · mg]) | 8.4 ± 2.8 | 17.6 ± 6.3 | 14.9 ± 4.7 | 15.2 ± 4.5 |
| Relative bioavailability (%) | — | 49.5 ± 14 | 58.9 ± 18 | 59.8 ± 29 |
| AUCLD/AUCCD | 9.58 ± 4.18 | 5.59 ± 2.33 | 6.93 ± 2.23 | 9.07 ± 3.13 |
ER, extended release; IR, immediate release; CR, sustained release; CD, carbidopa; LD, levodopa; Cmax, maximum observed plasma concentration; Cmax/D, dose‐normalized Cmax; Tmax, time to Cmax; t1/2, elimination half‐life; AUCinf, area under the concentration–time curve from time zero extrapolated to infinity; AUCinf/D, dose‐normalized AUC; C6 h, observed plasma concentration at hour 6; CV, coefficient of variation.
Pharmacokinetic parameter values are expressed as mean ± SD except for Tmax, which is expressed as median (range).
The mean peak LD plasma concentration after a single dose of 2 ER CD‐LD capsules (total LD dose, 390 mg) was approximately 21%, 55%, and 29% higher than the mean Cmax value for 25‐100 mg IR CD‐LD, 25‐100 mg CR CD‐LD, and 25‐100‐200 mg CD‐LD‐entacapone, respectively. The mean terminal half‐life of LD was comparable for the 4 treatments (1.6–1.9 hours). ER CD‐LD provided sustained LD concentrations as noted by the lower Cmax/C6 h ratio. This ratio, which reflects the fold decrease in LD concentration from the peak to 6 hours, provides an estimate of how sustained the levodopa concentrations are following the peak. The bioavailability of LD from ER CD‐LD relative to IR CD‐LD, CR CD‐LD and CD‐LD‐entacapone was 83.5%, 78.3%, and 58.8%, respectively. As expected, the dose‐normalized levodopa Cmax (Cmax/D) following ER CD‐LD was lower compared with the other CD‐LD products (30%–40% of Cmax compared with the other products; Table 1, Table 2).
Table 2.
Comparison of Dose‐Normalized Pharmacokinetic Parameters for Levodopa and Carbidopa Following Single Doses of ER CD‐LD, IR CD‐LD, CR CD‐LD, and CD‐LD‐Entacapone (n = 22)
| Test | Reference | Ratio of Geometric Least‐Squares Mean (90% Confidence Interval) | ||
|---|---|---|---|---|
| Cmax | AUCt | AUCinf | ||
| Levodopa | ||||
| ER CD‐LD | IR CD‐LD | 32.04 (28.51–36.02) | 77.23 (71.54–83.36) | 80.35 (74.30–86.89) |
| ER CD‐LD | CR CD‐LD | 40.83 (36.33–45.89) | 71.85 (66.56–77.56) | 75.11 (69.45–81.22) |
| ER CD‐LD | CD‐LD‐entacapone | 33.54 (29.84–37.70) | 53.60 (49.65–57.86) | 56.12 (51.90–60.69) |
| CR CD‐LD | IR CD‐LD | 78.48 (69.82–88.21) | 107.49 (99.57–116.03) | 106.98 (98.93–115.69) |
| CD‐LD‐entacapone | IR CD‐LD | 95.53 (84.99–107.37) | 114.08 (133.48–155.53) | 143.17 (132.39–154.83) |
| Carbidopa | ||||
| ER CD‐LD | IR CD‐LD | 36.63 (32.31–41.53) | 45.75 (40.50–51.69) | 47.53 (42.25–53.46) |
| ER CD‐LD | CR CD‐LD | 45.32 (39.97–51.38) | 55.20 (48.86–62.36) | 57.03 (50.69–64.15) |
| ER CD‐LD | CD‐LD‐entacapone | 40.92 (36.10–46.40) | 52.82 (46.76–59.68) | 54.95 (48.85–61.82) |
| CR CD‐LD | IR CD‐LD | 80.83 (71.36–93.63) | 82.88 (73.36–93.63) | 83.34 (74.08–93.75) |
| CD‐LD‐entacapone | IR CD‐LD | 89.50 (78.94–101.47) | 86.61 (76.66–97.85) | 86.49 (76.88–97.29) |
ER, extended release; IR, immediate release; CR, sustained release; CD, carbidopa; LD, levodopa.
