

Summary
Aim
It is now clear that insulin signaling has important roles in regulation of neuronal functions in the brain. Dysregulation of brain insulin signaling has been linked to neurodegenerative disease, particularly Alzheimer's disease (AD). In this regard, there is evidence that improvement of neuronal insulin signaling has neuroprotective activity against amyloid β (Aβ)‐induced neurotoxicity for patients with AD. Linagliptin is an inhibitor of dipeptidylpeptidase‐4 (DPP‐4), which improves impaired insulin secretion and insulin downstream signaling in the in peripheral tissues. However, whether the protective effects of linagliptin involved in Aβ‐mediated neurotoxicity have not yet been investigated.
Methods
In the present study, we evaluated the mechanisms by which linagliptin protects against Aβ‐induced impaired insulin signaling and cytotoxicity in cultured SK‐N‐MC human neuronal cells.
Results
Our results showed that Aβ impairs insulin signaling and causes cell death. However, linagliptin significantly protected against Aβ‐induced cytotoxicity, and prevented the activation of glycogen synthase kinase 3β (GSK3β) and tau hyperphosphorylation by restoring insulin downstream signaling. Furthermore, linagliptin alleviated Aβ‐induced mitochondrial dysfunction and intracellular ROS generation, which may be due to the activation of 5′ AMP‐activated protein kinase (AMPK)‐Sirt1 signaling. This upregulation of Sirt1 expression was also observed in diabetic patients with AD coadministration of linagliptin.
Conclusions
Taken together, our findings suggest linagliptin can restore the impaired insulin signaling caused by Aβ in neuronal cells, suggesting DPP‐4 inhibitors may have therapeutic potential for reducing Aβ‐induced impairment of insulin signaling and neurotoxicity in AD pathogenesis.
Keywords: Alzheimer's disease, AMP‐activated protein kinase, Amyloid‐β, Linagliptin, Sirtuin 1
Introduction
Impaired insulin signaling is a physiological condition that cells lost the ability to respond to insulin. This failure to respond is called insulin resistance and plays a central in the development of the metabolic disorders such as diabetes, obesity, hypertension, and dyslipidemia. Particularly, defective brain insulin signaling has been implicated in the development of Alzheimer's disease (AD), the most common cause of dementia 1. In fact, abnormal insulin sensitivity is shown to be associated with AD‐related pathological features. For example, type 2 diabetes (T2D) is identified as a major risk factor for AD, suggesting defective insulin signaling may account for pathogenesis of neurodegeneration 2. Moreover, patients with AD show significantly reduced expression of insulin receptors and insulin receptor substrate (IRS) in the brain that contributes to the severity of cognitive impairment 3. All these findings suggest that neuronal insulin signaling is altered in the AD brain resembling T2D 4. Actually, AD pathogenesis is initially triggered by the presence of extracellular amyloid‐β (Aβ) proteins, which are found to cause oxidative stress and neurotoxicity in the brain 5. It is well known that insulin and its receptors are widely expressed in neurons and glial cells throughout the brain 6, and evidence is also presented that insulin can be produced locally within the brain 7. In addition, Aβ has been reported to impair synaptic insulin sensitivity in cultured neurons, which may impair synaptic functions associated with pathogenesis of AD 8. This indicates insulin signaling may serve as an important regulatory role in neurons. However, the molecular basis that links between insulin signaling and Aβ‐induced neurotoxicity remains unclear.
At a molecular level, the serine phosphorylation of insulin receptor substrate (IRS) can block the insulin signaling. This results in the inhibition of IRS tyrosine phosphorylation, suppressing the downstream phosphatidylinositol 3‐kinase (PI3‐kinase) signaling and subsequent inactivation of the kinase Akt 9. It is known that activated Akt inactivates glycogen synthase kinase 3β (GSK3β) by phosphorylating its Ser9 residue, which is one of the important enzymes induce tau hyperphosphorylation and neurotoxicity 10. For this reason, impairment of insulin signaling may result in a high activity of GSK3β, which leads to an enhanced tau hyperphosphorylation, a crucial step in AD pathogenesis. Hyperphosphorylated tau and Aβ cooperatively impair mitochondrial membrane potential and further increase in accumulation of intracellular reactive oxygen species (ROS), which ultimately result in neurodegeneration 11. Therefore, it is not surprising that pharmaceuticals found to be effective treatment of impaired insulin signaling have also shown benefits in the prevention or reduction of AD 12.
The enzyme dipeptidyl peptidase‐4 (DPP‐4) is a ubiquitous membrane‐bound prolyl peptidase that was responsible for the degradation of incretin hormones 13. Incretins are a group of gut‐derived hormones that potentiate insulin secretion and related cellular signaling 14. However, incretins are rapidly metabolized and inactivated by DPP‐4. As a result, DPP‐4 inhibitors such as linagliptin have a relevant effect of increasing the half‐life in retaining the physiological effects of endogenous incretins 15. Interestingly, it has been recently shown that incretins may be good candidates for treating AD 16. For example, glucagon‐like peptide‐1 (GLP‐1), the major incretin in humans, has been shown to elicit neuroprotective properties against AD pathological processes 17. Similar to insulin, GLP‐1 is produced in the brain mediating many neuronal functions, including neuroprotection, improvement of learning and memory ability, and potentiation of insulin signaling 18. Therefore, GLP‐1 signaling have demonstrated the potential to serve as therapeutic or preventive strategies against diabetes‐related AD 19. As DPP‐4 inhibitor effectively increases GLP‐1 levels, it may also exert protective effects against AD‐related Aβ‐induced neurotoxicity. Linagliptin is a recently approved DPP‐4 inhibitor and widely considered as the first‐line treatment for T2D patients. It has been demonstrated greater inhibitory effects than other DPP‐4 inhibitors such as alogliptin, saxagliptin, sitagliptin, or vildagliptin 20. Moreover, linagliptin also significantly improves insulin secretory dysfunction and sensitivity in animal studies 21. This indicates linagliptin may have beneficial effects on impaired insulin signaling in neuronal cells. However, whether linagliptin is involved in Aβ‐induced neurotoxicity is still largely unknown. In this study, we postulated that neuronal insulin resistance may be one of the underlying neurotoxic mechanisms by Aβ, whereas linagliptin can protect neuronal cells by restoring impaired insulin signaling, and thereby contribute to the alleviation of Aβ‐induced neurotoxicity.
