

Abstract
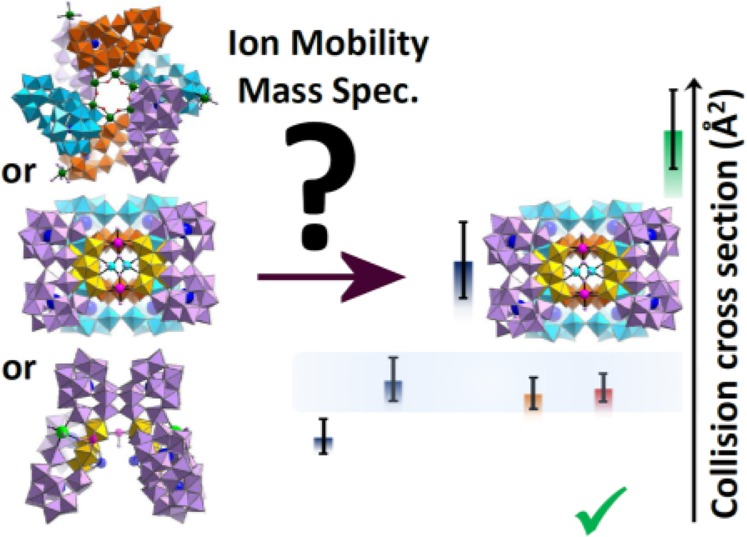
Ion mobility-mass spectrometry (IM-MS) is a powerful technique for structural characterization, e.g., sizing and conformation, particularly when combined with quantitative modeling and comparison to theoretical values. Traveling wave IM-MS (TW-IM-MS) has recently become commercially available to nonspecialist groups and has been exploited in the structural study of large biomolecules, however reliable calibrants for large anions have not been available. Polyoxometalate (POM) species—nanoscale inorganic anions—share many of the facets of large biomolecules, however, the full potential of IM-MS in their study has yet to be realized due to a lack of suitable calibration data or validated theoretical models. Herein we address these limitations by reporting DT-IM (drift tube) data for a set of POM clusters {M12} Keggin 1, {M18} Dawson 2, and two {M7} Anderson derivatives 3 and 4 which demonstrate their use as a TW-IM-MS calibrant set to facilitate characterization of very large (ca. 1–4 nm) anionic species. The data was also used to assess the validity of standard techniques to model the collision cross sections of large inorganic anions using the nanoscale family of compounds based upon the {Se2W29} unit including the trimer, {Se8W86O299} A, tetramer, {Se8W116O408} B, and hexamer {Se12W174O612} C, including their relative sizing in solution. Furthermore, using this data set, we demonstrated how IM-MS can be used to conveniently characterize and identify the synthesis of two new, i.e., previously unreported POM species, {P8W116}, unknown D, and {Te8W116}, unknown E, which are not amenable to analysis by other means with the approximate formulation of [H34W118X8M2O416]44–, where X = P and M = Co for D and X = Te and M = Mn for E. This work establishes a new type of inorganic calibrant for IM-MS allowing sizing, structural analysis, and discovery of molecular nanostructures directly from solution.
1. Introduction
Ion mobility spectrometry (IMS), combined with mass spectrometry (as IM-MS), is an analytical technique which is able to provide considerable information beyond that available from standalone MS experiments.1−3 A typical IMS experiment measures the time taken for ions to travel through a “drift tube”; in contrast with MS, this is partially filled with a gas (the “drift gas”) and the time taken for ions to pass through, the “drift time” (tD), is a function of both m/z (mass to charge ratio) and interaction with that drift gas. In this way, IMS can resolve isobaric (same m/z) species that would be indistinguishable in MS and provide structural information beyond simple mass/charge ratios (m/z), on converting drift time to a collision cross-section (CCS) parameter giving information about size and conformation. Until recently, the use of IM-MS has been a specialist activity, generally limited to groups in which bespoke instruments are constructed; this changed when traveling wave (TW) IM-MS instruments became commercially available, putting the technique in the hands of nonspecialists.4−9
Our interest in IM-MS lies in its use for the analysis of large polyoxometalate (POM) anions.10,11 POMs are a class of anionic metal oxide nanosized molecules, see Figure 1, with myriad potential applications in fields as diverse as electronic devices,12 medicine,13 and energy production and storage.14 IM-MS is promising for the analysis of POMs, both as it provides an extra dimension for their resolution (drift time) and promises a means for determination of the size and identity of new species (by assessing CCS). Structural information may be inferred by comparing with a model, and conformational and information about the supramolecular interactions may also be obtained.
Figure 1.
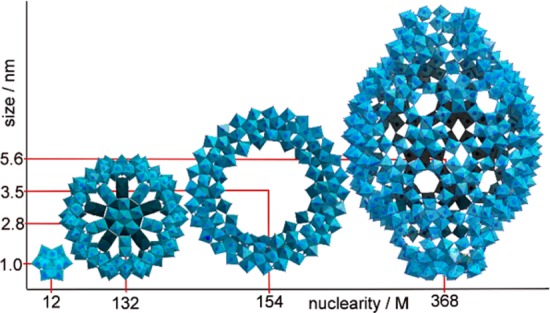
Structural representations of the archetypal polyoxometalate molecules that span a 1–6 nm diameter range. From left to right: {Mo12} Keggin, {Mo132} Keplerate, {Mo154} Wheel, and {Mo368} “lemon”.15−17 Actual crystal structures, to scale, are shown; the polyhedral are {MoOx} units (x = 4–7).
