

Abstract
Whole genome duplication (polyploidization) is a mechanism of “instantaneous” species formation that has played a major role in the evolutionary history of plants. Much of what we know about the early evolution of polyploids is based upon studies of a handful of recently formed species. A new polyploid hybrid (allopolyploid) species Mimulus peregrinus, formed within the last 140 years, was recently discovered on the Scottish mainland and corroborated by chromosome counts. Here, using targeted, high‐depth sequencing of 1200 genic regions, we confirm the parental origins of this new species from M. x robertsii, a sterile triploid hybrid between the two introduced species M. guttatus and M. luteus that are naturalized and widespread in the United Kingdom. We also report a new population of M. peregrinus on the Orkney Islands and demonstrate that populations on the Scottish mainland and Orkney Islands arose independently via genome duplication from local populations of M. x robertsii. Our data raise the possibility that some alleles are already being lost in the evolving M. peregrinus genomes. The recent origins of a new species of the ecological model genus Mimulus via allopolyploidization provide a powerful opportunity to explore the early stages of hybridization and genome duplication in naturally evolved lineages.
Keywords: Hybridization, invasive species, plant speciation, polyploidy, whole genome duplication
Whole genome duplication, or polyploidization, has been linked to major evolutionary transitions in the history of land plants including the diversification of angiosperms (Levin 1983; Cui et al. 2006; Soltis et al. 2014). Approximately 35% of angiosperm species are recent polyploids, and 15% of speciation events in angiosperms are directly associated with genome duplication (Wood et al. 2009). Polyploidization is often correlated with hybridization events (Abbott et al. 2013), and the combination of hybridization and polyploidy (allopolyploidy) is thought to contribute to the rapid evolution of genome structure and ecological novelty (Song et al. 1995; Wright et al. 1998; Osborn et al. 2003; Buggs et al. 2012a; Chao et al. 2013; Soltis et al. 2014). However, our ability to study the contribution of hybridization and polyploidy to evolutionary change in established allopolyploids is limited as it is not easy to distinguish the changes happening at the moment of hybridization and polyploidization from those accumulating after subsequent evolution. Recently formed allopolyploid species provide opportunities to investigate the earliest stages of speciation by genome duplication. Examples of recently formed allopolyploid species (< 200 years old) include only a handful of species in four families: Senecio cambrensis, S. eboracensis (Abbott and Lowe 2004), Tragopogon mirus and T. miscellus (Soltis et al. 2004) in the Asteraceae, Cardamine x insueta and C. x schulzii in the Brassicaceae (Mandakova et al. 2013), Spartina anglica in the Poaceae (Ainouche et al. 2004), and Salsola ryanii in the Amaranthaceae (Ayres et al. 2009). Generalizations of the evolutionary changes observed during polyploid speciation clearly will benefit from the study of additional young allopolyploids.
The recent discovery of an allopolyploid species of monkeyflower, Mimulus peregrinus (Phrymaceae), formed in the last 140 years provides an exciting opportunity to study the early stages of speciation by genome duplication (Vallejo‐Marín 2012). The monkeyflower genus, Mimulus L. (Phrymaceae), comprises approximately 120 species of herbaceous plants with centers of diversity in North America where the majority of species occur, and also the Andes, the Himalayas, and Australia (Grant 1924; Beardsley and Olmstead 2002; Wu et al. 2007). Mimulus has served as a model to study ecology, evolution, and speciation in plants for more than 50 years (Vickery 1959; Bradshaw and Schemske 2003; Wu et al. 2007; Lowry and Willis 2010), but only recently have studies begun investigating the ecological and evolutionary dynamics of Mimulus species in their nonnative range (van Kleunen 2007; Murren et al. 2009; Puzey and Vallejo‐Marín 2014).
Monkeyflowers were introduced into Europe from the Americas in the early 1800s, and detailed accounts of the history of colonization of this genus in Europe are provided elsewhere (Tokarska‐Guzik and Dajdok 2010; Vallejo‐Marín 2012; Puzey and Vallejo‐Marín 2014). The two most successful colonization events involved the North American taxon Mimulus guttatus, which was introduced into Europe in 1812, closely followed by taxa in the South American species‐aggregate M. luteus (Vallejo‐Marín and Lye 2013). In their native range, M. guttatus has a variable mating system from high to intermediate levels of outcrossing (e.g., Willis 1993), and M. luteus is also likely to be partially to predominantly outcrossed (Cooley et al. 2008). In the United Kingdom, M. guttatus is naturalized and widespread along the banks of streams and rivers, in water‐logged ground and other wet places (Preston et al. 2002; Truscott et al. 2008). Mimulus luteus inhabits similar environments, and also became naturalized and widespread soon after its introduction to the United Kingdom (BSBI 2013). However, M. luteus is currently much rarer than M. guttatus (Vallejo‐Marín and Lye 2013). The mating system of introduced Mimulus is unknown. Mimulus guttatus and M. luteus are closely related, belonging to the section Simiolus (Grant 1924), but, importantly, differ in ploidy level: while M. guttatus occurs mostly as a diploid (2n = 2x = 28), taxa in the M. luteus group are tetraploid (2n = 4x = 60–62; Vickery 1995). In the United Kingdom, M. guttatus and M. luteus hybridize to produce a sexually sterile, but vegetatively vigorous triploid taxon, M. x robertsii (2n = 3x = 44–46; Roberts 1964; Parker 1975; Silverside 1990). This sterile hybrid is found in approximately 40% of extant Mimulus populations throughout the United Kingdom, and can form very large populations of thousands of ramets thanks to its ability to reproduce from even small plant fragments (Vallejo‐Marín and Lye 2013).
Mimulus peregrinus is a newly described taxon recently found in mainland Scotland. It shares the morphological characteristics of the M. x robertsii triploid hybrids, but possesses twice the genome size and number of chromosomes (2n = 6x = 92), and is characterized by high (>90%) pollen fertility (Vallejo‐Marín 2012). Based on these phenotypic traits, and the fact that triploid hybrids have long been known to give rise to fertile species via whole genome duplication (e.g., Ainouche et al. 2004; Hegarty et al. 2012), it appears that M. peregrinus has arisen through allopolyploidization between introduced populations of M. guttatus and M. luteus (Vallejo‐Marín 2012). In addition to the originally described (Vallejo‐Marín 2012) population of M. peregrinus in mainland Scotland (Leadhills, South Lanarkshire), polyploid individuals of similar morphology have been found in a separate stream nearby (5 km away), and also 400 km to the north of the type location, on the Orkney Islands, which are separated from the Scottish mainland by 16 km of ocean. Populations of M. x robertsii are also present on Orkney. Given molecular evidence suggesting that multiple origins of several neoallopolyploid species (Soltis and Soltis 1999; Soltis et al. 2009) such as S. cambrensis in the United Kingdom (Ashton and Abbott 1992), and Tragopogon miscellus and T. mirus in Idaho and Washington (Symonds et al. 2010), we hypothesized that M. peregrinus populations on mainland Scotland and Orkney had independent origins. A genome‐level study has the potential to rigorously test these hypotheses on the identity and repeated origins of M. peregrinus.
Here, we use high‐depth targeted sequencing of 1200 genic regions to characterize the genomic composition of M. peregrinus individuals from two populations in Southern Scotland and the newly discovered population in the Orkney Islands. We compare these with the same regions sequenced at high coverage in the parental taxa, M. guttatus and M. luteus s. l., and in the M. x robertsii triploid hybrids. We address two specific questions: (1) Is the genome composition of M. peregrinus consistent with recent allopolyploidization from M. guttatus and M. luteus via M. x robertsii? (2) Has M. peregrinus originated multiple times?
Materials and Methods
SAMPLING
To elucidate the relationships between M. peregrinus and its putative parental taxa, we sampled four individuals of each of M. guttatus, M. luteus, M. x robertsii, and M. peregrinus, identified on the basis of morphology and genome size measured using flow cytometry (Table 1, Fig. 1A). For M. guttatus, we sampled four individuals from natural populations in the United Kingdom. For M. luteus, we included the only two extant naturalized populations of this taxon that we could find in the United Kingdom, plus two advanced generation inbred lines, M. luteus var. luteus and M. luteus var. variegatus, both of which are field‐collected varieties originating from Chile (Cooley and Willis 2009), and hypothesized to have contributed to naturalized populations of M. luteus in the United Kingdom (Stace 2010; Vallejo‐Marín 2012). Other related taxa such as M. naiandinus and M. cupreus are not naturalized in the United Kingdom (Stace 2010). For M. peregrinus, we obtained samples from two populations LED (sampled twice, Table 1) and GON occurring in close proximity to each other (∼5 km) but on different streams in South Lanarkshire, and another population (STR) found approximately 400 km to the North, near Stromness on the Orkney Islands. Both populations, LED and GON, also contain individuals of M. x robertsii which we sampled. The STR population is composed only of M. peregrinus so we sampled the nearest M. x robertsii population, 7 km distant, near Maeshowe. We sampled a further M. x robertsii population from Nenthall, England, which is far from all known M. peregrinus populations.
