

Abstract
High heparanase expression is associated with enhanced tumor growth, angiogenesis, and metastasis in many types of cancer. However, the mechanisms driving high heparanase expression are not fully understood. In the present study, we discovered that drugs used in the treatment of myeloma upregulate heparanase expression. Frontline anti-myeloma drugs, bortezomib and carfilzomib activate the nuclear factor-kappa B (NF-κB) pathway to trigger heparanase expression in tumor cells. Blocking the NF-κB pathway diminished this chemotherapy-induced upregulation of heparanase expression. Activated NF-κB signaling was also found to drive high heparanase expression in drug resistant myeloma cell lines. In addition to enhancing heparanase expression, chemotherapy also caused release of heparanase by tumor cells into the conditioned medium. This soluble heparanase was taken up by macrophages and triggered an increase in TNF-α production. Heparanase is also taken up by tumor cells where it induced expression of HGF, VEGF and MMP-9 and activated ERK and Akt signaling pathways. These changes induced by heparanase are known to be associated with the promotion of an aggressive tumor phenotype. Importantly, the heparanase inhibitor Roneparstat diminished the uptake and the downstream effects of soluble heparanase. Together, these discoveries reveal a novel mechanism whereby chemotherapy upregulates heparanase, a known promoter of myeloma growth, and suggest that therapeutic targeting of heparanase during anti-cancer therapy may improve patient outcome.
Keywords: Heparanase, Chemotherapy, Multiple Myeloma, Proteasome inhibitors, NF-κB, Roneparstat
1. Introduction
Heparanase, an endo-β-d-glucuronidase that trims the heparan sulfate chains of proteoglycans, is elevated in cancers and drives different cellular events to fuel tumor progression [1–5]. In the hematological malignancy multiple myeloma, data from our lab and others has demonstrated that heparanase drives angiogenesis, tumor growth, and metastasis [6–12], establishing it as a master regulator of aggressive tumor behavior. Soluble heparanase is present in the peripheral blood and plasma from bone marrow aspirates of myeloma patients, and high heparanase activity in the bone marrow strongly correlates with high microvessel density, consistent with the known role of heparanase in angiogenesis [9]. Despite clear evidence for high heparanase expression in cancers, molecular mechanisms that elevate heparanase expression or generate soluble heparanase in cancer patients are not fully understood.
Chemotherapy is the most widely used form of anti-cancer therapy [13]. In spite of several advances in the clinical use of chemotherapy, often it doesn’t eradicate cancer. Especially in an aggressive cancer like multiple myeloma, even after several rounds of chemotherapy involving concurrent administration of different drugs, tumor cells persist and regrow leading to relapse. In addition, chemotherapy often has undesirable side effects and in some cases chemotherapy has been shown to actually lead to enhanced tumor growth and resistance [14, 15]. We recently demonstrated that chemotherapy induces syndecan-1 shedding from tumor cells [16]. We also recently demonstrated that in myeloma patients, tumor cells that survive past chemotherapy had dramatically elevated levels of heparanase expression compared to cells harvested prior to therapy [17]. The above findings led us to speculate that treatment of myeloma cells with chemotherapeutic drugs may have the undesirable effect of stimulating the expression of heparanase. In the present work we find that anti-myeloma drugs activate the NF-κB pathway thereby elevating heparanase expression in tumor cells. In addition, chemotherapy leads to release of a soluble form of heparanase from tumor cells. Both macrophages and tumor cells endocytose the therapy-induced soluble heparanase in a heparan sulfate dependent manner. Uptake of therapy-induced soluble heparanase elevates TNF-α secretion from macrophages and stimulates the expression of pro-tumorigenic genes (HGF, MMP-9, and VEGF) and activates ERK and Akt signaling pathways in myeloma cells. We also demonstrate a novel use for the heparanase inhibitor Roneparstat in blocking the uptake of therapy-induced heparanase. Collectively, our above findings demonstrate a novel mechanism whereby heparanase induced by chemotherapy can contribute to the progression of cancer.
2. Results
2.1. Chemotherapy elevates heparanase expression by myeloma cells
We recently demonstrated that chemotherapy elevates shedding of syndecan-1 in myeloma [16]. Because heparanase (HPSE) is known to enhance syndecan-1 shedding, we investigated whether chemotherapy was inducing heparanase expression. Results revealed a robust increase in tumor cell heparanase level upon treatment with some chemotherapeutic drugs (Fig. 1A). While dexamethasone failed to affect heparanase expression in the myeloma cell lines tested, doxorubicin elevated heparanase expression in RPMI-8226 and CAG but not U266 cells while the proteasome inhibitor bortezomib (BTZ) stimulated heparanase expression in all three cell lines (Fig. 1A). Real-time PCR revealed a significant increase in heparanase transcript levels in BTZ and carfilzomib (CFZ) treated cells (Fig. 1B). Melphalan (Mel), an alkylating agent representing another class of myeloma therapeutics, also elevated heparanase transcript (Fig. 1B). Addition of cycloheximide (CHX) blocked the BTZ induced increase in heparanase expression confirming that BTZ was inducing synthesis of new heparanase protein (Fig. 1C).
Figure 1.

Anti-myeloma therapy elevates heparanase expression. (A) Heparanase protein was assessed by western blotting of total cell extracts from different myeloma cell lines after 14 h treatment with BTZ (25 nM), Dex (10 μM) or Doxorubicin (Dox, 5 μM). At these concentrations the drugs inhibited cell proliferation to a similar extent. Untreated cells served as controls. GAPDH is the loading control. (B) Heparanase transcript level in myeloma cells was assessed by real time PCR after 8 h treatment with BTZ (50 nM), CFZ (100 nM), Mel (20 μM). Transcript levels in vehicle treated cells served as control, *p<0.05 versus vehicle treated cells. (C) HPSE protein levels as revealed by western blotting after treatment with BTZ (25 nM) alone for 14 h or with BTZ after 2 h pretreatment with cycloheximide (CHX, 30 μg/ml).