Materials and Methods
Materials
Chemicals such as 3‐(4,5‐dimethylthiazol‐2‐yl)‐2,5‐diphenyltetrazolium bromide (MTT), 4′,6‐diamidino‐2‐phenylindole (DAPI), and JC‐1 were purchased from Sigma (München, Germany). Amyloid‐β (Aβ) 1‐42 was acquired from AnaSpec Inc. (San Jose, CA, USA). We purchased antibodies against Akt, p‐Akt, GSK3β, p‐GSK3β and IRS‐1, caspase 3, SOD1, and poly(ADP‐ribose) polymerase (PARP) from Santa Cruz Biotechnology (Santa Cruz, CA, USA), Sirt1 antibody from GeneTex (Irvine, CA, USA), β‐actin antibody from Novus Biologicals (Littleton, CO, USA), and p‐IRS‐1 antibodies from Cell Signaling Technology (Danvers, MA, USA). Primary antibodies were used at a dilution of 1:1000 in 0.1% Tween‐20 and secondary antibodies were used at 1:5000 dilutions. Pure linagliptin was provided by Boehringer Ingelheim Pharmaceuticals (Biberach, Germany). All the chemicals were prepared by dissolving phosphate buffer saline solutions stored at −20°C until needed for use in experiments.
Cell Culture and Viability Assay
Human neuroblastoma SK‐N‐MC cells were obtained from the American Type Culture Collection (Bethesda, MD, USA). Cells were maintained in minimal Eagle's medium (MEM; Gibco, Carlsbad, CA, USA), supplemented with 10% fetal calf serum, 100 units/mL penicillin, 100 μg/mL streptomycin, and 2 mM L‐glutamine at 37°C, 5% CO2. The Aβ solutions were prepared as described previously 22. Briefly, Aβ 1‐42 lyophilizates were dissolved at 10 mM in 10% 60 mM NaOH and 90% 10 mM phosphate buffer (pH 7.4) as a stock reagent, and stored at −78°C until use. For viability assay, cells were seeded in 96‐welled plates at a density of 1 × 104 cells/well overnight and then treated as indicated. After 24 h, the tetrazolium salt MTT was added to the medium following the manufacturer's instructions. Only viable cells could metabolize MTT into a purple formazan product, of which the color density (OD) was further quantified by an EZ Read 400 microplate reader (Biochrom, Holliston, MA, USA) at 550 nm. Cell viability was determined by the percentage of OD of the treated cells divided that of the untreated controls.
mRNA Expression Analysis by Reverse Transcription Quantitative PCR
Total mRNA was extracted from the samples after treatment for the indicated conditions by utilizing the kit Qiagen RNeasy Kit (Qiagen, Germantown, MD, USA), and was quantified spectrophotometrically. RNA reverse transcription was performed at 25°C for 10 m for primer binding, 37°C for 120 min for reverse transcriptase, and 85°C for reverse transcriptase denaturation using the TProfessional Thermocycler (Biometra). Real‐time quantitative PCR (qPCR) was performed for quantification of mRNA by using an ABI 7300 Sequence Detection System (Applied Biosystems, Foster City, CA, USA). PCR amplifications of target mRNA genes were carried out in conjunction with Power SYBR Green PCR Master Mix (Applied Biosystems) according to the manufacturer's instructions. Each cDNA sample was tested in triplicate. The following temperature parameters were 95°C/10 min, 40 cycles of 95°C/15 s, 60°C/1 min and dissociation stage was 95°C/15 s, 60°C/15 s and 95°C/15 s. The following primer pairs were used, forward 5′‐ACACCTGTGCGGCTCACA‐3′ and reverse 5′‐TCCCGGCGGGTCTTG‐3′ for insulin, forward 5′‐TGCTTCCGGAGCTGTGATCT‐3′ and reverse 5′‐ CGGACAGAGCGAGCTGACTT‐3′ for IGF‐1, forward 5′‐ ATTGCTTGGCTGGTGAAAGG‐3′ and reverse 5′‐ TGTCTGCGGCCAAGTTCTTC‐3′ for proglucagon, and forward 5′‐TGGTATCGTGGAAGGACTCATGAC‐3′ and reverse 5′‐ATGCCAGTGAGCTTCCCGTTCAGC‐3′ for GAPDH. Values of relative mRNA expression were obtained by using the software SDS (Sequence Detection Systems 7300 Real Time PCR System; Applied Biosystems), and the values were standardized by comparing with values from relative expression of GAPDH.
Western Blot Analysis
After treatment, cells were harvested and homogenized in a protein extraction lysis buffer (50 mM Tris‐HCl, pH 8.0; 5 mM EDTA; 150 mM NaCl; 0.5% Nonidet P‐40; 0.5 mM phenylmethylsulfonyl fluoride; and 0.5 mM dithiothreitol), and centrifuged at 12,000 g for 30 min at 4°C. The supernatants were used as cell extracts for immunoblotting analysis. SDS‐solubilized samples were then loaded onto SDS‐polyacrylamide gels. Equal protein amounts of total cell lysates were resolved by 10% SDS‐PAGE, transferred onto polyvinylidene difluoride membranes (Millipore, Bedford, MA, USA), and then probed with a primary antibody followed by a secondary antibody conjugated with horseradish peroxidase. The immunocomplexes were visualized with enhanced chemiluminescence kits (Millipore). The relative expression of proteins was quantified densitometrically by using the software QuantityOne (BioRad, Hercules, CA, USA) and was calculated according to the reference bands of β‐actin. Each blot represents at least in three independent experiments.
Microscopic Observation and Nucleus Morphology
Changes in cell nucleus morphology, characteristic of apoptosis, were examined in cells grown on coverslips, using a microscope. The cells were fixed in 4% paraformaldehyde after 24 h of treatment with the indicated compounds. For phase‐contrast inverted microscopy, images of cells were captured with no specific staining procedure. For nucleus morphology microscopy, cells were fixed in ice‐cold methanol, and incubated for 15 min at room temperature with 1 ng/mL of 4′,6‐diamidino‐2‐phenylindole (DAPI) stain, and observed under a fluorescence microscope (DP80/BX53; Olympus, Tokyo, Japan). Apoptosis was quantified by averaging cell counts in twenty random 400× fields. Values were expressed as the percentage of apoptotic cells relative to total number of cells.
