

Abstract
Dinoflagellates are one of the last major lineages of eukaryotes for which little is known about genome structure and organization. We report here the sequence and gene structure of a clone isolated from a cosmid library which, to our knowledge, represents the largest contiguously sequenced, dinoflagellate genomic, tandem gene array. These data, combined with information from a large transcriptomic library, allowed a high level of confidence of every base pair call. This degree of confidence is not possible with PCR‐based contigs. The sequence contains an intron‐rich set of five highly expressed gene repeats arranged in tandem. One of the tandem repeat gene members contains an intron 26,372 bp long. This study characterizes a splice site consensus sequence for dinoflagellate introns. Two to nine base pairs around the 3′ splice site are repeated by an identical two to nine base pairs around the 5′ splice site. The 5′ and 3′ splice sites are in the same locations within each repeat so that the repeat is found only once in the mature mRNA. This identically repeated intron boundary sequence might be useful in gene modeling and annotation of genomes.
Keywords: Alternative splicing, Crypthecodinium, identically repeated intron boundary, intron, splicing
DINOFLAGELLATES are biflagellate protists that can be found in most of the world's aquatic environments. Depending upon the species, they play the diverse environmental roles of predators, prey, parasites, symbionts, and primary producers, with many showing plasticity of nutritive mode.
Dinoflagellates possess such a wide variety of unique nuclear characteristics that they were, until the 1990s, widely regarded as a “missing‐link” between prokaryotes and eukaryotes. They were referred to as Dino‐ or Mesokaryota, and sometimes viewed as a fourth domain of life unto themselves (Dodge 1965). These perplexing nuclear characteristics include large genomes, modified DNA bases, permanently condensed liquid‐crystalline cholesteric‐like chromosomes, a lack of nucleosomes, highly duplicated genes found in tandem arrays, a gene organization lacking typical eukaryotic conserved motifs, and a massive transfer of plastid genes to the nuclear genome (Bachvaroff and Place 2008; Bachvaroff et al. 2004; Kim et al. 2011; LaJeunesse et al. 2005; Rae 1973; Rill et al. 1989; Rowan et al. 1996; Zhang et al. 2006). The application of phylogenetic methods and molecular systematic data revealed that dinoflagellates reside firmly in the crown of the eukaryotes, among the Alveolates rather than belonging to a unique domain of life or even a basal lineage of eukaryotes (Cavalier‐Smith 1993; Gajadhar et al. 1991).
Perhaps, the most striking feature of a dinoflagellate cell is the large nucleus containing permanently condensed chromosomes. Dinoflagellate genome sizes vary by as much as two orders of magnitude, but the smallest dinoflagellate genome yet measured belongs to the endosymbiotic Symbioninium spp. with 1.5 pg per haploid cell, approximately half the size of the human genome (LaJeunesse et al. 2005). The average size of a Dinoflagellate genome is more than 10X larger than the human genome, and some species can be as large as 100X the size of the human genome (LaJeunesse et al. 2005). These genomes are prohibitively large to analyze with limited resources using current sequencing and assembly technology. At present, the most complete genomic data published are from the diminutive Symbiodinium minutum genome, which represents approximately 41% of the genome in 33,815 contigs across 21,898 scaffolds. Despite its incompleteness and fragmentation, the genome survey represents the best look at a dinoflagellate genome to date. The intractability of completing an assembled dinoflagellate genome has meant that most dinoflagellate sequences have been generated primarily using two methods: shotgun sequencing of transcriptome libraries (e.g. EST sequencing) and PCR. Sequencing of mRNA is valuable, but by definition carries essentially no information about genome structure, and PCR‐based methods depend upon flanking conserved primers, which imposes constraints on the insights that can be obtained from them.
Crypthecodinium cohnii is a heterotrophic marine dinoflagellate with uncertain phylogenetic affinity; in some analyses, it is placed in the crown of the Gonyaulacoid lineage (Logares et al. 2007; Parrow et al. 2006; Saldarriaga and Cavalier‐Smith 2004; Saldarriaga et al. 2001), while other analyses find it placed with more basal dinoflagellate lineages (Harper et al. 2005; Lin et al. 2006; Zhang et al. 2005). Crypthecodinium cohnii has been used in the industrial manufacture of omega‐3 fatty acids for fortification of infant formula (Wynn et al. 2005). Among dinoflagellates, C. cohnii is a relatively facile organism, capable of culture in either liquid or solid media, axenic culture, and accelerated growth in a specialized medium such that cultures reach late log phase 4‐10X faster than other dinoflagellates. The genome size of C. cohnii is a third the size of the average dinoflagellate genome at 3.8 pg per haploid cell (Allen et al. 1975). Many important discoveries have been made using C. cohnii, including the discovery of rare bases in dinoflagellate DNA, mutagenesis, and breeding studies, and the low protein content of dinoflagellate chromosomes (Rae 1973; Rizzo and Noodén 1972; Tuttle and Loeblich 1974). The gene alcohol dehyrodgenase (ADH) was targeted in the present study because it was found to be highly expressed in a C. cohnii cDNA library (Xue et al. 1999).
Prior to a genome survey of S. minutum, understanding of dinoflagellate genomic organization was almost exclusively based on a small number of publications comprising just 11 sequences of six genes from eight species (Hiller et al. 2001; Le et al. 1997; Lee et al. 1993; Li and Hastings 1998; Machabee et al. 1994; Okamoto et al. 2001a; Reichman et al. 2003; Rowan et al. 1996; Sharples et al. 1996; Yoshikawa et al. 1996; Zhang and Lin 2003; Zhang et al. 2006). Despite their paucity, these data led to an understanding of dinoflagellate genomic organization that is different from that of other eukaryotes. While never specifically codified, it is possible to articulate an implicit model of dinoflagellate genome organization that is widely shared and has shaped the understanding of dinoflagellate genomes. Although there is a diversity of opinions regarding many aspects of this model, we believe that the general interpretation we present here is widespread, and refer to it as a “consensus model.”
