

Abstract
Velaglucerase alfa is a human β‐glucocerebrosidase approved for Gaucher disease type 1 (GD1) treatment. This report summarizes the 7‐year experience of the now‐completed phase I/II and extension studies of adult GD1 patients who received velaglucerase alfa. Ten patients who completed the 9‐month, phase I/II study entered the extension trial TKT025EXT, of which eight completed this study. Doses were reduced after a cumulative treatment period of 15 to 18 months. Although all patients experienced ≥1 adverse event, no patient withdrew due to a drug‐related adverse event or required premedication. No patient developed anti‐drug antibodies, compliance remained high (median 98%), and seven of eight eligible patients transitioned to home infusions under supervision by healthcare professionals. Statistically significant improvements were observed for efficacy parameters: mean percentage changes from baseline (95% confidence intervals) were 18% (12%, 24%) for hemoglobin concentration, 115% (66%, 164%) for platelet counts, and −42% (−53%, −31%) and −78% (−94%, −62%) for liver and spleen volumes, respectively. Improvements were also observed for secondary endpoints chitotriosidase and CCL18 levels and exploratory endpoints (bone mineral density [BMD], bone marrow burden [BMB] scores). Normalization to near‐normalization of individuals' hemoglobin concentrations, platelet counts, liver volumes, and BMB scores was observed, and there were marked improvements in spleen volumes, biomarkers, and BMD. TKT025EXT represents the longest, prospective clinical trial for GD1 treatment to date and suggests that, despite dose reduction within 18 months of initiating therapy, velaglucerase alfa was generally well tolerated and was associated with marked improvement, including near normalization and/or normalization of key GD1 disease parameters. Am. J. Hematol. 90:577–583, 2015. © 2015 The Authors. American Journal of Hematology published by Wiley Periodicals, Inc.
Introduction
Velaglucerase alfa (VPRIV [GA‐GCB], Shire, Lexington, MA) is a human recombinant β‐glucocerebrosidase that was designed and developed as an enzyme replacement therapy (ERT) for the treatment of Gaucher disease type 1 (GD1). Velaglucerase alfa is manufactured in a human cell line, HT‐1080, using gene activation technology® 1, 2. Following targeted recombination with a constitutively active promoter, the human wild type GBA gene is expressed. The intracellular post‐translational glycosylation of the expressed protein is modified somewhat by the addition to cell culture of the mannose I inhibitor, kifunensine, resulting in a secreted enzyme containing predominantly high‐mannose type glycans, which serve to enhance macrophage uptake of the highly purified enzyme 1.
The clinical development program for velaglucerase alfa was initiated with a phase I/II trial (TKT025). This trial was a 9‐month, single‐arm, single‐center study undertaken to evaluate the safety and clinical activity of velaglucerase alfa among adults with GD1. Twelve patients were enrolled and received velaglucerase alfa intravenously at a dose of 60 U/kg body weight every other week (EOW) 2. Ten patients completed the phase I/II trial and were eligible to participate in a phase I/II extension study (TKT025EXT). The extension study had many unique attributes, one being a step‐wise dose reduction at 15 to 18 months to 30 U/kg body weight EOW for patients who met two of the four goals for hematology and viscera 2.
On the basis of the phase I/II trial findings, two phase III trials were conducted in treatment‐naïve pediatric and adult patients with GD1. TKT032 was a 12‐month, two‐arm, multicenter trial designed to evaluate two doses of intravenous velaglucerase alfa (45 U/kg or 60 U/kg, EOW), whereas HGT‐GCB‐039 was a 9‐month, two‐arm noninferiority trial with imiglucerase (Cerezyme®, Genzyme Corp, Cambridge, MA) serving as an active comparator 3, 4. In addition, a phase II/III trial, TKT034, was conducted as a 12‐month, single‐arm, open‐label multicenter trial examining the safety and efficacy of velaglucerase alfa in a population of clinically stable pediatric and adult patients with GD1 switched from imiglucerase to velaglucerase alfa at their previously stable dose 5. All subjects completing the phase II/III and phase III trials were eligible to enroll into a combined extension trial (HGT‐GCB‐044). Lastly, following an FDA request, a treatment protocol (HGT‐GCB‐058) was undertaken to enable the provision of velaglucerase alfa to patients with GD1 who were faced with imiglucerase dose reductions or treatment interruption due to imiglucerase supply constraints 6.
In total, these clinical trials enrolled 322 patients; 77 were treatment‐naïve and 245 were GD1 patients switched from imiglucerase. Collectively, these studies demonstrated that velaglucerase alfa was generally well tolerated among both treatment‐naïve and switch patients including children, adults, pregnant patients, and geriatric patients 2, 3, 4, 5, 6. There were few drug‐related serious adverse events (AEs) and the vast majority of AEs were classified by the investigators as infusion related and considered to be mild to moderate and transient in nature. One case of anaphylactoid reaction was reported (<1%) 5. Overall, <1% of patients developed anti‐drug antibodies following exposure to velaglucerase alfa and no patients developed IgE antibodies 2, 3, 4, 5, 6.
Among treatment‐naïve patients in the phase III trials, as in the seminal report of the phase I/II trial, improvements were seen in key clinical parameters for GD1 (hemoglobin concentration, platelet count, spleen volume and liver volume). Moreover, among patients who participated in the phase II/III switch trial, hemoglobin concentrations, platelet counts as well as liver and spleen volumes remained stable following the transition to velaglucerase alfa.