Table 1.
Summary characteristics of 16 individuals from four Mimulus taxa sampled for the targeted genome re‐sequencing analysis
| Expected | ||||||||
|---|---|---|---|---|---|---|---|---|
| genomic | Percentage | Mean | Fraction | |||||
| Sample ID | Population | Location | Latitude | Longitude | composition | of mapped | depth | heterozygous |
| M. guttatus (2x) | ||||||||
| gut‐1 | DBL | Dunblane, Scotland | 56.187 | −3.965 | GG | 94.5 | 65 | 0.018 |
| gut‐2 | AYR | Ayr, Scotland | 55.461 | −4.625 | GG | 94.3 | 74 | 0.019 |
| gut‐3 | CER | Cerrigydrudion, Wales | 53.006 | −3.549 | GG | 94 | 83 | 0.021 |
| gut‐4 | HOU | Houghton Lodge, England | 51.097 | −1.508 | GG | 93.8 | 111 | 0.018 |
| M. luteus (4x) | ||||||||
| lut‐1 | COL | Coldstream, Scotland | 55.655 | −2.240 | LLLL | 88.2 | 90 | 0.047 |
| lut‐2 | EY | El Yeso, Chile | −33.4 | −70.0 | LLLL | 87.9 | 96 | 0.044 |
| lut‐3 | EVI | Evie, Orkney, Scotland | 59.112 | −3.108 | LLLL | 88.6 | 74 | 0.051 |
| lut‐4 | RC | Río Cipreses, Chile | −34.2 | −70.3 | LLLL | 87.5 | 90 | 0.045 |
| M. x robertsii (3x) | ||||||||
| rob‐1 | TOR | Tormiston, Orkney, Scotland | 58.996 | −3.183 | GLL | 89.3 | 112 | 0.067 |
| rob‐2 | LED | Leadhills, Scotland | 55.424 | −3.735 | GLL | 90.1 | 98 | 0.065 |
| rob‐3 | NEN | Nenthall, England | 54.806 | −2.376 | GLL | 89.8 | 106 | 0.066 |
| rob‐4 | GON | Glen Gonnar, Scotland | 55.467 | −3.738 | GLL | 89.7 | 102 | 0.066 |
| M. peregrinus (6x) | ||||||||
| per‐1 | LED | Leadhills, Scotland | 55.423 | −3.735 | GGLLLL | 88.9 | 90 | 0.064 |
| per‐2 | LED | Leadhills, Scotland | 55.424 | −3.735 | GGLLLL | 89.8 | 117 | 0.067 |
| per‐3 | STR | Stromness, Orkney, Scotland | 58.969 | −3.283 | GGLLLL | 90.8 | 82 | 0.064 |
| per‐4 | GON | Glen Gonnar, Scotland | 55.467 | 3.738 | GGLLLL | 90.3 | 109 | 0.067 |
Taxon names are followed by estimates of ploidy level obtained using flow cytometry and/or chromosome counts. For chromosome counts and details on taxon identification, see Vallejo‐Marín (2012). Latitude and Longitude are expressed in decimal degrees in the WGS84 coordinate system. Expected genomic composition indicates the number of haploid complements expected from each of the putative parental taxa: M. guttatus (G) and M. luteus (L). Percentage of mapped: percent of total number reads mapped; mean depth: average read depth of genotyped sites; fraction heterozygous: fraction of heterozygous sites. Values for number sites, mean depth, and fraction het, were calculated from sites genotyped in all 16 individuals when running GATK in polyploidy mode allowing up to four alternate alleles.
Figure 1.
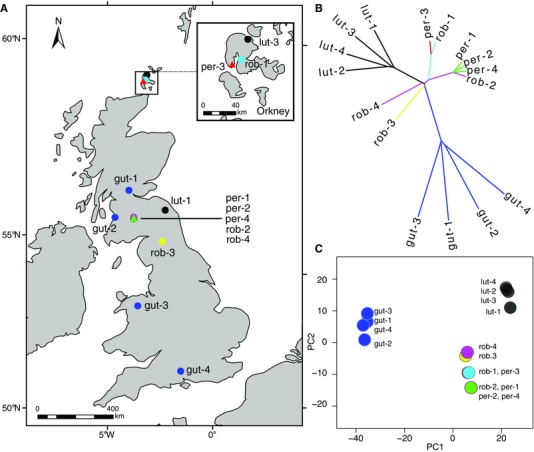
Geographic location and genomic relationships between 16 individuals of Mimulus spp. in each of four taxa: M. guttatus (gut), M. luteus (lut), M. x robertsii (rob), and M. peregrinus (per). (A) Map of the United Kingdom showing the geographic location of the samples analyzed here. Two individuals of M. luteus (lut‐2 and lut‐4) were collected in Chile and are not shown in this map. (B) Neighbor joining tree showing monophyletic clades for the parental taxa (M. guttatus and M. luteus), and clustering of the allopolyploid M. peregrinus with geographically proximate populations of M. x robertsii. Bootstrap support for all nodes is shown in Figure S3. (C) Principal component analysis (PCA) plot showing clustering of individuals along the first two principle components. Mimulus peregrinus and M. robertsii fall between parental species, M. guttatus and M. luteus and next to geographically proximate M. x robertsii.
SELECTION AND SEQUENCING OF SEQUENCE CAPTURE BAITS
To carry out targeted genomic sequencing, we designed probes using the M. guttatus genome version 1.1 (www.phytozome.net, based on a North American accession of M. guttatus). We randomly chose coding genic and untranslated regions in a ratio of 3:1 for a total of 1198 regions of 450 bp each (540 kbp total) across the whole genome. For these target regions, we designed Agilent SureSelect RNA baits using the SureDesign software (Agilent Sure Select XT Enrichment Protocol version 1.6; Agilent Technologies, Stockport, Cheshire, UK). The final design yielded 20,385 probes with a total size of 488,076 bp. We extracted DNA from dried leaf tissue using DNeasy Plant Mini Kits (Qiagen, Crawley, West Sussex, UK) and used 1.5–7.9 μg of DNA for hybridization with the SureSelect RNA baits. The captured DNA was sequenced in a 100 bp pair‐end run in a MiSeq desktop sequencer (Illumina, Little Chesterford, Essex, UK) at the NERC Biomolecular Analysis Facility‐Edinburgh (Genepool, Edinburgh, UK). Fastq files were obtained using the Illumina pipeline CASAVA version 1.8.3.
ANALYSES
Read alignment and genotyping
We aligned raw reads to the newest available version of the M. guttatus genome (version 2.0; www.phytozome.net) using bowtie2. Following initial alignment and conversion to binary format (SAMtools, Li et al. 2009), we used Picard‐tools (http://broadinstitute.github.io/picard) to validate mates, sort alignments, remove duplicates, and add read groups. After Picard‐tool filtering, we used the Genome Analysis Toolkit Unified Genotyper to call genotyping with base quality (BQ ≥ 25) and mapping quality (MQ ≥ 25) filters in place. After genotyping, we discarded indels and bases with genotype quality scores less than 30. Sites with two or more alternate alleles (as determined using GATK in polyploidy mode with four alternate alleles allowed) were discarded. Detailed commands and pipelines are provided in the Supplementary Information.
To map M. guttatus probes (designed with version 1 of the genome) to version 2 locations, we used blastn (NCBI C++ Toolkit). From all designed probes, 64% (772/1198) mapped uniquely to the version 2 genome, 7.7% probes (92/1198) had two hits with identity greater than 90%, and 13% (156/1198) mapped twice or more throughout the genome with identity greater than 72%. Figure S1 provides a genome‐wide plot showing the location of the probes. Only probes with one unique and perfect hit in the version 2 genome were used for subsequent analyses.
Genomic relationships between individuals
To reconstruct the relationships among the 16 sampled individuals, variable loci were selected from the sequenced reads. Heterozygotes were defined as those sites in which the frequency of the reference allele was between 0.1 and 0.90. Only SNPs (Single Nucleotide Polymorphisms) with a read depth of at least 50×, and which were amplified in all 16 individuals and polymorphic in at least one individual were retained for this analysis. Our genotype calling did not distinguish between different allele dosage types of heterozygotes in polyploid individuals (e.g., guttatus‐guttatus‐luteus vs. guttatus‐luteus‐luteus), but enabled us to directly compare presence/absence of alleles across taxa.