The consensus model suggests that: (1) dinoflagellate genes are highly duplicated and organized in tandem repeats, (2) genes of the tandem repeat (hereon referred to as tandem repeat members or “members” for short) are found in long arrays, encoding isoforms of the same protein, with synonymous substitutions at nearly every available site and rare amino acid substitutions, (3) Traditional eukaryotic promoter, terminator, and intron boundary sequences are thought to be absent, (4) introns are rare and tend to be small when present, (5) a 22 base pair (bp) sequence, encoded in a separate gene, is trans‐spliced to the 5′ end of pre‐mRNA transcripts to form a mature mRNA.
Prior to 2007, discussion of this unusual feature set was, to our knowledge, always couched as applying only to the specific genes under discussion. A general consensus was reached when three publications in 2007 and 2008 all described these unusual features as being broadly representative of dinoflagellate genomic organization (Bachvaroff and Place 2008; Lidie and van Dolah 2007; Zhang et al. 2007). Since then, the features of this consensus model have been found listed as general features of dinoflagellates in most publications on dinoflagellate biology. While no author has apparently felt sufficiently confident in the model to codify it, the model has nevertheless shaped discussion, analysis, and understanding of dinoflagellates for nearly a decade.
Dinoflagellate introns are also unusual. Intron splice site consensus sequences in most eukaryotes conform to the consensus sequence MAG¦GTRAGT at the 5′ splice site and CAG¦G at the 3′ splice site (Mount et al. 1992; Zhang 1998). The most common, noncanonical, splice site consensus sequence uses GC at the 5′ splice site rather than the canonical GT, but otherwise conforms well to the remaining consensuses and is spliced by the same spliceosomal complex as canonical introns (Thanaraj and Clark 2001; Wu and Krainer 1999). A rare class of introns, spliced by a separate spliceosomal complex, conforms to the consensus sequence RTATCCTY at the 5′ splice site. These introns account for a small percentage of introns in a variety of eukaryotes including Homo sapiens, Drosophila melanogaster, and Arabadopsis thaliana (Burge et al. 1998; Tarn and Steitz 1997). Dinoflagellate introns, when observed, have been noted to not conform to any known splice site consensus sequence, nor to have the secondary structure characteristic of self‐splicing introns (Bachvaroff and Place 2008; Okamoto et al. 2001b; Rowan et al. 1996; Shoguchi et al. 2013; Yoshikawa et al. 1996).
We report here the map‐based Sanger sequence of a 39,500 bp cosmid containing three entire and two partial copies of genes encoding members of the ADH superfamily, derived from the C. cohnii genome. Although labor‐intensive, this approach has neither the advantage of requiring neither conserved PCR primer sites nor the assumptions of short‐read sequence assembly. These data provide unique insights into genome structure of C. cohnii.
Materials and methods
Nucleic acid isolation
Genomic DNA was isolated from C. cohnii Seligo strain “KO”, an axenic, monoclonal isolate from the nonclonal culture ATCC #30340. The KO culture was grown to a concentration of approximately 107 cells/ml in a medium containing 50 g/l glucose, 6 g/l yeast extract, 32‐ppt artificial seawater, pH 6.7 at 27 °C shaking at 200 rpm. Cells were harvested by centrifugation at 4 °C and 3,000 g for 20 min. Cell pellets were transferred to plastic bags and flash frozen in liquid nitrogen. The frozen pellets were ground to a fine powder with a liquid nitrogen cooled mortar and pestle. Thirty grams of frozen‐powdered biomass was mixed with 100 ml extraction buffer (100 mM Tris, 1.5 M NaCl, 50 mM EDTA, 2% w/v cetrimonium bromide, 50 mM dithiothreitol, 100 U RNAse) and warmed to room temperature in a water bath. When the frozen pellet had thawed and was resuspended in extraction buffer, lysis was allowed to continue at room temperature for an additional 5 min. DNA was extracted twice with an equal volume of a phenol–chloroform–isoamyl alcohol mixture (25:24:1, v:v:v). Residual phenol was then removed with a chloroform–isoamyl alcohol (24:1, v:v) solution. DNA was precipitated with 2 volumes of 95% ethanol and 0.3 M sodium acetate at −20 °C for 1 h. DNA was pelleted by centrifugation at 3,000 g for 30 min at 4 °C, washed with 70% ethanol and resuspended in 10 mM Tris to a concentration of 1 μg/μl. Cells for RNA extractions were collected and ground using a mortar and pestle as previously described for DNA extraction. RNA isolation from the frozen‐powdered pellet was performed using Ambion's RNAqueous Kit (Life Technologies, Grand Island, NY).