Measurement of Reactive Oxygen Species
To evaluate the levels of intracellular ROS, cells were seeded onto glass coverslips and incubated with 10 μM of 2′, 7′‐dichlorodihydrofluorescin diacetate (DCFH‐DA, a general oxidative stress indicator) for 0.5 h at 37°C under 5% CO2 after treatment. After incubation, the staining medium was discarded and cells were washed twice with immediately with PBS, after which the intensity of fluorescence was imaged by a fluorescence microscopy (DP72/CKX41; Olympus) using an excitation wavelength of 488 nm and an emission wavelength of 525 nm. One representative image of three different experiments is shown.
Analysis of Mitochondrial Membrane Potential
The vital mitochondrial cationic dye JC‐1, which exhibits potential‐dependent accumulation in mitochondria, was used to investigate mitochondrial function. Cells were treated in fresh medium containing 1 μM JC‐1 and were incubated at 37°C for 30 min. The staining medium was then discarded and the cells were washed. Cells then imaged using an inverted fluorescence microscope (DP72/CKX41; Olympus) excited at 488 nm. In normal cells, JC‐1 continues to exist as aggregates and produces a red fluorescence (~590 nm). During the induction of apoptosis, the mitochondrial potential collapses and JC‐1 forms a monomer producing green fluorescence (~525 nm).
Blood Samples from Patients
A total of 14 Chinese patients with non‐diabetic AD or diabetic AD were recruited from Chung Shan Medical University Hospital, Taichung, Taiwan. T2D was diagnosed according to the 1985 World Health Organization criteria using diagnostic values of fasting plasma glucose ≥7.0 mmol/L, and/or 2 h plasma glucose ≥11.1 mmol/L with or without 75 g oral glucose tolerance test, depending on the presence or absence of symptoms. AD was diagnosed according to the Diagnostic and Statistical Manual of Mental Disorders IV (DSM‐IV) criteria. From each subject, 20 mL of venous peripheral ethylenediamine tetra‐acetic acid (EDTA) blood was obtained, and total RNA was isolated by utilizing the kit Qiagen RNeasy Kit (Qiagen) which was quantified spectrophotometrically in accordance with the manufacturer's instructions. The aforementioned protocol was approved by the Chung Shan Medical University Hospital Institutional Review Board (IRB) protocols (CSMUH No: CS13233). Informed consent was obtained from all participants according to the Declaration of Helsinki and was approved by the IRB.
Statistical Analysis
All data are presented as means ± standard error of the means (SEM). Statistical analysis of data was performed using analysis of variance (ANOVA), followed by Dunnett's post hoc test for multiple comparisons with SPSS statistical software (SPSS, Inc., Chicago, IL, USA). Differences were considered statistically significant at P < 0.05.
Results
Effects of Linagliptin on Viability of SK‐N‐MC Neuronal Cells
The influence of DPP‐4 inhibitor linagliptin treated in neuronal cells is largely unknown. To evaluate the effects of linagliptin on cell viability, SK‐N‐MC neuronal cells were exposed to 10 to 100 μM of linagliptin for 24 h, and the cytotoxic effects were determined by MTT assay (Figure 1A). The results showed that linagliptin concentrations ranging from 10 to 50 μM did not induce significant cytotoxicity, whereas treatment with 100 μM linagliptin slightly reduced cell viability. In accordance, treated with 50 μM of linagliptin displays no significant time‐dependent change within a 48‐h period (Figure 1B). This indicates no detectable toxic effect is present at a concentration <50 μM of linagliptin. Thus, in subsequent experiments, we investigated the mode of action of linagliptin at a concentration of 50 μM. It is known that the main action of linagliptin is to stimulate insulin action by the incretin hormones such as glucagon‐like peptide 1 (GLP‐1). To determine the effects of linagliptin on insulin signaling‐related gene expression in SK‐N‐MC cells, we performed relative expression qPCR assays to measure levels of mRNA transcripts. As shown in Figure 1C, adding linagliptin to SK‐N‐MC cells resulted in upregulation of insulin and insulin‐like growth factor‐1 (IGF‐1) but no significant effects in proglucagon (the pro‐hormonal precursor mRNA of GLP‐1), indicating linagliptin may exert its pharmacological action by stimulating insulin/IGF‐related signaling to neuronal cells.
Figure 1.
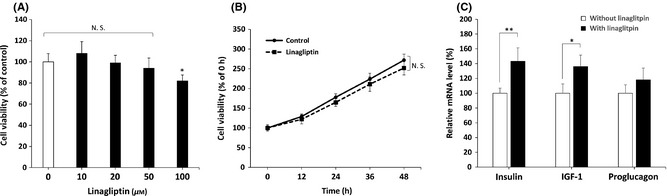
Effects of linagliptin on cell viability and incretin‐related mRNA expression in SK‐N‐MC neuronal cells. (A) Dose effects of linagliptin on SK‐N‐MC cells by MTT assay. Linagliptin shows no significant cytotoxicity <50 μM. (B) 50 μM of linagliptin causes no significant alteration of cell viability within a 48‐h period. (C) SK‐N‐MC cells are treated with or without 50 μM of linagliptin for 24 h. The mRNA levels of incretin‐related target genes including insulin, insulin‐like growth factor‐1 (IGF‐1), and glucagon‐like peptide 1 (GLP‐1) are measured by using real‐time qPCR, and the results are presented as means ± standard error of the means (SEM) of three independent experiments. *P < 0.05 and **P < 0.01 by using multiple comparisons of Dunnett's post‐hoc test. N. S., no significant difference.
