

Abstract
Impairing the division of cancer cells with genotoxic small molecules has been a primary goal to develop chemotherapeutic agents. However, DNA mismatch repair (MMR)-deficient cancer cells, are resistant to most conventional chemotherapeutic agents. Here we have identified baicalein as a small molecule that selectively kills MutSα-deficient cancer cells. Baicalein binds preferentially to mismatched DNA and induces a DNA damage response in a mismatch repair-dependent manner. In MutSα-proficient cells, baicalein binds to MutSα to dissociate CHK2 from MutSα leading to S phase arrest and cell survival. In contrast, continued replication in the presence of baicalein in MutSα-deficient cells results in a high number of DNA double-strand breaks and ultimately leads to apoptosis. Consistently, baicalein specifically shrinks MutSα-deficient xenograft tumors and inhibits the growth of AOM-DSS-induced colon tumors in colon-specific MSH2 knockout mice. Collectively, baicalein offers the potential of an improved treatment option for patients with tumors with a DNA MMR deficiency.
Keywords: Lynch Syndrome, Baicalein, DNA mismatch repair, Modulation of DNA repair
Introduction
The DNA mismatch repair (MMR) pathway maintains genome stability by removing base-base mismatches and insertion/deletion loops that arise during DNA replication or as a result of DNA damage(1,2). These DNA lesions are recognized by MutSα (a heterodimer of MSH2/MSH6) or MutSβ (a heterodimer of MSH2/MSH3), which recruit the MutLα (MLH1/PMS2) or MutLβ (MLH1/PMS1) proteins. The MutS/MutL complex then loads exonuclease I to degrade the strand containing the mispaired nucleotide, and the resulting gap is filled in by Polymerase δ(3,4). The MMR proteins can also affect cell cycle control and apoptosis in response to certain types of DNA damage(4,5). However, the precise mechanism by which MMR proteins regulate the cell cycle is not clearly understood.
Germline loss-of-function mutations in genes involved in the DNA MMR pathway cause the autosomal dominant Lynch Syndrome, also known as Hereditary Nonpolyposis Colorectal Cancer (HNPCC). Individuals with Lynch Syndrome have an 80% risk of developing colorectal cancer in their lifetime, with the average age of onset being 45 years(6). Lynch Syndrome accounts for approximately 2% of all colorectal cancers diagnosed each year(7), and patients with Lynch Syndrome are at an increased risk of developing a range of other tumor types, including gastric, endometrial, ovarian, and bladder cancers(8–10). Although some standard chemotherapeutic regiments as well as immunotherapy PD-1 may be beneficial to Lynch Syndrome patients(11–13), the underlying mechanisms of these treatment options for Lynch Syndrome tumors are not clear. And some studies also pointed out that MMR deficient cells may be resistant to the standard chemotherapeutic regimens for colorectal cancer(14).
We have recently developed an ATAD5 (ATPase family AAA domain containing protein 5)-luciferase assay that allows high-throughput screening for potential chemotherapeutic compounds that act by inducing DNA replication stress or DNA damage. In this assay, the stabilization of the ATAD5-luciferase protein, which is measured by an increase in luciferase activity, serves as a biomarker of DNA replication stress or DNA damage(15). Using the ATAD5-luciferase assay, we screened 344,385 small molecules from the NIH Molecular Library Probe production Centers Network (MLPCN) collection, and selected 289 compounds that induced an increase in luciferase activity, indicating that they caused DNA damage. Hypothesizing that these compounds could provide a better treatment option for Lynch Syndrome tumors, we tested the ability of the compounds to selectively kill cells with mutations in the MSH2 gene. From this screen, we identified baicalein as a small molecule capable of inducing apoptosis in MSH2-deficient cells both in vitro and in vivo. Baicalein is a flavone derived from the roots of Scutellaria baicalensis and Scutellaria lateriflora. Previously, baicalein was used in some Asian counties as herbal supplements to enhance liver health. Recent years, more and more studies of baicalein have proceeded on many human diseases related areas, such as cancer and diabetes(16–20). Although baicalein has previously been reported to have anti-tumor activities(18,19,21), this manuscript identifies a novel mechanism of action for baicalein in which the compound acts on the MutSα/CHK2/ATM pathway as well as preferentially binding to the mismatched DNA.