Cosmid library construction and screening
The cosmid library was constructed according to Sambrook (Sambrook and Russell 2001) using Agilent's SuperCos1 Cosmid Vector Kit (Agilent, Santa Clara, CA), XL1 Blue Escherichia coli cells, and Gigapack III XL (Agilent) packaging kit. The library was plated, 60,000 colony forming units (CFU) per plate, on Whatman Nytran N (Whatman, Maidstone, Kent, UK) filters layered atop an LB ampicillin plate. Replica filters were produced and allowed to grow overnight before being stored at 4 °C. Cell lysis and nucleic acid crosslinking was performed per Whatman's protocol, with the modification that cell debris was vigorously scraped off the filters in the 2X SSPE bath followed by a brief rinse in 2X SSPE. An alcohol dehydrogenase gene with a highly abundant transcript was selected from an existing EST library for isolation. Probes for screening the library were made using a previously isolated cDNA clone (GenBank accession KJ831651) by restriction digest and radiolabeled using Promega Prime‐a‐Gene Labeling System (Promega, Madison, WI). Probes were hybridized to primary filters according to Sambrook and Russell (2001) using Church buffer in thermal‐sealed plastic bags in a shaking water bath, and washed according to Sambrook and Russell (2001). Positive colonies were identified following overnight exposure of the filters to a phosphor imaging screen and imaged with a phosphorimager. Images produced by the phosphorimager were used to correlate positive signals to colonies on the replica plates. Putative positive colonies were picked and rescreened by the same process until pure colonies were isolated. Cosmids were isolated from 500‐ml broth cultures of positive colonies using Qiagen Plasmid Maxi Kit (Qiagen, Venlo, the Netherlands).
DNA sequencing and analysis
The cosmid was sequenced by Eurofin's MWG Operon transposon‐based sequencing service (Eurofin, Luxenberg). Analysis of the sequence was performed in Biomatters Ltd Geneious software package (Biomatters, Auckland, New Zealand). Alignments of the cosmid insert sequence to previously identified cDNAs were used to identify gene repeats, intergenic spacers, exons, and introns. The open reading frame of each member, including putative start and stop codons and splice leader acceptor sites were identified by comparison to previously identified cDNAs and manual examinations of the alignments.
Analysis of the HCc gene previously published involved a BLAST search of a proprietary EST library using the HCc sequence as a query term and alignment of all matching ESTs to identify the putative reading frame and splice sites.
Results
Analysis of cosmid sequence
A C. cohnii cosmid library was screened using a probe for the gene ADH. One colony on nearly every initial plate that was screened, hybridized to the ADH probe (~1.5 in every 60,000 CFU). Upon re‐screening, fewer than half of these signals were confirmed. Northern blots were also performed for ADH to establish that we were not observing a polycistronic mRNA (data not shown).
Map of cosmid sequence
Of several clones eliciting a confirmed ADH hybridization signal, one was selected for scale‐up and full DNA sequence analysis. This cosmid (GenBank accession KJ831652) was found to contain an insert of 39,500 bp. The probe sequence and other ADH sequences from the EST database were compared to the cosmid sequence to map gene boundaries. One region of the cosmid was found to be a perfect match in 10 discreet exons to a cDNA (GenBank accession KJ831649) while the 3′ end of the cosmid sequence clipped by the insert ligation point matched another cDNA (GenBank accession KJ831650). Using this approach, a total of five similar but nonidentical ADH gene copies could be annotated in the cosmid sequence, designated here A through E (Fig. 1). The center three copies appear to be complete, and two copies are truncated by the ligation points of the insert to the cosmid vector, one at each end of the insert.
Figure 1.

Schematic of alcohol dehydrogenase (ADH) cosmid insert. The exons, poly‐adenylation signals, splice leader acceptor sites, and start and stop codons are all indicated. The transcripts as they align to genomic sequences are indicated. Exactly 24,000 bp from the middle of the eighth intron of member ADH‐C has been removed for illustrative purposes.
These initial predictions were modified to take into account the putative locations of splice leader acceptor sites and poly‐adenylation signal sites (Fig. 1). The SL‐acceptor site was identified as the first AG upstream of the aligned EST sequence. This change extended the 5′ UTR of ADH‐B and ADH‐E 16 and 14 bp further upstream of the aligned ESTs KJ831649 and EST KJ831650, respectively. While a variety of potential alternative SL‐acceptor sites can be observed in the 5′ UTR of all the observed gene repeats, the first available AG upstream of the previously predicted 5′ UTR was used, which corresponded to the same location in all the gene repeats. The poly‐adenylation signal was marked as 5′‐AAAAACAAAAA‐3′ or 5′‐AAAAACAACAA‐3′. This extends the 3′ UTR of each gene 11bp past where the aligned EST begins a poly‐adenylation sequence. Start and stop codons were identified by aligning all available ADH ESTs with the ADH gene repeats from the cosmid. A sharp drop‐off in sequence conservation marked the extremities of the coding sequences and allowed start and stop codons to easily be identified.
Sequence similarity analysis
A total of 34 introns were identified, all but one of which ranged in length between 83 and 371 bp, with a median size of 209 bp. The exception was a large intron measuring 26,372 bp located between the seventh and eighth predicted exons of ADH‐C. The coding regions of the complete exons ranged in size from 6 to 246 bp with a median size of 92 bp. The coding region of the first exon of each member was particularly small; just 6 bp. The coding regions of the exons comprise just 9.9% of the cosmid insert, and just 29.8% of the insert when the particularly large intron is removed from the calculation.
The coding regions of the members were highly conserved. The pairwise nucleotide identity of the exons in the five paralogs observed in the cosmid insert is 97.7%. The pairwise identity of the amino acid translation is 99.8%, differing in just one amino acid where the codon TTT, for phenylalanine in ADH‐C is TCT, for serine, in the other members. Noncoding regions were less conserved, but are still very similar (Fig. S1). Indels in noncoding regions significantly lowered calculations of pairwise identity. The pairwise identities of the introns range from 1.8% to 84.4%. The fourth introns of ADH‐A and ADH‐B are identical and the sixth introns of ADH‐B and ADH‐C are identical. Alignment of intron 8 revealed the large intron from ADH‐C has a 26,073 bp insertion compared to the other copies, accounting for 66.0% of the entire cosmid insert. The intergenic spacers range in size from 86 to 108 bp. The pairwise identity of the intergenic spacers is 81.0%, with 9.5% of the consensus sequence composed of gaps.