Resolution of isobaric signals, without quantitative assessment of cross-section, is in itself useful. This is because we may confirm that signals are derived from single large species, rather than multiple species of similar m/z (e.g oligomeric species or experimental artifacts aggregation of anions in spray),10,18 and we hypothesize it should be possible to observe new species in otherwise intractable reaction mixtures, as well as probe conformational changes,19 and explore the mechanism of self-assembly.20,21 This potential is of great interest as very few methods exist to confirm the structure of new POM species, other than X-ray diffraction of crystal samples; this dramatically limits the field to slow discovery of species which can be isolated as diffraction-quality single crystals.
Most notable applications of TW-IM-MS reported thus far have been in biology, although it is increasingly applied to supramolecular chemistry4−9 and noble metal clusters.22 In most cases, where structural characterization is the aim (rather than simply resolving/distinguishing species identical in m/z), a collision cross-section is obtained for ions of interest. This CCS can be informative in itself as a measure of size or be compared to those observed for other species. Most powerfully, however, CCS measurements can be compared to theoretically derived values for different putative structures (themselves derived from either theory, such as molecular dynamics calculations, or experiment, such as X-ray diffraction or NMR), allowing the end-user to infer the structure/conformation of the ion in question.
The primary limitation of TW-IM-MS, notwithstanding some concerns about the heating of ions causing distortions in their conformation,23 is the need for a sample-specific CCS calibration to obtain CCS values from experimental observations, due to the nonlinear nature of the drift time response. Calibration requires reference data: published, reliable data obtained from a (generally bespoke) linear drift tube (DT) IM-MS instrument, which is able to directly measure an ion’s CCS without external calibration.24 Such data is available for many applications and has been collated in various publications25 and Web sites; principal among these is the Clemmer group’s online CCS database.26 Significantly, the vast majority pertains to positive ions, whereas our interest is in anions; very little data is available to use in calibrating for negative ions, particularly those that are of intermediate size (between that of small molecules and very large proteins).27 Other calibrations have been reported, sometimes using data from unpublished sources (which may nonetheless be reliable) or employing data derived from theoretical calculations.28,29 In previously published work we have calibrated TW-IM-MS using a collection of data from these sources.10,18 The calibrants used in these cases were all biomolecules and as such imposed some difficulties: samples required careful storage and preparation (e.g., annealing of DNA), multiple conformations may be observed for several of the species under investigation (thus requiring deconvolution, or rejection of the data when reliable deconvolution was not possible), and furthermore this set covers only a relatively narrow size range.
Herein we report the establishment and application of a new calibration set consisting of POM-based anions for TW-IM-MS measurements, using DTCCSHe (CCS obtained with He as a drift gas) values determined by DT-IM-MS. We report the use of this calibration set to determine the TWCCSN2→He of several large POM species and compare these values to those derived from theoretical calculations. Furthermore, we demonstrate the inference of structure of new POM species for which diffraction-quality single crystals could not be obtained, by quantitative CCS comparison with a range of potential structures.
2. Results and Discussion
2.1. Calibration Set and DT-IM-MS Measurements
Potential POM calibrants were chosen to cover a range of sizes and charge densities and, additionally, were all required to be readily synthesized and stable over prolonged periods of time (in contrast with biomolecule calibrants). Figure 2 depicts the structures of the four POM calibrants chosen, incorporating species from three of the archetypal POM families including: 1, {PW12O40} Keggin ion;302, {P2W18O62} Dawson ion,31 and 3/4, two {MnMo6} Anderson derivatives capped with tris-based ligands (3 = MnMo6O24(C4H8N)2 tris32 and 4 = MnMo6O24(C20H38NO)2).33 Observed peaks corresponding to intact ions were chosen, based on their being reliably observed in both TW-IM-MS and DT-IM-MS instruments, across a range of conditions.
Figure 2.
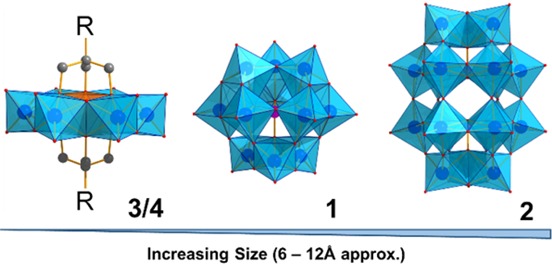
Structural representations of the four POM calibrants in this study. The above include three of the major POM archetypes, namely, the Anderson, left, where R = tris(alkoxo) ligand, (3) or “aliphatic” ligand-bound (4) {MnMo6O24}), Keggin (1, {PW12O40}) and Dawson (2, {P2W18O62}) clusters. [Metal oxide units represented as polyhedra, organic ligands as “ball and stick”; atom colors: O = red; W and Mo = blue; C = black.].
DT-IM-MS of this calibrant set yields robust DTCCSHe values for 29 different negative ions: five from 1 (a “Keggin” structure), eight from 2 (a Wells–Dawson structure), nine from 3 (a “Tris” Mn-Anderson structure), and seven from 4 (an “aliphatic” Mn-Anderson). Ionization parameters were set to maintain as soft an ionization as possible, while each analyte’s drift time was measured in response to a range (3−9 Td) of reduced electric field strengths. Of these, some were disregarded, as not being observed in other instruments (vide infra), those used in calibration are tabulated in the Supporting Information.