Materials and Methods
Compounds
Baicalein, netropsin, colochicine and CHK2 inhibitor hydrate (2-(4-(4-Chlorophenoxy)phenyl)-1H-benzimidazole-5-carboxamide hydrate) were all purchased from Sigma. AOM and DSS (MW 40,000–50,000) were purchased from Sigma-Aldrich and USBAffymetrix, respectively.
Control and baicalein-supplemented diets
All diets were made by Harlan Laboratories using baicalein purchased from Sigma. The control mice were fed a Global 18% Protein Rodent Diet, and the experimental group was fed a Global 18% Protein Rodent Diet containing 0.05% baicalein.
Cell culture
HEC59, HEC59-2, LoVo (Gift from Dr. Peggy Hsieh (NIH)’s laboratory), HeLa, HEK293T (Gift from Dr. Pamela Schwartzberg (NIH)’s laboratory), and MRC5 (Gift from Dr. Roger Woodgate (NIH)’s laboratory) cells were cultured in Dulbecco’s Minimum Eagle Medium (DMEM)+ GlutaMAX (Invitrogen) supplemented with 10% FBS (Hyclone Laboratories) and 50 U/mL penicillin and 50 μg/mL streptomycin (Invitrogen). HEC59-2 cells were also maintained in 400 μg/mL G418 (Invitrogen). HT-29 cells (purchased from ATCC) were cultured in McCoy’s 5A Medium (Invitrogen) supplemented with 10% FBS and 50 U/mL penicillin and 50 μg/mL streptomycin (Invitrogen). All cells were maintained at 37°C under a humidified atmosphere and 5% CO2. To authenticate cells used in the study, we frequently checked the expression of DNA repair proteins, especially MMR-related proteins to make sure cells used in the study did (HEC59-2, HeLa, MRC5, HEK293T, HCT116-3) or did not (HEC59, HCT116, LoVo) express proteins having mutations. In addition, we checked the morphology of cell lines with microscopy to make sure they did not change their original shapes. We checked all cells for mycoplasma test to make sure that there was no contamination. Lastly, we checked chromosome numbers by karyotyping of cell lines used to make sure cells did not produce abnormal chromosome numbers. We did not use cell lines more than a month from original frozen stocks in culture to avoid potential accumulation of unnecessary mutations.
Cell viability assay
Cells were plated in white, solid-bottom 96-well plates at a final density of 15,000 cells/well. After the cells had attached to the plate, 1:2 dilutions of the indicated compounds were added. 24 hours following treatment, cell viability was determined using Cell Titer-Glo (Promega) according to the manufacturer’s protocol. Viability was quantified on a Fluoroskan Ascent Luminometer (Thermo Scientific). Dose-response curves and EC50 values were generated using GraphPad Prism.
Western Blot and Coimmunoprecipitation
Anti-PARP was purchased from BD Biosciences. Anti-caspase 3 and anti-p-CHK2 (Thr68) were purchased from Cell Signaling. Anti-CHK2, anti-ATM, HRP-conjugated-anti-PCNA and anti-MSH2 were purchased from Santa Cruz Biotechnology. Anti-MSH2, anti-MSH6 and HRP-conjugated-anti-β tubulin were purchased from Abcam. Anti-histone H3 was purchased from Upstate. Anti-phospho-histone H2AX was purchased from GeneTex. Anti-CDC25A was purchased from Thermo Scientific. Anti-SHPRH was purchased from Origene. Anti-Digoxigenin was purchased from Roche.
For Western immunoblotting, proteins from cells or mice colon were separated by SDS–polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to PVDF membrane filter using bio-Rad mini, midi transfer packs and Transblot turbo. After incubation with the desired antibodies, the blots were developed with Pierce ECL plus western blotting substrate. For Coimmunoprecipitation, cells lysed with the lysis/wash buffer [0.025 M Tris, 0.15 M NaCl, 0.001 M EDTA, 1% NP-40, 5% glycerol, pH 7.4 with Protease Inhibitor Cocktail (Roche)] for 0.5 hour at 4°C. After incubation with the desired primary antibody for 18 hours at 4°C via gentle rocking, immune complexes were captured by mixing with a final concentration of 2.5% protein G Sepharose beads (GE) for 2 hours at 4°C on a rotator. Beads were subsequently washed three times with the lysis/wash buffer, followed by SDS-PAGE and immunoblotting analysis after elution by boiling in 2 × SDS loading buffer.