Linagliptin Protects SK‐N‐MC Cells Against Aβ‐induced Neurotoxicity
Recent studies have demonstrated the DPP‐4 inhibitory properties indicating neuroprotective effects against AD pathological hallmarks such as Aβ accumulation, tau phosphorylation and neuroinflammation 23. To assess whether linagliptin exerts these similar beneficial effects, cell viability assay was conducted to determine the neuroprotective effect of linagliptin on Aβ‐induced cell death. As shown in Figure 2A, incubation of SK‐N‐MC cells with 2.5 μM of Aβ for 24 h markedly underwent a ~50% decrease of MTT reduction. However, linagliptin alleviated cell death at concentrations ranging from 10 to 50 μM of SK‐N‐MC cells in a dose‐dependent manner during Aβ treatment. To precisely determine which mode of cell death is induced by Aβ, we examined the expressions of cleaved caspase 3 and poly (ADP‐ribose) polymerase (PARP), two typical markers of apoptosis by western blotting. As shown in Figure 2B, Aβ markedly increased cleavage of caspase 3 and PARP, indicating the enhanced apoptosis occurs mainly in Aβ treatment. On the contrary, co‐treated with linagliptin was shown to effectively inhibit caspase 3 and PARP activation by Aβ. These results were also confirmed by DAPI staining that treatment with linagliptin significantly reduced nuclei fragmentation as shown in Figure 2C. Taken together, this support the idea that the addition of linagliptin may effectively attenuate Aβ‐induced apoptosis in neuronal cells.
Figure 2.
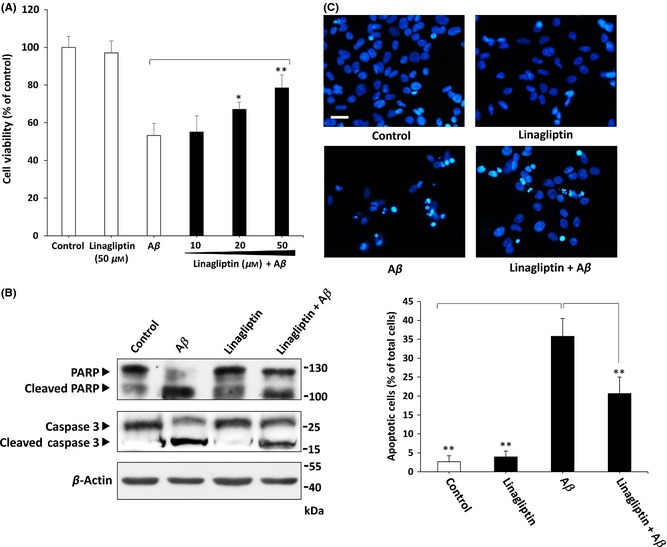
Linagliptin protects against Aβ‐induced SK‐N‐MC cell death. (A) MTT assays indicate 2.5 μM of Aβ markedly induces cell death after 24 h of incubation. However, linagliptin significantly prevents Aβ‐induced neurotoxic effects in a dose‐dependent manner. (B) Western blotting results demonstrate that linagliptin (50 μM) treatment suppresses both caspase 3 and PARP activation induced by Aβ (2.5 μM). (C) Linagliptin (50 μM) markedly reduces 2.5 μM of Aβ‐induced nucleus fragmentation. Apoptosis is determined by fragmented morphology in the nucleus for DAPI fluorescence. The numbers of apoptotic cells are quantified by averaging cell counts in twenty random 400× fields. Other data were performed in three independent experiments, and values are presented as mean ± SEM. Significant differences was determined by using the multiple comparisons of Dunnett's post‐hoc test for *P < 0.05 and **P < 0.01 compared to Aβ only groups. Scale bar represents 50 μm.
Linagliptin Restores Aβ‐induced Insulin Signaling Blockade in Neuronal Cells
Because the mechanism of DPP‐4 inhibitors is to increase endogenous incretin levels, we proposed that linagliptin might exert neuroprotective effects by enhancing GLP‐1 downstream signaling. To determine whether linagliptin‐mediated neuroprotection operates via alteration of GLP‐1 receptor expression in the SK‐N‐MC cells, we performed immunocytochemical staining. As shown in Figure 3A, both Aβ and linagliptin treatment did not markedly alter the cellular distribution and expression of GLP‐1 receptor in SK‐N‐MC cells. Similarly, the constant expression of GLP‐1 receptor was also confirmed by western blotting (Figure 3B). Several lines of evidence have indicated that linagliptin can enhance insulin action and plays an important role in improving insulin sensitivity in peripheral tissues 24. To further elucidate the molecular mechanism of linagliptin‐mediated neuroprotection, Western blot analysis was conducted to detect the levels of phospho insulin receptor substrate‐1 (IRS‐1) at residue Ser307 and Tyr in SK‐N‐MC cells. As shown in Figure 3C, Aβ treatment for 24 h caused a significant increase in Ser307 IRS‐1 phosphorylation, which is recognized as a hallmark of insulin resistance. Accordingly, Aβ also prevented IRS‐1 phosphotyrosine (4G10 clone) expression. By contrast, co‐treated with linagliptin returned the Tyr phosphorylation of IRS‐1 and its downstream target Akt to basal levels, showing that the neuronal insulin signaling can be activated by linagliptin during Aβ treatment. To gain insight into the downstream effects of Akt in the presence of linagliptin, we investigated the glycogen synthase kinase 3β (GSK3β), a direct phosphorylation target of Akt. After 24 h of Aβ treatment, the Ser9 phosphorylation of GSK3β was markedly inhibited, which indicates the Aβ‐suppressed Akt pathway leads to activation of GSK3β (Figure 3D). However, linagliptin could reduce GSK3β activity by increasing Akt‐mediated GSK3β Ser9 phosphorylation during Aβ treatment. This linagliptin‐mediated neuroprotection was also confirmed by inhibiting Thr231 phosphorylation of one GSK3β's downstream substrate tau, which was recognized as one of the crucial pathological hallmarks of AD. To further investigate the role of insulin signaling in linagliptin‐mediated neuroprotection, the PI3‐kinase inhibitor LY294002 was used as negative control. As shown is Figure 3D and 3E, LY294002 significantly blocked the linagliptin‐restored Akt signaling and cell viability during Aβ treatment. This indicates that Aβ‐impaired insulin signaling may trigger neuronal apoptosis; however, linagliptin effectively reduces Aβ‐induced cytotoxicity by returning the blocked neuronal insulin signaling.
Figure 3.