2.2. Heparanase expression is elevated in chemoresistant myeloma cell lines
Because chemotherapy elevated heparanase expression, we next examined heparanase levels in variants of RPMI-8226 cells selected for their resistance to doxorubicin (Dox1V) or melphalan (LR5) [18, 19]. In both of these resistant lines, heparanase protein and transcript were elevated compared to the chemosensitive parental cells (Fig. 2A, B). To confirm that the elevation in heparanase expression was having an impact on the chemoresistant cells that could affect their behavior, we examined syndecan-1 size and HGF expression in these cells. Because heparanase trims the heparan sulfate chains on syndecan-1, an increase in heparanase expression reduces the size of syndecan-1 and this can be visualized on western blots [20]. Results revealed that syndecan-1 present in the chemoresistant cell lines was more polydisperse and smaller in size compared to syndecan-1 in parental cells (Fig. 2A) (note that we previously demonstrated that the amount of syndecan-1 present is similar in all three lines [16]). While the SDC1 size is not remarkably different between the parental and Dox1V (where only low levels of HPSE are present), it differs substantially in LR5 cells which have high levels of active HPSE. Hepatocyte growth factor (HGF), a cytokine elevated by heparanase [6], was significantly elevated in LR5 cells compared to RPMI-8226 cells (Fig. 2C).
Figure 2.
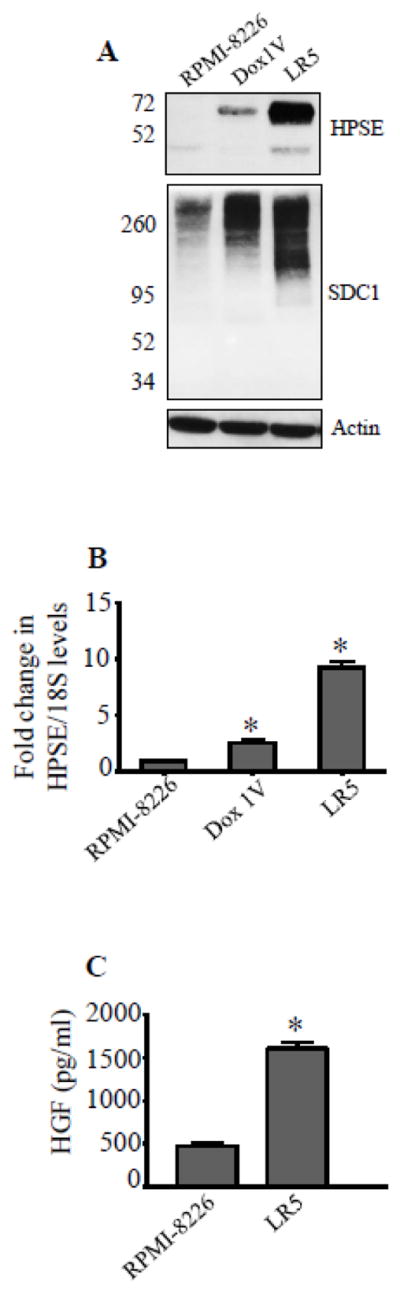
Heparanase expression is elevated in drug resistant myeloma cells. (A) Western blotting for heparanase (HPSE) and syndecan-1 (SDC1) in chemosensitive (RPMI-8226) cells and chemoresistant RPMI-8226 clones (Dox1V – doxorubicin resistant clone, LR5-melphalan resistant clone). (B) Heparanase transcript levels were determined by real time PCR and normalized against 18S rRNA levels in the above cell lines, *p<0.05 versus RPMI-8226. (C) Level of soluble HGF in conditioned media from RPMI-8226 and LR5 cells as determined by ELISA, *p<0.01 versus RPMI-8226.
2.3. Activation of the NF-κB pathway by chemotherapy drives heparanase expression
A series of experiments were performed to pinpoint the molecular mechanism leading to therapy-induced heparanase expression. We examined the involvement of nuclear factor NF-κB because it is a known regulator of heparanase expression in different pathologies [21–23]. Moreover, proteasome inhibitors like bortezomib are shown to activate the NF-κB pathway in many cancers [24–26] including myeloma [27]. Increasing doses of BTZ and CFZ clearly elevated heparanase protein expression in a dose-dependent manner in both RPMI-8226 and U266 cells (Fig. 3A). It is known that BTZ triggers the phosphorylation of IκB kinase (IKKβ) which in turn phosphorylates IκBα in these cell lines. This leads to degradation of IκBα, the inhibitor of NF-κB thereby leading to activation of the NF-κB pathway and expression of NF-κB – inducible genes [27]. Fig. 3A demonstrates that the increase in heparanase upon BTZ treatment correlated with the loss of IκBα expression (top panel – RPMI-8226, bottom panel – U266). Consistent with a role for NF-κB, blocking IκB kinase (IKKβ) by addition of specific IKKβ inhibitors, BMS345541 or BAY 11-7082 alongside BTZ diminished heparanase increase and partially restored IκBα (Fig. 3B). Additionally, BMS345541 or BAY 11-7082 also diminished BTZ-induced heparanase increase in myeloma cells with high heparanase expression (CAG HPSE-high) (Fig. 3C). Targeting reactive oxygen species (ROS) using L-N-acetylcysteine (L-NAC) is shown to diminish BTZ-induced NF- κB activation in RPMI-8226 [27]. Consistent with this finding, adding L-NAC alongside BTZ also decreased heparanase up-regulation (Fig. 3D), confirming that NF-κB regulates heparanase expression in response to therapy. Consistent with our finding of NF-κB elevating heparanase expression during therapy, high NF-κB binding activity is reported in chemoresistant LR5 cells and is shown to be affected by the IKKβ inhibitor (BMS345541) [28]. Addition of BMS345541 diminished the high heparanase expression in LR5 cells (Fig. 3E) demonstrating that NF-κB regulates high heparanase expression in drug resistant myeloma cells.
Figure 3.

Chemotherapy activates NF-κB to stimulate heparanase expression. (A) Levels of HPSE, total IκB, and GAPDH protein in RPMI-8226 (top panel) and U266 (bottom panel) cell extracts after treatment with increasing concentrations of BTZ or CFZ for 14 h. (B) Myeloma cell lines were pretreated with different inhibitors of IKKβ, BMS-345541 (BMS, 5 μM) or BAY11-7082 (BAY, 4 μM) for 2 h prior to incubation with BTZ (25 nM) for 12 h. At end of the incubation, cell extracts were probed by western blotting for HPSE, total IκB, and GAPDH. (C) CAG HPSE-high cells were pretreated with BMS-345541 (5 μM) or BAY11-7082 (4 μM) for 2 h and then incubated with BTZ for 12 h. After incubation, HPSE was probed by western blotting. Arrow points to pro-HPSE (~65 kDa), arrowhead points to active HPSE (~50 kDa). (D) Western blots for HPSE in extracts of myeloma cells treated with BTZ (25 nM) alone or with increasing concentrations of ROS inhibitor, N-acetyl L-cysteine (LNAC) for 14 h. (E) Western blot for heparanase in extracts from LR5 cells treated for 14 h with BMS-345541 (5 μM) or DMSO.