Based on the clinical trial findings, the FDA approved velaglucerase alfa in 2010 for “long‐term ERT for pediatric and adult patients with type 1 Gaucher disease” 7. Velaglucerase alfa has subsequently been approved in more than 40 countries globally and it is the only commercially available ERT with the wild type amino acid sequence.
Since initial drug approval, additional post hoc analyses of the phase I/II extension study have been published. An analysis of therapeutic goals following 48 months of velaglucerase alfa treatment demonstrated improvements across five therapeutic goals (hemoglobin concentration, platelet counts, liver volume, spleen volume, and bone mineral density [BMD]). In addition, an analysis of bone response through 69 months of velaglucerase alfa therapy suggested improvements in two exploratory endpoints, BMD and bone marrow burden (BMB) 8, 9.
As the phase I/II extension study has now been completed, this report summarizes the 7‐year experience of the phase I/II trial and phase I/II extension study combined (TKT025 and TKT025EXT) and represents the longest, prospective, ERT clinical trial for GD1.
Methods
Ethics and trial conduct
The phase I/II trial TKT025 and the extension study TKT025EXT were single‐center clinical studies conducted at the Gaucher Clinic, Shaare Zedek Medical Center in Jerusalem, Israel. Approvals from the Institutional Helsinki (Ethics) Committee and the Israeli Ministry of Health were obtained for both these studies, which also complied with US FDA regulations. The phase I/II trial started in April 2004, before the requirement for clinical study registration that was introduced in July 2005. The extension study was registered with the National Institutes of Health (registry number NCT00391625; www.clinicaltrials.gov/ct/show/NCT00391625) 2.
Patients
The 12 patients who enrolled in the phase I/II trial were symptomatic adults (≥18 years of age) with biochemically confirmed diagnoses of GD1 (deficient enzyme activity). Inclusion criteria included GD‐related anemia (hemoglobin concentration ≥1 g/dL below the lowest value of the sex‐specific normal range used by the local laboratory) and thrombocytopenia (platelet count below the normal range) as well as negative serology for hepatitis B and C antigens and human immunodeficiency virus. Patients who were not naïve to (imiglucerase) ERT were only eligible if they tested negative for anti‐imiglucerase antibodies and were last treated >12 months before study enrollment. Prior splenectomy, an inability to comply with the protocol, and treatment with an investigational therapy in the 30 days before enrollment were key exclusion criteria. The extension study was open to patients who completed the 9‐month phase I/II trial 2.
In both studies, the use of a medically acceptable form of contraception was required of all female patients of child‐bearing potential, as well as a negative serum pregnancy test on enrollment and continually negative tests at set intervals throughout the studies 2.
Velaglucerase alfa preparation and dosing
As detailed previously, Shire (Lexington, MA) supplied the enzyme, which was administered intravenously at the clinical trial site 2. In the phase I/II trial, the dose administered to patients was 60 U/kg body weight over 60 min EOW. In the extension study, a step‐wise dose reduction from 60 to 30 U/kg was implemented after a cumulative study period of 15 to 18 months (after the phase I/II trial and 6 to 9 months in the extension study) in patients in whom at least two of four therapeutic goals were achieved as previously described 10. Patients enrolled in the extension study who did not experience a drug‐related serious AE or an infusion‐related AE were eligible to receive home‐based infusions under the supervision of a healthcare professional. The option for home therapy was added to the protocol after the extension study start date.
Assessments of safety
Evaluating the safety of velaglucerase alfa was the primary objective of the initial trial and the extension study and the safety assessments were: AE assessments, recording concomitant medications, measurement of vital signs (blood pressure, pulse, respirations, temperature), a complete organ system physical examination and evaluation by a medical doctor, 12‐lead electrocardiograms with interpretation by a senior cardiologist when necessary, standard echocardiograms plus Doppler, and pulmonary function tests (PFTs). The tricuspid incompetence (TI) gradient was measured with echocardiograms and the PFTs included the following standard parameters: forced vital capacity, forced expiratory volume, expiratory reserve volume, total lung capacity, residual volume, uncorrected and corrected diffusion capacity (DLCO), alveolar volume (VA), and DL/VA, and interpretation by a senior pulmonologist. Clinical laboratory testing (serum chemistry, hematology, urinalysis, blood pregnancy tests, and serology for anti‐velaglucerase alfa antibodies) was done at a central laboratory 2.
Clinical activity (efficacy) measurements
Evaluation of clinical activity was a secondary objective of this study. The primary clinical activity parameters were hemoglobin concentration, platelet count, liver volume, and spleen volume. The biomarkers plasma chitotriosidase and CCL18 comprised the secondary clinical activity parameters. BMD, BMB score, and a summary of skeletal abnormalities from a plain film skeletal survey were included as exploratory clinical activity measures.
Hemoglobin concentration and platelet counts were measured at baseline and then at least every 3 months in the phase I/II and extension studies. Baseline was defined as before the first dose in the phase I/II trial and all time points reported are relative to baseline. In the analysis of the results, normal hemoglobin concentration was defined as ≥11 g/dL for women and ≥12 g/dL for men. Any values below these limits indicated anemia (this was different from the definition of anemia used to determine eligibility for the phase I/II trial). Platelet counts were considered normal if ≥150 × 109/L and pathologic if <100 × 109/L 11.