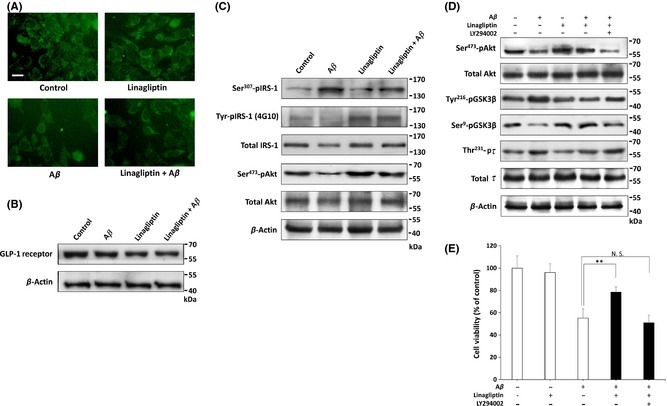
Linagliptin alleviates Aβ‐impaired insulin downstream signaling in SK‐N‐MC neuronal cells. (A) Immunofluorescence images show that the cellular distribution of GLP‐1 receptor is not altered by treatment with Aβ (2.5 μM), linagliptin (50 μM) or in combination for 24 h. (B) Western blotting also reveals that the expression of GLP‐1 receptor is not altered by Aβ (2.5 μM) or linagliptin (50 μM) treatment for 24 h in SK‐N‐MC cells. (C) Immunoblotting reveals that phosphorylation of Tyr‐IRS‐1 and Ser473‐Akt are inhibited when cells are exposed to Aβ (2.5 μM) for 24 h, and this inhibition is effectively restored by linagliptin (50 μM). (D) Western blotting shows that 50 μM of linagliptin‐activated Akt leads to the Ser9 phosphorylation of GSK3β, resulting in the inhibition of tau Thr231 phosphorylation by Aβ (2.5 μM) for 24 h. (E) Cell viability is determined by MTT assay, and the linagliptin‐mediated neuroprotective effects are abolished by the co‐treatment of LY294002 (20 μM), a specific inhibitor of PI3‐kinase. All data were performed in three independent experiments, and values are presented as mean ± SEM. Significant differences was determined by using the multiple comparisons of Dunnett's post‐hoc test for *P < 0.05 and **P < 0.01. Scale bar represents 20 μm.
Linagliptin Protects Cells Against Aβ‐Induced Intracellular ROS Accumulation and Mitochondria Dysfunction
Previous studies show strong evidence that Aβ‐induced ROS accumulation and mitochondrial dysfunction are both potential pathogenic markers in AD 25. To determine whether linagliptin protects cells from Aβ‐induced oxidative stress, we measured intracellular ROS levels by a 2′,7′‐dichlorofluorescin diacetate (DCFH‐DA) fluorometric method. As expected, our results showed that linagliptin suppresses Aβ‐induced ROS intracellular accumulation, which in turn protects cells from oxidative stress (Figure 4A). It is also suggested that increased ROS levels may inhibit AMP‐activated protein kinase (AMPK) activity, which is likely to promote the development of insulin resistance 26. To test whether such mechanism is also involved in SK‐N‐MC cells, the phosphorylation of AMPK was determined by immunoblotting in Figure 4B. Our results showed that Aβ significantly downregulates the Thr172 phosphorylation of AMPK, whereas this effect was counteracted by co‐treatment of linagliptin. Furthermore, western blot analysis of AMPK downstream target sirtuin 1 (Sirt1) and superoxide dismutase 1 (SOD1) protein expressions also provided evidence that levels of these antioxidative pathways are increased significantly by linagliptin compared to Aβ only groups. As discussed previously, the pathogenic role of Aβ in mediating ROS accumulation was often accompanied by mitochondrial dysfunction. To further examine the details of linagliptin‐mediated neuroprotection, we performed JC‐1 staining to assess the mitochondrial membrane potential. As shown in Figure 4C, JC‐1 aggregates were found in healthy mitochondria by a red fluorescence in nontreated controls. However, exposure of cells to Aβ resulted in significant increases in green fluorescence, indicating a loss of mitochondrial membrane potential. On the contrary, co‐treatment with linagliptin reduced the deteriorating effects of Aβ on mitochondrial membrane potential. We further confirmed that insulin signaling inhibition results in an oxidative stress damage by Aβ, as the PI3‐kinase inhibitor LY294002 significantly attenuated linagliptin‐mediated antioxidative effects, suggesting these benefits may depend on linagliptin‐improved insulin sensitivity in neuronal cells.
Figure 4.
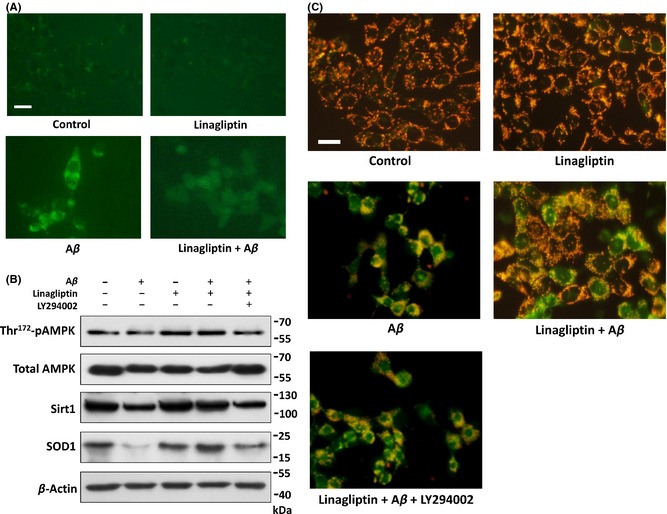
Linagliptin reduces Aβ‐induced intracellular ROS accumulation and improves mitochondria dysfunction. (A) Effects of linagliptin (50 μM) in reducing 2.5 μM of Aβ‐induced intracellular ROS accumulation determined by dichlorofluorescin diacetate (DCFH‐DA) staining under microscope. (B) Effects of linagliptin (50 μM), Aβ (2.5 μM), and LY294002 (20 μM) on Thr172 phosphorylation of AMPK, and the protein levels of AMPK, Sirt1 and SOD1 by immunoblotting. (C) JC‐1 immunofluorescent staining. Green fluorescence represents Aβ‐induced mitochondrial dysfunction by dissipation of mitochondrial membrane potential. Red fluorescence indicates that co‐treatment with linagliptin (50 μM) preserved an intact mitochondrial membrane potential compared with the group treated with Aβ (2.5 μM) alone. LY294002 (20 μM), a specific inhibitor of PI3‐kinase. Scale bar represents 20 μm.