Intron border repeat
The intron junctions of ADH‐B were closely examined for potential splice site consensus sequences. A unique pattern was discovered in which the last 2–9 bp on the 3′ end of each exon exactly matched the 3′ end of the immediately downstream adjacent intron (Fig. 2A). This identical repeated intron boundary (IRIB) sequence is found only once in the corresponding cDNA; therefore, one could annotate the sequence so that one or the other IRIB is annotated as being exonic or intronic, or even that a part of each IRIB contributes to the translated sequence (Fig. 2A). Here, the splice sites are always placed between the conserved GG as found in canonical U2 splice site consensus sequences. When the exons are defined in this manner, the exons from each member fall in the same locations. If a different convention is used, the exons may differ in length by several base pairs. The repeat always contains a GG and often an AGG at the 3′ splice site and always has an AGG at the 5′ splice site (Fig. 2B). The rest of the repeated sequence was unique to each splice site.
Figure 2.
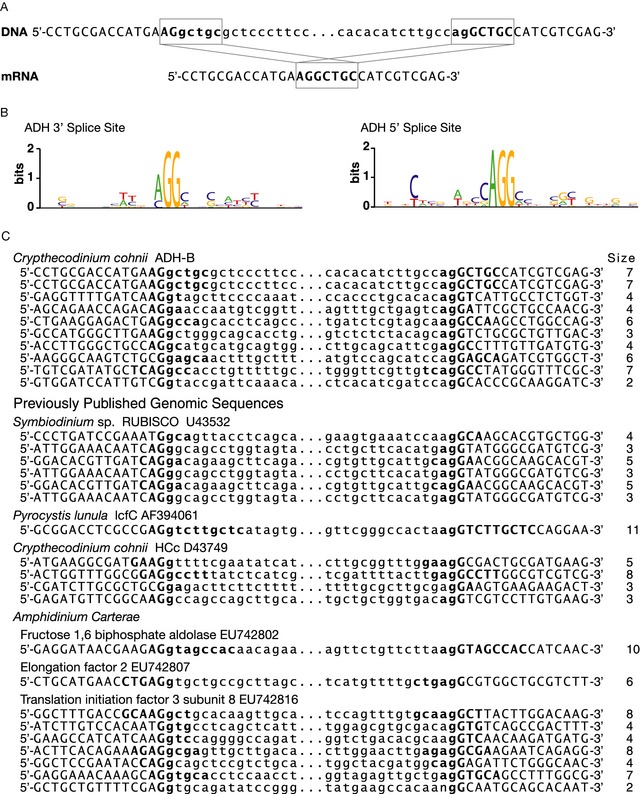
Nucleotide sequences surrounding the intron splice sites of alcohol dehydrogenase (ADH) from Crypthecodinium cohnii as well as previously published genes of various dinoflagellates. A. Genomic and mRNA sequences of the intron splice sites of ADH‐B intron 1 indicating the identical repeated intron boundary (IRIB) sequence is present at either end of the intron twice, but is present in the mRNA only once. B. Sequence logos, generated by WebLogo, using the intron splice sites from all introns present in the ADH cosmid sequence. C. Intron splice sites of ADH‐B and previously published dinoflagellate genomic sequences indicating the IRIB sequence and the size of the IRIB. Each IRIB sequence is indicated in bold and their corresponding sizes are listed at right. Portions of each internal intron sequence have been replaced by ellipses as shown.
Evidence of IRIB sequences was sought in previously published data from other researchers. RUBISCO from Symbiodinium sp. and LCF from Pyrocystis both show IRIB sequences in their published introns as annotated by their authors (Fig. 2C). The introns from the gene HCc from C. cohnii do not show an IRIB sequence as published; however, the intron exon boundaries annotated by the authors could not be established with confidence as the authors lacked a mRNA sequence with 100% identity. Using the HCc gene as a query sequence against our own C. cohnii EST library revealed several ESTs that allowed confident re‐annotation of the exon/intron boundaries of the previously published HCc genomic sequence and revealed IRIB sequences at every intron (Fig. 2C). Analysis of genes from the Amphidinium carterae survey revealed many introns with IRIB sequences, a subset of which are pictured in Fig. 2C.
Discussion
The consensus model
To the best of our knowledge, other than the Symbiodinium genome survey, these data represent the longest contiguous dinoflagellate genomic tandem array yet published, and has the advantage of being sequentially sequenced. While dinoflagellate genes are known to be present in tandem repeats and it has been inferred that many copies exist in tandem arrays, this sequence is unique in containing three complete gene duplicates and two more flanking gene duplicates for a total of five tandem genes. Previous evidence of multiple genes in tandem from dinoflagellates included very small genes, in the case of the gene encoding the splice leader, alignments that accept a small amount of mismatch in overlapping sequence and thus do not necessarily represent sequences that were physically adjacent to each other, or assembly from very short reads (Bachvaroff and Place 2008; Hiller et al. 2001; Le et al. 1997; Lee et al. 1993; Li and Hastings 1998; Machabee et al. 1994; McEwan et al. 2008; Okamoto et al. 2001b; Reichman et al. 2003; Sharples et al. 1996; Yoshikawa et al. 1996; Zhang and Lin 2003; Zhang et al. 2006). This longer contiguous copy set provides additional evidence that genes arranged in a common array all encode the same protein, consistent with the consensus model.