We used allele presence/absence calls at 20,749 SNP loci that were biallelic within our samples to calculate a pairwise Nei's genetic distance matrix among all individuals in the software adegenet (Jombart and Ahmed 2011) in R (R Development Core Team 2014), and used these data to generate a neighbor joining (NJ) tree in ape (Paradis et al. 2004). We calculated bootstrap values for the nodes in the NJ tree using 1000 replicates. For comparison, we also used these calls to conduct a principal component analysis (PCA) using the function glPca in adegenet. As PCA is a simple method to reduce dimensionality of multivariate data, it does not assume a population genetic or inheritance model, and collapses correlated information in linked SNP loci, providing a comparison for our NJ analysis with few assumptions.
Estimating allele dosage using loci with reciprocally fixed alleles in M. guttatus and M. luteus
We identified loci that are fixed for alternative alleles in M. guttatus and M. luteus using biallelic SNPs with a read depth of at least 50× sequenced in all 16 individuals. Reads from M. x robertsii and M. peregrinus that contain these SNPs can therefore be characterized according to which taxon they were derived from. For each locus/individual combination in M. x robertsii and M. peregrinus, we calculated the ratio of “guttatus” alleles to “luteus” alleles by counting reads for each allele at each locus. To ascertain whether these results might be influenced by hybridization bias by the probes for sequences from the reference genome that the probes were designed from (a North American M. guttatus), we analyzed separately the SNP loci in which our “luteus” allele is the same as the allele in the reference genome (and consequently our “guttatus” allele is the variant relative to the reference genome), and those loci in which our “luteus” allele is different from the allele in the reference genome (and hence our guttatus allele is the same as the reference genome).
Allelic variants unique to M. x robertsii or M. peregrinus
We searched for alleles that were unique to either M. x robertsii or M. peregrinus, that is, not present in either M. guttatus or M. luteus. To do this, we focused on biallelic SNP loci with a read depth of at least 50×, and which were sequenced in all 16 individuals. We looked for loci in which a given allele was observed in any individual of M. x robertsii or M. peregrinus at a frequency of at least q > 0.167 (the frequency of one out of six allele copies in a hexaploid), but which was not detected in any individual of M. guttatus and M. luteus (i.e., the frequency of this allele was less than q < 0.1 in the putative parental taxa).
Allelic variants differentiating M. peregrinus from M. x robertsii
We then searched for SNP alleles that are found in M. peregrinus but not in M. x robertsii, again focusing on biallelic SNP loci with a read depth of at least 50×, and sequenced in all 16 individuals. Among these, we selected sites that were fixed in all individuals of M. x robertsii (q > 0.9), and in which reads containing the alternative allele were found in any individual of M. peregrinus at a frequency of at least 1 − q ≥ 0.167 (i.e., equal or higher than the frequency of one out of six allele copies in a hexaploid). Finally, we looked for alleles that are found in M. x robertsii but not M. peregrinus. We searched for heterozygote sites (0.167 < q < 0.833) in M. x robertsii, and selected those that occurred as homozygotes in M. peregrinus using a slightly more conservative threshold (0.1 > q > 0.9).
Results
The average sequencing depth at probed regions ranged from 113 to 202× per individual, with an overall average of 164× coverage (Table 1). Individuals from M. guttatus had slightly more aligned reads (94%) than other taxa (M. luteus: 88%, M. robertsii: 90%, and M. peregrinus: 90%; Table 1). On a per individual basis, the number of sites within probes confidently genotyped ranged from 338166 to 347182 with an average of 334419 sites (Table 1).
GENOME‐WIDE HETEROZYGOSITY
Observed levels of genome‐wide heterozygosity varied extensively among the four taxa as indicated by the fraction of heterozygous sites (number of heterozygote sites/total sites genotyped; Table 1), and illustrated in Figure S2 using a subset of 20,749 SNPs with read depth >50× and genotyped in all individuals. Individuals of M. guttatus collected in naturalized populations in the United Kingdom had the lowest fraction of heterozygous sites, ranging from 0.018 to 0.021 (mean = 0.019 ± 0.001; mean ± SD; Table 1). This level of heterozygosity is consistent with a previous study of 10 U.K. populations of M. guttatus, which showed that the overall genome‐wide pairwise nucleotide diversity was π = 0.015 (Puzey and Vallejo‐Marín 2014). In contrast, individuals of the tetraploid M. luteus had over twice as many heterozygous bases (mean = 0.047 ± 0.003). This high level of heterozygosity was observed in both wild‐collected individuals of M. luteus in the United Kingdom (lut‐1 and lut‐3; fraction heterozygous sites = 0.047 and 0.051, respectively), as well as in two individuals from fifth generation inbred lines: M. luteus var. luteus (lut‐2; fraction heterozygous sites = 0.044) and M. luteus var. variegatus (lut‐4; fraction heterozygous sites = 0.0458; Table 1; Fig. S2). However, the highest levels of heterozygosity were observed in individuals of M. x robertsii and M. peregrinus (0.066 ± 0.001 and 0.065 ± 0.002, respectively; Table 1; Fig. S2), as would be expected if they are derived from a recent hybridization event between two distinct taxa.
RELATIONSHIPS BETWEEN TAXA
The NJ analysis of the SNP data showed that M. guttatus and M. luteus form monophyletic clades clearly separated by relatively deep branches with very strong bootstrap support (Figs. 1B, S3). This analysis also supports a close association between M. x robertsii and M. peregrinus. The three individuals of M. peregrinus sampled from Southern Scotland (per‐1, per‐2, and per‐4; LED and GON populations) are most closely associated with M. x robertsii from the same geographic area (rob‐2; LED; Fig. 1B). Interestingly, although per‐4 (GON) co‐occurs with M. x robertsii (rob‐4; GON), it is actually more closely related to individuals from the Leadhills population located 5 km away (LED; per‐1, per‐2, and rob‐2; 100% bootstrap support for this node; Figs. 1B, S3). Furthermore, the M. peregrinus individual sampled in the Orkney Islands (per‐3; STR population) is most closely related to M. x robertsii from the same island (rob‐1; TOR population; Table 1, Fig. 1B).
The PCA grouped the populations in a manner similar to the relationships suggested by the NJ tree (Fig. 1C). The first principal component (PC) separated M. guttatus, M. luteus, and the hybrids (M. x robertsii and M. peregrinus), whereas the second PC provided some resolution within taxa. Importantly, the PCA shows that M. robertsii and M. peregrinus fall between M. guttatus and M. luteus, and indicates that M. peregrinus from each of the two main regions in Southern Scotland and Orkney are most closely associated to geographically proximate M. x robertsii (Fig. 1C).
GENOMIC COMPOSITION OF M. X ROBERTSII AND M. PEREGRINUS
In our sample of 20,749 SNP loci sequenced in all individuals, 93.78% loci only contained alleles in M. x robertsii and M. peregrinus that were also observed in M. guttatus and/or M. luteus, consistent with the hypothesis that M. guttatus and M. luteus are the parental species. To explore the contributions of these two species to the genomes of M. x robertsii and M. peregrinus, we identified 881 sites that were fixed for alternative alleles in all individuals of M. guttatus and M. luteus analyzed. These 881 informative sites were distributed in 370 different probes and included all 14 major linkage groups of the reference genome (Fig. S4). Figure 2 shows the distribution of read depth across these SNP loci across all individuals of M. x robertsii and M. peregrinus. The average frequency of the M. guttatus alleles estimated at each SNP loci separately was 0.418 ± 0.155 (mean ± SD) in M. x robertsii, and 0.420 ± 0.161 in M. peregrinus. The average frequency of the M. guttatus alleles when calculated as the mean frequency in each probe was 0.415 ± 0.076 and 0.413 ± 0.074 in M. x robertsii and M. peregrinus, respectively. If the tetraploid species M. luteus contributed twice as many genomes as the diploid M. guttatus to M. x robertsii, and M. peregrinus, the expected frequency of M. guttatus alleles in both M. x robertsii and M. peregrinus is 1/3. In this case, our results seem to indicate a slight overall deficit of luteus‐like reads (or an excess of guttatus‐like reads; Fig 2A).
Figure 2.