2.4. Chemotherapy stimulates the release of heparanase by myeloma tumor cells
Next we examined whether any of the heparanase being induced by chemotherapy was being released from cells. Western blots of medium conditioned by myeloma cells for 24 h after treatment with BTZ or CFZ revealed the presence of high levels of soluble heparanase (sHPSE) whereas cells treated with vehicle (control) released very low or no detectable levels of heparanase (Fig. 4A). Interestingly upon drug treatment both RPMI-8226 and CAG wild type cells primarily released the ~50kDa active heparanase molecule, whereas CAG HPSE-high cells released high levels of the 65 kDa pro form of heparanase (Fig. 4A).
Figure 4.

Anti-myeloma therapy causes release of high levels of soluble heparanase from tumor cells. (A) Western blotting for HPSE in concentrated conditioned media from different myeloma cell lines after 24 h exposure to BTZ (0.5 μM) or CFZ (0.5 μM). (B) Western blotting for HPSE in RPMI-8226 cell extracts after 2 h incubation with concentrated conditioned media from BTZ treated HPSE-high cells (BTZ CM) or concentrated conditioned media from vehicle treated HPSE-high cells (Con CM). Controls included RPMI-8226 cells treated with heparinase III enzyme (10 μg/ml) or cytochalasin D (10 μg/ml) for 2 h prior to incubation with BTZ CM. (C) Blotting for HPSE in total cell extracts from ARH-77 cells expressing either full length human syndecan-1 (ARH-77 SDC-1 WT) or mutated syndecan-1 lacking glycosaminoglycan attachment sites (ARH-77 SDC1 Δ GAG) following their incubation with BTZ CM for 0–120 min. (D) RPMI-8226, J774A.1 cells (murine macrophage cells), and HS-5 (human bone marrow stromal cells) were treated with 6.75μM of Roneparstat (Rone) prior to incubation for 2 h with BTZ CM. Cells were extracted and probed for HPSE. Control included cells incubated with Con CM. (E) Western blot for HPSE in cell extracts from RPMI-8226 cells after treatment with Con CM, BTZ CM immunodepleted with Con IgG (Con IP) or with anti-HPSE antibody (HPSE IP). The absence of HPSE in RPMI-8226 cells incubated with HPSE immunodepleted BTZ CM confirms that HPSE detected in RPMI-8226 extracts is supplied by the BTZ CM.
It has been well-described that cells can take up and activate exogenously supplied heparanase [29, 30]. To determine if sHPSE can be taken up by cells, RPMI-8226 cells were exposed to medium obtained from HPSE-high cells after BTZ treatment (BTZ conditioned medium, BTZ CM). Western blots of extracts from these RPMI-8226 cells revealed they take up the pro-from of sHPSE present in BTZ CM and process it into the active 50 kDa heparanase molecule (Fig. 4B). Digesting heparan sulfates on RPMI-8226 cells using bacterial heparinase III enzyme (that extensively degrades heparan sulfate) or addition of cytochalasin D (inhibitor of actin mediated endocytosis) prior to addition of BTZ CM, both diminished sHPSE uptake and processing demonstrating a role for heparan sulfate mediated endocytosis (Fig. 4B). To further confirm the role for heparan sulfates in sHPSE uptake, BTZ CM was added to ARH-77 (human lymphoblastoid) cells expressing either wild-type human syndecan-1 (ARH-77 SDC1 WT) or a mutated form of human syndecan-1 lacking all glycosylation attachment sites including glycosaminoglycan attachment sites (ARH-77 SDC1 ΔGAG). Blots revealed uptake of sHPSE was diminished in ARH-77 SDC1 ΔGAG cells (Fig. 4C). Because uptake of sHPSE was heparan sulfate dependent, we speculated that use of heparanase inhibitor, Roneparstat (Rone), a modified heparin that is 100% N-acetylated and 25% glycol split would block sHPSE uptake. In RPMI-8226, J774A.1 (murine macrophage cell line), and HS-5 (human bone marrow stromal cell line) uptake and processing of sHPSE was inhibited by Roneparstat (Fig. 4D). Immunodepletion of sHPSE from BTZ CM before addition to RPMI-8226 yielded no heparanase in RPMI-8226 cell extracts (Fig. 4E), confirming that heparanase detected in cells extracts was supplied by BTZ CM.
2.5. Soluble heparanase induced by chemotherapy upregulates proteins and signaling pathways known to promote aggressive tumor behavior
Addition of recombinant heparanase was previously shown to elevate tumor necrosis factor alpha (TNF-α) production by macrophages [31]. Because TNF-α is known to promote myeloma progression [32], we examined whether sHPSE present in BTZ CM could elevate TNF-α secretion by macrophage cell line J774 A.1. Addition of the BTZ CM significantly upregulated TNF-α secretion and this effect was lost if sHPSE was immunodepleted from the BTZ CM prior to its addition to the J774A.1 cells (Fig. 5A). We previously demonstrated that high expression of heparanase or addition of recombinant heparanase elevates expression of HGF, MMP-9, and VEGF in myeloma [6, 8, 33], proteins that promote aggressive tumor behavior. Similarly, we find that sHPSE also elevates MMP-9, VEGF, and HGF expression in myeloma cells (Fig. 5B). Immunodepletion of sHPSE or addition of heparanase inhibitor Roneparstat diminished the increase in gene expression triggered by BTZ CM (Fig. 5B). Activation of ERK and Akt are two major signaling events triggered by heparanase in different cancers [34]. To test the effect of sHPSE on ERK and Akt, we used media conditioned by CAG wild-type cells that were treated with bortezomib (BTZ CAG CM) or with vehicle (Con CAG CM) as a control. Results revealed that both ERK and Akt in different myeloma cell lines were activated upon addition of BTZ CAG CM and this was blocked by Roneparstat (Fig. 5C).
Figure 5.