Quantitative abdominal magnetic resonance imaging (MRI) was used to measure the volumes of the liver and spleen at baseline and 6, 9, 24, 33, 45, 57, 69, and 81 months. The senior radiologist assessing the images was blinded concerning the patients' identities and the chronological order of the MRI assessments.
Blood was sampled also to determine plasma chitotriosidase and CCL18 levels (samples analyzed at the Academic Medical Center in Amsterdam, The Netherlands). BMD and BMB were evaluated using dual‐energy X‐ray absorptiometry (DXA) and MRI, respectively, of the lumbar spine and femora as previously described 8, 9. Each patient enrolled into the TKT025 trial underwent an annual full‐skeletal survey (plain X‐rays) with a clinical reading by a radiologist who had experience in GD.
Treatment compliance
If a patient was outside of the 17‐day maximum visit window allowed, the patient was to receive the overdue infusion as soon as possible after approval from the Shire Medical Monitor to continue in the study. It was acceptable to give the next “catch up” infusion as early as 7 days later. The subsequent infusions would then return to the original schedule.
The proportion of actual infusions received relative to the number of infusions that were scheduled over the patient's participation in the study was calculated and presented as the compliance rate.
Statistical analyses
Both the safety population and the intention‐to‐treat (ITT) population comprised all patients who were enrolled in the extension study and in it received at least one full or partial velaglucerase alfa infusion (n = 10). The completer population, a supportive analysis population for clinical activity parameters, was defined as the subset of patients who completed the extension study (n = 8).
Mean within‐patient changes and percentage changes from baseline to annual protocol‐defined time points were calculated for hemoglobin concentration, platelet count, liver volume, spleen volume, chitotriosidase and CCL18, with associated two‐sided 95% confidence intervals around the means. In ITT analyses, intermittent missing postbaseline values were imputed using last observation carried forward. There was no data imputation for analyses in the completer population.
Patients with a baseline chitotriosidase level <5 nmol/mL/h (lower limit of detection) were excluded from the analysis of chitotriosidase. This threshold for deficiency was recommended and provided by the Academic Medical Center (Amsterdam, The Netherlands).
Analyses of BMD measurements, BMB scores, and therapeutic goal attainment are detailed elsewhere 8, 9, 12.
Results
Of the 10 patients enrolled into TKT025EXT, eight (80%) completed the trial at 7 years: one female patient withdrew consent because of a desire to become pregnant and another female patient was withdrawn by the sponsor following a positive pregnancy test. Demographic characteristics of the ITT and completer cohorts are presented in Table 1.
Table 1.
Baseline Characteristics
| ITT population (n = 10) | Completer population (n = 8) | |
|---|---|---|
| Median agea, years (range) | 36.0 (19–63) | 39.5 (19–63) |
| Male sex, n (%) | 4 (40.0) | 4 (50.0) |
| Gaucher disease genotype, n (%) | ||
| N370S/N370S | 4 (40.0) | 3 (37.5) |
| N370S/L444P | 1 (10.0) | 1 (12.5) |
| N370S/Other | 5 (50.0) | 4 (50.0) |
| Chitotriosidaseb, n (%) | ||
| Not deficient | 9 (90.0) | 7 (87.5) |
| Deficient | 1 (10.0) | 1 (12.5) |
| Clinical activity variables, median (range) | ||
| Hemoglobin concentration, g/dL | 11.10 (9.8–12.9) | 12.00 (10.1–12.9) |
| Platelet count ×109/L | 62.25 (37–98.5) | 64.00 (37–98.5) |
| Spleen volumec, MN | 19.00 (11–32.5) | 19.00 (11–28) |
| Liver volume, MN | 1.760 (1.04–2.32) | 1.620 (1.04–2.32) |
| Chitotriosidase activityd, nmol/mL/h | 23,375 (7391–68,552) | 23,375 (12,191–66,804) |
| CCL18 level, ng/mL | 1978.5 (1837–5247) | 1923.0 (1837–5247) |
Baseline is defined as data collected at the baseline visit in TKT025.
Age at the time TKT025 informed consent was obtained.
Chitotriosidase deficient is defined as values below 5 nmol/mL/h.
One patient was excluded due to an intravascular metallic device which precluded accurate spleen evaluation.
One chitotriosidase deficient patient was excluded.
MN: multiple of normal.
The 10 patients enrolled in TKT025EXT received a median of 76.9 months' exposure to velaglucerase alfa in the extension study. The median cumulative exposure (TKT025 + TKT025EXT) was 86.1 months, with a median number of infusions of 185. No infusions were only partially administered. In addition to the 20 bi‐weekly infusions received by the 10 patients completing TKT025, the eight completer patients received a median number of 165 bi‐weekly infusions during TKT025EXT.
Over the course of 7 years, the percentage compliance was high: the median percentage compliance over the entire study period (including TKT025) was 98% in both the ITT population and the completer population with only one patient (who travelled bi‐weekly from Eastern Europe) missing the most infusions, 7 of 194 (3.6%).