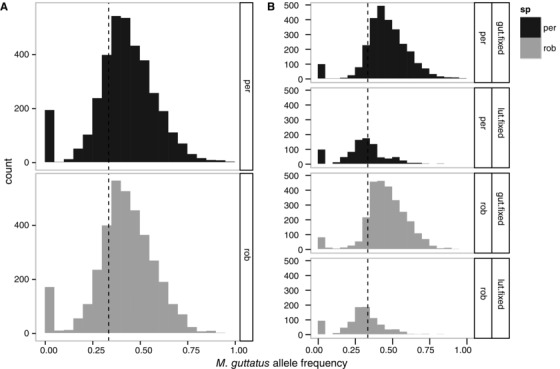
Histograms showing combined read depth frequency (allelic dosage) in four individuals of M. peregrinus (per) and four of M. x robertsii (rob) for SNP alleles that are fixed for alternative variants in the parental taxa M. guttatus and M. luteus. Only biallelic SNPs with a read depth of >50× and genotyped in all individuals were included in this analysis. The allelic frequency depicted in the x‐axis is for the allele coming from the diploid parent (M. guttatus). The dotted line indicates 1:2 ratio, expected in a hybrid product of the combination of diploid and tetraploid parental genomes. (A) All loci that are alternatively fixed in M. guttatus and M. luteus. (B) The same data but separated by whether SNPs alleles from M. guttatus are fixed for the reference allele and M. luteus are fixed for the alternate allele (gut‐fixed); or whether SNP alleles are fixed in M. luteus for the reference allele and M. guttatus is fixed for the alternate allele (lut‐fixed). Histogram bin size = 0.1.
To test whether a hybridization bias by our probes might be contributing to the apparent deficit of luteus‐like alleles in M. x robertsii and M. peregrinus, we analyzed separately the SNP loci in which the “guttatus” allele was the same as the allele in the reference genome and those loci in which the “guttatus” allele was different from the allele in the reference genome (Fig. 2B). We found that for loci in which the “guttatus” allele did not match the reference genome (n = 212 SNP loci in 118 probes), the allelic frequency of the guttatus‐like allele was 0.295 ± 0.136 and 0.300 ± 0.146 in M. x robertsii and M. peregrinus, respectively. In contrast, for loci in which the “guttatus” allele matched the reference genome (n = 669 SNP loci in 317 probes), the allelic frequency of the guttatus‐like allele was 0.458 ± 0.140 and 0.458 ± 0.147 for M. x robertsii and M. peregrinus, respectively. This finding suggests that there was a hybridization bias in our probes for alleles matching our reference genome; in general this inflated the observed frequency of guttatus‐like reads (Fig. 2).
Due to the bias in our quantitative measures of allelic dosage, we focused our analysis of genome evolution during hybridization and polyploidization on clear cases of allelic loss (i.e., when one of the parental subgenomes was completely lost). We used the 889 sites (see above) where M. guttatus and M. luteus were fixed for different alleles in our samples (Fig. 3). For these sites, we found 110 were homozygous in at least one of the four individuals of M. x robertsii, and 87 were homozygous in at least one of the four M. peregrinus individuals (Fig. 3; Table S1). These sites were distributed in 54 (M. x robertsii) and 45 (M. peregrinus) probed regions on all the major linkage groups, except LG12 (Table S1). The majority of these sites were homozygous for the luteus‐like allele, that is, they were missing the guttatus‐like allele (something unlikely to be due to hybridization bias). In M. x robertsii, only one site (in LG5) was homozygous for the guttatus‐like allele, whereas 109 (in 53 probed regions in 13 LGs) were homozygous for the luteus‐like allele (Table S1). In M. peregrinus, three sites in two probes (in LG2 and LG14) were homozygous for the guttatus‐like allele, and 84 sites in 43 probes (in 13 LGs) were homozygous for the luteus‐like allele. The number and identity of these homozygous loci varied among individuals within taxon, with each individual having between 32 and 55 homozygous sites (in 15–29 probes; 7–11 LGs) in M. x robertsii, and between 48 and 57 sites (in 25–30 probes; 10–11 LGs) in M. peregrinus (Table S1). Therefore, from the 881 informative SNP sites we identified across the genome, between 3.6 and 6.5% are missing one of the expected parental alleles in any one individual of M. x robertsii or M. peregrinus.
Figure 3.
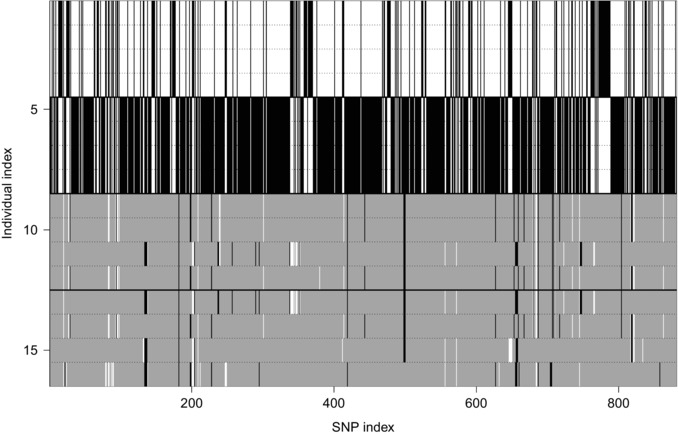
Genotype plot of 881 biallelic SNPs that are fixed for alternate alleles in M. guttatus and M. luteus. Only SNPs with a read depth of >50× and genotyped in all individuals were included. Color depicts the genotype at a specific SNP locus (columns), with white and black designating homozygosity, and gray indicating heterozygosity. Each row represents a single individual of M. guttatus (rows 1–4; gut‐1 to gut‐4), M. luteus (rows 5–8; lut‐1 to lut‐4), M. peregrinus (rows 9–12; per‐1 to per‐4), or M. x robertsii (rows 13–16; rob‐1 to rob‐4). The four taxa are separated by thick horizontal lines. The x‐axis indicates the relative order of SNPs along linkage groups. Notice the very high level of fixed heterozygosity in the SNP genotypes of M. x robertsii and M. peregrinus.
ALLELE GAIN AND LOSS BETWEEN M. X ROBERTSII AND M. PEREGRINUS
By comparing allelic composition between M. x robertsii and M. peregrinus, we were able to search for changes in read frequency that could reflect gain and loss of individual alleles as well as entire sections of the subgenomes. For instance, if M. x robertsii is homozygous at a given SNP site, the presence of a heterozygote at that SNP site in M. peregrinus could indicate an allele gain in any one of the duplicated chromosomes. Similarly, a heterozygous SNP site in M. x robertsii that is observed as homozygous in M. peregrinus could represent a case of allele loss in the allopolyploid. Our search for allelic variants in any M. peregrinus individual that were not present in any of the four M. x robertsii analyzed here uncovered nine such loci (0.119%, 9/7535 sites; Table S2). We then made this comparison between locally paired taxa of M. peregrinus and M. robertsii. In the Orkney pair (rob‐1 and per‐3), we found 25 loci with alleles in M. peregrinus that were absent in the local M. x robertsii (out of 10,612 potentially informative loci). The average frequency of the putatively new alleles in M. peregrinus was 0.229 ± 0.125 (mean ± SD). In a similar comparison in Leadhills (rob‐2 and per‐1), we found 21 loci in M. peregrinus with an allele not seen in the corresponding M. x robertsii. The average frequency of the putatively new alleles was 0.186 ± 0.019. The values for the comparison for a second LED pair (rob‐2 and per‐2) were 16 loci with putatively new alleles (out of 10,611), with an average frequency of the alleles of 0.182 ± 0.013.
The analysis of allele absence in M. peregrinus compared to M. x robertsii revealed that 40 of the 6434 sites that were heterozygous in all individuals of M. x robertsii, were observed as homozygotes (0.1 < q > 0.9) in at least one individual of M. peregrinus (Table S2). The majority of these sites (24/40) were located in a single linkage group (LG14) of the M. peregrinus individual from Orkney (per‐3), and these sites were not shared with the other three allopolyploid individuals (Fig. 4; Table S2). The remaining 18 homozygous sites were observed across M. peregrinus individuals and linkage groups (Table S2).
Figure 4.
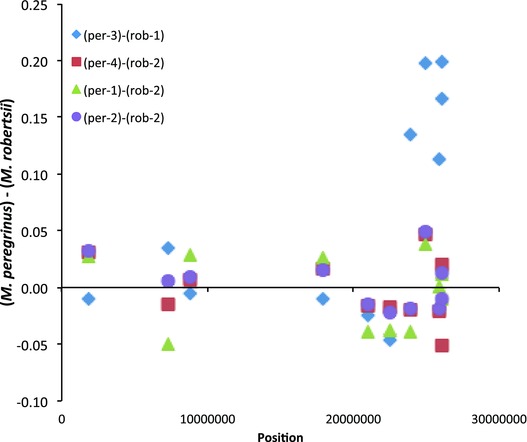
Pairwise difference in allele frequency between individuals of M. peregrinus (per‐1 to per‐4) and their closest M. x robertsii relative (rob‐1 or rob‐2) at linkage group 14 (LG14). The pairwise allele frequency difference was calculated using a 10 SNP moving average. The horizontal dashed line indicates no allele frequency difference. The x‐axis denotes the position of the informative SNP loci along LG14 in number of base pairs. The individual of M. peregrinus from Orkney (per‐3) shows a large allele frequency difference toward one end of LG14.