Therapy-generated soluble heparanase activates pro-tumorigenic gene expression and cell signaling. (A) ELISA measuring the level of TNF-α secreted by J774 A.1 cells after incubation with Con CM or BTZ CM. Controls included BTZ CM immunodepleted of HPSE using HPSE IgG (HPSE IP) or Con IgG (Con IP). *p<0.001 versus Con CM, # p<0.01 versus Con IP. (B) Real-time PCR for VEGF, MMP-9, and HGF in RPMI-8226 cells after incubation with Con CM, BTZ CM immunodepleted with anti-heparanase antibody (HPSE IP) or BTZ CM immunodepleted with isotype matching control antibody (Con IP). Controls included Roneparstat (6.75uM) added along with BTZ CM (BTZ CM + Roneparstat). $p<0.05 versus Con CM, *p<0.05 versus BTZ CM + HPSE IP, # p<0.05 versus BTZ CM + Con IP. (C) For testing effect of sHPSE on cell signaling, we used concentrated conditioned media from BTZ treated CAG cells (BTZ CAG CM) and concentrated conditioned media from vehicle treated CAG cells (Con CAG CM). Western blot for different signaling molecules from myeloma cell lines after 2 h incubation; 1 - Con CAG CM, 2 - BTZ CAG CM, and 3 - BTZ CAG CM + Roneparstat (Rone, 6.75 μM).
3. Discussion
The present study demonstrates that chemotherapy can stimulate expression and release of heparanase, an enzyme known to promote aggressive tumor growth and metastasis. We found that: i) in tumor cells expressing little or no heparanase, anti-myeloma drugs can induce the synthesis and accumulation of heparanase, ii) mechanistically, this chemotherapy-induced increase in heparanase expression is traced to activation of the NF-kB pathway, iii) the heparanase induced by chemotherapy is released by the tumor cells and can be taken up by tumor and host (macrophage) cells and, iv) following uptake of heparanase it is activated and can promote the expression of pro-tumorigenic genes and stimulate cell signaling, and these effects are blocked by the anti-heparanase drug Roneparstat. Together our findings demonstrate how chemotherapy, counter to its intended purpose, could potentially fuel aggressive tumor behavior by stimulating heparanase expression. Moreover, these results are consistent with reports that other types of cancer therapy can induce heparanase. For example, radiotherapy enhances expression of the transcription factor Egr1 leading to elevation of heparanase in pancreatic cancer [35]. Also, treatment of prostate cancer cells with the methylation inhibitor 5′-aza-2-deoxycytidine upregulates heparanase expression [36].
In many cancer patients and in essentially all myeloma patients, chemotherapy reduces tumor burden but does not completely eliminate the tumor. In addition, it is likely that due to varying rates of diffusion, areas within tumor lesions are exposed to different concentrations of drug and thus death of tumor cells will occur at different rates leaving some cells alive for a period of time after first exposure to drug. In an attempt to simulate this scenario, in the present study we treated cells in vitro with concentrations of drugs that would kill a majority (~60–70%) of tumor cells within 14 hours. We then analyzed the remaining intact cells (Fig. 1). Although these cells were marked for apoptosis and would eventually die, we found that they had upregulated, and were actively synthesizing, heparanase. We believe this is due to the tumor cell response to stress, stress being previously demonstrated to upregulate heparanase expression [37]. We found upregulated heparanase expression was linked to activation of NF-κB (Fig. 3). NF-κB has been shown to regulate heparanase expression in different pathologies [21, 22, 38] and direct evidence of NF-κB regulation of heparanase promoter activity was also recently demonstrated [23]. Also, it has been shown that NF-κB drives pathogenesis and progression of multiple myeloma and is aberrantly activated in primary myeloma tumor cells [39, 40]. Thus, it is likely that in patients undergoing chemotherapy, NF-κB gets activated leading to upregulation of heparanase. This is consistent with our finding that in eight of nine myeloma patients examined; the tumor cells surviving chemotherapy had elevated heparanase expression compared to baseline levels measured prior to therapy. We have also found that cells having high heparanase expression are more resistant to chemotherapy than cells expressing low level of heparanase [17]. These findings together indicate that either therapy induces heparanase expression and this promotes survival, or that the populations of cells having high heparanase expression prior to therapy are able to survive the drug treatment.
Regardless of the role heparanase plays in the initial survival of tumor cells during chemotherapy, our finding that soluble heparanase is released by these cells is a highly significant observation. It is well-documented that soluble recombinant heparanase can be taken up by cells and alter their behavior [6, 29, 30, 33, 41]. We confirmed in the present study that the heparanase induced by chemotherapy and released by cells behaves similarly. This demonstrates a paracrine effect of the enzyme, the result being increased expression of pro-tumorigenic genes and stimulation of cell signaling. This could have a substantial impact on tumor regrowth because heparanase liberated during chemotherapy would likely accumulate at high levels within the tumor microenvironment via binding to heparan sulfate located on cell surfaces or within the extracellular matrix. We have previously demonstrated that chemotherapy stimulates shedding of syndecan-1 from tumor cells [16] and that large amounts of syndecan-1 accumulate within the marrow of myeloma patients post-chemotherapy [42]. This marrow-bound syndecan-1 could act to create reservoirs of heparanase that would be available to facilitate growth of the tumor cells that survived the therapy. This growth would be supported by the impact of heparanase on gene expression and cell signaling (Fig. 5) or, for example, via heparanase promotion of angiogenesis [33] and osteolysis [6, 41]. Interestingly, the type of HPSE, latent or active generated upon therapy is dependent on the cell type. For example, in wild type myeloma cells like RPMI-8226 and U266, exposure to therapy generates and releases the active form of HPSE (Fig. 1, 4). The chemoresistant cells (Dox 1V and LR5) express predominantly the latent HPSE (Fig. 2). Interestingly, myeloma cells engineered to express high levels of heparanase (CAG HPSE-high) express and release the latent heparanase when treated with therapy (Fig. 3, 4). Please note, both the latent and active forms of HPSE are detected only when the soluble latent form of HPSE generated from CAG-HPSE high cells is added to any other cell type (Fig. 4A, B, C). Such differences between the level of latent and active HPSE could be attributed to the amount of heparanase activating proteases like Cathepsin L, which may vary between different cell types.