2.2. Calibration of TW-IM-MS with the New Calibration Set
As outlined above, it is not practical to directly determine CCS values using TW-IM-MS instruments; instead they must be obtained through calibration with known species. A further complication to this is that the bespoke DT IM-MS instruments tend to be operated with helium as the buffer gas, whereas the TW-IM-MS instruments by default use nitrogen and are then converted to apparent helium CCS values. To denote the method and the buffer gas used, we have adopted a notation system IMSCCSgas where both the method (TW or DT and the buffer gas used are indicated); in the special case that TW-IM-MS data (using N2 as a drift gas) is used to estimate CCSHe values, we use the notation TWCCSN2→He.
Using drift time values from TW-IM-MS measurements of the calibration set, along with the corresponding DTCCSHe values derived from DT-IM-MS experiments, we were able to construct calibration curves. In cases where ions were not robustly and reproducibly observable in both classes of instruments, they were disregarded and not used further as calibrants. An example calibration curve can be seen in (see Figure 3), following as per the Ashcroft/Williams method;34 R2 values of around were 0.982 typical over a number of repetitions.
Figure 3.
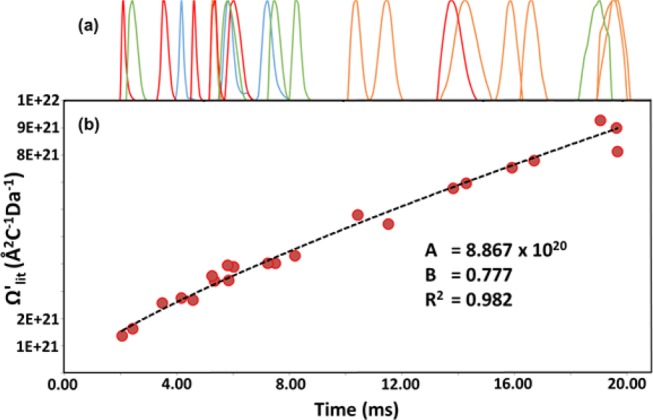
(a) Arrival time distribution (ATD), showing clear peaks in normalized plots of raw drift time (tD) yielded by the POM calibration set (green = 1, red = 2, orange = 3, and blue = 4; intensities normalized; x-axis values are tD). (b) From these data, modified drift time values (tD′) are calculated and fit to DTCCSHe data using a y = AxB curve, using the approach and notation set out in ref (34).
To validate the use of this calibrant set, we then used such a calibration curve obtained to estimate TWCCSN2→He values for two of the only negatively charged structures which have been extensively studied in DT-IM-MS: cytochrome c(14) and decathymidine.35 The experimental values obtained were largely in agreement with the reported values, displaying high linearity over multiple charge states. The gradient for this linear relationship was found to be ca. 1.15 (see Figure 4), however: this may well result from conformational differences in the biomolecules (e.g., combinations of multiple conformations have been observed for cytochrome c) between DT-IM-MS and TW-IM-MS instruments, and frequent difficulty in assigning a clear peak maximum where many conformations are present. This small discrepancy highlights the importance of conformational regularity/reproducibility in TW-IM-MS calibrant species, when faced with unavoidable variance in the “hardness” of the ionization conditions, ion–gas interaction potentials, and of ion optic arrangements/effective ion temperature in different instruments.23
Figure 4.
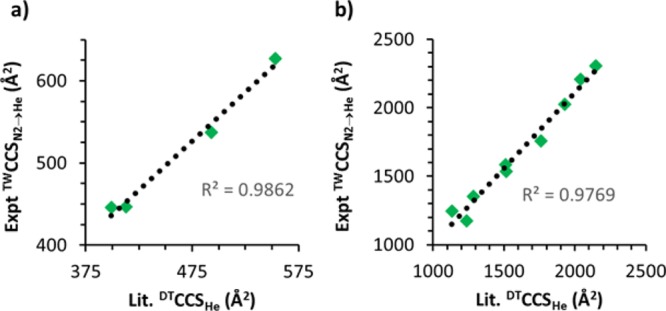
Correlation between previously published literature DTCCSHe values and experimentally determined TWCCSN2→He values for (a) decathymidine, gradient of the trend line = 1.19, and (b) cytochrome c, gradient of the trend line = 1.11.
2.3. CCS Determination for Large Inorganic Anions
Having validated the use of our new calibrant set using species of known DTCCSHe, we then used it to estimate TWCCSN2→He values for a related series of large polyoxotungstate clusters. To achieve this, we chose a set of very large polyoxotungstate clusters that are built from the building block {Se2W29} found in the cluster anion [H10Se2W29O103]14–, see Figure 5.36,37
Figure 5.
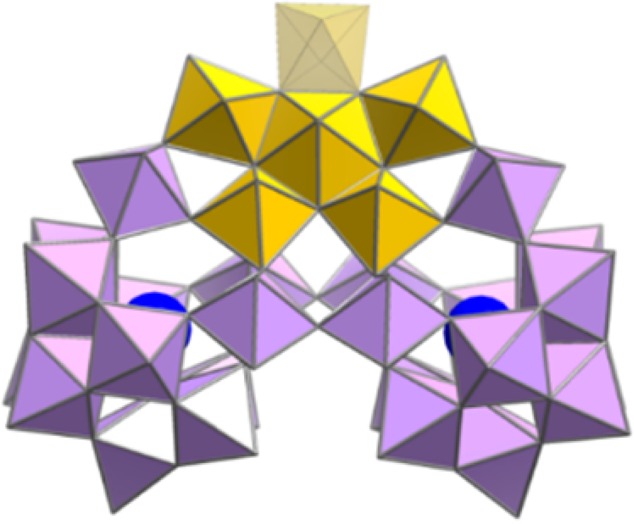
Representation of the structure of the {Se2W29} building block. Purple polyhedra, {WO6}; yellow polyhedra, pentagonal unit {(WO7)W4} with the lacunary position shown with semitransparent polyhedron; and blue spheres: Se.