Some evidence suggests that the consensus model does not best describe all dinoflagellate genes (Bachvaroff and Place 2008; Shoguchi et al. 2013). There may in fact be two models of genes that are organized and transcribed differently from one another (Bachvaroff and Place 2008). The first model is typified by genes that are organized in tandem repeats, present in high copy number, highly expressed, trans‐spliced with a conserved leader sequence, and have low intron density (Bachvaroff and Place 2008). These genes can be considered the consensus model group. The second model of genes is not well studied, but seems to be organized like classic eukaryotic genes (Bachvaroff and Place 2008). These genes may have eluded initial detection because they are found in low copy number and are transcribed at much lower levels than the genes in tandem arrays. These genes are intron‐rich, are not trans‐spliced, are transcribed at low levels, and contain common eukaryotic motifs for transcription and RNA processing (Bachvaroff and Place 2008). Testing the comprehensiveness and accuracy of the consensus model is beyond the scope of this study, but it is important to note that our data are difficult to reconcile with the either of these models. The consensus model was shaped largely by data collected using PCR: of the 11 gene sequences that our research indicates have contributed to the consensus model, 10 have been isolated using PCR methods (Hiller et al. 2001; Le et al. 1997; Lee et al. 1993; Li and Hastings 1998; Machabee et al. 1994; Okamoto et al. 2001b; Reichman et al. 2003; Sharples et al. 1996; Yoshikawa et al. 1996; Zhang and Lin 2003; Zhang et al. 2006). The consensus model also seems to be in conflict with data from the S. minutum genome survey. Two major disagreements between the consensus model and the S. minutum genome survey are the paucity of genes arranged in tandem and the high frequency of introns in the S. minutum data. The data we present here deviate from the consensus model principally in the high frequency of introns. Whether the organization of this gene cluster is representative of the rest of the C. cohnii genome and whether C. cohnii's genome is broadly representative of dinoflagellate genomes is unknown, but it is notable that the kind of biases expected from a model developed using PCR data, happen to be the very areas that conflict with sequences collected via cosmid library screening. Selection of genes that are short enough to amplify by PCR could have resulted in a model built upon a nonrepresentative set which lack introns or have unusually few and small introns. How the S. minutum genome survey fits in is unclear. The scarcity of genes in tandem could be the result of incomplete assembly and gene modeling or could be real and simply the result of an endosymbiotic lifestyle. Whether the S. minutum genome is representative of broader dinoflagellate genomic organization or not, it highlights the need to vigorously test the consensus model with broader datasets.
Conservation of noncoding regions
The observation that dinoflagellate genomes are often organized into tandem gene arrays has led to speculation on the evolutionary processes underlying this organization (Kim et al. 2011; Reichman et al. 2003; Slamovits and Keeling 2008). The presence of what appear to be vestigial splice leader sequences in several dinoflagellate transcriptomes led to the inference that dinoflagellate genes could be duplicated via reverse transcription and reintegration into the genome of mature, trans‐spliced transcripts (Slamovits and Keeling 2008). Because introns would regularly be purged via such reintegration of reverse‐transcribed mature transcripts, this hypothesis predicts that introns would be rare, and when present would have been inserted relatively recently. Our observations conflict with that hypothesis, providing evidence for a gene duplication mechanism that preserves intron/exon structure. Assuming our splicing inferences are accurate, the relative intron positions of all five ADH members are perfectly conserved. Furthermore, the sequences of corresponding introns are also conserved. These observations are inconsistent with an mRNA intermediary and reintroduction of introns after duplication. Nor was there any evidence of vestigial splice leader sequences in any of the ADH members. If reverse transcription does play a role in the duplication of dinoflagellate genes, it is unlikely to have been the process that created the gene cluster described here. The conservation of intron splice sites and sequences suggests a genome‐level duplication mechanism, as well as either relatively recent duplication or concerted evolution (or both).
Whether the gene duplication of members of a tandem array in dinoflagellates has arisen due to concerted evolution or whether it represents a birth–death model has been examined in detail for both actin and peridinin‐chlorophyll a‐binding protein genes of A. carterae and Symbiodinium, respectively (Kim et al. 2011; Reichman et al. 2003). In most eukaryotes, the sequence uniformity in tandem arrays of rRNA genes is thought to be maintained by concerted evolution. In concerted evolution, uneven crossing‐over and gene conversion result in high sequence similarity between members of an array. In a birth–death model, sequence similarity is maintained via purifying selection of the encoded proteins. In an array functioning under a birth–death model, only nonsynonymous substitutions will be homogenized. As the process underlying concerted evolution affects synonymous and nonsynonymous substitutions equally, a comparison of the number of synonymous substitutions to nonsynonymous substitutions between members of an array can reveal the dominant contributing model. Analysis of members of a PCP tandem array led Reichman et al. to conclude that similarity was maintained via low levels of concerted evolution. The analysis of 142 members of actin by Kim et al., however, indicated that a birth–death model best explained the similarity of members. Our data are consistent with the findings of Kim et al. of the birth–death model. Duplication events via uneven crossing‐over and gene conversion cannot account for the differences in sequence similarity between coding and noncoding regions and the prevalence of synonymous substitutions in the coding regions. The birth–death model of gene duplication best explains the observed similarity of tandem array members, where the majority of changes occur in regions that do not affect protein structure. The similarities of noncoding regions are, however, still striking. It is possible that while most of the similarity between array members is maintained by purifying selection, low levels of concerted evolution are still at work.