Figure 2 presents the time course of Cmax/Ct (Figure 2A) and percent deviation from Cavg(Figure 2B) for LD for the CD‐LD treatments. The mean Cmax/Ct and percent deviation from Cave for LD were generally lower for ER CD‐LD compared with the other CD‐LD products. The mean LD Cmax/Ct ratio–time profile for ER CD‐LD was below 2 for up to 4 hours, whereas for IR CD‐LD, CR CD‐LD, and CD‐LD‐entacapone the durations averaged 1.5, 2.1, and 2.1 hours, respectively (Table 1). The steady‐state LD plasma concentration profile for ER CD‐LD dosed every 6 hours and the other CD‐LD products dosed very 3 hours was predicted based on superposition of the observed data following the single‐dose administrations. The predicted maximum absolute deviation from Cave at steady state (Figure 2C) was 32% for ER CD‐LD dosed every 6 hours compared with 72% to 87% for the other CD‐LD products dosed every 3 hours.
Figure 2.
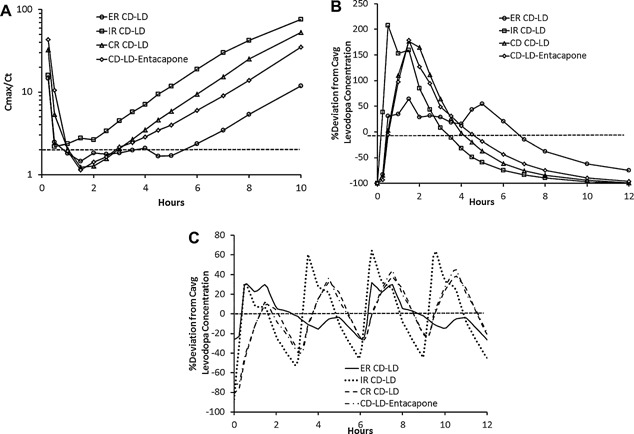
(A) Mean ratio of levodopa Cmax/Ct. (B) Mean percent deviation from LD Cavg following a single dose. (C) Predicted mean percent deviation from LD Cavg at steady state. Cmax, maximum concentration, Cavg, average concentration.
Similar to observations made for LD, peak plasma concentrations of CD occurred later with ER CD‐LD than with IR CD‐LD, CR CD‐LD, and CD‐LD‐entacapone. The terminal half‐life was approximately 2 hours and was comparable for all 4 treatments. The bioavailability of CD from ER CD‐LD was 49.5%, 58.9%, and 59.8% relative to IR CD‐LD, CR CD‐LD, and CD‐LD‐entacapone, respectively (Table 1).
Clinical Observations
Following ER CD‐LD treatment, 6 of 24 subjects (25%) had at least 1 adverse event (AE). Following the IR CD‐LD, CR CD‐LD, and CD‐LD‐entacapone treatments, 4 (17%), 2 (9%), and 2 (9%) subjects, respectively, had at least 1 AE. All AEs were characterized as mild with the exception of an ankle fracture, which was characterized as severe and unrelated to study treatment. No serious AEs were reported. Common AEs (experienced by ≥2 subjects) were nausea, vomiting, and headache (Table 3). The incidence of nausea and vomiting appeared higher during the ER CD‐LD treatment period than during the other treatment periods in this healthy volunteer population. No notable patterns in hematology, chemistry, urinalysis, ECGs, or vital signs were noted for any subject.