Seven of eight eligible patients received home infusions under medical supervision in TKT025EXT, and all seven patients remained on home therapy and completed the study. The eighth patient, though eligible for home therapy, continued to travel to Israel for her infusions as no authorized home infusion site was available locally. One patient who experienced an infusion‐related AE was not considered eligible and one patient discontinued from the study before home infusions were offered.
Safety outcomes
All patients who participated in TKT025EXT experienced at least one AE, with only two study drug‐related AEs (bone pain and fatigue), which occurred in one patient. The bone pain was also considered an infusion‐related AE. Bone pain in another patient prompted a dose increase from 30 U/kg to 60 U/kg (this patient was withdrawn because of pregnancy). None of the nine serious AEs in four patients (which included repair of an inguinal hernia, repair of an atrophic scar, ankle fracture, and worsening of pre‐existing osteonecrosis) was considered drug‐related; the remaining AEs were mild to moderate and all were transient (such as arthralgia, fever, and abdominal pain). The incidence of AEs decreased over time with the peak occurring during the first year. None of the patients discontinued from the study because of an AE.
No clinically significant abnormal tracings on electrocardiograms were seen.
Echocardiography and PFTs, undertaken to rule out possible onset of pulmonary hypertension 13, 14, revealed no pathology or safety concerns throughout the observation period and mean values remained within the normal range for age, ethnicity, and height 15.
No patient developed anti‐velaglucerase alfa antibodies over the 7‐year period. No patient required premedication or a decrease in their infusion rate.
Efficacy outcomes in key disease parameters
The clinically meaningful and/or statistically significant improvements at 9 months in TKT025 key disease parameters, hemoglobin concentration, platelet count, liver volume, spleen volume, and plasma biomarkers chitotriosidase and CCL18, were in some cases achieved as early as 3 to 6 months following intravenous velaglucerase alfa 60 U/kg. They were improved upon or sustained over 75 additional months of velaglucerase alfa EOW in the extension study (Fig. 1), which included a dose reduction to 30 U/kg at 15 to 18 months cumulative ERT.
Figure 1.
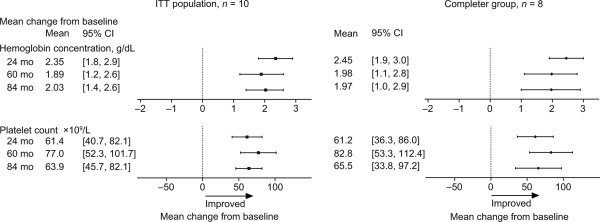
Mean changes from baseline in the ITT (n = 10) and completer (n = 8) cohorts in hemoglobin concentration and platelet count. Baseline was defined as before the first dose in the phase I/II trial. For the ITT analysis, imputation was applied to missing data. For the completer group analysis, imputation was not applied to missing data. In the completer group, n = 6 at 84 months.
In addition to the statistical improvement in the key clinical activity endpoints, these improvements were also found to be clinically relevant. All patients achieved normal hemoglobin levels within 24 months of initiation of therapy while the therapeutic goal for anemia at 1 year 10 was achieved in all patients by 9 months. Although all patients who entered the phase I/II trial exhibited moderate to severe thrombocytopenia at baseline, all eight completers achieved platelet counts in the nonpathologic range (five achieved near‐normal values, and three achieved normal values at their final assessment). The 1‐year therapeutic goal for increased platelet counts 10 was achieved by three (30%) patients at 6 months. The therapeutic goal for liver volume reduction of 1 year 10 was seen in four (40%) patients at 6 months, and normalization of hepatomegaly was observed by 45 months among all seven completers whose assessments were consistent with hepatomegaly at baseline. The therapeutic goal of 1 year for reduction of spleen volume 10 was seen at 6 months in nine (90%) patients. Although no patient exhibited normalization in spleen volume, a marked reduction (>74%) from baseline splenomegaly was nevertheless observed among all completers. In addition, four completers who exhibited a combination of anemia, moderate or severe thrombocytopenia, hepatomegaly, and splenomegaly at baseline demonstrated normalization of hemoglobin concentration and liver volumes, normalization or near normalization of their platelet counts, as well as marked improvements in splenomegaly (>74% reduction in spleen volumes).
In a combined analysis of therapeutic goals, 50% and 90% of patients achieved four of four (hematologic and organ volume) goals by 9 months and 24 months of velaglucerase alfa exposure, respectively. With a fifth therapeutic goal (BMD) having been added at 36 months, 100% of patients achieved five of five therapeutic goals by 48 months of exposure 12.
Laboratory tests and urinalysis
The only observed biochemical abnormalities were fluctuations (including within the normal range) in blood glucose levels towards the end of the trial extension in a 63‐year‐old patient with a strong family history of diabetes.
Skeletal survey
The youngest female patient (18 years old at baseline) and a 57‐year‐old male patient had no clinically significant findings at baseline; the other patients' baseline images demonstrated radiologic features consistent with GD1. Over the course of the 7 years, one patient had a traumatic ankle fracture at age 26 years (DXA showed osteopenia at both lumbar spine and femoral neck at the advent of the trial, which improved into the normal range by 69 months); the patient's ankle had evidence of sclerosis before the advent of the trial, the bone pain for which an increase of dose was allowed. There were also two rib fractures (traumatic) and a broken ankle in a single patient (aged 62 years at advent of the trial with signs of osteoporosis on a pretrial DXA scan and baseline skeletal survey, who was, therefore, receiving bisphosphonates from before advent of the trial). Finally, one patient underwent total hip replacement for osteonecrosis (at age 38 years, but whose DXA bone density was in the normal range at the contralateral hip), which had been documented in the skeletal survey and other imaging before the advent of the trial.