We chose this building block as it is the smallest lacunary polyoxotungstate unit isolated so far which contains a pentagonal unit (such pentagonal units are common in nanoscale polyoxomolybdates,15 but rarely observed in large polyoxotungstates), is very stable in solution allowing analysis by electrospray mass spectrometry, and has a very well-defined crystal structure. Furthermore, this unit can be connected to a range of gigantic molecular nanostructures that are solution stable. For this work we chose to explore the trimer, {Se8W86O299} A, tetramer, {Se8W116O408} B, and hexamer {Se12W174O612} C of the {Se2W29} building block. The structures of A–C are very large, ranging from ca. 22 to 45 kDa.36,37 Finally, it is important to note that all these clusters are stable under the electrospray mass spectrometry conditions and hence are perfect candidates to be investigated by IM-MS, see Figure 6.
Figure 6.
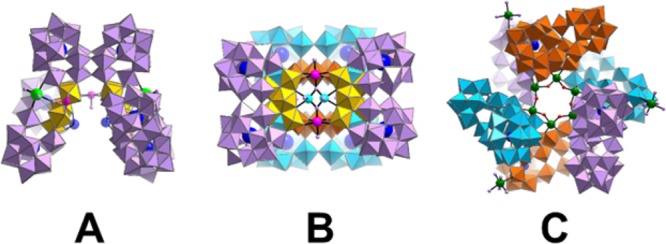
Structures of large POM structures A–C, with average diameters of 2.2, 2.5, and 3.4 nm, respectively (estimated from single crystal data with cations omitted by encapsulating each structure with an ellipsoid). [Metal oxide units represented as polyhedral; light-purple polyhedra, {WO6}; yellow polyhedra, pentagonal unit {(WO7)W4}; cyan spheres, W; blue spheres, Se; light-purple spheres, O; red spheres, disordered O and Cl; pink and dark-green spheres, heterometal ions.].
An example of the resulting spectra can be seen in Figure 7: a series of intense peaks covering a range of charge states (−12 to −7) corresponds to the core {Se2W29}4 structure, B; as with other large POM species, this resembles the spectrum of a large protein. It is worth noting that in previous conventional ESI-MS analyses,36,37 a series of low-intensity peaks which did not form part of the main series could only be tentatively assigned to the dimeric association of these clusters during electrospray ionization (clustering is common in large POM polyanions).10,18 By contrast, IM-MS can resolve these species at shorter drift times across several charge states (−17 to −14) and confirm this assignment.
Figure 7.
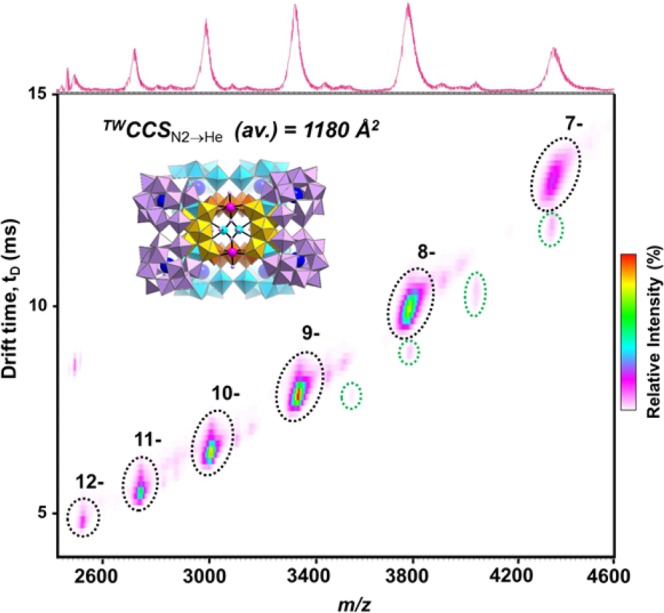
IM-MS spectrum of structure B, where the major series of peaks corresponds to the core {Se2W29}4 cluster, and the minor/less-intense series (at shorter tD drift times, peaks highlighted in green) is assigned to the aggregation of two such clusters. The TWCCSN2→He value quoted is an average across many charge states of the dominant cluster (see Supporting Information for individual values).
TWCCSN2→He values could then be estimated for all the large species observed (see Supporting Information). Since these vary slightly over multiple charge states, averages are taken to allow comparison between clusters (while counterions are alkali metals/protons, their size is treated as negligible). Determination of average TWCCSN2→He values was possible for the full series A–C; all spectra are available in the Supporting Information, while the average values are presented in Table 1. Inspection of these values permits a broad assessment of large species, not necessarily just resulting from the analysis of pure crystalline products, as in this case, but potentially also species present in a reaction solution.29 This is important since it was possible, using the crystal data as guide, to show that the TWCCSN2→He values correlate well with the crystallographic dimensions. This indicates that it is possible to crudely “size” the clusters from solution using IM-MS.