Table 3.
Common Adverse Events in Healthy Subjects Under Fasting Conditions
| Number of Subjects With AEs (%) | ||||
|---|---|---|---|---|
| ER CD‐LD (97.5 mg‐390 mg) | IR CD‐LD (25 mg‐100 mg) | CR CD‐LD (25 mg‐100 mg) | CD‐LD‐Entacapone (25 mg‐100 mg‐200 mg) | |
| n a | 24 | 23 | 22 | 22 |
| At least 1 AE | 6 (25.0) | 4 (17.4) | 2 (9.1) | 2 (9.1) |
| Nausea | 5 (20.8) | 2 (8.7) | 1 (4.5) | 1 (4.5) |
| Vomiting | 2 (8.3) | 1 (4.3) | 0 | 0 |
| Headache | 2 (8.3) | 1 (4.3) | 0 | 0 |
Common AEs, any AE experienced by 2 or more subjects; ER, extended release; IR, immediate release; CR, sustained release; CD, carbidopa; LD, levodopa; AE, adverse event.
Number of subjects.
Discussion
The need to develop an improved oral extended‐release LD product that can rapidly achieve and sustain constant LD plasma concentrations has been recognized as an unmet need in the treatment of Parkinson's disease.23 Various approaches including adding enzyme inhibitors (such as COMT inhibitors), gastric‐retention formulations, duodenal infusion delivery systems, and prodrug approaches have been explored to extend the duration of effect. Gastric retention formulations generally require concomitant administration of food to retain the dosage form in the stomach after swelling. This constantly fed state may not be feasible for Parkinson's disease patients. Drawbacks of intraduodenal infusion systems include the invasive surgical procedure, risk of infections, the inconvenience of carrying and refilling the pump, and issues related to device malfunction. For this reason, this surgical option is typically reserved for the most severe patients. At this time, LD prodrugs have not shown an advantage over immediate‐release formulations.24, 25
The present study was conducted in healthy adults and focused on comparing LD and CD pharmacokinetics of ER CD‐LD with those of currently marketed CD‐LD products. This single‐dose study showed that ER CD‐LD provides an initial increase in LD concentration comparable to that with IR CD‐LD and sustains the concentration (defined as >50% of Cmax) for 1.9 to 2.5 hours longer than the other CD‐LD products. These results are consistent with the pharmacokinetic and efficacy results reported by Hauser et al (2011) in an open‐label phase 2 study in patients with advanced Parkinson's disease. In that study, ER CD‐LD resulted in a rapid onset and longer duration of motor control compared with IR CD‐LD.26
The CR matrix formulations of CD‐LD have reduced drug availability, and because of the slower absorption, peak plasma concentrations are delayed compared with the IR CD‐LD formulation. As a result, time to an “on” state is delayed with the CR product, particularly for the first morning dose5, 27, 28, 29 and supplemental doses of IR CD‐LD may be required for onset of effect.30 ER CD‐LD would not require supplementation with an IR dose because the ER CD‐LD capsule formulation includes an immediate‐release portion designed to provide the initial rapid increase in LD concentration, followed by delayed and extended release to provide the sustained LD concentrations. Although the LD dose with ER CD‐LD was 3.9‐fold higher compared with IR and CR CD‐LD, it should be noted that the LD peak concentration with ER CD‐LD is only 20%–30% higher and the dose‐normalized Cmax is considerably lower (Table 1, Table 2). Thus, the rapid initial increase in LD concentration with ER CD‐LD is a result of the immediate‐release portion of the formulation and not due to the higher LD dose.