Protein preparations
Human wild-type MutSα were expressed in baculovirus and purified as previously described(22). SHPRH protein was expressed in baculovirus and purified (unpublished data).
H&E staining, TUNEL and immunohistochemistry
H&E staining, TUNEL and immunohistochemistry were commercially performed by the Histoserv Company (Tumors or tissues were collected, and fixed in formalin, then were picked up by Histoserv Company). Detailed procedures can be found on the company website (http://www.histoservinc.com/). To quantify the images, the number of positive signals was manually counted in multiple fields of view. The number of total positive signals was then divided by the number of fields visualized for counting.
Results
Baicalein selectively kills MutSα-deficient cells
The human endometrial adenocarcinoma cell line HEC59 carrying two different loss-of-function MSH2 nonsense mutations(23), and HEC59-2 cells, which are HEC59 cells into which human chromosome 2 was transferred restoring wild type MSH2 expression and functional MMR(24), were treated with serial dilutions of the 289 compounds, and dose-response viability curves were used to calculate the concentration of each compound required to induce 50% cell death (EC50). The screen revealed 9 compounds that were at least 2-fold more potent in the HEC59 cells than in the HEC59-2 cells (Fig. 1A). A secondary viability screen using a luciferase-based ATP assay and a colony formation assay showed that one of these compounds, baicalein (Fig. 1A pale gray dot, B), generated the most reproducible results (Fig. 1C and Supplementary Fig. S1A). Baicalein treatment resulted in PARP1 and caspase 3 cleavages in HEC59 cells, but not in the MMR-proficient HEC59-2 cells (Fig. 1E), indicating that the loss of viability observed upon MSH2 deficiency occurs via apoptosis. We confirmed the specificity of baicalein’s activity for MMR-deficiency by reducing MSH2 protein levels by siRNA in HT29 (colorectal adenocarcinoma) cells and HeLa (cervical cancer) cells (Fig. 1D, Supplementary Fig. S1B, S1C and S1D). Furthermore, we measured the cell viability of LoVo cell, which is a colorectal carcinoma cell defective in MSH2. Due to the lack of LoVo WT cells, we used MMR-proficient MRC5 (human lung fibroblast) cells and HT29 cells for comparison. Consistent with other cell viability results, LoVo cells were more sensitive to baicalein than MRC5 cells and HT29 cells (Supplementary Fig. S1E). In addition, we tested baicalein in another pair of cell lines, HCT116, which harbors a homozygous mutation of the MLH1 gene, and HCT116-3 cell line, where the transfer of human chromosome 3 into HCT116 cells restores wild type MLH1 expression(25)(Fig. S1F). Baicalein also exhibited selective sensitivity on HCT116 cells, which suggests a general activity of baicalein targeting mismatch repair deficient cells. In contrast to MMR-deficiency, there was no selective killing of other DNA repair deficient cells including PARP1, p53, RAD54b, FANCA, FANCG, FANCD2, ATM, ATR, NBS, Ku70/Rad54, FANCC, XPA, PolB, UBC13, FEN1, ATG5, Pol η/ζ, and Rev3 by baicalein (data not shown).
Figure 1. MutSα deficient cells are sensitive to baicalein.