Table 1. Comparison of Simulated and Experimental CCSHe Dataa.
| compd | Rcrys (Å) | TWCCSN2→He (Å2) | PA (Å2) | EHS (Å2) | TM (Å2) |
|---|---|---|---|---|---|
| A | 10.8 | 893 | 645 | 760 | 744 |
| B | 12.5 | 1180 | 774 | 926 | 910 |
| C | 17.1 | 1784 | 1049 | 1295 | 1286 |
Rcyst is the crystallographically determined radius in Å (estimated from single crystal data with cations omitted by encapsulating each structure with an ellipsoid); TWCCSN2→He values are averages across the measured charge states; PA, EHS, and TA denote CalcCCSHe values simulated using Mobcal, employing the “projection approximation”, “exact hard spheres” and “trajectory method” methods, respectively.
2.4. Usefulness of Modeling for Structural Elucidation
As outlined earlier, orthodox use of IM-MS often employs a comparison of simulated CCS values (CalcCCSHe) for putative structures (most commonly derived from the MobCal software) with experimental data, to assign observations to a putative structure. This approach would be extremely attractive as a means to infer the structure of unknown/newly discovered POM species, but this first requires the validation of suitable modeling/simulation routines. We have previously published some preliminary results suggesting that this may be possible,10,16 but surprisingly, the CalcCCSHe values produced using standard (MobCal) models and standard parameters (produced for the study of carbon-based species) correlated well with experimental TWCCSN2→He data obtained for a large Mo-based POM wheel.
To investigate these possibilities further, CalcCCSHe values were simulated for structures A–C, inputting structural data derived from their published X-ray diffraction structures into Mobcal and simulating CCS values using each of the models available within Mobcal (“projection method”, “exact hard spheres”, and “trajectory approximation”); the results are set out alongside the corresponding experimentally derived values in Table 1. It is clear that in all cases no model (using standard parameters) yields a reasonable correlation with experimental measurements. Contrary to our initial observations, it appears that this approach is unlikely to allow useful structural inferences for this class of compounds, without some considerable improvement or refinement. Such refinement may take the form of more appropriate parameter sets for currently routine models (e.g., Lennard-Jones parameters for O atoms bound directly to metal ions, rather than carbon; parameters for long-range interaction of atoms not currently described in Mobcal) or the use of more advanced models, in particular the treatment and calculation of atom centered point charges and more thorough consideration of counter-cations.38,39
2.5. IM-MS As a Tool for Structural Assignment of New POM Species
Most reaction systems producing POMs can yield a number of different products, depending on precise control of reaction conditions. For example, reaction of WO42– and SeO32– salts in acidic aqueous solution is known to produce {Se2W29} “building blocks”; these form a range of large structures, depending on reaction conditions structures A–C can then be isolated from reaction mixtures (other observed products are smaller). Much exploration of POM synthesis and function involves systematic synthesis of series of analogous structures from such systems, incorporating small changes to internally located heteroatoms (e.g., Se in {Se2W29} units) to alter clusters’ properties.40,41 We propose that, since changes in the heteroatoms do not affect the rigid POM framework structure, comparison of TWCCSN2→He values for known structures with different heteroatoms may be substituted in the orthodox IM-MS workflow, replacing comparison with simulated values and allowing the rapid structural assignment of newly discovered POM species.
To assess the usefulness of this approach, we chose to investigate the substitution of the Se heteroatom with P and Te in the WO42–/SeO32– reaction system which produces structures A–C. Substitution of SeO32– salts with the corresponding HPO33– or TeO32– salts, in conditions which with Se would yield structure B, yielded unknown compounds D and E, respectively. Despite some effort, it was not possible to produce diffraction-quality crystals of D or E, underlining the acute need for alternative tools to characterize POM structures.
TW-IM-MS spectra of unknown structures D and E were readily obtained (see Supporting Information); in the case of D, a single charge series was observed, in the case of E, a second series (denoted E2) was observed at shorter drift times, similar to that assigned to aggregates of structure B (see Figure 3). TWCCSN2→He values were then estimated from this experimental data, using the calibration approach described earlier, and are set out in Table 2. Comparison to the TWCCSN2→He values obtained for these new, unknown structures with those already established for structures A–C reveal a very good fit with structure B (Figure 8) and allow us to assign an analogous {X2W29}4 structure to D and E (with X = P or Te, respectively). As such we can tentatively suggest not only that the compounds could be formulated approximately as [H34W118X8M2O416]44– X = P and M = Co for D and X = Te and M = Mn for E, respectively, but also that the four {X2W29}4 building blocks in each case are arranged in a structure analogous to B. Since so many arrangements of 119 tungsten oxide units are possible, such confident structural assignments would not be possible with conventional MS. Such rapid structural analysis from solution is of enormous value to POM exploration and discovery efforts and shows that the {Se2W29} unit can be expanded to include both {P2W29} and {Te2W29} building blocks.
Table 2. Average TWCCSN2→He Values Determined for Structures A–C and Unknown Structures D and E.
| structure | TWCCSN2→He (Å2) | std dev |
|---|---|---|
| A | 893 | 87 |
| B | 1180 | 110 |
| C | 1784 | 191 |
| D | 1111 | 80 |
| E | 1140 | 72 |
| E2 | 2444 | 199 |
Figure 8.