Peripheral Blood Leukocyte Sirt1 mRNA Expression is Partially Returned by Linagliptin in Diabetic Patients with AD
Sirt1 is an important modulator in humans in the protection against oxidative events. A previous study reported that the Sirt1 mRNA expression level is suppressed in blood samples obtained from patients with AD or T2D 27, 28. However, it remains unclear whether the reduction of Sirt1 mRNA is more susceptible to diabetic patients with AD. To evaluate Sirt1 inhibition in diabetic AD patients, peripheral leukocytes were isolated and Sirt1 mRNA levels were determined with fourteen human subjects with clinically diagnosed AD (six pure AD, four diabetic AD without linagliptin treatments, and four diabetic AD with linagliptin treatments at least for 6 months). A detailed overview of the patient's characteristics is summarized in Table 1. In line with our preliminary expectation, both mini‐mental state examination (MMSE) scores and Sirt1 mRNA expressions were lower in patients with diabetic AD as compared to pure patients with AD. However, the MMSE scores and expressions of Sirt1 mRNA were significantly restored in diabetic patients with AD treated with linagliptin (Table 1). This observation consists the idea that patients with diabetic AD expressed reduced Sirt1 by inhibition of incretin signaling, which may contribute to pathogenesis of neurodegeneration.
Table 1.
Characteristics of patients with AD, and diabetic AD treated with or without linagliptin
| Characteristics | Non‐diabetic AD (n = 6) | Diabetic AD no linagliptin (n = 4) | Diabetic AD treated with linagliptin (n = 4) |
|---|---|---|---|
| Age, years | 81.3 ± 5.4 | 83.5 ± 4.5 | 79.5 ± 3.4 |
| Sex (male/female) | 3/3 | 2/2 | 2/2 |
| MMSE score | 20.6 ± 2.0a | 15.5 ± 3.4 | 20.3 ± 4.1 |
| Sirt1 mRNA (folds) | 2.6 ± 0.66a | 1.0 ± 0.29 | 1.88 ± 0.28a |
AD, Alzheimer's disease; MMSE, mini‐mental status examination.
Values shown are means ± SD.
P < 0.05 compared to diabetic AD groups.
Discussion
Interestingly, accumulating evidence indicates a strong link between T2D and AD, highlighting the key role of insulin signaling in the pathogenesis of these diseases. Although the underlying mechanism remains largely unknown, now the evidence for it has become very significant 29. In fact, Steen et al. have firstly proposed a connection between increased insulin resistance in the brain with AD and termed it as “type 3 diabetes” 30, hinting that insulin‐based therapies may be useful in the treatment of AD. Traditionally, the majority of insulin in the brain is generated from pancreatic β‐cells and transported across the blood–brain barrier (BBB). However, insulin can be locally synthesized and released by neurons 31. Moreover, GLP‐1 and its receptor are also known to ubiquitously express in central nervous system (CNS), particularly in hypothalamus, cortex and hippocampus that typically vulnerable in patients with AD 32. Therefore, it is not surprising that the locally produced GLP‐1 may be upregulated by treatment with a DPP‐4 inhibitor which stimulates insulin downstream effects related to neuronal functions. Regarding the insulin protective effects in CNS, we provided evidence that linagliptin can protect neuronal cells against Aβ‐induced neurotoxicity likely by blocking DPP‐4 makes GLP‐1 levels rise, which increases insulin release and restores insulin signaling impairment. As brain GLP‐1 has been suggested to be neuroprotective 33, it is possible that DPP‐4 inhibitors such as linagliptin may represent a promising strategy against Aβ‐induced neurodegeneration.
Insulin resistance and mitochondrial dysfunction are the two common features both in AD and T2D 34. As previous mentioned, mitochondrial dysfunction leads to impairment of insulin sensitivity by reduced activity of AMPK, an important cellular fuel sensor and regulator 35. Aβ was found to cause ROS accumulation and oxidative damage in the brain, which is believed to play a pivotal role in the development of insulin resistance 36. Interestingly, GLP‐1 has been reported to stimulate AMPK activation in preventing the ROS production and vice versa 37. Additionally, AMPK activation has also been suggested to enhance insulin sensitivity by GLP‐1 agonist liraglutide 38. This indicates that AMPK may play a key role in response to Aβ exposure by DPP‐4 inhibition. In accordance with these findings, we found that the Thr172 phosphorylation of AMPK could be reduced during the incubation of cells with Aβ, and this inhibition was prevented by linagliptin co‐treatment. Moreover, we also observed that linagliptin protects mitochondrial function and suppresses intracellular ROS accumulation depends on insulin signaling pathways. These observations were further confirmed by Sirt1, a well‐known longevity factor, is in fact upregulated by linagliptin. By linagliptin treatment, AMPK can trigger its downstream target Sirt1, which was reported previously in triggering antioxidant pathways such as SOD 39. Considering the important roles of the Aβ‐induced oxidative stress in AD pathogenesis, our research unveils a new neuroprotective mechanism by which linagliptin suppresses oxidative damage and preserves mitochondria function through restoration of neuronal insulin signaling.
Recently, Kosaraju et al. 40, 41 observed that inhibition of DPP‐4 ameliorates streptozotocin‐induced memory loss and neuronal death in rats, indicating the possibility of using of these agents for the treating diabetes‐associated AD. Their results revealed a significant improvement in a dose‐dependent attenuation of Aβ production, tau hyperphosphorylation and cognitive deficits by upregulation of GLP‐1 signaling. These robust therapeutic effects of DPP‐4 inhibitors demonstrate a unique mechanism for Aβ‐related pathology observed in AD. However, previous study has demonstrated that linagliptin does not pass through the BBB easily 42, whereas GLP‐1 could be able to effectively penetrate into the brain 43. Because linagliptin has been suggested to have direct neuroprotective effects, we postulate that linagliptin treatment may increase levels of brain blood GLP‐1 and confers its neuroprotection. This is further supported by the fact that linagliptin‐mediated neuroprotection occurs directly at the neuronal level because the brain expression of GLP‐1 receptors is exclusively in neurons 44. However, further evaluation is necessary to confirm the neuroprotective effect of linagliptin in patients with AD. Collectively, in the present study we provided evidence for the view that linagliptin inhibits neurotoxicity induced by Aβ. This protection appears to be associated with the insulin signaling‐dependent AMPK activation and the Sirt1‐elicited antioxidant pathways such as SOD1. To our knowledge, this is the first report demonstrating the AMPK‐Sirt1 molecular mechanism of linagliptin against Aβ‐induced insulin signaling impairment and oxidative damage. Our report therefore provides new insights that incretin‐based agents such as linagliptin may be a potential useful therapeutic approach to AD.