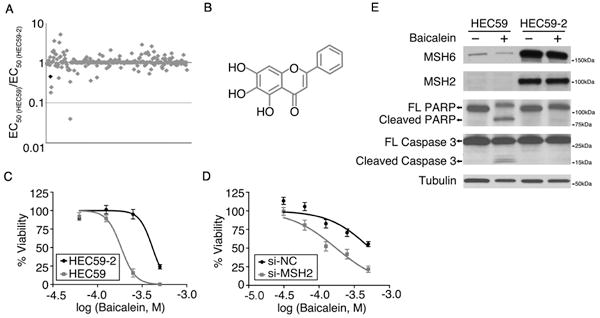
A, HEC59 and HEC59-2 cells were treated with 289 positive hit compounds from the ATAD5-luc screen. EC50 values were calculated using GraphPad Prism. B, Chemical structure of baicalein (5,6,7-trihydroxyflavone). C, D, HEC59 and HEC59-2 cells (C), and HT29 cells transfected with negative control (NC) siRNA or siRNA targeting MSH2 (D) were treated with baicalein (0–500 μM final concentration) for 24 hours. Cell viability was determined using Cell Titer-Glo. Data are shown as mean ± S.E. EC50 values in (C) is 184 μM for HEC59 and 412 μM for HEC59-2. EC50 values in (E) is 530 μM for si-NC and 175 μM for si-MSH2. E, HEC59 and HEC59-2 cells were treated with 250 μM baicalein for 72 hours. Total lysate from these cells was subjected to western blot analysis.
Baicalein-treated HEC59 cells are deficient in CHK2-regulated S phase checkpoint arrest
Given the influence of MutSα on S phase cell cycle checkpoint activation(5), we tested the effect of baicalein on cell cycle progression in cells synchronized with hydroxyurea (HU). We found that baicalein-treated HEC59-2 cells remained largely in the S phase of the cell cycle, whereas a large percentage of baicalein-treated HEC59 cells were able to progress to the G2/M phase (Fig. 2A). In addition, we monitored DNA replication using an EdU incorporation assay. Baicalein inhibited DNA replication in MutSα-proficient HEC59-2 cells, but not in the MutSα-deficient HEC59 cells (Supplementary Fig. S2A and S2B). We hypothesized that there would be MMR-dependent checkpoint activation in response to baicalein. Consistent with this hypothesis, we found an induction of CHK2 phosphorylation and subsequent CDC25A degradation predominately in baicalein-treated MutSα-proficient HEC59-2 cells, although baicalein treatment resulted in similar chromatin-bound mono-ubiquitylated PCNA regardless of MMR activity (Fig. 2B). Low levels of CHK2 phosphorylation were also observed in baicalein-treated HEC59 cells, most likely due to the MutSα–independent activity of ATM Consistent with the activation of CHK2 by baicalein in a MutSα-dependent manner, the MutSα-proficient cells became sensitive to baicalein following treatment with a CHK2 inhibitor (Fig. 2C).
Figure 2. Baicalein-treated HEC59 cells are deficient in CHK2-regulated S phase checkpoint arrest.
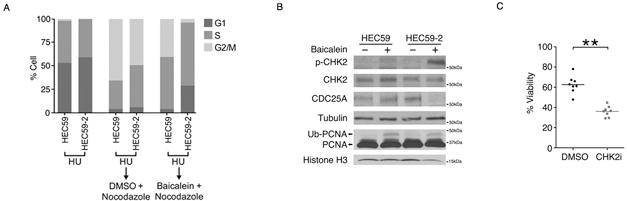
A, Cell cycle analysis of HEC59 and HEC59-2 cells synchronized with hydroxyurea (HU) and then released into fresh media containing nocodazole plus DMSO or 100 μM baicalein. B, HEC59 and HEC59-2 cells were treated with 100 μM baicalein for 24 hours. Total lysate from the cells was subjected to western blot analysis for CHK2, CDC25A, and tubulin. Chromatin-bound lysate from the cells was subjected to western blot analysis for PCNA and histone H3. C, HEC59-2 cells were treated with 125 μM baicalein with or without CHK2 inhibitor (10 μM) for 24 hours. Cell viability was determined using Cell Titer-Glo. Data is shown as mean ± S.E. **p < 0.01 as determined by two-tailed Student’s t-test.