Lastly, the finding that heparanase inhibitor Roneparstat blocks heparanase-induced stimulation of gene expression and cell signaling indicates this inhibitor could be used in combination with anti-myeloma drugs to improve their efficacy. In recent preclinical studies we tested this concept by treating animals harboring established myeloma tumors with Roneparstat in combination with either bortezomib or melphalan. We found that the combination inhibited tumor growth significantly better than did the use of these drugs as single agents [17]. Similar results were recently found employing a model of brain metastatic breast cancer where Roneparstat was found to overcome resistance to lapatinib [43]. Together with our current finding that chemotherapy upregulates heparanase expression and release, these studies provide a strong rationale for a clinical trial in myeloma patients designed to test Roneparstat in combination with anti-myeloma drugs.
4. Experimental procedures
4.1. Cell Lines and Reagents
Myeloma cell lines were grown in RPMI 1640 medium supplemented with 10% fetal bovine serum (FBS). Murine macrophage cell line, J774A.1 was cultured in DMEM supplemented with 10% FBS. Human bone marrow stromal cells (HS-5) was cultured in MEM supplemented with 10% FBS. The cell lines and the investigators who kindly provided the lines were: MM1.S (Drs. Nancy Krett and Steven Rosen, Northwestern University), RPMI-8226/LR5 (Dr. William Dalton, Moffitt Cancer Center, Tampa), RPMI-8226/Dox1V (Dr. Erming Tian, University of Arkansas for Medical Sciences, Little Rock), CAG (Dr. Joshua Epstein, University of Arkansas for Medical Sciences, Little Rock), and J774A.1 (Dr. David Briles, University of Alabama at Birmingham). HS-5, RPMI-8226 and U266 were obtained from the American Type Culture Collection (Manassas, VA). Generation of ARH-77 cells expressing either full length human syndecan-1 or syndecan-1 lacking glycosaminoglycan attachment sites has been previously described in [44]. CAG cells engineered to express high heparanase (CAG HPSE-high) have been described in [20]. Bortezomib and carfilzomib were purchased from SelleckChem; doxorubicin, dexamethasone, cycloheximide, L-N-acetylcysteine (L-NAC), and cytochalasin D from Sigma-Aldrich. BMS345541, and BAY 11-7082 were purchased from Calbiochem, and antibodies against total IκBα, P-ERK, total ERK, P-Akt, total Akt, and GAPDH were from Cell Signaling. Polyclonal anti-heparanase antibody (#1453) has been described previously [30]. Roneparstat is a proprietary drug of sigma-tau Research Switzerland and is currently in Phase I trials in advanced multiple myeloma patients (ClinicalTrials.gov Identifier: NCT01764880).
4.2. Western blotting
Cells were washed in PBS and extracted for 30 min in lysis buffer (50 mM Tris, pH 7.5, 150 mM NaCl, 0.5% Triton X-100) containing 1× HALT protease and phosphatase inhibitor mixture (Pierce) on ice. Cell lysates were then centrifuged at 12,000 × g at 4 °C for 15 min and supernatants were removed from the pellets. Total protein concentration of supernatant was determined by BCA assay (Pierce). Equal amounts of total protein were loaded onto 4–20% gradient SDS-polyacrylamide gels (Bio-Rad), transferred to either positively charged nylon membrane or nitrocellulose membranes (Schleicher & Schuell). Blots were incubated overnight with specific primary antibodies followed by horseradish peroxidase-conjugated secondary anti-mouse antibody (GE Healthcare). Either β-actin or GAPDH were used as loading controls in different experiments. Immunoreactive bands were detected using either enhanced chemiluminescence (GE Healthcare) or SuperSignal™ West Femto Maximum Sensitivity Substrate (Pierce).
4.3. RNA Extraction and Real-time PCR
After different treatments, cells were washed once with PBS and RNA was extracted using RNeasy columns (Qiagen). 1μg of total RNA was reverse transcribed using Maxima® First Strand cDNA synthesis kit for RT-qPCR (Thermo scientific). 25 ng of diluted cDNA was then mixed with 2X IQ™ SYBR® green supermix (Bio-Rad) together with gene specific primers (200 nM final concentration). The primers used were HGF 5′-CAATAGCATGTCAAGTGGAG-3′ (forward) and 5′-CTGTGTTCGTGTGGTATCAT-3′ (reverse), and HPSE 5′-TACCTTC ATTGCACAAACACTG-3′ (forward), 5′-ACTTGGTGACATTATGGAGGTT-3′(reverse), MMP-9 (F) TGACAGCGACAA GAAGTG, MMP-9 (R) CAGTGAAGCGGTACATAGG, VEGF (F) CTTGCCTTGCTGCTCTAC, VEGF (R) TGGCTTGAAGATGTACTCG), 5′-CGGC GACGACCCATTCGAAC-3′ (forward), 5′-GAATCGAACCCTGATTCCCCGTC-3′ (reverse) for 18S rRNA. The cycle parameters and analyses of real- time PCR have been described [6].
4.4. Preparation of heparanase monoclonal antibody and immunoprecipitation
Anti-human heparanase monoclonal antibody H1023 was generated in Balb/c mice at Eli Lilly and Company laboratories. Five 6 week old female mice were immunized subcutaneously with 75 μg of human heparanase protein in complete Freund’s adjuvant. The primed mice received 50 μg heparanase in incomplete Freund’s adjuvant subcutaneously every 3 weeks. Ten days after the third boost, animals were bled using a retro-orbital procedure and serum was extracted for antibody titer testing. Spleen cells were isolated and fused with a myeloma cell line (P3X63Ag8.653, ATCC® CRL-1580) at a ratio (1:4) using polyethylene glycol (PEG). Fused cells were seeded and after 1 week in selection media containing HAT (Hypoxanthine-aminopterin-thymidine) supernatants were tested for binding to heparanase by ELISA. Positives supernatants were tested for blocking activity using a high-throughput heparanase activity assay. Hybridoma cultures with inhibitory activity were subcloned by serial dilutions and the antibodies were purified by affinity chromatography. A recent study demonstrated the use of H1023 for targeting heparanase function in vivo [45]. All immunizations and bleeding procedures were performed following approved protocols. In the present study, soluble heparanase in the conditioned media was immunodepleted using the above anti-heparanase monoclonal antibody (6 μg/ml) bound to protein G-Sepharose beads (GE Healthcare).