Completer set (n = 8)
A completer analysis was performed to evaluate the response of those patients who remained within the clinical trial throughout its duration. Figure 2 presents individual graphs for all patients for the efficacy endpoints and biomarkers and BMD Z‐scores at the lumbar spine and femoral neck. Figure 3 presents the completer group mean change by disease parameter over time.
Figure 2.
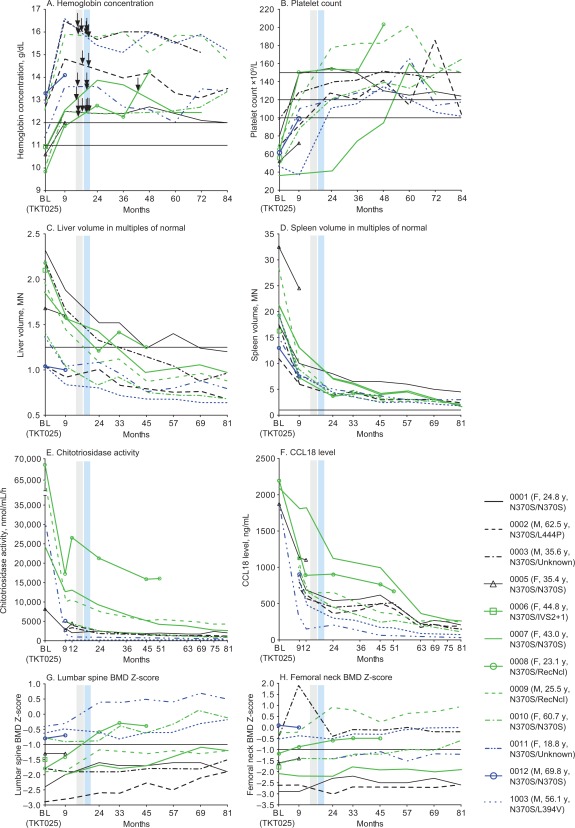
Individual patient data, all patients (n = 12) for key disease parameters (hemoglobin concentration, platelet count, liver volume, spleen volume) and plasma biomarkers (chitotriosidase and CCL18). Grey and blue vertical bands denote the two phases of dose reduction: 60 to 45 U/kg, 45 to 30 U/kg. The arrows indicate the times when individual patients had a dose adjustment; patient 8 had a dose increase after initiation of the reduced dose. Horizontal lines in panels A to D, G, and H indicate thresholds for normal values: in panel A, 11 g/dL for women and 12 g/dL for men; in panel B, 100, 120, and 150 × 109/L, denoting normal and “low‐normal” thresholds; in panel C, 1.25 MN; and in panel D, 1 MN. BL: baseline; MN: multiple of normal; BMD: bone mineral density.
Figure 3.
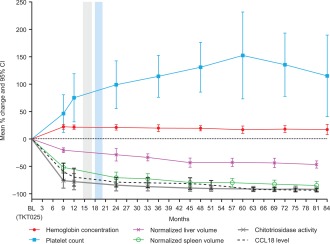
Results of the completer group, mean percentage changes by disease parameter over time; n = 8 except for hemoglobin and platelets at 84 months (where n = 6), for spleen volume at months 24, 33, 45, 57, 69, and 81 (where n = 7), and for chitotriosidase activity at all months (where n = 7). Grey and blue vertical bands denote the two phases of dose reduction that were implemented: 60 to 45 U/kg and 45 to 30 U/kg. BL: baseline.
Discussion
In Guide to Drug Development, Bert Spilker writes, “One of the most important clinical principles is that good data in a few patients are far better than mediocre or poor data in many patients” 16. This is particularly relevant to orphan drug development where few patients are available for well‐designed and well‐controlled trials.
Phase I and early phase II studies, in contrast with phase III trials, are often smaller, time‐limited, single‐arm trials that focus on safety and therapeutic potential. The velaglucerase alfa phase I/II and extension trials offer multiple unique features beyond the 9‐month assessment of safety and efficacy of velaglucerase alfa for GD1. Importantly, the full extension resulted in a 7‐year follow‐up under clinical trial conditions and thus unique features that could be documented include: the implementation of therapeutic goals 10 as the criteria for therapeutic dosing decisions such that dose reduction at 15 to 18 months related to achievement of therapeutic goals; the option for home therapy of an intravenous investigational drug (under medical supervision) within the constraints of a clinical trial; and the inclusion of two different bone disease parameters, BMD and BMB scoring 8, 9, as exploratory efficacy endpoints.
The importance of this report is the quality and quantity of the safety and efficacy results from the seminal trial and the extension trial, and the importance of a dose reduction at 15 to 18 months of cumulative exposure to velaglucerase alfa that did not result in diminution of response: this too was an important feature because it underscores the unimpeded safety while documenting the continuous trajectory for amelioration of disease‐specific parameters.