Baicalein binds to MutSα
It has been demonstrated that MutSα interacts with CHK2, and that it forms a complex with ATM (5,26). We hypothesized that baicalein would affect the interaction between MutSα and CHK2/ATM that is required for cell cycle arrest. In MutSα proficient HEC59-2 cells, immunoprecipitation of MutSα with a MSH6 antibody also pulled down ATM and CHK2 (Fig. 3A). Similarly, immunoprecipitation of CHK2 co-precipitated MutSα and ATM (Fig. 3B). Baicalein treatment inhibited the interaction between MutSα and CHK2/ATM (Fig. 3A and 3B). The interactions were mediated by MutSα, because cells not expressing MSH2 (either by mutation or silencing by siRNA) had significantly reduced interactions between MutSα and CHK2/ATM (Fig. 3A, B and Supplementary Fig. S3A) and between ATM and CHK2 (Fig. 3B and Supplementary Fig. S3A). The MutSα/CHK2/ATM interactions were not mediated through DNA because the interactions between proteins remained intact even when cell extracts were pre-treated with ethidium bromide (EB), which disrupts the interaction between proteins and DNA (Supplementary Fig. S3B). Thus, MutSα, acting as a docking platform for ATM and CHK2, is dissociated from ATM and CHK2 by baicalein, which in turn, activates the S-phase checkpoint through CHK2 phosphorylation by ATM. To study the dynamics of the interaction among these proteins, we used siRNA targeting CHK2 or ATM. The absence of CHK2 had little effect on the interaction between MutSα and ATM in untreated cells and in cells treated with baicalein, and absence of ATM also had little effect on the interaction between MutSα and CHK2 in untreated cells and in cells treated with baicalein (Supplementary Fig. S3C). In addition, we used a double-thymidine block to synchronize HeLa cells to compare the interaction pattern in different cell cycle phases. Immunoprecipitation of MSH6 co-precipitated a similar amount of ATM or CHK2 in both G1/S and G2 phases, suggesting the interaction patterns are similar in G1/S and G2 (Supplementary Fig. S3D).
Figure 3. Baicalein binds to MutSα.
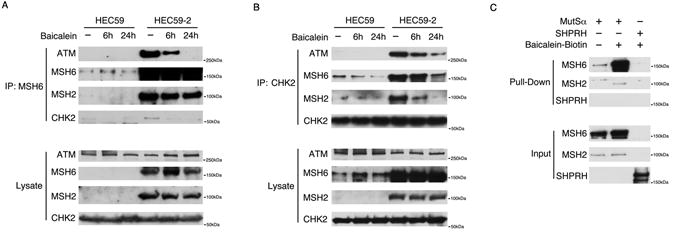
A, B, HEC59 and HEC59-2 cells were treated with 100 μM baicalein for 6 or 24 hours. The indicated proteins were immunoprecipitated with antibodies against MSH6 (A) or CHK2 (B). C, Purified MutSα or SHPRH was incubated with baicalein-biotin and bound proteins were detected by western blotting following a pull-down with streptavidin.
The baicalein-induced dissociation of MutSα from CHK2/ATM suggested that baicalein could directly bind to MutSα and inhibit its interaction with CHK2/ATM. To test this hypothesis, we used the Drug Affinity Responsive Target Stability (DARTS) assay that detects the interaction between the target protein and a small molecule by measuring resistance to proteolysis (27,28) (Supplementary Fig. S4A). As a control we used SHPRH, a nuclear protein with a molecular weight similar to MSH6. DARTS analysis revealed that baicalein protects MutSα, but not SHPRH, from degradation by pronase, suggesting that MutSα interacts with baicalein (Supplementary Fig. S4B and C). To confirm the direct interaction, biotin-tagged baicalein (baicalein-biotin) (Supplementary Fig. S4D), which was shown to be as active in MMR-dependent cell viability assays as the untagged molecule (Supplementary Fig. S4E), was incubated with MutSα or SHPRH and pulled down with streptavidin beads. MutSα, but not SHPRH, was co-precipitated suggesting that baicalein directly binds to MutSα (Fig. 3C). In addition, we used a Biacore assay to measure the interaction between baicalein and MutSα as well as baicalein and SHPRH, and calculate the KD between baicalein and MutSα, which was about 32.2 μM. In contrast, SHPRH did not show interaction with baicalein(Supplementary Fig. S4F and S4G). Collectively, these data suggest that baicalein directly binds to MutSα, which in turn dissociates CHK2/ATM from MutSα.