Conflict of Interest
The authors declare no conflict of interest.
Acknowledgments
This work was supported by grants from the Chung Shan Medical University Hospital (CSH‐2014‐C‐015) and from the Ministry of Science and Technology (101‐2320‐B‐040‐015‐MY3 and 103‐2314‐B‐040‐011). Pure linagliptin was a kind gift from Boehringer Ingelheim Pharmaceuticals (Biberach, Germany). The fluorescence microscope and imaging analyzer were performed in the Instrument Center of Chung Shan Medical University, which is supported by Ministry of Science and Technology, Ministry of Education and Chung Shan Medical University.
The first two authors contributed equally to this work.
References
- 1. Spielman LJ, Little JP, Klegeris A. Inflammation and insulin/IGF‐1 resistance as the possible link between obesity and neurodegeneration. J Neuroimmunol 2014;273:8–21. [DOI] [PubMed] [Google Scholar]
- 2. de la Monte SM, Tong M. Brain metabolic dysfunction at the core of Alzheimer's disease. Biochem Pharmacol 2014;88:548–559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Zemva J, Schubert M. Central insulin and insulin‐like growth factor‐1 signaling: Implications for diabetes associated dementia. Curr Diabetes Rev 2011;7:356–366. [DOI] [PubMed] [Google Scholar]
- 4. Kleinridders A, Ferris HA, Cai W, Kahn CR. Insulin action in brain regulates systemic metabolism and brain function. Diabetes 2014;63:2232–2243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Butterfield DA, Swomley AM, Sultana R. Amyloid beta‐peptide (1‐42)‐induced oxidative stress in Alzheimer disease: Importance in disease pathogenesis and progression. Antioxid Redox Signal 2013;19:823–835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Neumann KF, Rojo L, Navarrete LP, Farias G, Reyes P, Maccioni RB. Insulin resistance and Alzheimer's disease: Molecular links & clinical implications. Curr Alzheimer Res 2008;5:438–447. [DOI] [PubMed] [Google Scholar]
- 7. Rulifson EJ, Kim SK, Nusse R. Ablation of insulin‐producing neurons in flies: Growth and diabetic phenotypes. Science 2002;296:1118–1120. [DOI] [PubMed] [Google Scholar]
- 8. Heras‐Sandoval D, Ferrera P, Arias C. Amyloid‐beta protein modulates insulin signaling in presynaptic terminals. Neurochem Res 2012;37:1879–1885. [DOI] [PubMed] [Google Scholar]
- 9. Bhat NR, Thirumangalakudi L. Increased tau phosphorylation and impaired brain insulin/IGF signaling in mice fed a high fat/high cholesterol diet. J Alzheimers Dis 2013;36:781–789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Tokutake T, Kasuga K, Yajima R, et al. Hyperphosphorylation of Tau induced by naturally secreted amyloid‐beta at nanomolar concentrations is modulated by insulin‐dependent Akt‐GSK3beta signaling pathway. J Biol Chem 2012;287:35222–35233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Quintanilla RA, Dolan PJ, Jin YN, Johnson GV. Truncated tau and Abeta cooperatively impair mitochondria in primary neurons. Neurobiol Aging 2012;33:619 e25–619 e35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Alagiakrishnan K, Sankaralingam S, Ghosh M, Mereu L, Senior P. Antidiabetic drugs and their potential role in treating mild cognitive impairment and Alzheimer's disease. Discov Med 2013;16:277–286. [PubMed] [Google Scholar]
- 13. Drucker DJ, Nauck MA. The incretin system: Glucagon‐like peptide‐1 receptor agonists and dipeptidyl peptidase‐4 inhibitors in type 2 diabetes. Lancet 2006;368:1696–1705. [DOI] [PubMed] [Google Scholar]
- 14. Grigoropoulou P, Eleftheriadou I, Zoupas C, Diamanti‐Kandarakis E, Tentolouris N. Incretin‐based therapies for type 2 diabetes mellitus: Effects on insulin resistance. Curr Diabetes Rev 2013;9:412–417. [DOI] [PubMed] [Google Scholar]
- 15. Deeks ED. Linagliptin: A review of its use in the management of type 2 diabetes mellitus. Drugs 2012;72:1793–1824. [DOI] [PubMed] [Google Scholar]
- 16. Holscher C. Incretin analogues that have been developed to treat type 2 diabetes hold promise as a novel treatment strategy for Alzheimer's disease. Recent Pat CNS Drug Discov 2010;5:109–117. [DOI] [PubMed] [Google Scholar]
- 17. Holscher C. Central effects of GLP‐1: New opportunities for treatments of neurodegenerative diseases. J Endocrinol 2014;221:T31–T41. [DOI] [PubMed] [Google Scholar]
- 18. Xiong H, Zheng C, Wang J, et al. The neuroprotection of liraglutide on Alzheimer‐like learning and memory impairment by modulating the hyperphosphorylation of tau and neurofilament proteins and insulin signaling pathways in mice. J Alzheimers Dis 2013;37:623–635. [DOI] [PubMed] [Google Scholar]
- 19. Holscher C. Potential role of glucagon‐like peptide‐1 (GLP‐1) in neuroprotection. CNS Drugs 2012;26:871–882. [DOI] [PubMed] [Google Scholar]
- 20. Thomas L, Eckhardt M, Langkopf E, Tadayyon M, Himmelsbach F, Mark M. (R)‐8‐(3‐amino‐piperidin‐1‐yl)‐7‐but‐2‐ynyl‐3‐methyl‐1‐(4‐methyl‐quinazolin‐2‐ylm ethyl)‐3,7‐dihydro‐purine‐2,6‐dione (BI 1356), a novel xanthine‐based dipeptidyl peptidase 4 inhibitor, has a superior potency and longer duration of action compared with other dipeptidyl peptidase‐4 inhibitors. J Pharmacol Exp Ther 2008;325:175–182. [DOI] [PubMed] [Google Scholar]