Although the present study was conducted in healthy volunteers and thus did not include assessment of efficacy, published reports indicate a good concentration–effect relationship for LD.31, 32, 33 The similar initial increase in LD plasma concentrations for ER CD‐LD and IR CD‐LD in healthy subjects correlates with the onset of effect in patients with advanced Parkinson disease (PD). The time to onset of effect (defined as an increase of more than 15% from baseline in the rate of finger tapping) for IR CD‐LD and ER CD‐LD in advanced PD patients was 0.39 and 0.36 hours, respectively.34
The pharmacokinetics of the reference drugs in the present study are comparable to published reports (Table 4). With IR CD‐LD (100 mg LD), peak levodopa plasma concentrations were approximately 1 μg/mL and were noted 1 hour after dosing in the present study (Table 1). Previous investigators have reported peak levodopa concentrations between 0.85 and 2.0 μg/mL (Table 4).19, 20, 35, 36, 37 There is a paucity of pharamacokinetic data with CR CD‐LD, but LD peak concentrations noted in the present study compare favorably with published data (Table 4). Levodopa Cmax and AUC from CD‐LD‐entacapone noted in the present study were comparable to those reported by Keränen et al (1993)19 and higher than those noted by Rouru et al (1999)38 and Heikkinen et al (2002).36 Importantly, all studies, including the present study, show that the time to maximum LD concentration for CD‐LD‐entacapone is later than with IR CD‐LD.
Table 4.
Summary of Single‐Dose Levodopa Pharmacokinetic Parameters for Sinemet, Sinemet CR, and Stalevo (100‐mg Dose) From Published Reports
| Study | n | Cmax (ng/mL) | Tmax (h) | AUCinf (ng · h/mL) | AUCL/AUCC a |
|---|---|---|---|---|---|
| Sinemet (IR CD‐LD) | |||||
| Kaakkola 1985 | 11 | 1091.8 ± 242.4 | 0.9 ± 0.2 | 1648 ± 124 | 2.20 |
| Heikkinen 2002 | 14–16 | 850 ± 310 | 0.58 ± 0.25 | 1430 ± 340 b | 4.27 |
| Keränen 1993 | 12 | 1210 ± 579 | 0.94 ± 0.49 | 2340 ± 438 | 1.19 |
| Myllylä 1993 e | 8 | 2080 | 0.78 | 3620 | 6.44 |
| Liang 2006 | 18 | 1040 ± 260 | 0.75 | 1950 ± 960 | — |
| Sinemet CR (CR CD‐LD) c | |||||
| Hammerstad 1994 e | 9 | 887 ± 355 | 1.3 ± 0.6 | — | — |
| Liang 2006 | 18 | 770 ± 310 | 2 | 2020 ± 600 | — |
| Stalevo (CD‐LD‐Entacapone) | |||||
| Rouro 1999 | 12 | 701 ± 243 | 0.8 ± 0.4 | 1704 ± 319 d | 10.3 |
| Heikkinen 2002 | 14–16 | 720 ± 250 | 1.16 ± 0.59 | 1930 ± 350 b | 6.17 |
| Keränen 1993 | 12 | 1040 ± 141 | 1.27 ± 0.66 | 3330 ± 580 | 1.71 |
| Myllylä 1993 e | 8 | 1490 | 1.17 | 5280 | 11.4 |
Cmax, maximum concentration; Tmax, time to Cmax; AUCinf, area under the curve to infinity; IR, immediate release; CR, sustained release; CD, carbidopa; LD, levodopa.
Ratio of AUClevodopa to AUCcarbidopa. Calculated based on a ratio of the reported means for individual drugs.
AUC0–12 is reported.
Data are scaled proportionally to allow comparison to a LD dose of 100 mg.
AUC0–24 is reported.
Data reported in patients.