Discussion
PARENTAGE OF M. PEREGRINUS
Our study represents the first analysis of genome composition in the polyploid Scottish monkeyflower, M. peregrinus. Across genome‐wide loci, we found M. peregrinus genomes to be highly similar to the genomes of individuals of the widespread sterile triploid hybrid M. x robertsii. Almost all the alleles present in M. peregrinus and M. x robertsii were found in our sampled individuals of the diploid species M. guttatus and the tetraploid species M. luteus. We found higher levels of heterozygosity per base genotyped in M. peregrinus and M. x robertsii than in M. guttatus and M. luteus. For those loci that were fixed for different alleles in M. guttatus and M. luteus parent, we found that 94.3% showed fixed heterozygosity in M. peregrinus and 94.9% in M. x robertsii (Fig. 3). Thus, our results corroborate the origin of M. peregrinus via whole genome duplication of M. x robertsii, itself derived via hybridization from M. guttatus and the tetraploid species M. luteus. Given that M. guttatus and M. luteus were introduced into the United Kingdom from the American continent in the 19th century (Roberts 1964; Vallejo‐Marín 2012; Vallejo‐Marín and Lye 2013), M. peregrinus is one of a small number of known species (see Introduction) that have arisen via allopolyploid speciation in the last 200 years, and provides the first such study system in the order Lamiales.
Although the triploid M. x robertsii suffers from acute sexual sterility (Roberts 1964; Parker 1975), it is not an evolutionary dead‐end. Its ability to propagate via clonal reproduction has allowed this taxon to become part of the ecological landscape of riparian habitats in the United Kingdom (Vallejo‐Marín and Lye 2013), and from it the fertile allopolyploid M. peregrinus has emerged. Triploid taxa may give rise to polyploids through a variety of mechanisms including somatic doubling or via mating of unreduced gametes (Mable 2003; Mason and Pires 2015). Although these are improbable events, the ecological success of the triploid hybrid M. x robertsii as clonal populations seems to have allowed this probabilistic barrier to have been overcome, and a new allopolyploid to have formed. Other recently formed allopolyploids seem to have emerged from a similar route: for example, the allohexaploid S. cambrensis emerged from triploid hybrid, S. x baxteri whose parental species are S. vulgaris (tetraploid) and S. squalidus (diploid; Hegarty et al. 2005). The allopolyploid S. anglica (2n = 120–124) formed in around 1890 from S. x townsendii which is a sterile hybrid of S. maritima (2n = 60) and S. alterniflora (2n = 62; Baumel et al. 2002).
MULTIPLE ORIGINS OF THE ALLOPOLYPLOID
Our data show that M. peregrinus has formed at least twice, independently, from local populations of M. x robertsii, both on the Scottish mainland and on the Orkney Islands. Populations of M. peregrinus are very similar in the genic regions that we sequenced to their local populations of M. x robertsii, providing strong evidence for local origins from nearby populations of the sterile hybrid. The large distance between the two areas currently known to contain M. peregrinus, including 16 km of ocean, make a very strong cumulative case for the independent origins of this allopolyploid. Multiple origins of allopolyploids have been found in other recently formed allopolyploid species (Soltis and Soltis 1999; Soltis et al. 2009) including T. mirus and T. miscellus (Symonds et al. 2010) and S. cambrensis (Ashton and Abbott 1992).
It is likely that M. peregrinus is underreported in the field as many of the diagnostic characteristics that differentiate it from other Mimulus taxa in the United Kingdom, such as pollen and stomata size (Vallejo‐Marín 2012), can only be assessed in laboratory settings. Moreover, the presence of viable pollen and floral characteristics that are superficially similar to some forms of M. guttatus, represent a challenge to recognize M. peregrinus as a hybrid taxon (Silverside 1998; Stace 2010). The widespread distribution of its parental taxon, M. x robertsii, throughout the British Isles (Preston et al. 2002) may provide ample opportunities for additional origins of M. peregrinus, but this remains to be established in future field surveys.
POLYPLOIDIZATION IN MIMULUS
Chromosomal change, including aneuploidy and polyploidization, is considered to be an important mechanism of speciation in many plant groups (Otto and Whitton 2000; Levin 2002), including Mimulus (Vickery 1995). In Mimulus, phylogenetic analyses have revealed at least 13 polyploidization and 15 aneuploidization events within western North American species (Beardsley et al. 2004). However, genetic evidence in support of this view is only recently beginning to accumulate. For example, genetic analyses of three chloroplast and six nuclear loci revealed that the North American allotetraploid M. sookensis (2n = 4x = 56), evolved via hybridization and genome duplication between two diploid taxa, M. guttatus and M. nasutus (Benedict et al. 2012). As in M. peregrinus, genetic analysis show elevated heterozygosity and multiple origins for M. sookensis (11 origins in this case), but unlike the case of M. x robertsii and M. peregrinus, triploid hybrids between M. guttatus and M. nasutus, expected if tetraploids arise via a triploid bridge stage (Ramsey and Schemske 1998; Husband 2004), are not known from nature (Modliszewski and Willis 2012). Importantly, experimental crosses indicate a lack of reproductive barriers between populations of M. sookensis stressing the potential for multiple origins to contribute to genetic diversity of nascent polyploids (Modliszewski and Willis 2012; Soltis et al. 2014). The fact that M. sookensis and M. peregrinus share a parent in common provides an exciting opportunity to compare closely related allopolyploid taxa of different ages, and which have arisen via hybridization between M. guttatus and taxa with similar (M. nasutus) or different (M. luteus) chromosome numbers (Buggs 2012).
Fixed heterozygosity in M. luteus
Mimulus luteus, one of the parents of M. peregrinus, is tetraploid (Mukherjee and Vickery 1962). Our findings of very high levels of heterozygosity in M. luteus, including in the two advanced generation inbred lines studied here (lut‐2 and lut‐4), indicate that heterozygosity is fixed at many loci in M. luteus (Fig. S2), supporting a previous hypothesis that this species is itself an allopolyploid (Mukherjee and Vickery 1962). Further studies are needed to establish the parentage and age of this allopolyploid, and in particular, whether M. guttatus is one of its parental species.
GENOMIC COMPOSITION OF A YOUNG ALLOPOLYPLOID
We found that overall allele dosage ratios within M. peregrinus and M. x robertsii at loci that have alternatively fixed alleles in M. guttatus and M. luteus departed slightly from the expected genomic composition of a 1:2 ratio within a diploid × tetraploid hybrid. Overall, a bias was found in favor of M. guttatus alleles. This discrepancy is partly due to hybridization bias in favor of alleles found in the reference genome used to design the probes, which mainly favored M. guttatus alleles. Using alternative approaches such as whole genome sequencing or RADseq (Buggs et al. 2012b) could reduce biases against sequencing divergent copies of duplicated loci by bypassing the hybridization step used in sequence capture. In addition, future analyses of the M. luteus genome could indicate the extent to which M. luteus has diploidized at some loci (diploidization defined here as the process where duplicated gene copies are lost, Conant et al. 2014). The absence of one of the homeologous loci, for example, due to deletion, in M. luteus could result in an increase in the observed read depth of M. guttatus alleles in their hybrid offspring. In this regard, elucidating the origin and genomic structure of M. luteus will help refine the hypotheses for the expected genomic composition of M. peregrinus.
The allele dosage ratio analysis further uncovered a relatively large number of loci that were missing one of the expected parental alleles (Fig. 2, Table S1), which could represent either candidates for allele loss in M. peregrinus or unsampled genetic variation in the parental taxa. The confounding effect of unsampled variation in the current analysis may be particularly acute in the high incidence of “subgenome loss” observed in both M. x robertsii and M. peregrinus (Fig. 1) relative to M. guttatus and M. luteus. We found more than 100 loci in which the M. guttatus allele appears to be missing. It is possible that the diploid subgenome (M. guttatus) is more likely to be lost during hybridization than the tetraploid subgenome (M. luteus). However, a more likely explanation is simply that our limited sample of M. guttatus individuals did not capture some of the variation segregating within this species, and thus that the alleles used to depict luteus‐specific reads were also present in (unsampled) M. guttatus. Evidence in support of the hypothesis that unsampled variation could create the pattern of deletions observed here, includes that no probed regions showed a complete loss of one of the parental copies—only individual SNPs or subsets of SNPs within a given probe showed signs of deletion—and that most loci with missing alleles were observed in both M. peregrinus and M. x robertsii (Fig. 3), implying that missing alleles were not exclusively associated with genome duplication.