4.5. Preparation of conditioned medium
Cells were seeded in complete growth medium 24 h prior to being tested. On the day of the experiment, cells were washed in serum-free medium (SFM) twice and incubated in SFM containing different chemotherapeutic agents. Conditioned medium was collected, cells and cellular debris separated by centrifugation and clarified media was concentrated 10 fold using Centriplus columns – 10 kDa cutoff value (Millipore Corp). Heparan sulfate chains in the concentrated media were degraded by incubating with 10 μg/ml heparinase III enzyme (generously provided by Dr. Jian Liu, University of North Carolina, Chapel Hill) at 37°C for 4 h. The media was then diluted 10 fold with SFM and concentrated as described above. This procedure was repeated twice. Complete removal of BTZ in concentrated media was confirmed by testing for its effect on accumulation of ubiquitinated proteins (marker of proteasome activity) in myeloma cells.
4.6. TNF-α bioassay
2.5 × 105 J774A.1 cells were seeded in 12 well plates. After overnight incubation with SFM, monolayers were washed once and incubated with conditioned media mixed with an equal volume of SFM for 24 h. At the end of the incubation, supernatant was collected, centrifuged to remove cell debris, and levels of TNF-α were determined using murine TNF-α DuoSet ELISA (R&D Systems).
4.7. Statistical Analyses
Data from experiments are expressed as mean ± SEM. Statistical analyses used for comparing different groups were performed using GraphPad Prism 5 (GraphPad Prism Software Inc., San Diego, CA).
Highlights.
Anti-myeloma drugs, bortezomib and carfilzomib elevate heparanase expression in an NF-κB dependent manner.
Upon treatment with chemotherapy, myeloma cells generate a soluble form of heparanase molecule.
Soluble heparanase generated by therapy activated pro-tumorigenic gene expression and signaling pathways in stromal and tumor cells.
Targeting heparanase alongside chemotherapy may improve the outcome of anti-cancer therapy and patient outcome.
Acknowledgments
This work was supported by commercial research grants from sigma-tau Research Switzerland (to RDS), and NIH CA138340 (to RDS), and United States-Israel Binational Science Foundation (BSF 2009230) to IV and RDS. We thank Dr. Jian Liu (The University of North Carolina at Chapel Hill) for kindly providing the heparinase III enzyme and Drs. Nancy Krett and Steven Rosen (Northwestern University, Chicago), Dr. William Dalton (Moffitt Cancer Center, Tampa), Dr. Erming Tian (University of Arkansas for Medical Sciences, Little Rock), Dr. Joshua Epstein (University of Arkansas for Medical Sciences, Little Rock), and Dr. David Briles (University of Alabama at Birmingham) for generously providing the cell lines used in the study. We also thank Juan Carcamo and Carla Bernard for assistance in preparing the H1023 monoclonal antibody against heparanase.
List of abbreviations
- HPSE
Heparanase
- NF-κB
Nuclear factor-kappa B
- SDC1
syndecan-1
- BTZ
bortezomib
- CFZ
carfilzomib
- HGF
hepatocyte growth factor
- VEGF
vascular endothelial growth factor
- CM
conditioned media
- ROS
reactive oxygen species
- WT
wild type
- GAG
glycosaminoglycan
- sHPSE
chemotherapy-generated soluble heparanase
- TNF-α
Tumor necrosis factor alpha
Footnotes
6. Author contributions
VCR and RDS conceived and coordinated the study, designed the experiments, and wrote the paper. VCR performed and analyzed the experiments. IV, MN, YZ developed, provided, and advised on use of reagents for figures 1–4. BP and AN provided and advised on the use of the anti-heparanase drug. All authors reviewed the results and approved the final version of the manuscript.
5. Competing interests
RDS received commercial grant support from sigma-tau Research Switzerland S.A. and is a member of their Scientific Advisory Board. Roneparstat (Rone) is a proprietary drug of sigma-tau Research Switzerland S.A. PB and AN are employees of sigma-tau Research Switzerland S.A. No potential conflicts of interest were disclosed by the other authors.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Vishnu C. Ramani, Email: vpramani@uab.edu.
Israel Vlodavsky, Email: vlodavsk@mail.huji.ac.il.
Mary Ng, Email: maryng00@gmail.com.
Yi Zhang, Email: yi.zhang@lilly.com.
Paola Barbieri, Email: paola.barbieri@st-research.ch.
Alessandro Noseda, Email: alessandro.noseda@st-research.ch.
References
- 1.Barash U, Cohen-Kaplan V, Dowek I, Sanderson RD, Ilan N, Vlodavsky I. Proteoglycans in health and disease: new concepts for heparanase function in tumor progression and metastasis. FEBS J. 2010;277:3890–3903. doi: 10.1111/j.1742-4658.2010.07799.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ramani VC, Purushothaman A, Stewart MD, Thompson CA, Vlodavsky I, Au JL, Sanderson RD. The heparanase/syndecan-1 axis in cancer: mechanisms and therapies. FEBS J. 2013;280:2294–2306. doi: 10.1111/febs.12168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Iozzo RV, Sanderson RD. Proteoglycans in cancer biology, tumour microenvironment and angiogenesis. J Cell Mol Med. 2011;15:1013–1031. doi: 10.1111/j.1582-4934.2010.01236.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Theocharis AD, Skandalis SS, Neill T, Multhaupt HA, Hubo M, Frey H, Gopal S, Gomes A, Afratis N, Lim HC, Couchman JR, Filmus J, Sanderson RD, Schaefer L, Iozzo RV, Karamanos NK. Insights into the key roles of proteoglycans in breast cancer biology and translational medicine. Biochim Biophys Acta Rev Cancer. 2015;1855:276–300. doi: 10.1016/j.bbcan.2015.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Iozzo RV, Schaefer L. Proteoglycan form and function: A comprehensive nomenclature of proteoglycans. Matrix Biol. 2015;42:11–55. doi: 10.1016/j.matbio.2015.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ramani VC, Yang Y, Ren Y, Nan L, Sanderson RD. Heparanase plays a dual role in driving hepatocyte growth factor (HGF) signaling by enhancing HGF expression and activity. J Biol Chem. 2011;286:6490–6499. doi: 10.1074/jbc.M110.183277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yang Y, Macleod V, Bendre M, Huang Y, Theus AM, Miao HQ, Kussie P, Yaccoby S, Epstein J, Suva LJ, Kelly T, Sanderson RD. Heparanase promotes the spontaneous metastasis of myeloma cells to bone. Blood. 2005;105:1303–1309. doi: 10.1182/blood-2004-06-2141. [DOI] [PubMed] [Google Scholar]