In presenting the completer data i.e., without imputing values and without carrying forth the last observation in the four patients who withdrew before the end of the extension trial, there is further evidence of the robustness of the response over time. In other words, the completer data was approximately comparable to that of the entire ITT cohort and as such, no individual withdrawal or single completer was the driver for the ultimate group values on any of the continua measured.
The safety observations reported herein are remarkable for the following. Throughout the 7‐year study period, compliance remained high; there were no study discontinuations because of an AE; there were no drug‐related serious or severe AEs; no patient exhibited any signs or symptoms suggestive of a drug‐related allergic reaction and consequently no patient required premedication. There were no drug‐related laboratory abnormalities, and all electrocardiographic, echocardiographic, and PFT studies remained essentially unchanged from baseline. Although electrocardiography is clearly a universal safety measure, the choice of the other two measures was Gaucher‐specific. At the time of initiation of this trial, there was concern about the possibility of either primary‐like 13 or secondary 14 pulmonary hypertension among patients with GD who may have been respectively treated with imiglucerase 13 or insufficiently treated with imiglucerase 14. Beyond the very mild natural diminution because of age in PFTs 15, there was no clinical evidence of pulmonary hypertension following prolonged exposure to velaglucerase alfa.
Among the noteworthy features in the safety profile is the fact that none of the patients developed anti‐velaglucerase alfa antibodies over the 7 years.
Given these assurances of safety, a benchmark decision was made to allow for home infusions within this clinical trial, for all infusions that were not protocol‐mandated in‐hospital infusions (i.e., those at quarterly or annual monitoring visits). Of eight patients eligible for home therapy, seven received medically‐supervised, home infusions and all seven remained on home therapy and completed the study. No drug‐related AEs were observed among the home‐infused patients. Furthermore, the tolerability of velaglucerase alfa combined with the availability of home therapy may have contributed to the high degree of protocol compliance observed, as home therapy has been reported to be less stressful and more convenient for patients compared to hospital based infusions 17. Beyond the implications to this study, the success of home therapy in this trial afforded the option of home therapy in the subsequent velaglucerase alfa clinical trials including the phase II/III study (TKT034), the phase III trials (TKT032, HGT‐GCB‐039), the treatment protocol (HGT‐GCB‐058), and the HGT‐GCB‐044 extension study.
In short, all the measures of safety and tolerability observed in the initial 9‐month trial were maintained throughout the 7 years, and no new concerns were raised of belated AEs and/or immunological responses including new anti‐drug antibody formation.
In addition to the reassuring safety profile, the 7‐year clinical results observed in this study are impressive. In the present study, statistically significant improvements from baseline in four key disease parameters (hemoglobin concentration, platelet counts, liver, and spleen volumes) were observed within the phase I/II study and no patient exhibited deterioration from baseline in any of these clinical parameters during the combined 7‐year study period. The results of the clinical activity endpoints were also clinically meaningful with all completers achieving normal hemoglobin levels, nonpathologic to normal platelet counts, normal liver volumes and marked reductions in spleen volumes. Most impressively, four completers who exhibited a combination of anemia, moderate or severe thrombocytopenia, hepatomegaly and splenomegaly at baseline demonstrated normalization of hemoglobin concentration and liver volumes, normalization or near normalization of their platelet counts, as well as marked improvements in splenomegaly (>74% reduction in spleen volumes), suggesting a comprehensive, therapeutic response. Finally, these results were observed despite a dose reduction between 15 and 18 months.
Patients did not all respond at the same rate. In particular, two patients had low platelet counts that did not improve in the first months, one of whom had a platelet count of 37 to 42 × 109/L until 24 months, whereas other patients had a definitive platelet response by 9 months. However, near‐normal platelet counts were achieved even in these apparently slow responders.
Regarding the trajectory of improvement in the major Gaucher‐specific parameters, one can appreciate that for the most part, each patient, despite moderate‐severe disease manifestations at baseline achieved or approached normal values, albeit the rate of improvements for each parameter and for each patient varied considerably. The concept of approaching or achieving normal values in the key disease parameters is in contrast to the benchmark of therapeutic goals 10 which has heretofore been applied to management of Gaucher‐specific signs and symptoms. Moreover, it could therefore be suggested that even at lower doses (but not lower than 30 U/kg), velaglucerase alfa appears to induce good responses even in platelet counts and even among patients who are slower to initiate responsiveness. However, identifying demographic or other correlates of patients with a slower response is beyond the scope of this trial with a relatively small cohort.
Improved outcomes in the exploratory endpoints for Gaucher‐related bone disease in this trial and patient cohort have been published 8, 9. Statistically significant improvements in BMD as assessed by the mean changes from baseline in lumbar spine and femoral neck Z‐scores were observed at 24 and 33 months, respectively. In that there was evidence of baseline bone disease in most patients, it was gratifying that the response was both robust and was observed earlier than the 3‐ to 5‐year time period expected among adults 18.
Similarly, a statistically significant improvement in lumbar spine BMB was observed as early as 9 months in the TKT025 trial, along with continued improvement over time in the extension study. Although all patients at baseline demonstrated moderate to severe bone marrow involvement, all but one demonstrated a normal lumbar spine BMB score by the final BMB evaluation 9. This was an equally impressive response to therapy, as the time frame seemed to be shorter and more dramatic than previously reported 19. Due to the small sample size, this warrants further investigation.