However, our data on allelic presence/absence in the specific comparison between the genomes of M. x robertsii and M. peregrinus raise the possibility that a small level of genome evolution is occurring in neoallopolyploids. In this comparison, we found evidence consistent with either gain (10/9362 sites) or loss/conversion (42/6625) of alleles following polyploidization (Table S2). In contrast to the allele dosage ratio analysis, these changes must have occurred principally during or after whole genome duplication. The evidence for genome evolution in M. peregrinus is particularly compelling in the pattern of allele loss/conversion observed in linkage group 14 (LG14) in the individual from the Orkney Islands (per‐3; Fig. 4). The pairwise comparison of allele frequencies between per‐3 and the closest M. x robertsii sample (rob‐1) show a consistent pattern of differences in allele frequencies in 24 SNP loci across seven different probes in the terminal portion of LG14, in a region spanning approximately 2,500,000 bp. (Fig. 4, Table S2). The average allele frequency difference between per‐3 and rob‐1 is 0.18 ± 0.05 (mean ± SD), which is very close to the expected change in frequency caused by the loss/conversion of a single allele in a hexaploid (1/6 = 0.167). Although we cannot completely rule out that some of this “gain” and “loss/conversion” of alleles represents unsampled genetic variation in the exact parents of the studied polyploids, the assumption that the studied individuals of M. x robertsii reflect the genomic composition of the direct ancestors of M. peregrinus, seems reasonable given that we included in our analyses the local M. x robertsii for the two origins of M. peregrinus (Fig. 1).
Ultimately, studying synthetic polyploids may provide a more direct estimation of genome restructuring following whole genome duplication (Hegarty et al. 2013). For example, a study of synthetic individuals of M. sookensis found 10 instances of fragment loss among 48 individuals from a polyploid backcross (BC1N) between M. guttatus and M. nasutus genotyped at seven fragment‐length polymorphism markers (Modliszewski and Willis 2014). This number of fragment‐loss events is lower than the expectation if loss occurs via homeologous recombination (Modliszewski and Willis 2014), but highlights the potential of genome restructuring to occur relatively rapidly. In synthetic M. sookensis, there is little evidence that this level of fragment loss results in phenotypic variation among synthetic polyploids, but our understanding of the link between genome restructuring in polyploids and phenotypic variation is still in its infancy.
In tetraploid species of Tragopogon, which have arisen via allopolyploidization of two diploid species within the last century, substantial losses of parental alleles have been found (Buggs et al. 2009, 2012a; Tate et al. 2009; Soltis et al. 2012). Evidence from cytogenetic studies strongly suggests that some of these parental alleles have been removed from the genome via nonhomeologous recombination (Chester et al. 2012, 2013, 2014). However, not all documented cases of parent allele loss can be accounted for in this way, suggesting that smaller scale genomic rearrangement or gene conversion events may also play a role (Chester et al. 2012; Buggs et al. 2012aa). In contrast, other allopolyploid alleles appear to have much more stable genomes (Liu et al. 2001; Mandáková et al. 2014). In the long term, we do not know if genomic changes in allopolyploids provide a source of variation that may contribute to their success, or whether genomic variants tend to be of low fitness and only individuals with stable genomes ultimately survive (Soltis et al. 2010).
It is therefore hard to predict the long‐term evolutionary trajectory of M. peregrinus. On the one hand, each population currently has limited genetic diversity, which as with all neoallopolyploids may represent a serious challenge to the ability of these taxa to respond to changing environments (Levin 2002). On the other hand, its multiple origins and apparent rapid genome evolution could provide the variation that it needs to become a successful species (Modliszewski and Willis 2012; Soltis et al. 2014). Whether it will remain a short‐lived scientific curiosity like the allotetraploid Senecio eboracensis (Lowe and Abbott 2004), which is now extinct in the wild, or spread well beyond its place of origin like allopolyploid S. anglica (Ainouche et al. 2009) remains to be seen.
Supporting information
Figure S1. Positions of probes used in the sequence capture experiment mapped on the 14 major linkage groups (scaffolds) of M. guttatus (genome version 2.0, www.phytozome.net).
Figure S2. Heterozygosity plot of 16 Mimulus spp. individuals across 20,749 biallelic SNPs genotyped at a minimum read depth of 50× in all individuals.
Figure S3. Neighbor joining tree of 16 Mimulus spp. showing bootstrap support for all nodes.
Figure S4. Allele frequency for 881 SNPs in four individuals of M. x robertsii (A) and four of M. peregrinus (B) mapped against the 14 major linkage groups of the M. guttatus reference genome.
Table S1. List of SNP loci showing a departure from expected heterozygosity in M. x robertsii and M. peregrinus based on expectation from parental genotypes.
Table S2. Location and identity of the SNP sites in which a loss or gain of an allele was detected between M. x robertsii and M. peregrinus.
Additional Supplementary Material: Bioinformatic commands for alignment and SNP genotyping.
ACKNOWLEDGMENTS
We would like to thank J. Willis, J. Modliszewski, Y.‐W. Lee, P. Edger, and the Willis Lab for advice and discussion in the earlier stages of this project and for sharing unpublished data, and G. Vallejo and A. Taylor for help during fieldwork. Two anonymous reviewers provided helpful comments in a previous version of this manuscript. This study was partly supported by a Natural Environment Research Council grant (NERC, NE/J012645/1) to MVM, and a NERC Biomolecular Analysis Facility grant (NBAF‐710) to MVM and RJAB. MVM, RJAB, and JRP designed the experiment. MVM and AMC collected the samples. MVM and JRP performed all analyses. MVM, JRP, RJAB, and AMC wrote the article.
DATA ARCHIVING
The doi for our data is 10.5061/dryad.gg273.
LITERATURE CITED
Associate Editor: K. Bomblies
Handling Editor: J. Conner
- Abbott, R. J. , and Lowe A. J.. 2004. Origins, establishment and evolution of new polyploid species: Senecio cambrensis and S. eboracensis in the British Isles. Biol. J. Linn. Soc. 82:467–474. [Google Scholar]
- Abbott, R. , Albach D., Ansell S., Arntzen J. W., Baird S. J. E., Bierne N., Boughman J. W., Brelsford A., Buerkle C. A., Buggs R., et al. 2013. Hybridization and speciation. J. Evol. Biol. 26:229–246. [DOI] [PubMed] [Google Scholar]
- Ainouche, M. , Baumel A., Salmon A., and Yannic G.. 2004. Hybridization, polyploidy and speciation in Spartina (Poaceae). New Phytol. 161:165–172. [Google Scholar]
- Ainouche, M. , Fortune P., Salmon A., Parisod C., Grandbastien M., Fukunaga K., Ricou M., and Misset M.. 2009. Hybridization, polyploidy and invasion: lessons from Spartina (Poaceae). Biol. Invasions 11:1159–1173. [Google Scholar]
- Ashton, P. A. , and Abbott R. J.. 1992. Multiple origins and genetic diversity in the newly arisen allopolyploid species, Senecio cambrensis Rosser (Compositae). Heredity 68:25–32. [Google Scholar]
- Ayres, D. , Ryan F. J., Grotkopp E., Bailey J., and Gaskin J.. 2009. Tumbleweed (Salsola, section Kali) species and speciation in California. Biol. Invasions 11:1175–1187. [Google Scholar]
- Baumel, A. , Ainouche M., Kalendar R., and Schulman A.. 2002. Retrotransposons and genomic stability in populations of the young allopolyploid species Spartina anglica CE Hubbard (Poaceae). Mol. Biol. Evol. 19:1218–1227. [DOI] [PubMed] [Google Scholar]
- Beardsley, P. M. , and Olmstead R. G.. 2002. Redefining Phrymaceae: the placement of Mimulus, tribe Mimuleae and Phryma . Am. J. Bot. 89:1093–1102. [DOI] [PubMed] [Google Scholar]
- Beardsley, P. M. , Schoenig S. E., Whittall J. B., and Olmstead R. G.. 2004. Patterns of evolution in Western North American Mimulus (Phrymaceae). Am. J. Bot. 91:474–489. [DOI] [PubMed] [Google Scholar]
- Benedict, B. G. , Modliszewski J. L., Sweigart A. L., Martin N. H., Ganders F. R., and Willis J. H.. 2012. Mimulus sookensis (Phrymaceae), a new allotetraploid species derived from Mimulus guttatus and Mimulus nasutus . Madroño 59:29–43. [Google Scholar]
- Bradshaw, H. D. , and Schemske D. W.. 2003. Allele substitution at a flower colour locus produces a pollinator shift in monkeyflowers. Nature 426:176–178. [DOI] [PubMed] [Google Scholar]
- BSBI . 2013. Botanical Society of the British Isles. Available at http://www.bsbi.org.uk.