- 8.Purushothaman A, Chen L, Yang Y, Sanderson RD. Heparanase stimulation of protease expression implicates it as a master regulator of the aggressive tumor phenotype in myeloma. J Biol Chem. 2008;283:32628–32636. doi: 10.1074/jbc.M806266200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kelly T, Miao HQ, Yang Y, Navarro E, Kussie P, Huang Y, MacLeod V, Casciano J, Joseph L, Zhan F, Zangari M, Barlogie B, Shaughnessy J, Sanderson RD. High heparanase activity in multiple myeloma is associated with elevated microvessel density. Cancer Res. 2003;63:8749–8756. [PubMed] [Google Scholar]
- 10.Barash U, Zohar Y, Wildbaum G, Beider K, Nagler A, Karin N, Ilan N, Vlodavsky I. Heparanase enhances myeloma progression via CXCL10 downregulation. Leukemia. 2014;28:2178–2187. doi: 10.1038/leu.2014.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sanderson RD, Iozzo RV. Targeting heparanase for cancer therapy at the tumor-matrix interface. Matrix Biol. 2012;31:283–284. doi: 10.1016/j.matbio.2012.05.001. [DOI] [PubMed] [Google Scholar]
- 12.Jung O, Trapp-Stamborski V, Purushothaman A, Jin H, Wang H, Sanderson RD, Rapraeger AC. Heparanase-induced shedding of syndecan-1/CD138 in myeloma and endothelial cells activates VEGFR2 and an invasive phenotype: prevention by novel synstatins. Oncogenesis. 2016;5:e202. doi: 10.1038/oncsis.2016.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.DeVita VT, Jr, Chu E. A history of cancer chemotherapy. Cancer Res. 2008;68:8643–8653. doi: 10.1158/0008-5472.CAN-07-6611. [DOI] [PubMed] [Google Scholar]
- 14.Bruchard M, Mignot G, Derangere V, Chalmin F, Chevriaux A, Vegran F, Boireau W, Simon B, Ryffel B, Connat JL, Kanellopoulos J, Martin F, Rebe C, Apetoh L, Ghiringhelli F. Chemotherapy-triggered cathepsin B release in myeloid-derived suppressor cells activates the Nlrp3 inflammasome and promotes tumor growth. Nat Med. 2013;19:57–64. doi: 10.1038/nm.2999. [DOI] [PubMed] [Google Scholar]
- 15.Sun Y, Campisi J, Higano C, Beer TM, Porter P, Coleman I, True L, Nelson PS. Treatment-induced damage to the tumor microenvironment promotes prostate cancer therapy resistance through WNT16B. Nat Med. 2012;18:1359–1368. doi: 10.1038/nm.2890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ramani VC, Sanderson RD. Chemotherapy stimulates syndecan-1 shedding: a potentially negative effect of treatment that may promote tumor relapse. Matrix Biol. 2014;35:215–222. doi: 10.1016/j.matbio.2013.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ramani VC, Zhan F, He J, Barbieri P, Noseda A, Tricot G, Sanderson RD. Targeting heparanase overcomes chemoresistance and diminishes relapse in myeloma. Oncotarget. 2015 doi: 10.18632/oncotarget.6408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Futscher BW, Foley NE, Gleason-Guzman MC, Meltzer PS, Sullivan DM, Dalton WS. Verapamil suppresses the emergence of P-glycoprotein-mediated multi-drug resistance. Int J Cancer. 1996;66:520–525. doi: 10.1002/(SICI)1097-0215(19960516)66:4<520::AID-IJC16>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- 19.Bellamy WT, Dalton WS, Gleason MC, Grogan TM, Trent JM. Development and characterization of a melphalan-resistant human multiple myeloma cell line. Cancer Res. 1991;51:995–1002. [PubMed] [Google Scholar]
- 20.Yang Y, Macleod V, Miao HQ, Theus A, Zhan F, Shaughnessy JD, Jr, Sawyer J, Li JP, Zcharia E, Vlodavsky I, Sanderson RD. Heparanase enhances syndecan-1 shedding: a novel mechanism for stimulation of tumor growth and metastasis. J Biol Chem. 2007;282:13326–13333. doi: 10.1074/jbc.M611259200. [DOI] [PubMed] [Google Scholar]
- 21.Wu W, Pan C, Meng K, Zhao L, Du L, Liu Q, Lin R. Hypoxia activates heparanase expression in an NF-kappaB dependent manner. Oncol Rep. 2010;23:255–261. [PubMed] [Google Scholar]
- 22.Cao HJ, Fang Y, Zhang X, Chen WJ, Zhou WP, Wang H, Wang LB, Wu JM. Tumor metastasis and the reciprocal regulation of heparanase gene expression by nuclear factor kappa B in human gastric carcinoma tissue. World J Gastroenterol. 2005;11:903–907. doi: 10.3748/wjg.v11.i6.903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hadigal SR, Agelidis AM, Karasneh GA, Antoine TE, Yakoub AM, Ramani VC, Djalilian AR, Sanderson RD, Shukla D. Heparanase is a host enzyme required for herpes simplex virus-1 release from cells. Nat Commun. 2015;6:6985. doi: 10.1038/ncomms7985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dolcet X, Llobet D, Encinas M, Pallares J, Cabero A, Schoenenberger JA, Comella JX, Matias-Guiu X. Proteasome inhibitors induce death but activate NF-kappaB on endometrial carcinoma cell lines and primary culture explants. J Biol Chem. 2006;281:22118–22130. doi: 10.1074/jbc.M601350200. [DOI] [PubMed] [Google Scholar]
- 25.Nemeth ZH, Wong HR, Odoms K, Deitch EA, Szabo C, Vizi ES, Hasko G. Proteasome inhibitors induce inhibitory kappa B (I kappa B) kinase activation, I kappa B alpha degradation, and nuclear factor kappa B activation in HT-29 cells. Mol Pharmacol. 2004;65:342–349. doi: 10.1124/mol.65.2.342. [DOI] [PubMed] [Google Scholar]