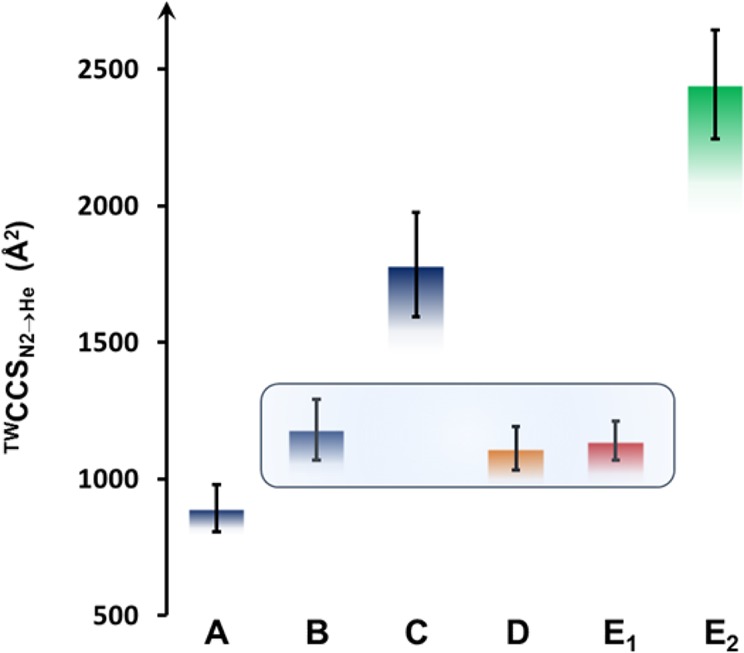
Comparison of average TWCCSN2→He values (see Table 2, error bars represent standard deviations) of structures A–C and unknown structures D and E. The close correlation of the values of D and E with those for B is highlighted.
3. Conclusions
IM-MS can be a valuable technique to assess the structure of POM species in solution and is highly complementary to existing approaches. To facilitate its use in the quantitative assessment of the size of POM clusters in solution, we have presented new DT-IM-MS data for calibration, applied that data to calibrate a TW-IM-MS instrument, and done further investigation of large inorganic species known from X-ray data. In addition, we have shown that the calibration set is suitable to quantitatively assess the size of large POM species—known and unknown—allowing useful structural inferences. This approach is particularly significant, as it may be accomplished quickly and from solution, circumventing the need to produce diffraction-quality crystals in order to discover new POM structures, which is a major limitation to the development of the field. This means that we not only have filled a gap due to the lack of anion-calibrants in IM-MS but also have shown that it is possible to explore the structure/confirmation/supramolecular aggregation as well as size the clusters from solution. Finally, by use of a comparative approach we have been able to use IM-MS to discover two hitherto unknown POM clusters without the need of producing single crystals.
In future work we aim to expand the set of calibrants across a whole range of molecular nanostructures to investigate if IM-MS may become a general tool for the sizing of molecular species in solution as well as more firmly establishing the potential for identifying new architectures and exploring structures. Finally, we hope that this work will stimulate further activity to develop more appropriate models or parameter sets, to allow researchers in other fields access to the structural inferences possible in biomolecular IM-MS.
4. Experimental Procedures
The details are explained in the Supporting Information in more detail, but briefly, the Keggin (1) and Dawson (2) clusters were synthesized according to well-established literature procedures, while the “tris” Anderson (3), “aliphatic” Anderson (4) and the series of polyoxotungstate structures A–C were provided by our colleagues Drs. Mali Rosnes, Carine Yvon and Jing Gao, respectively. Negative mode high-resolution and ion-mobility mass spectrometry measurements were performed on a bespoke DT-IM-MS instrument42 and a Synapt G2 HDMS instrument from Waters, and all analysis was performed using the supplied software suite MassLynx v4.1. All calibrant samples were prepared to a concentration of 10–5 M in analytical grade acetonitrile, while the polyoxotungstate samples were dissolved in the minimum required water and diluted with acetonitrile to a final concentration of 10–5 M. All analyte solutions were passed through to 0.22 μm filter before injection via a syringe pump at 5 μL·min–1. DT-IM-MS mobilities were determined from a drift time vs P/V slope at reduced field strength range 3–9 Td.43DTCCSHe were then calculated from the measured K using the fundamental ion mobility equation.44 Capillary source voltages were adjusted for optimum ionization of each sample in HRES-MS mode and maintained during subsequent IM-MS measurements. In contrast, IM-MS drift tube settings amenable to all samples were determined and kept constant, regardless of optimization, throughout to allow comparison between drift times.
Acknowledgments
We thank the EPSRC grants (nos. EP/J015156/1; EP/L023652/1; EP/I033459/1; EP/J015156/1; EP/K023004/1; EP/L023652/1) and EC grant 318671 MICREAGENTS, and L.C. thanks the Royal Society/Wolfson Foundation for a Merit Award and the ERC for an Advanced Grant (ERC-ADG, 670467 SMART-POM). The authors would like to thank Drs. Mali Rosnes, Carine Yvon, and Jing Gao for supplying the samples mentioned above; Dr. Johannes Thiel for the initial suggestion of the calibrant set; and Dr. Trevor Hinkley for assistance with Mobcal and Linux. We would also like to thank the EPSRC and University of Glasgow for support.
Supporting Information Available
crystallographic parameters (CIF). The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.6b00070.
Experimental details, characterization data, and detailed mass spectrometry analysis (PDF)
Author Contributions
⊥ These authors contributed equally.
The authors declare no competing financial interest.