- Buggs, R. J. A. 2012. Monkeying around with ploidy. Mol. Ecol. 21:5159–5161. [DOI] [PubMed] [Google Scholar]
- Buggs, R. J. A. , Doust A. N., Tate J. A., Koh J., Soltis K., Feltus F. A., Paterson A. H., Soltis P. S., and Soltis D. E.. 2009. Gene loss and silencing in Tragopogon miscellus (Asteraceae): comparison of natural and synthetic allotetraploids. Heredity 103:73–81. [DOI] [PubMed] [Google Scholar]
- Buggs, R. J. A. , Chamala S., Wu W., Tate J. A., Schnable P. S., Soltis D. E., Soltis P. S., and Barbazuk W. B.. 2012a. Rapid, repeated, and clustered loss of duplicate genes in allopolyploid plant populations of independent origin. Curr. Biol. 22:248–252. [DOI] [PubMed] [Google Scholar]
- Buggs, R. J. A. , Renny‐Byfield S., Chester M., Jordon‐Thaden I. E., Viccini L. F., Chamala S., Leitch A. R., Schnable P. S., Barbazuk W. B., Soltis P. S., et al. 2012b. Next‐generation sequencing and genome evolution in allopolyploids. Am. J. Bot. 99:372–382. [DOI] [PubMed] [Google Scholar]
- Chao, D. Y. , Dilkes B., Luo H., Douglas A., Yakubova E., Lahner B., and Salt D. E.. 2013. Polyploids exhibit higher potassium uptake and salinity tolerance in Arabidopsis . Science 341:658–659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chester, M. , Riley R., Soltis P., and Soltis D.. 2014. Patterns of chromosomal variation in natural populations of the neoallotetraploid Tragopogon mirus (Asteraceae). Heredity. 114:309–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chester, M. , Gallagher J. P., Symonds V. V., Cruz da Silva A. V., Mavrodiev E. V., Leitch A. R., Soltis P. S., and Soltis D. E.. 2012. Extensive chromosomal variation in a recently formed natural allopolyploid species, Tragopogon miscellus (Asteraceae). Proc. Natl. Acad. Sci. USA 109:1176–1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chester, M. , Lipman M. J., Gallagher J. P., Soltis P. S., and Soltis D. E.. 2013. An assessment of karyotype restructuring in the neoallotetraploid Tragopogon miscellus (Asteraceae). Chrom. Res. 21:75–85. [DOI] [PubMed] [Google Scholar]
- Conant, G. C. , Birchler J. A., and Pires J. C.. 2014. Dosage, duplication, and diploidization: clarifying the interplay of multiple models for duplicate gene evolution over time. Curr. Opin. Plant Biol. 19:91–98. [DOI] [PubMed] [Google Scholar]
- Cooley, A. M. , and Willis J. H.. 2009. Genetic divergence causes parallel evolution of flower color in Chilean Mimulus . New Phytol. 183:729–739. [DOI] [PubMed] [Google Scholar]
- Cooley, A. M. , Carvallo G., and Willis J. H.. 2008. Is floral diversification associated with pollinator divergence? Flower shape, flower colour and pollinator preference in Chilean Mimulus . Ann. Bot. 101:641–650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui, L. , Wall P. K., Leebens‐Mack J. H., Lindsay B. G., Soltis D. E., Doyle J. J., Soltis P. S., Carlson J. E., Arumuganathan K., Barakat A., et al. 2006. Widespread genome duplications throughout the history of flowering plants. Genome Res. 16:738–749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fishman, L. , Willis J., Wu C., and Lee Y.. 2014. Comparative linkage maps suggest that fission, not polyploidy, underlies near‐doubling of chromosome number within monkeyflowers (Mimulus; Phrymaceae). Heredity. 112:562–568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grant, A. L. 1924. A monograph of the genus Mimulus . Ann. Mo. Bot. Gard. 11:99–380. [Google Scholar]
- Hegarty, M. , Jones J., Wilson I., Barker G., Coghill J., Sanchez‐Baracaldo P., Liu G., Buggs R., Abbott R., Edwards K., et al. 2005. Development of anonymous cDNA microarrays to study changes to the Senecio floral transcriptome during hybrid speciation. Mol. Ecol. 14:2493–2510. [DOI] [PubMed] [Google Scholar]
- Hegarty, M. J. , Abbott R. J., and Hiscock S. J.. 2012. Allopolyploid speciation in action: the origins and evolution of Senecio cambrensis Pp. 245–270 in Soltis P. S. and Soltis D. E., eds. Polyploidy and genome evolution. Springer‐Verlag, Berlin. [Google Scholar]
- Hegarty, M. , Coate J., Sherman‐Broyles S., Abbott R., Hiscock S., and Doyle J.. 2013. Lessons from natural and artificial polyploids in higher plants. Cytogenet. Genome Res. 140:204–225. [DOI] [PubMed] [Google Scholar]
- Husband, B. C. 2004. The role of triploid hybrids in the evolutionary dynamics of mixed‐ploidy populations. Biol. J. Linn. Soc. 82:537–546. [Google Scholar]
- Jiao, Y. , Wickett N. J., Ayyampalayam S., Chanderbali A. S., Landherr L., Ralph P. E., Tomsho L. P., Hu Y., Liang H., Soltis P. S., et al. 2011. Ancestral polyploidy in seed plants and angiosperms. Nature 473:97–U113. [DOI] [PubMed] [Google Scholar]
- Jombart, T. , and Ahmed I.. 2011. adegenet 1.3‐1: new tools for the analysis of genome‐wide SNP data. Bioinformatics 27:3070–3071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levin, D. A. 1983. Polyploidy and novelty in flowering plants. Am. Nat. 122:1–25. [Google Scholar]
- Levin, D. A. 2002. The role of chromosomal change in plant evolution. Oxford Univ. Press, Oxford , U . K. [Google Scholar]
- Li, H. , Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., and Durbin R.. 2009. The sequence alignment/map format and SAMtools . Bioinformatics 25:2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, B. , Brubaker C., Mergeai G., Cronn R., and Wendel J.. 2001. Polyploid formation in cotton is not accompanied by rapid genomic changes. Genome 44:321–330. [PubMed] [Google Scholar]
- Lowe, A. , and Abbott R. J.. 2004. Reproductive isolation of a new hybrid species, Senecio eboracensis Abbott & Lowe (Asteraceae). Heredity 92:386–395. [DOI] [PubMed] [Google Scholar]
- Lowry, D. B. , and Willis J. H.. 2010. A Widespread chromosomal inversion polymorphism contributes to a major life‐history transition, local adaptation, and reproductive isolation. PLoS Biol. 8:e1000500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mable, B. K. 2003. Breaking down taxonomic barriers in polyploidy research. Trends Plant Sci. 8:582–590. [DOI] [PubMed] [Google Scholar]
- Mandakova, T. , Kovarik A., Zozomova‐Lihova J., Shimizu‐Inatsugi R., Shimizu K. K., Mummenhoff K., Marhold K., and Lysak M. A.. 2013. The more the merrier: recent hybridization and polyploidy in Cardamine . Plant Cell 25:3280–3295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandáková, T. , Marhold K., and Lysak M. A.. 2014. The widespread crucifer species Cardamine flexuosa is an allotetraploid with a conserved subgenomic structure. New Phytol. 201:982–992. [DOI] [PubMed] [Google Scholar]
- Mason, A. S. , and Pires J. C.. 2015. Unreduced gametes: meiotic mishap or evolutionary mechanism? Trends Genet. 31:5–10. [DOI] [PubMed] [Google Scholar]
- Modliszewski, J. L. , and Willis J. H.. 2012. Allotetraploid Mimulus sookensis are highly interfertile despite independent origins. Mol. Ecol. 21:5280–5298. [DOI] [PubMed] [Google Scholar]
- Modliszewski, J. L. 2014. Near‐absent levels of segregational variation suggest limited opportunities for the introduction of genetic variation via homeologous chromosome pairing in synthetic neoallotetraploid Mimulus . G3 4:509–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukherjee, B. B. , and Vickery R. K.. 1962. Chromosome counts in the section Simiolus of the genus Mimulus (Scrophulariaceae). V. The chromosomal homologies of M. guttatus and its allied species and varieties. Madroño 16:141–172. [Google Scholar]