Baicalein preferentially binds to mismatched DNA
Since CHK2 activation is often linked to DNA double-strand break (DSB) damage, we hypothesized that baicalein would generate DSBs, and that MutSα deficiency would further increase the level of DSBs due to the absence of proper checkpoint activation through CHK2. Consistent with the hypothesis, pulsed-field gel electrophoresis (PFGE) revealed that there were more DSBs produced in MutSα mutant cells than wild type cells following baicalein treatment (Fig. 4A and 4B). To determine whether baicalein intercalates in DNA, we conducted an assay that measures the decrease in fluorescence due to the displacement of DNA-bound EB by a DNA-binding compound(29). Similar to netropsin, which is known to bind to the minor groove of DNA, but in contrast to colchicine, which does not interact with DNA, the incubation of baicalein with EB-bound DNA resulted in a dose-dependent decrease in fluorescence (Supplementary Fig. S5A). However, baicalein does not form inter-strand crosslinks since denatured baicalein-bound DNA only produced single stranded DNA (Supplementary Fig. S5B). To determine whether baicalein has a preference for binding to matched or mismatched double-stranded DNA, we compared one dimensional proton NMR spectra of both free and baicalein-bound matched and mismatched DNA. Baicalein resulted in much higher chemical shift perturbation of mismatched DNA compared to the matched DNA (Fig. 4C). The resonances of the imino group generated by the mismatched bases disappeared upon baicalein binding, suggesting that baicalein binds to the mismatched bases. Consistently, baicalein-biotin preferentially pulled down mismatched DNA (Supplementary Fig. S5C). In addition, we used a Biacore assay to measure the interaction between baicalein and DNA, and calculate the KD between baicalein and DNA, which was about 210 μM for matched DNA and about 31.7 μM for mismatched DNA (Supplementary Fig. S5D). To elucidate how baicalein interacts with both MutSα and mismatched DNA, mismatched DNA and baicalein-biotin were mixed together and baicalein was pulled down with streptavidin beads. The double bindings were examined separately (Supplementary Fig. S5E and S5F), and revealed that baicalein interacts with both mismatched DNA and MutSα. Interestingly, the binding of baicalein to mismatched DNA reduced the interaction of MutSα with mismatched DNA (Supplementary Fig. S5G). Consistently, the dose-dependent interaction between MutSα and mismatched DNA measured by the biacore assay (Supplementary Fig. S5H) was blocked when baicalein was preincubated with MutSα (Supplementary Fig. S5I). Such inhibition of the interaction between MutSα and mismatched DNA by baicalein was baicalein dose dependent (Supplementary Fig. S5J). Collectively, these data indicate that baicalein binds to mismatched DNA and MutSα, which releases MutSα from DNA and dissociates it from CHK2/ATM.
Figure 4. Baicalein binds to mismatched DNA and induced DSB in MMR deficient cells.
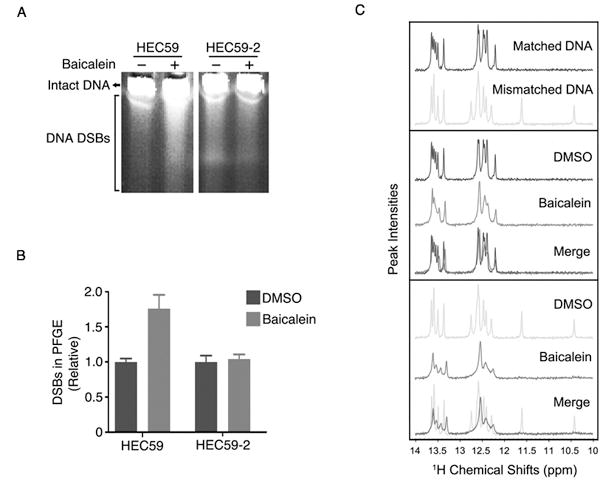
A, HEC59 and HEC59-2 cells were treated with 100 μM baicalein for 24 hours and DNA DSBs were visualized by pulsed-field gel electrophoresis. B, The intensity of the smears produced by DNA containing DSBs in (A) was quantified using ImageJ and normalized to the DMSO control. The graph represents the average of two independent experiments ± S.E. C, Upper panel: Comparison of the NMR spectra of matched double stranded DNA (Matched DNA) and A/G base mismatched DNA (Mismatched DNA). Middle panel: Comparison of the chemical shifts upon the binding of drug to the matched DNA. Bottom panel: Comparison of the chemical shifts upon the binding of drug to the mismatched DNA. Only the resonances of the imino groups (10–14 ppm) are shown here.