The smoothness of the LD concentration–time curve is an important consideration in the treatment of patients with advanced Parkinson's disease. Continuous levodopa administration is associated with reduced motor complications compared with pulsatile delivery.27 The CR CD‐LD formulation has a slower initial absorption, with a mean time to peak concentration of approximately 1.5–2 hours compared with 1 hour for the IR formulation. This often requires supplementation of a CR dose with an IR tablet, particularly for the first morning dose. The ER CD‐LD capsule formulation includes both an IR and ER components and would not require supplementation with an IR tablet. If CD‐LD can be delivered orally in a manner that is similar to the plasma profile of an infusion, it may result in reduced motor complications. The variability in LD plasma concentration as measured by the %CV was lower with ER CD‐LD (64.9%) than with IR CD‐LD (121.4%), CR CD‐LD (101.7%), and CD‐LD‐entacapone (92.4%). Similarly, the decline in LD plasma concentration from the peak to 6 hours, calculated as the ratio of Cmax/C6 h, was 2.36 for ER CD‐LD. The reference drugs all had much steeper declines: Cmax/C6 h ratios ranged from about 6 to 19. Notably, ER CD‐LD also had a smaller deviation from average LD concentration (∼32%) compared with the other CD‐LD products (72% to 87%).
ER CD‐LD was well tolerated in this single‐dose pharmacokinetic study in healthy subjects. The incidence of nausea following ER CD‐LD dosing was higher compared with the other CD‐LD products, which may be attributed to the higher LD dose or longer LD exposure in a population (healthy subjects) that is typically not accustomed to dopaminergic effects.
In summary, the sustained LD concentrations with reduced peak‐trough fluctuations achieved with ER CD‐LD may result in consolidation of “on” periods without an increase in the incidence of dyskinesia compared with the frequent cycles of “on” and “off' periods with current IR and CR CD‐LD therapy.
Funding
This study was funded by Impax Laboratories (Impax), and all authors are employees of Impax and own stock or stock options in the company.
Acknowledgments
All authors actively participated in the conception, design, and analysis of the study. The authors acknowledge the assistance of Jeanette Gates, BS, in the editing the article.
References
- 1. Jankovic J. Levodopa strengths and weaknesses. Neurology. 2002; 58(suppl 1):S19–S32. [DOI] [PubMed] [Google Scholar]
- 2. LeWitt PA. Levodopa for the treatment of Parkinson's disease. N Engl J Med. 2008; 359:2468–2476. [DOI] [PubMed] [Google Scholar]
- 3. Schapira AH, Emre M, Jenner P, Poewe W. Levodopa in the treatment of Parkinson's disease. Eur J Neurol. 2009; 16:982–989. [DOI] [PubMed] [Google Scholar]
- 4. Tsouli S, Konitsiotis S. How should we treat a patient with early Parkinson's disease? Int J Clin Pract. 2010; 64(9):1210–1219. [DOI] [PubMed] [Google Scholar]
- 5. Nyholm D. Pharmacokinetic optimization in the treatment of Parkinson's disease: an update. Clin Pharmacokinet. 2006; 45:109–136. [DOI] [PubMed] [Google Scholar]
- 6. Jankovic J. Motor fluctuations and dyskinesias in Parkinson's disease: clinical manifestations. Mov Disord. 2005; 11:11–16. [DOI] [PubMed] [Google Scholar]
- 7. Chase TN. The significance of continuous dopaminergic stimulation in the treatment of Parkinson's disease. Drugs. 1998; 55(Suppl 1): 1–9. [DOI] [PubMed] [Google Scholar]