- Murren, C. J. , Chang C. C., and Dudash M. R.. 2009. Patterns of selection of two North American native and nonnative populations of monkeyflower (Phrymaceae). New Phytol. 183:691–701. [DOI] [PubMed] [Google Scholar]
- Osborn, T. C. , Chris Pires J., Birchler J. A., Auger D. L., Jeffery Chen Z., Lee H., Comai L., Madlung A., Doerge R., and Colot V.. 2003. Understanding mechanisms of novel gene expression in polyploids. Trends Gen. 19:141–147. [DOI] [PubMed] [Google Scholar]
- Otto, S. , and Whitton J.. 2000. Polyploid incidence and evolution. Annu. Rev. Genet. 34:401–437. [DOI] [PubMed] [Google Scholar]
- Paradis, E. , Claude J., and Strimmer K.. 2004. APE: analyses of phylogenetics and evolution in R language. Bioinformatics 20:289–290. [DOI] [PubMed] [Google Scholar]
- Parker, P. F. 1975. Mimulus in Great Britain: a cytotaxonomic note. New Phytol. 74:155–160. [Google Scholar]
- Preston C. D., Pearman D. A., and Dines T. D., eds. 2002. New Atlas of the British and Irish Flora. Oxford Univ. Press, Oxford , U.K. [Google Scholar]
- Puzey, J. , and Vallejo‐Marín M.. 2014. Genomics of invasion: diversity and selection in introduced populations of monkeyflowers (Mimulus guttatus). Mol. Ecol. 23:4472–4485. [DOI] [PubMed] [Google Scholar]
- R Development Core Team . 2014. R. A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria: Available at: http://www.R‐project.org/. Accessed July 10, 2014. [Google Scholar]
- Ramsey, J. , and Schemske D. W.. 1998. Pathways, mechanisms, and rates of polyploid formation in flowering plants. Annu. Rev. Ecol. Syst. 29:467–501. [Google Scholar]
- Roberts, R. H. 1964. Mimulus hybrids in Britain. Watsonia 6:70–75. [Google Scholar]
- Silverside, A. J. 1990. A new binomial in Mimulus . Watsonia 18:210–212. [Google Scholar]
- Silverside, A. J. 1998. Mimulus section Simiolus. Pp. 259–261 in Rich T. and Jermy A., eds. Plant crib. Botanical Society of the British Isles, London. [Google Scholar]
- Soltis, D. E. , and Soltis P. S.. 1999. Polyploidy: recurrent formation and genome evolution. Trends Ecol. Evol. 14:348–352. [DOI] [PubMed] [Google Scholar]
- Soltis, D. E. , Soltis P. S., Pires J. C., Kovarik A., Tate J. A., and Mavrodiev E.. 2004. Recent and recurrent polyploidy in Tragopogon (Asteraceae): cytogenetic, genomic and genetic comparisons. Biol. J. Linn. Soc. 82:485–501. [Google Scholar]
- Soltis, D. E. , Buggs R. J., Barbazuk W. B., Schnable P. S., and Soltis P. S.. 2009. On the origins of species: does evolution repeat itself in polyploid populations of independent origin? Cold Spring Harb. Symp. Quant. Biol. 74:215–223. [DOI] [PubMed] [Google Scholar]
- Soltis, D. E. , Buggs R. J. A., Doyle J. J., and Soltis P. S.. 2010. What we still don't know about polyploidy. Taxon 59:1387–1403. [Google Scholar]
- Soltis, D. E. , Buggs R. J., Barbazuk W. B., Chamala S., Chester M., Gallagher J. P., Schnable P. S., and Soltis P. S.. 2012. The early stages of polyploidy: rapid and repeated evolution in Tragopogon Pp. 271–292 in Soltis P. and Soltis D. E. eds. Polyploidy and genome evolution. Springer, Berlin. [Google Scholar]
- Soltis, D. E. , Visger C. J., and Soltis P. S.. 2014. The polyploidy revolution then…and now: Stebbins revisited. Am. J. Bot. 101:1057–1078. [DOI] [PubMed] [Google Scholar]
- Song, K. , Lu P., Tang K., and Osborn T. C.. 1995. Rapid genome change in synthetic polyploids of Brassica and its implications for polyploid evolution. Proc. Natl. Acad. Sci. USA 92:7719–7723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stace, C. 2010. New flora of the British Isles. Cambridge Univ. Press, Cambridge. [Google Scholar]
- Symonds, V. V. , Soltis P. S., and Soltis D. E.. 2010. Dynamics of polyploid formation in Tragopogon (Asteraceae): Rrecurrent formation, gene flow, and population structure. Evolution 64:1984–2003. [DOI] [PubMed] [Google Scholar]
- Tate, J. A. , Joshi P., Soltis K. A., Soltis P. S., and Soltis D. E.. 2009. On the road to diploidization? Homoeolog loss in independently formed populations of the allopolyploid Tragopogon miscellus (Asteraceae). BMC Plant Biol. 9:1–10. doi:10.1186/1471‐2229‐9‐80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tokarska‐Guzik, B. , and Dajdok Z.. 2010. NOBANIS. Invasive alien species fact sheet: Mimulus guttatus . Online Database of the North European and Baltic Network on Invasive Alien Species, NOBANIS. Available at: www.nobanis.org. Accessed February 2, 2012.
- Truscott, A. M. , Palmer S. C., Soulsby C., Westaway S., and Hulme P. E.. 2008. Consequences of invasion by the alien plant Mimulus guttatus on the species composition and soil properties of riparian plant communities in Scotland. Persp. Plant Ecol. Evol. Syst. 10:231–240. [Google Scholar]
- Vallejo‐Marín, M. 2012. Mimulus peregrinus (Phrymaceae): a new British allopolyploid species. PhytoKeys 14:1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vallejo‐Marín, M. , and Lye G. C.. 2013. Hybridisation and genetic diversity in introduced Mimulus (Phrymaceae). Heredity 110:111–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Kleunen, M. 2007. Adaptive genetic differentiation in life‐history traits between populations of Mimulus guttatus with annual and perennial life‐cycles. Evol. Ecol. 21:185–199. [Google Scholar]
- Vickery, R. K. 1959. Barriers to gene exchange within Mimulus guttatus (Scrophulariaceae). Evolution 13:300–310. [Google Scholar]
- Vickery, R. K. 1995. Speciation by aneuploidy and polyploidy in Mimulus (Scrophulariaceae). Great Basin Nat. 55:174–176. [Google Scholar]
- Willis, J. H. 1993. Partial self‐fertilization and inbreeding depression in two populations of Mimulus guttatus . Heredity 71:145–154. [Google Scholar]
- Wood, T. E. , Takebayashi N., Barker M. S., Mayrose I., Greenspoon P. B., and Rieseberg L. H.. 2009. The frequency of polyploid speciation in vascular plants. Proc. Natl. Acad. Sci. USA 106:13875–13879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright, R. J. , Thaxton P. M., El‐Zik K. M., and Paterson A. H.. 1998. D‐subgenome bias of Xcm resistance genes in tetraploid Gossypium (cotton) suggests that polyploid formation has created novel avenues for evolution. Genetics 149:1987–1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu, C. A. , Lowry D. B., Cooley A. M., Wright K. M., Lee Y. W., and Willis J. H.. 2007. Mimulus is an emerging model system for the integration of ecological and genomic studies. Heredity 100:220–230. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Positions of probes used in the sequence capture experiment mapped on the 14 major linkage groups (scaffolds) of M. guttatus (genome version 2.0, www.phytozome.net).
Figure S2. Heterozygosity plot of 16 Mimulus spp. individuals across 20,749 biallelic SNPs genotyped at a minimum read depth of 50× in all individuals.
Figure S3. Neighbor joining tree of 16 Mimulus spp. showing bootstrap support for all nodes.
Figure S4. Allele frequency for 881 SNPs in four individuals of M. x robertsii (A) and four of M. peregrinus (B) mapped against the 14 major linkage groups of the M. guttatus reference genome.
Table S1. List of SNP loci showing a departure from expected heterozygosity in M. x robertsii and M. peregrinus based on expectation from parental genotypes.
Table S2. Location and identity of the SNP sites in which a loss or gain of an allele was detected between M. x robertsii and M. peregrinus.
Additional Supplementary Material: Bioinformatic commands for alignment and SNP genotyping.