Noncanonical splicing of introns
Intron splice sites in eukaryotes consist of a CAG¦G at the 3′ acceptor site and MAG¦GTRAGT at the 5′ donor site. While few dinoflagellate genes with introns have been sequenced, the unusual lack of the canonical GT‐AG consensus sequencing denoting intron splice sites in dinoflagellates has been noted in every case (Bachvaroff and Place 2008; Okamoto et al. 2001a; Rowan et al. 1996; Shoguchi et al. 2013; Yoshikawa et al. 1996). Two of these authors noted a repeat at the ends of introns, but did not fully describe the pattern (Bachvaroff and Place 2008; Yoshikawa et al. 1996). Within our data, there is a consistent splicing pattern that is also consistent with most other published splice sites from dinoflagellates. The AG¦G of the 3′ and 5′ splice site is usually conserved, and the splice donor and acceptor sites have a duplicate 2–11 bp sequence flanking the intron which remains in the mature mRNA only once (Fig. 2). Consequently, this creates ambiguity in the exact annotation of splice sites anywhere within the IRIB. We believe that the splice site we have designated here is correct and consistent with other studies, but this ambiguity has resulted in understandable variation in the annotation of exact intron boundaries in other studies. Intron splice sites in HCc from C. cohiii, lcfC from Pyrocystis lunula, and sequences from the survey of A. cartera were all consistent with splicing as inferred here, although annotated slightly differently (Bachvaroff and Place 2008; Okamoto et al. 2001b; Shoguchi et al. 2013; Yoshikawa et al. 1996). Analysis of previously published dinoflagellate genomic sequences containing introns reveals that the IRIB sequence is present in all dinoflagellates for which there are available data, but the inherent flexibility of IRIB annotation makes the pattern difficult to recognize (Bachvaroff and Place 2008; Okamoto et al. 2001a; Rowan et al. 1996; Shoguchi et al. 2013; Yoshikawa et al. 1996). Interestingly, the splice site logo generated in the S. minutum genome survey looks very similar to the one generated here. The major difference between the S. minutum logo and our C. cohnii ADH logo is in the nature of the GG at the 5′ donor site. This GG is conserved in C. cohnii ADH sequences and all other published dinoflagellate introns, but is not well conserved in the splice site logo generated for the S. minutum genome survey. With the continued improvement of sequencing technology, more dinoflagellate genomic data are surely forthcoming; hopefully discovery of the dinoflagellate IRIB will improve the automated gene modeling necessary in such large scale sequencing projects.
Supporting information
Figure S1. Alignments and mean pairwise identity graphs for the intergenic spacers between alcohol dehydrogenase (ADH) members and the nine introns of the five members of ADH. Length and pairwise identity is displayed to the right of each alignment. Gene sequences are indicated by black bars, and indels are indicated by intervening black lines. Each column of the alignment has a bar graph above it indicating the mean pairwise identity of all pairs in the alignment. The height and color of each bar in the graph is proportional to the mean pairwise identity of all pairs in that column ‐ green at 100%, yellow between 30 and less than 100%, and red when less than 30%.
Acknowledgments
The authors thank Tsvetan R. Bachvaroff of the Institute of Marine and Environmental Technology for his insights and attention during the extended preparation of this manuscript. This work was supported in part by NSF Grant #0629624 to C.F.D. This paper is dedicated to the memory of Kirk Apt, who was the primary driving force behind the project.
Literature Cited
- Allen, J. , Roberts, T. & Loeblich, A. 1975. Characterization of the DNA from the dinoflagellate Crypthecodinium cohnii and implications for nuclear organization. Cell, 6:161–169. [DOI] [PubMed] [Google Scholar]
- Bachvaroff, T. , Concepcion, G. , Rogers, C. , Herman, E. & Delwiche, C. 2004. Dinoflagellate expressed sequence tag data indicate massive transfer of chloroplast genes to the nuclear genome sequence. Protist, 155:65–78. [DOI] [PubMed] [Google Scholar]
- Bachvaroff, T. R. & Place, A. R. 2008. From stop to start: tandem gene arrangement, copy number and trans‐splicing sites in the dinoflagellate Amphidinium carterae . PLoS ONE, 3:e2929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burge, C. B. , Padgett, R. A. & Sharp, P. A. 1998. Evolutionary fates and origins of U12‐type introns. Mol. Cell, 2:773–785. [DOI] [PubMed] [Google Scholar]
- Cavalier‐Smith, T. 1993. Kingdom protozoa and its 18 phyla. Microbiol. Rev., 57:953–994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dodge, J. D. 1965. Chromosome structure in the dinoflagellate and the problem of the mesokaryotic cell. Int. Congress Ser., 91:264–265. [Google Scholar]