Supplementary Material
References
- Lanucara F.; Holman S. W.; Gray C. J.; Eyers C. E. Nat. Chem. 2014, 6, 281–294. 10.1038/nchem.1889. [DOI] [PubMed] [Google Scholar]
- Wilkins C. L.; Trimpin S.. Ion Mobility-Mass Spectrometry: Theory and Applications. CRC Press: Boca Raton, FL, 2010. [Google Scholar]
- Jurneczko E.; Barran P. E. Analyst 2011, 136 (1), 20–28. 10.1039/C0AN00373E. [DOI] [PubMed] [Google Scholar]
- Lu X. C.; Li X. P.; Cao Y.; Schultz A.; Wang J. L.; Moorefield C. N.; Wesdemiotis C.; Cheng S. Z. D.; Newkome G. R. Angew. Chem., Int. Ed. 2013, 52 (30), 7728–7731. 10.1002/anie.201302362. [DOI] [PubMed] [Google Scholar]
- Li X. P.; Chan Y. T.; Casiano-Maldonado M.; Yu J.; Carri G. A.; Newkome G. R.; Wesdemiotis C. Anal. Chem. 2011, 83 (17), 6667–6674. 10.1021/ac201161u. [DOI] [PubMed] [Google Scholar]
- Ujma J.; De Cecco M.; Chepelin O.; Levene H.; Moffat C.; Pike S. J.; Lusby P. J.; Barran P. E. Chem. Commun. 2012, 48 (37), 4423–5. 10.1039/c2cc30778b. [DOI] [PubMed] [Google Scholar]
- Brocker E. R.; Anderson S. E.; Northrop B. H.; Stang P. J.; Bowers M. T. J. Am. Chem. Soc. 2010, 132 (38), 13486–94. 10.1021/ja105702y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson S. E.; Bleiholder C.; Brocker E. R.; Stang P. J.; Bowers M. T. Int. J. Mass Spectrom. 2012, 330, 78–84. 10.1016/j.ijms.2012.08.024. [DOI] [Google Scholar]
- Chepelin O.; Ujma J.; Barran P. E.; Lusby P. J. Angew. Chem., Int. Ed. 2012, 51 (17), 4194–7. 10.1002/anie.201108994. [DOI] [PubMed] [Google Scholar]
- Xuan W. M.; Surman A. J.; Miras H. N.; Long D. L.; Cronin L. J. Am. Chem. Soc. 2014, 136 (40), 14114–14120. 10.1021/ja5062483. [DOI] [PubMed] [Google Scholar]
- Long D. L.; Burkholder E.; Cronin L. Chem. Soc. Rev. 2007, 36 (1), 105–121. 10.1039/B502666K. [DOI] [PubMed] [Google Scholar]
- Busche C.; Vila-Nadal L.; Yan J.; Miras H. N.; Long D. L.; Georgiev V. P.; Asenov A.; Pedersen R. H.; Gadegaard N.; Mirza M. M.; Paul D. J.; Poblet J. M.; Cronin L. Nature 2014, 515 (7528), 545–549. 10.1038/nature13951. [DOI] [PubMed] [Google Scholar]
- Gao N.; Sun H. J.; Dong K.; Ren J. S.; Duan T. C.; Xu C.; Qu X. G. Nat. Commun. 2014, 5, 3422. 10.1038/ncomms4422. [DOI] [PubMed] [Google Scholar]
- Rausch B.; Symes M. D.; Chisholm G.; Cronin L. Science 2014, 345 (6202), 1326–1330. 10.1126/science.1257443. [DOI] [PubMed] [Google Scholar]
- Müller A.; Krickemeyer E.; Meyer J.; Bö gge H.; Peters F.; Plass W.; Diemann E.; Dillinger S.; Nommenbruch F.; Randerath M.; Menke C. Angew. Chem., Int. Ed. Engl. 1995, 34, 2122–2123. 10.1002/anie.199521221. [DOI] [Google Scholar]
- Müller A.; Krickemeyer E.; Bögge H.; Schmidtmann M.; Peters F. Angew. Chem., Int. Ed. 1998, 37, 3360–3363. . [DOI] [PubMed] [Google Scholar]
- Müller A.; Beckmann E.; Bö gge H.; Schmidtmann M.; Dress A. Angew. Chem., Int. Ed. 2002, 41, 1162–1166. . [DOI] [PubMed] [Google Scholar]
- Robbins P. J.; Surman A. J.; Thiel J.; Long D. L.; Cronin L. Chem. Commun. 2013, 49 (19), 1909–1911. 10.1039/c3cc38615e. [DOI] [PubMed] [Google Scholar]
- Thiel J.; Yang D.; Rosnes M. H.; Liu X.; Yvon C.; Kelly S. E.; Song Y.-F.; Long D.-L.; Cronin L. Angew. Chem. 2011, 123 (38), 9033–9037. 10.1002/ange.201102340. [DOI] [PubMed] [Google Scholar]
- Xu F.; Miras H.; Scullion R.; Long D.-L.; Thiel J.; Cronin L. Proc. Natl. Acad. Sci. U. S. A. 2012, 109, 11609–11612. 10.1073/pnas.1202981109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scullion R. A.; Surman A. J.; Xu F.; Mathieson J. S.; Long D. L.; Haso F.; Liu T. B.; Cronin L. Angew. Chem., Int. Ed. 2014, 53 (38), 10032–10037. 10.1002/anie.201404621. [DOI] [PubMed] [Google Scholar]
- Angel L. A.; Majors L. T.; Dharmaratne A. C.; Dass A. ACS Nano 2010, 4 (8), 4691–4700. 10.1021/nn1012447. [DOI] [PubMed] [Google Scholar]
- Merenbloom S. I.; Flick T. G.; Williams E. R. J. Am. Soc. Mass Spectrom. 2012, 23 (3), 553–562. 10.1007/s13361-011-0313-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- It should be noted that while many commercially available TW-IM-MS instruments are run with N2 as a drift gas, historically most linear field drift tube instruments are operated using He as a drift gas. In order to compare with published data and the output of much modelling software, TW-IM-MS calibration is generally carried out against CCS values measured with helium as a drift gas, formally yielding an estimate of CCSHe rather than calculating CCSN2 (denoted TWCCSN2→He, see Section 2.2).