- 21. Kern M, Kloting N, Niessen HG, et al. Linagliptin improves insulin sensitivity and hepatic steatosis in diet‐induced obesity. PLoS One 2012;7:e38744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ono K, Yamada M. Antioxidant compounds have potent anti‐fibrillogenic and fibril‐destabilizing effects for alpha‐synuclein fibrils in vitro. J Neurochem 2006;97:105–115. [DOI] [PubMed] [Google Scholar]
- 23. Kosaraju J, Gali CC, Khatwal RB, et al. Saxagliptin: A dipeptidyl peptidase‐4 inhibitor ameliorates streptozotocin induced Alzheimer's disease. Neuropharmacology 2013;72:291–300. [DOI] [PubMed] [Google Scholar]
- 24. Fruci B, Giuliano S, Mazza A, Malaguarnera R, Belfiore A. Nonalcoholic Fatty liver: A possible new target for type 2 diabetes prevention and treatment. Int J Mol Sci 2013;14:22933–22966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Pinho CM, Teixeira PF, Glaser E. Mitochondrial import and degradation of amyloid‐beta peptide. Biochim Biophys Acta 2014;1837:1069–1074. [DOI] [PubMed] [Google Scholar]
- 26. Ruderman NB, Carling D, Prentki M, Cacicedo JM. AMPK, insulin resistance, and the metabolic syndrome. J Clin Invest 2013;123:2764–2772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Furuya TK, da Silva PN, Payao SL, et al. SORL1 and SIRT1 mRNA expression and promoter methylation levels in aging and Alzheimer's Disease. Neurochem Int 2012;61:973–975. [DOI] [PubMed] [Google Scholar]
- 28. de Kreutzenberg SV, Ceolotto G, Papparella I, et al. Downregulation of the longevity‐associated protein sirtuin 1 in insulin resistance and metabolic syndrome: Potential biochemical mechanisms. Diabetes 2010;59:1006–1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Vagelatos NT, Eslick GD. Type 2 diabetes as a risk factor for Alzheimer's disease: The confounders, interactions, and neuropathology associated with this relationship. Epidemiol Rev 2013;35:152–160. [DOI] [PubMed] [Google Scholar]
- 30. Steen E, Terry BM, Rivera EJ, et al. Impaired insulin and insulin‐like growth factor expression and signaling mechanisms in Alzheimer's disease–is this type 3 diabetes? J Alzheimers Dis 2005;7:63–80. [DOI] [PubMed] [Google Scholar]
- 31. Duarte AI, Moreira PI, Oliveira CR. Insulin in central nervous system: More than just a peripheral hormone. J Aging Res 2012;2012:384017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Hamilton A, Patterson S, Porter D, Gault VA, Holscher C. Novel GLP‐1 mimetics developed to treat type 2 diabetes promote progenitor cell proliferation in the brain. J Neurosci Res 2011;89:481–489. [DOI] [PubMed] [Google Scholar]
- 33. Talbot K, Wang HY. The nature, significance, and glucagon‐like peptide‐1 analog treatment of brain insulin resistance in Alzheimer's disease. Alzheimers Dement 2014;10:S12–S25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. De Felice FG, Ferreira ST. Inflammation, defective insulin signaling, and mitochondrial dysfunction as common molecular denominators connecting type 2 diabetes to Alzheimer disease. Diabetes 2014;63:2262–2272. [DOI] [PubMed] [Google Scholar]
- 35. Corbi G, Conti V, Russomanno G, et al. Adrenergic signaling and oxidative stress: A role for sirtuins? Front Physiol 2013;4:324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Butterfield DA, Di Domenico F, Barone E. Elevated risk of type 2 diabetes for development of Alzheimer disease: A key role for oxidative stress in brain. Biochim Biophys Acta 2014;1842:1693–1706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Balteau M, Van Steenbergen A, Timmermans AD, et al. AMPK activation by glucagon‐like peptide‐1 prevents NADPH oxidase activation induced by hyperglycemia in adult cardiomyocytes. Am J Physiol Heart Circ Physiol 2014;307:H1120–H1133. [DOI] [PubMed] [Google Scholar]
- 38. Yamazaki S, Satoh H, Watanabe T. Liraglutide enhances insulin sensitivity by activating AMP‐activated protein kinase in male Wistar rats. Endocrinology 2014;155:3288–3301. [DOI] [PubMed] [Google Scholar]
- 39. Kitada M, Koya D. SIRT1 in type 2 diabetes: Mechanisms and therapeutic potential. Diabetes Metab J 2013;37:315–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kosaraju J, Madhunapantula SV, Chinni S, et al. Dipeptidyl peptidase‐4 inhibition by Pterocarpus marsupium and Eugenia jambolana ameliorates streptozotocin induced Alzheimer's disease. Behav Brain Res 2014;267:55–65. [DOI] [PubMed] [Google Scholar]
- 41. Kosaraju J, Murthy V, Khatwal RB, et al. Vildagliptin: An anti‐diabetes agent ameliorates cognitive deficits and pathology observed in streptozotocin‐induced Alzheimer's disease. J Pharm Pharmacol 2013;65:1773–1784. [DOI] [PubMed] [Google Scholar]
- 42. Fuchs H, Binder R, Greischel A. Tissue distribution of the novel DPP‐4 inhibitor BI 1356 is dominated by saturable binding to its target in rats. Biopharm Drug Dispos 2009;30:229–240. [DOI] [PubMed] [Google Scholar]
- 43. Kastin AJ, Akerstrom V, Pan W. Interactions of glucagon‐like peptide‐1 (GLP‐1) with the blood‐brain barrier. J Mol Neurosci 2002;18:7–14. [DOI] [PubMed] [Google Scholar]
- 44. Darsalia V, Ortsater H, Olverling A, et al. The DPP‐4 inhibitor linagliptin counteracts stroke in the normal and diabetic mouse brain: A comparison with glimepiride. Diabetes 2013;62:1289–1296. [DOI] [PMC free article] [PubMed] [Google Scholar]