- Gajadhar, A. , Marquardt, W. , Hall, R. , Gunderson, J. , Ariztia‐Carmona, E. & Sogin, M. 1991. Ribosomal RNA sequences of Sarcocystis muris, Theileria annulata and Crypthecodinium cohnii reveal evolutionary relationships among apicomplexans, dinoflagellates, and ciliates. Mol. Biochem. Parasitol., 45:147–154. [DOI] [PubMed] [Google Scholar]
- Harper, J. T. , Waanders, E. & Keeling, P. J. 2005. On the monophyly of chromalveolates using a six‐protein phylogeny of eukaryotes. Int. J. Syst. Evol. Microbiol., 55:487–496. [DOI] [PubMed] [Google Scholar]
- Hiller, R. G. , Crossley, L. G. , Wrench, P. M. , Santucci, N. & Hofmann, E. 2001. The 15‐kDa forms of the apo‐peridinin‐chlorophyll a protein (PCP) in dinoflagellates show high identity with the apo‐32 kDa PCP forms, and have similar N‐terminal leaders and gene arrangements. Mol. Genet. Genomics, 266:254–259. [DOI] [PubMed] [Google Scholar]
- Kim, S. , Bachvaroff, T. R. , Handy, S. M. & Delwiche, C. F. 2011. Dynamics of actin evolution in dinoflagellates. Mol. Biol. Evol., 28:1469–1480. [DOI] [PubMed] [Google Scholar]
- LaJeunesse, T. C. , Lambert, G. , Andersen, R. A. , Coffroth, M. A. & Galbraith, D. W. 2005. Symbiodinium (Pyrrhophyta) genome sizes (DNA content) are smallest among dinoflagellates. J. Phycol., 41:880–886. [Google Scholar]
- Le, Q. H. , Markovic, P. , Hastings, J. W. , Jovine, R. V. & Morse, D. 1997. Structure and organization of the peridinin‐chlorophyll a‐binding protein gene in Gonyaulax polyedra . Mol. Gen. Genet., 255:595–604. [DOI] [PubMed] [Google Scholar]
- Lee, D. H. , Mittag, M. , Sczekan, S. , Morse, D. & Hastings, J. W. 1993. Molecular cloning and genomic organization of a gene for luciferin‐binding protein from the dinoflagellate Gonyaulax polyedra . J. Biol. Chem., 268:8842–8850. [PubMed] [Google Scholar]
- Li, L. & Hastings, J. W. 1998. The structure and organization of the luciferase gene in the photosynthetic dinoflagellate Gonyaulax polyedra . Plant Mol. Biol., 36:275–284. [DOI] [PubMed] [Google Scholar]
- Lidie, K. & van Dolah, F. 2007. Spliced leader RNA‐mediated trans‐splicing in a dinoflagellate, Karenia brevis . J. Eukaryot. Microbiol., 54:427–435. [DOI] [PubMed] [Google Scholar]
- Lin, S. , Zhang, H. & Jiao, N. 2006. Potential utility of mitochondrial cytochrome B and its mRNA editing in resolving closely related dinoflagellates: a case study of Prorocentrum (Dinophyceae). J. Phycol., 42:646–654. [Google Scholar]
- Logares, R. , Shalchian‐Tabrizi, K. , Boltovskoy, A. & Rengefors, K. 2007. Extensive dinoflagellate phylogenies indicate infrequent marine‐freshwater transitions. Mol. Phylogenet. Evol., 45:887–903. [DOI] [PubMed] [Google Scholar]
- Machabee, S. , Wall, L. & Morse, D. 1994. Expression and genomic organization of a dinoflagellate gene family. Plant Mol. Biol., 25:23–31. [DOI] [PubMed] [Google Scholar]
- McEwan, M. , Humayun, R. , Slamovits, C. H. & Keeling, P. J. 2008. Nuclear genome sequence survey of the dinoflagellate Heterocapsa triquetra . J. Eukaryot. Microbiol., 55:530–535. [DOI] [PubMed] [Google Scholar]
- Mount, S. M. , Burks, C. , Hertz, G. , Stormo, G. D. , White, O. & Fields, C. 1992. Splicing signals in Drosophila: intron size, information content, and consensus sequences. Nucleic Acids Res., 20:4255–4262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamoto, O. K. , Liu, L. , Robertson, D. L. & Hastings, J. W. 2001a. Members of a dinoflagellate luciferase gene family differ in synonymous substitution rates. Biochemistry, 40:15862–15868. [DOI] [PubMed] [Google Scholar]
- Okamoto, O. K. , Robertson, D. L. , Fagan, T. F. , Hastings, J. W. & Colepicolo, P. 2001b. Different regulatory mechanisms modulate the expression of a dinoflagellate iron‐superoxide dismutase. J. Biol. Chem., 276:19989–19993. [DOI] [PubMed] [Google Scholar]
- Parrow, M. , Elbrächter, M. , Krause, M. , Burkholder, J. , Deamer, N. , Htyte, N. & Allen, E. 2006. The taxonomy and growth of a Crypthecodinium species (Dinophyceae) isolated from a brackish‐water fish aquarium. Afr. J. Mar. Sci., 28:185–191. [Google Scholar]
- Rae, P. 1973. 5‐Hydroxymethyluracil in the DNA of a dinoflagellate. Proc. Natl Acad. Sci. USA, 70:1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reichman, J. R. , Wilcox, T. P. & Vize, P. D. 2003. PCP gene family in Symbiodinium from Hippopus hippopus: low levels of concerted evolution, isoform diversity, and spectral tuning of chromophores. Mol. Biol. Evol., 20:2143–2154. [DOI] [PubMed] [Google Scholar]
- Rill, R. L. , Livolant, F. , Aldrich, H. C. & Davidson, M. W. 1989. Electron microscopy of liquid crystalline DNA: direct evidence for cholesteric‐like organization of DNA in dinoflagellate chromosomes. Chromosoma, 98:280–286. [DOI] [PubMed] [Google Scholar]
- Rizzo, P. J. & Noodén, L. D. 1972. Chromosomal proteins in the dinoflagellate alga Gyrodinium cohnii . Sci. New Ser., 176:796–797. [DOI] [PubMed] [Google Scholar]