- Chawner R.; McCullough B.; Giles K.; Barran P. E.; Gaskell S. J.; Eyers C. E. J. Proteome Res. 2012, 11 (11), 5564–5572. 10.1021/pr3005327. [DOI] [PubMed] [Google Scholar]
- Clemmer D. E. Clemmer Group Cross Section Database. http://www.indiana.edu/~clemmer/Research/Cross%20Section%20Database/cs_database.php (accessed December 22, 2015).
- Baker E. S.; Bernstein S. L.; Gabelica V.; De Pauw E.; Bowers M. T. Int. J. Mass Spectrom. 2006, 253, 225–237. 10.1016/j.ijms.2006.03.016. [DOI] [Google Scholar]
- Although the polymer calibration set report in ref (14) was purchased, it was not available from the same source without contracting bespoke synthesis (>U.S. $1000, November, 2012; Polymer Source Inc.); as a result and as the calibration values in that work are derived from calculation not experiment, we did not feel they were an appropriate calibration set for our purposes.
- Hamilton J. V.; Renaud J. B.; Mayer P. M. Rapid Commun. Mass Spectrom. 2012, 26 (14), 1591–1595. 10.1002/rcm.6266. [DOI] [PubMed] [Google Scholar]
- Gabb D.; Pradeep C. P.; Boyd T.; Mitchell S. G.; Miras H. N.; Long D.-L.; Cronin L. Polyhedron 2013, 52, 159–164. 10.1016/j.poly.2012.10.015. [DOI] [Google Scholar]
- Long D.-L.; Streb C.; Song Y.-F.; Mitchell S.; Cronin L. J. Am. Chem. Soc. 2008, 130, 1830–1832. 10.1021/ja075940z. [DOI] [PubMed] [Google Scholar]
- Hasenknopf B.; Delmont R.; Herson P.; Gouzerh P. Eur. J. Inorg. Chem. 2002, 2002, 1081–1087. . [DOI] [Google Scholar]
- Song Y.-F.; McMillan N.; Long D.-L.; Thiel J.; Ding Y. L.; Chen H. S.; Gadegaard N.; Cronin L. Chem. - Eur. J. 2008, 14, 2349–2354. 10.1002/chem.200701629. [DOI] [PubMed] [Google Scholar]
- Smith D. P.; Knapman T. W.; Campuzano I.; Malham R. W.; Berryman J. T.; Radford S. E.; Ashcroft A. E. Eur. Mass Spectrom. 2009, 15 (2), 113–130. 10.1255/ejms.947. [DOI] [PubMed] [Google Scholar]
- Hoaglund C. S.; Liu Y. S.; Ellington A. D.; Pagel M.; Clemmer D. E. J. Am. Chem. Soc. 1997, 119 (38), 9051–9052. 10.1021/ja970652w. [DOI] [Google Scholar]
- Gao J.; Yan J.; Beeg S.; Long D. L.; Cronin L. J. Am. Chem. Soc. 2013, 135 (5), 1796–1805. 10.1021/ja309237x. [DOI] [PubMed] [Google Scholar]
- Yan J.; Gao J.; Long D.-L.; Miras H. N.; Cronin L. J. Am. Chem. Soc. 2010, 132, 11410–11411. 10.1021/ja104043s. [DOI] [PubMed] [Google Scholar]
- Bleiholder C.; Contreras S.; Do T. D.; Bowers M. T. Int. J. Mass Spectrom. 2013, 345, 89–96. 10.1016/j.ijms.2012.08.027. [DOI] [Google Scholar]
- Bleiholder C.; Johnson N. R.; Contreras S.; Wyttenbach T.; Bowers M. T. Anal. Chem. 2015, 87 (14), 7196–7203. 10.1021/acs.analchem.5b01429. [DOI] [PubMed] [Google Scholar]
- Zheng Q.; Vila-Nadal L.; Busche C.; Mathieson J. S.; Long D. L.; Cronin L. Angew. Chem., Int. Ed. 2015, 54 (27), 7895–7899. 10.1002/anie.201502295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vila-Nadal L.; Peuntinger K.; Busche C.; Yan J.; Luders D.; Long D. L.; Poblet J. M.; Guldi D. M.; Cronin L. Angew. Chem., Int. Ed. 2013, 52 (37), 9695–9699. 10.1002/anie.201303126. [DOI] [PubMed] [Google Scholar]
- McCullough B. J.; Kalapothakis J.; Eastwood H.; Kemper P.; MacMillan D.; Taylor K.; Dorin J.; Barran P. E. Anal. Chem. 2008, 80 (16), 6336–6344. 10.1021/ac800651b. [DOI] [PubMed] [Google Scholar]
- Kemper P. R.; Bowers M. T. J. Am. Soc. Mass Spectrom. 1990, 1 (3), 197–207. 10.1016/1044-0305(90)85036-L. [DOI] [Google Scholar]
- Mason E.; McDaniel E.. Transport properties of Ions in Gases. Wiley-VCH: Weinheim, Germany, 1988. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.