We hypothesized that the baicalein-induced DSBs would be generated by endonuclease(s). We silenced the expression of APE1, XPF, XPG and MUS81 endonucleases with siRNA to determine their role in DSB formation upon baicalein treatment. Only XPF expression was required for baicalein-induced H2AX phosphorylation (γ-H2AX) in the MMR-deficient HEC59 cells (Fig. 5A). Moreover, baicalein-induced cell death in HEC59 cells was partially rescued by silencing XPF expression (Fig. 5B). To further confirm this observation, we transfected HT29 cells with negative control siRNA, siRNA targeting MSH2, and/or siRNA targeting XPF. Upon the silencing of XPF expression, baicalein-induced γ-H2AX was reduced in MSH2-deficient HT29 cells (Supplementary Fig. S6A). In addition, baicalein-induced cell death was rescued by silencing XPF expression in MSH2-deficient HT29 cells (Supplementary Fig. S6B).
Figure 5. XPF generates DSB in MMR deficient cells upon baicalein treatment.
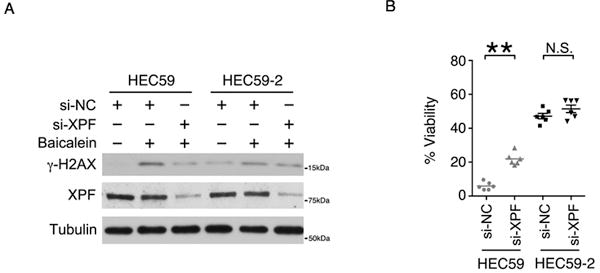
A, The level of DNA DSBs, as indicated by the phosphorylation of H2AX (γ–H2AX), was determined by western blotting in HEC59 and HEC59-2 cells lysates after treatment with 100 μM baicalein for 24 hours and after silencing endonuclease XPF expression with siRNA. B, HEC59 and HEC59-2 cells were transfected with siRNA targeting XPF, and cell survival was determined following treatment with 250 μM baicalein for 24 hours. Data are shown as mean ± S.E. **p < 0.01 as determined by two-tailed Student’s t-test.
Baicalein reduces the size of tumors formed by MutSα-deficient cells
MutSα-deficient cancer cells are not sensitive to radiation, alkylating agents or most chemotherapeutic treatments(14). Thus, we investigated whether baicalein would be an improved chemotherapy option for MutSα-deficient tumors. We generated xenograft tumors with MutSα-deficient HEC59 cells and MMR-proficient HT29 cells in nude mice. When tumors grew to ~1cm, Matrix-Driven Delivery Pellets were implanted (day 0) to deliver baicalein at a constant dose for two weeks (Fig. 6A and Supplementary Fig. S7A–S7C). During this process, the body weight of these mice was not significantly changed in either the placebo- or baicalein-treated groups (Supplementary Fig. S7D and S7E). Baicalein significantly shrank the MutSα-deficient xenografted HEC59 tumors (Fig. 6B and Supplementary Fig. S7F). Apoptotic cell death and DNA damage, as measured by the TUNEL assay and by γ-H2AX, respectively, were induced approximately two-fold by baicalein in MutSα-deficient HEC59-xenografted tumors (Fig. 6D, 6E and 6F). However, CHK2 phosphorylation was not changed by baicalein (Fig. 6D and 6G). Baicalein had no significant effect on the growth of MutSα-expressing HT29-xenografted tumors (Fig. 6C and Supplementary Fig. S7G) and there was no induction of the TUNEL signal or γ-H2AX (Fig. 6H, 6I and 6J). Consistent with in vitro results, CHK2 phosphorylation was induced about two-fold by baicalein in MutSα-expressing HT29-xenografted tumors (Fig. 6H and 6K). Thus, baicalein can selectively inhibit the growth of MutSα-deficient tumors in vivo.
Figure 6. Baicalein inhibits the growth of MSH2-deficient xenograft tumors in nude mice.