Reduction in biomarker levels of chitotriosidase and CCL18 continued throughout the 7‐year study period. Particularly remarkable, was the absence of plateauing in the response of either biomarker.
Various approaches have been proposed to assess the clinical significance of the therapeutic response observed in GD1 patients. These include the 2‐ and 5‐year therapeutic goals published by Pastores et al. 10 and quantitative characterization of the therapeutic response as suggested by the European Medicines Agency (modified from Hollak et al.) 20. Applying the therapeutic response criteria of Hollak et al. 20 to all patients in this study would lead us to conclude that all completers achieved a good or complete response in their hematologic and visceral parameters at their last study visit. Based on retrospective clinical and registry data, it was suspected that most patients could achieve these therapeutic, albeit, subnormal goals with ERT. The current study implies that normalization and/or near normalization could be achieved in most Gaucher‐specific parameters even in the context of dose reduction. This is a new observation and so a re‐evaluation of benchmarks regarding therapeutic response in GD, to include expectation of near‐normalization of all disease‐specific parameters with continued use of disease‐specific treatment, may be appropriate.
This study has several limitations. As with many phase I/II studies, the single‐center TKT025/TKT025EXT study had a small population and there is a risk that the population was homogenous and not representative of the broader group of GD1 patients. To mitigate the potential effect of a small homogenous population, a phenotypically and genotypically diverse study population was recruited. In addition, there is a risk that the clinical and safety profile observed in a small study may not be representative of a response in a much larger population, particularly as it relates to rare events. Two patients did not complete the study due to pregnancy, possibly impacting the results, particularly given the small study size. However, the similarity in outcomes between the ITT group and the completer group suggests that the two patients who did not complete the study did not drive the clinical outcomes. In subsequent trials of velaglucerase alfa, there were patients who became pregnant during the extension phase and were able to continue receiving ERT (data on file), and several cases from clinical trials and an early access program indicate that velaglucerase alfa may be used safely in pregnancy 21. Finally, as this was a single‐arm study, there was no control or active comparator group. Nonetheless, the observed safety profile for this study group is supported by subsequent observations among more than 300 GD1 patients recruited in later‐phase clinical trials of velaglucerase alfa. Similarly, the clinical response observed in the first 9 months is supported by the efficacy demonstrated in two larger, randomized, double‐blind phase III trials.
Despite these limitations, the TKT025/TKT025EXT data represents the longest, most complete, and most comprehensive longitudinal clinical trial evidence (level 2b evidence) for the treatment of GD1 published to date. This study contrasts other longitudinal publications of GD1 which have generally reflected registry data (level 2c evidence) and whose limitations have recently been reviewed 22.
Conclusion
TKT025EXT is the longest prospective, longitudinal clinical trial for GD1 conducted to date. Though further study is needed to corroborate these 7‐year findings, the TKT025 results have been confirmed by the subsequent phase III trials. The 7‐year data suggest that velaglucerase alfa was generally well tolerated, did not require premedication, and was successfully administered at home under healthcare professional supervision. This report is the first to suggest that despite a marked reduction in dose during the second year of this 7‐year study, continued improvement of several disease parameters including normalization, near normalization and/or marked improvement of key disease parameters (hemoglobin concentrations, platelet counts, splenomegaly, hepatomegaly and lumbar spine BMD and BMB) may be feasible for most adult GD1 patients. It is provocative to think that more rigorous therapeutic expectations may be warranted and that these may one day guide treatment decisions for patients with GD1.
Acknowledgments
The authors gratefully acknowledge the work of David Zahrieh, MS, formerly of Shire (Lexington, Massachusetts), Irith Hadas‐Halpern, MD, of Shaare Zedek Medical Center (Jerusalem, Israel), Andrew H. Haims, MD, of Yale University School of Medicine, (New Haven, Connecticut), A. Joseph Foldes, MD, of The Jerusalem Osteoporosis Center, Hadassah University Hospital (Jerusalem, Israel), Hans Aerts, PhD, of the Academic Medical Center (Amsterdam, The Netherlands), Yune Kunes, PhD, of Shire (Lexington, Massachusetts), and all the experts and nurses who have been part of the studies, including Anat Oz, RN, and Naama Arbel, RN, who provided home therapy. The authors thank Clare Guni, BMBS, of Excel Scientific Solutions, who provided editorial support funded by Shire.
Conflicts of interest: AZ receives consultancy fees from Protalix Biotherapeutics, has options in Protalix Biotherapeutics, and is a member of its Scientific Advisory Board. AZ receives honoraria from Shire, Genzyme/Sanofi and Pfizer, and his institution receives support from Genzyme/Sanofi for participation in the International Collaborative Gaucher Group Gaucher Registry and from Shire for participation in the Gaucher Outcome Survey. NW and GMC are employees of Shire. EC and CO are former employees of Shire. DE was a consultant during the clinical trial and currently receives travel expenses and honoraria for presentations at professional meetings from Shire.
Contract grant sponsor: Shire.