- 8. Chase TN, Baronti F, Fabbrini G, Heuser IJ, Juncos JL, Mouradian MM. Rationale for continuous dopaminomimetic therapy of Parkinson's disease. Neurology. 1989; 39(suppl 2):7–10. [PubMed] [Google Scholar]
- 9. Thanvi BR, Lo TCN. Long term motor complications of levodopa: clinical features, mechanisms, and management strategies. Postgrad Med J. 2004; 80:452–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. LeWitt PA, Nyholm D. New developments in levodopa therapy. Neurology. 2004; 62(suppl 1):S9–S16. [DOI] [PubMed] [Google Scholar]
- 11. Quinn N, Parkes JD, Marsden CD. Control of on/off phenomenon by continuous intravenous infusion of levodopa. Neurology. 1984; 34:1131–1136. [DOI] [PubMed] [Google Scholar]
- 12. Shoulson I, Glaubinger GA, Chase TN. On‐off response: clinical and biochemical correlations during oral and intravenous levodopa administration in parkinsonian patients. Neurology. 1975; 25:1144–1148. [DOI] [PubMed] [Google Scholar]
- 13. Kurlan R, Rubin AJ, Miller C, Rivera‐Calimlin L, Clarke A, Shoulson I. Duodenal delivery of levodopa for on‐off fluctuations in parkinsonism: preliminary observations. Ann Neurol. 1986; 20:262–265. [DOI] [PubMed] [Google Scholar]
- 14. Syed N, Murphy J, Zimmerman T Jr, Mark MH, Sage JI. Ten years' experience with enteral levodopa infusions for motor fluctuations in Parkison's disease. Mov Disord. 1998; 13:336–338. [DOI] [PubMed] [Google Scholar]
- 15. Yeh KC, August TF, Bush DF, et al. Pharmacokinetics and bioavailability of Sinemet CR: a summary of human studies. Neurology. 1989; 39(suppl 2):25–38. [PubMed] [Google Scholar]
- 16. LeWitt PA. Clinical studies with and pharmacokinetic considerations of sustained‐release levodopa. Neurology. 1992; 42(suppl 1):S29–S32. [PubMed] [Google Scholar]
- 17. Hauser RA. Levodopa: past present, and future. Eur Neurol. 2009; 62:1–8. [DOI] [PubMed] [Google Scholar]
- 18. Ahtila S, Kaakkola S, Gordin A, et al. Effect of entacapone, a COMT inhibitor, on the pharmacokinetics and metabolism of levodopa after administration of controlled‐release levodopa‐carbidopa in volunteers. Clin Neuropharmacol. 1995; 18:46–57. [DOI] [PubMed] [Google Scholar]
- 19. Keränen T, Gordin A, Harjola VP, et al. The effect of cathecol‐O‐methyltransferease inhibition by entacapone on the pharmacokinetics and metabolism of levodopa in healthy volunteers. Clinical Neuropharmacol. 1993; 16:145–156. [DOI] [PubMed] [Google Scholar]
- 20. Myllylä VV, Sotaniemi KA, Suominen K, Keränen T. Effect of entacapone, a COMT inhibitor, on the pharmacokinetics of levodopa and on cardiovascular responses in patients with Parkinson's disease. Eur J Clin Pharmacol. 1993; 45:419–423. [DOI] [PubMed] [Google Scholar]
- 21. Di Stefano A, Sozio P, Iannitelli A, Cerasa LS. New drug delivery strategies for improved Parkinson's disease therapy. Expert Opin Drug Deliv. 2009; 6:389–404. [DOI] [PubMed] [Google Scholar]
- 22. Stocchi F, Rascol O, Kieburtz K, et al. Initiating levodopa/carbidopa therapy with and without entacapone in early Parkinson disease: the STRIDE‐PD study. Ann Neurol. 2010; 68:18–27. [DOI] [PubMed] [Google Scholar]
- 23. Stocchi F, Vacca L, Ruggieri S, Olanow CW. Intermittent vs continuous levodopa administration in patients with advanced Parkinson disease: a clinical and pharmacokinetic study. Arch Neurol. 2005; 62:905–910. [DOI] [PubMed] [Google Scholar]