- 26.Li C, Chen S, Yue P, Deng X, Lonial S, Khuri FR, Sun SY. Proteasome inhibitor PS-341 (bortezomib) induces calpain-dependent IkappaB(alpha) degradation. J Biol Chem. 2010;285:16096–16104. doi: 10.1074/jbc.M109.072694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hideshima T, Ikeda H, Chauhan D, Okawa Y, Raje N, Podar K, Mitsiades C, Munshi NC, Richardson PG, Carrasco RD, Anderson KC. Bortezomib induces canonical nuclear factor-kappaB activation in multiple myeloma cells. Blood. 2009;114:1046–1052. doi: 10.1182/blood-2009-01-199604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yarde DN, Oliveira V, Mathews L, Wang X, Villagra A, Boulware D, Shain KH, Hazlehurst LA, Alsina M, Chen DT, Beg AA, Dalton WS. Targeting the Fanconi anemia/BRCA pathway circumvents drug resistance in multiple myeloma. Cancer Res. 2009;69:9367–9375. doi: 10.1158/0008-5472.CAN-09-2616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Vreys V, Delande N, Zhang Z, Coomans C, Roebroek A, Durr J, David G. Cellular uptake of mammalian heparanase precursor involves low density lipoprotein receptor-related proteins, mannose 6-phosphate receptors, and heparan sulfate proteoglycans. J Biol Chem. 2005;280:33141–33148. doi: 10.1074/jbc.M503007200. [DOI] [PubMed] [Google Scholar]
- 30.Gingis-Velitski S, Zetser A, Kaplan V, Ben-Zaken O, Cohen E, Levy-Adam F, Bashenko Y, Flugelman MY, Vlodavsky I, Ilan N. Heparanase uptake is mediated by cell membrane heparan sulfate proteoglycans. J Biol Chem. 2004;279:44084–44092. doi: 10.1074/jbc.M402131200. [DOI] [PubMed] [Google Scholar]
- 31.Blich M, Golan A, Arvatz G, Sebbag A, Shafat I, Sabo E, Cohen-Kaplan V, Petcherski S, Avniel-Polak S, Eitan A, Hammerman H, Aronson D, Axelman E, Ilan N, Nussbaum G, Vlodavsky I. Macrophage activation by heparanase is mediated by TLR-2 and TLR-4 and associates with plaque progression. Arterioscler Thromb Vasc Biol. 2013;33:e56–65. doi: 10.1161/ATVBAHA.112.254961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hideshima T, Chauhan D, Schlossman R, Richardson P, Anderson KC. The role of tumor necrosis factor alpha in the pathophysiology of human multiple myeloma: therapeutic applications. Oncogene. 2001;20:4519–4527. doi: 10.1038/sj.onc.1204623. [DOI] [PubMed] [Google Scholar]
- 33.Purushothaman A, Uyama T, Kobayashi F, Yamada S, Sugahara K, Rapraeger AC, Sanderson RD. Heparanase-enhanced shedding of syndecan-1 by myeloma cells promotes endothelial invasion and angiogenesis. Blood. 2010;115:2449–2457. doi: 10.1182/blood-2009-07-234757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fux L, Ilan N, Sanderson RD, Vlodavsky I. Heparanase: busy at the cell surface. Trends Biochem Sci. 2009;34:511–519. doi: 10.1016/j.tibs.2009.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Meirovitz A, Hermano E, Lerner I, Zcharia E, Pisano C, Peretz T, Elkin M. Role of heparanase in radiation-enhanced invasiveness of pancreatic carcinoma. Cancer Res. 2011;71:2772–2780. doi: 10.1158/0008-5472.CAN-10-3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ogishima T, Shiina H, Breault JE, Tabatabai L, Bassett WW, Enokida H, Li LC, Kawakami T, Urakami S, Ribeiro-Filho LA, Terashima M, Fujime M, Igawa M, Dahiya R. Increased heparanase expression is caused by promoter hypomethylation and up-regulation of transcriptional factor early growth response-1 in human prostate cancer. Clin Cancer Res. 2005;11:1028–1036. [PubMed] [Google Scholar]
- 37.Ostrovsky O, Shimoni A, Baryakh P, Morgulis Y, Mayorov M, Beider K, Shteingauz A, Ilan N, Vlodavsky I, Nagler A. Modification of heparanase gene expression in response to conditioning and LPS treatment: strong correlation to rs4693608 SNP. J Leukoc Biol. 2014;95:677–688. doi: 10.1189/jlb.0313147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Andela VB, Schwarz EM, Puzas JE, O’Keefe RJ, Rosier RN. Tumor metastasis and the reciprocal regulation of prometastatic and antimetastatic factors by nuclear factor kappaB. Cancer Res. 2000;60:6557–6562. [PubMed] [Google Scholar]
- 39.Demchenko YN, Kuehl WM. A critical role for the NFkB pathway in multiple myeloma. Oncotarget. 2010;1:59–68. doi: 10.18632/oncotarget.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Demchenko YN, Glebov OK, Zingone A, Keats JJ, Bergsagel PL, Kuehl WM. Classical and/or alternative NF-kappaB pathway activation in multiple myeloma. Blood. 2010;115:3541–3552. doi: 10.1182/blood-2009-09-243535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yang Y, Ren Y, Ramani VC, Nan L, Suva LJ, Sanderson RD. Heparanase enhances local and systemic osteolysis in multiple myeloma by upregulating the expression and secretion of RANKL. Cancer Res. 2010;70:8329–8338. doi: 10.1158/0008-5472.CAN-10-2179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bayer-Garner IB, Sanderson RD, Dhodapkar MV, Owens RB, Wilson CS. Syndecan-1 (CD138) immunoreactivity in bone marrow biopsies of multiple myeloma: shed syndecan-1 accumulates in fibrotic regions. Mod Pathol. 2001;14:1052–1058. doi: 10.1038/modpathol.3880435. [DOI] [PubMed] [Google Scholar]
- 43.Zhang L, Ngo JA, Wetzel MD, Marchetti D. Heparanase mediates a novel mechanism in lapatinib-resistant brain metastatic breast cancer. Neoplasia. 2015;17:101–113. doi: 10.1016/j.neo.2014.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ramani VC, Pruett PS, Thompson CA, DeLucas LD, Sanderson RD. Heparan sulfate chains of syndecan-1 regulate ectodomain shedding. J Biol Chem. 2012;287:9952–9961. doi: 10.1074/jbc.M111.330803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Weissmann M, Arvatz G, Horowitz N, Feld S, Naroditsky I, Zhang Y, Ng M, Hammond E, Nevo E, Vlodavsky I, Ilan N. Heparanase-neutralizing antibodies attenuate lymphoma tumor growth and metastasis. Proc Natl Acad Sci U S A. 2016;113:704–709. doi: 10.1073/pnas.1519453113. [DOI] [PMC free article] [PubMed] [Google Scholar]