References
- 1. Brumshtein B, Salinas P, Peterson B, et al. Characterization of gene‐activated human acid‐beta‐glucosidase: Crystal structure, glycan composition, and internalization into macrophages. Glycobiology 2010;20:24–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Zimran A, Altarescu G, Philips M, et al. Phase 1/2 and extension study of velaglucerase alfa replacement therapy in adults with type 1 Gaucher disease: 48‐month experience. Blood 2010;115:4651–4656. [DOI] [PubMed] [Google Scholar]
- 3. Ben Turkia H, Gonzalez DE, Barton NW, et al. Velaglucerase alfa enzyme replacement therapy compared with imiglucerase in patients with Gaucher disease. Am J Hematol 2013;88:179–184. [DOI] [PubMed] [Google Scholar]
- 4. Gonzalez DE, Ben Turkia H, Lukina EA, et al. Enzyme replacement therapy with velaglucerase alfa in Gaucher disease: Results from a randomized, double‐blind, multinational, phase 3 study. Am J Hematol 2013;88:166–171. [DOI] [PubMed] [Google Scholar]
- 5. Zimran A, Pastores GM, Tylki‐Szymanska A, et al. Safety and efficacy of velaglucerase alfa in Gaucher disease type 1 patients previously treated with imiglucerase. Am J Hematol 2013;88:172–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Pastores GM, Rosenbloom B, Weinreb N, et al. A multicenter open‐label treatment protocol (HGT‐GCB‐058) of velaglucerase alfa enzyme replacement therapy in patients with Gaucher disease type 1: Safety and tolerability. Genet Med 2014;16:359–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Shire Human Genetic Therapies, Inc. VPRIV™ (velaglucerase alfa for injection): Highlights of prescribing information. 2010. Accessed June 9, 2014. Available at: http://www.accessdata.fda.gov/drugsatfda_docs/label/2010/022575lbl.pdf.
- 8. Elstein D, Foldes AJ, Zahrieh D, et al. Significant and continuous improvement in bone mineral density among type 1 Gaucher disease patients treated with velaglucerase alfa: 69‐month experience, including dose reduction. Blood Cells Mol Dis 2011;47:56–61. [DOI] [PubMed] [Google Scholar]
- 9. Elstein D, Haims AH, Zahrieh D, et al. Impact of velaglucerase alfa on bone marrow burden score in adult patients with type 1 Gaucher disease: 7‐year follow‐up. Blood Cells Mol Dis 2014;53:56–60. [DOI] [PubMed] [Google Scholar]
- 10. Pastores GM, Weinreb NJ, Aerts H, et al. Therapeutic goals in the treatment of Gaucher disease. Semin Hematol 2004;41:4–14. [DOI] [PubMed] [Google Scholar]
- 11. Stasi R. How to approach thrombocytopenia. Hematol Am Soc Hematol Educ Program 2012;2012:191–197. [DOI] [PubMed] [Google Scholar]
- 12. Elstein D, Cohn GM, Wang N, et al. Early achievement and maintenance of the therapeutic goals using velaglucerase alfa in type 1 Gaucher disease. Blood Cells Mol Dis 2011;46:119–123. [DOI] [PubMed] [Google Scholar]
- 13. Elstein D, Klutstein MW, Lahad A, et al. Echocardiographic assessment of pulmonary hypertension in Gaucher's disease. Lancet 1998;351:1544–1546. [DOI] [PubMed] [Google Scholar]
- 14. Mistry PK, Sirrs S, Chan A, et al. Pulmonary hypertension in type 1 Gaucher's disease: Genetic and epigenetic determinants of phenotype and response to therapy. Mol Genet Metab 2002;77:91–98. [DOI] [PubMed] [Google Scholar]
- 15. Quanjer PH, Stanojevic S, Cole TJ, et al. Multi‐ethnic reference values for spirometry for the 3‐95‐yr age range: The global lung function 2012 equations. Eur Respir J 2012;40:1324–1343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Spilker B. Guide to drug development: A comprehensive review and assessment. Philadelphia: Wolters Kluwer Health/Lippincott Williams & Wilkins; 2009. [Google Scholar]
- 17. Milligan A, Hughes D, Goodwin S, et al. Intravenous enzyme replacement therapy: Better in home or hospital? Br J Nurs 2006;15:330–333. [DOI] [PubMed] [Google Scholar]
- 18. Sims KB, Pastores GM, Weinreb NJ, et al. Improvement of bone disease by imiglucerase (Cerezyme) therapy in patients with skeletal manifestations of type 1 Gaucher disease: Results of a 48‐month longitudinal cohort study. Clin Genet 2008;73:430–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Robertson PL, Maas M, Goldblatt J. Semiquantitative assessment of skeletal response to enzyme replacement therapy for Gaucher's disease using the bone marrow burden score. AJR Am J Roentgenol 2007;188:1521–1528. [DOI] [PubMed] [Google Scholar]
- 20. Hollak CE, Aerts JM, Goudsmit R, et al. Individualised low‐dose alglucerase therapy for type 1 Gaucher's disease. Lancet 1995;345:1474–1478. [DOI] [PubMed] [Google Scholar]
- 21. Elstein D, Hughes D, Goker‐Alpan O, et al. Outcome of pregnancies in women receiving velaglucerase alfa for Gaucher disease. J Obstet Gynaecol Res 2014;40:968–975. [DOI] [PubMed] [Google Scholar]
- 22. Hollak CE, Aerts JM, Aymé S, et al. Limitations of drug registries to evaluate orphan medicinal products for the treatment of lysosomal storage disorders. Orphanet J Rare Dis 2011;6:16 [DOI] [PMC free article] [PubMed] [Google Scholar]