- 24. Juncos JL, Mouradian MM, Fabbrini G, Serrati C, Chase TN. Levodopa methyl ester treatment of Parkinson's disease. Neurology. 1987; 37:1242–1245. [DOI] [PubMed] [Google Scholar]
- 25. Blindauer K, Shoulson I, Oakes D, et al. A randomized controlled trial of etilevodopa in patients with Parkinson disease who have motor fluctuations. Arch Neurol. 2006; 63:210–216. [DOI] [PubMed] [Google Scholar]
- 26. Hauser RA, Ellenbogen AL, Metman LV, et al. Crossover comparison of IPX066 and a standard levodopa formulation in advanced Parkinson's disease. Mov Disord. 2011; 26;2246–2252. [DOI] [PubMed] [Google Scholar]
- 27. Poewe WH, Lees AJ, Stern GM. Treatment of motor fluctuations in Parkinson's disease with an oral sustained‐release preparation of L‐dopa: clinical and pharmacokinetic observations. Clin Neuropharmacol. 1986; 9:430–439. [DOI] [PubMed] [Google Scholar]
- 28. Hammerstad JP, Woodward WR, Nutt JG, Gancher ST, Block GA, Cyhan G. Controlled release levodopa/carbidopa 25/100 (Sinemet CR 25/100): pharmacokinetics and clinical efficacy in untreated parkinsonian patients. Clin Neuropharmacol. 1994; 17:429–434. [DOI] [PubMed] [Google Scholar]
- 29. LeWitt PA, Nelson MV, Berchou RC, et al. Controlled‐release carbidopa/ levodopa (Sinemet 50/200 CR4): clinical and pharmacokinetic studies. Neurology. 1989; 39(suppl 2):45–53. [PubMed] [Google Scholar]
- 30. Ahlskog JE, Muenter MD, McManis PG, Bell GN, Bailey PA. Controlled‐release Sinemet (CR‐4): a double‐blind crossover study in patients with fluctuating Parkinson's disease. Mayo Clin Proc. 1988; 63:876–886. [DOI] [PubMed] [Google Scholar]
- 31. Nutt JG, Woodward WR. Levodopa pharmacokinetics and pharmacodynamics in fluctuating parkinsonian patients. Neurology 1986; 36:739–744 [DOI] [PubMed] [Google Scholar]
- 32. Nelson MV, Berchou RC, Lewitt PA, et al. Pharmacokinetic and pharmacodynamic modeling of L‐dopa plasma concentrations and clinical effects in Parkinson's disease after Sinemet. Clin Neuropharmacol. 1989; 12:91–97. [DOI] [PubMed] [Google Scholar]
- 33. Nelson MV, Berchou RC, LeWitt PA, Kareti D, Galloway MP. Pharmacodynamic modeling of concentration‐effect relationships after controlled‐release carbidopa/levodopa (Sinemet CR4) in Parkinson's disease. Neurology. 1990; 40:70–74. [DOI] [PubMed] [Google Scholar]
- 34. Mao Z, Hsu A, Gupta S, Modi NB. Population pharmacodynamics of IPX066: an oral extended–release capsule formulation of carbidopa‐levodopa, and immediate‐release carbidopa‐levodopa in patients with advanced Parkinson's disease. J Clin Pharmacol. 2013; 53:523–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kaakkola S, Männistö PT, Nissinen E, Vuorela A, Mäntylä R. The effect of an increased ratio of carbidopa to levodopa on the pharmacokinetics of levodopa. Acta Neurol Scand. 1985; 72:385–391. [DOI] [PubMed] [Google Scholar]
- 36. Heikkinen H, Varhe A, Laine T, et al. Entacapone improves the availability of L‐dopa in plasma by decreasing its peripheral metabolism independent of L‐dopa/carbidopa dose. Br J Clin Pharmacol. 2002; 54:363–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Khor S‐P., Hsu A. The pharmacokinetics and pharmacodynamics of levodopa in the treatment of Parkinson's disease. Curr Clin Pharmacol. 2007; 2:234–243. [DOI] [PubMed] [Google Scholar]
- 38. Rouru J, Gordin A, Huupponen R, et al. Pharmacokinetics of oral entacapone after frequent multiple dosing and effects on levodopa disposition. Eur J Clin Pharmcol. 1999; 55:461–467. [DOI] [PubMed] [Google Scholar]