- Rowan, R. , Whitney, S. , Fowler, A. & Yellowlees, D. 1996. Rubisco in marine symbiotic dinoflagellates: form II enzymes in eukaryotic oxygenic phototrophs encoded by a nuclear multigene family. Plant Cell, 8:539–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saldarriaga, J. F. & Cavalier‐Smith, T. 2004. Molecular data and the evolutionary history of dinoflagellates. Eur. J. Protistol., 40:85–111. [Google Scholar]
- Saldarriaga, J. F. , Taylor, F. J. , Keeling, P. J. & Cavalier‐Smith, T. 2001. Dinoflagellate nuclear SSU rRNA phylogeny suggests multiple plastid losses and replacements. J. Mol. Evol., 53:204–213. [DOI] [PubMed] [Google Scholar]
- Sambrook, J. & Russell, D. W. 2001. Molecular Cloning, 3rd ed Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [Google Scholar]
- Sharples, F. P. , Wrench, P. M. , Ou, K. & Hiller, R. G. 1996. Two distinct forms of the peridinin‐chlorophyll a‐protein from Amphidinium carterae . Biochim. Biophys. Acta, 1276:117–123. [DOI] [PubMed] [Google Scholar]
- Shoguchi, E. , Shinzato, C. , Kawashima, T. , Gyoja, F. , Mungpakdee, S. , Koyanagi, R. , Takeuchi, T. , Hisata, K. , Tanaka, M. , Fujiwara, M. , Hamada, M. , Seidi, A. , Fujie, M. , Usami, T. , Goto, H. , Yamasaki, S. , Arakaki, N., Suzuki, Y. , Sugano, S. , Toyoda, A. , Kuroki, Y. , Fujiyama, A. , Medina, M. , Coffroth, M. A. , Bhattacharya, D. & Satoh, N. 2013. Draft assembly of the Symbiodinium minutum nuclear genome reveals dinoflagellate gene structure. Curr. Biol., 23:1399–1408. [DOI] [PubMed] [Google Scholar]
- Slamovits, C. H. & Keeling, P. J. 2008. Widespread recycling of processed cDNAs in dinoflagellates. Curr. Biol., 18:R550–R552. [DOI] [PubMed] [Google Scholar]
- Tarn, W. Y. & Steitz, J. A. 1997. Pre‐mRNA splicing: the discovery of a new spliceosome doubles the challenge. Trends Biochem. Sci., 22:132–137. [DOI] [PubMed] [Google Scholar]
- Thanaraj, T. A. & Clark, F. 2001. Human GC‐AG alternative intron isoforms with weak donor sites show enhanced consensus at acceptor exon positions. Nucleic Acids Res., 29:2581–2593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuttle, R. C. & Loeblich III, A. R. 1974. Genetic recombination in the dinoflagellate Crypthecodinium cohnii . Sci. New Ser., 185:1061–1062. [DOI] [PubMed] [Google Scholar]
- Wu, Q. & Krainer, A. R. 1999. AT‐AC pre‐mRNA splicing mechanisms and conservation of minor introns in voltage‐gated ion channel genes. Mol. Cell. Biol., 19:3225–3236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wynn, J. , Behrens, P. , Sundararajan, A. , Hansen, J. & Apt, K. 2005. Production of Single Cell Oils by Dinoflagellates. In: Cohen, Z. & Ratledge, C. (eds.), Single Cell Oils. AOCS Press, Urbana, IL: p. 86–98. [Google Scholar]
- Xue, L. , Lippmeier, J. C. , Bingham, S. & Apt, K. E . 1999. ESTs from the dinoflagellate Crypthecodinium cohnii. Poster session presented at: Plant Biology'99. American Society of Plant Biologists, Baltimore, MD. [Google Scholar]
- Yoshikawa, T. , Uchida, A. & Ishida, Y. 1996. There are 4 introns in the gene coding the DNA‐binding protein HCc of Crypthecodinium cohnii (Dinophyceae). Fisheries Sci., 62:204–209. [Google Scholar]
- Zhang, M. Q. 1998. Statistical features of human exons and their flanking regions. Hum. Mol. Genet., 7:919–932. [DOI] [PubMed] [Google Scholar]
- Zhang, H. , Bhattacharya, D. & Lin, S. 2005. Phylogeny of dinoflagellates based on mitochondrial Cytochrome b and nuclear small subunit rDNA sequence comparisons. J. Phycol., 41:411–420. [Google Scholar]
- Zhang, H. , Hou, Y. & Lin, S. 2006. Isolation and characterization of proliferating cell nuclear antigen from the dinoflagellate Pfiesteria piscicida . J. Eukaryot. Microbiol., 53:142–150. [DOI] [PubMed] [Google Scholar]
- Zhang, H. , Hou, Y. , Miranda, L. , Campbell, D. A. , Sturm, N. R. , Gaasterland, T. & Lin, S. 2007. Spliced leader RNA trans‐splicing in dinoflagellates. Proc. Natl Acad. Sci. USA, 104:4618–4623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, H. & Lin, S. 2003. Complex gene structure of the form ii rubisco in the dinoflagellate Prorocentrum minimum (dinophyceae). J. Phycol., 39:1160–1171. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Alignments and mean pairwise identity graphs for the intergenic spacers between alcohol dehydrogenase (ADH) members and the nine introns of the five members of ADH. Length and pairwise identity is displayed to the right of each alignment. Gene sequences are indicated by black bars, and indels are indicated by intervening black lines. Each column of the alignment has a bar graph above it indicating the mean pairwise identity of all pairs in the alignment. The height and color of each bar in the graph is proportional to the mean pairwise identity of all pairs in that column ‐ green at 100%, yellow between 30 and less than 100%, and red when less than 30%.