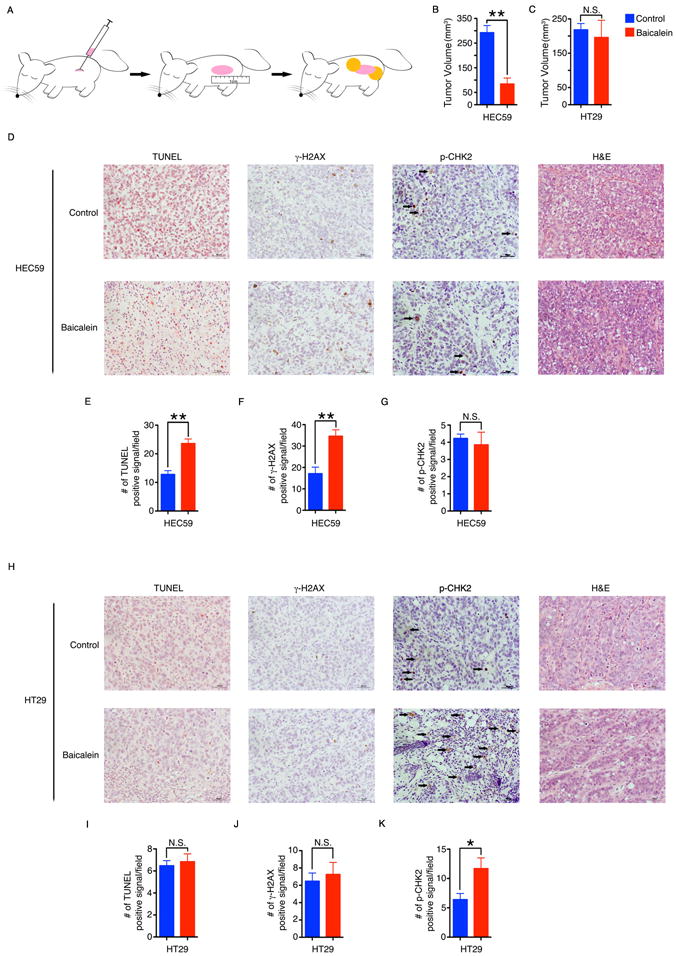
A, HEC59 or HT29 tumor cells were injected subcutaneously into the flanks of Nu/Nu mice and allowed to form tumors. When the tumor size reached approximately 1 cm in length, two pellets containing baicalein or placebo were implanted besides each tumor. After two weeks, the tumors were harvested. B, C, The volume of the tumors formed by HEC59 (B) or HT29 cells (C) was determined by the water displacement method. D, Representative images of hematoxylin-eosin (H&E), TUNEL, γ-H2AX and p-CHK2 staining of HEC59 tumor sections. E–G, Graphs showing the number of cells per field that were positive for TUNEL (E), γ-H2AX (F) and p-CHK2 (G) staining in (D). H, Representative images of hematoxylin-eosin (H&E), TUNEL, γ-H2AX and p-CHK2 staining of HT29 tumor sections. I–K, Graphs showing the number of cells per field that were positive for TUNEL (I), γ-H2AX (J) and p-CHK2 (K) staining in (H). The scale bar indicates 50 μm. Data in all panels are shown as mean ± S.E. *p < 0.05, **p < 0.01 versus placebo by two-tailed Student’s t-test.
To assess the effect of baicalein on tumor size over the course of time, we repeated the xenograft experiment, implanting the pellets for one week or two weeks before harvest. The tumor size before implantation was similar in all treatment groups as shown in Supplementary Fig. S8A and the body weight of the mice in both the placebo- and baicalein-treated groups remained relatively constant throughout the study (Supplementary Fig. S8B). Again, baicalein significantly shrank the MutSα-deficient xenografted HEC59 tumors in a time-dependent manner (Supplementary Fig. S8C and S8D).
To further confirm the ability of baicalein to shrink MSH2-deficient tumors in vivo, we used another MSH2-deficient colorectal carcinoma cell line, LoVo. The tumor size before implantation was similar in all treatment groups (Supplementary Fig. S9A) and the body weight of the mice did not change during the course of the study (Supplementary Fig. S9B). Consistent with what we observed with the HEC59 tumors, baicalein significantly shrank the MutSα-deficient LoVo tumors (Supplementary Fig. S9C and S9D).
Baicalein prevents the growth of AOM-DSS-induced colon tumors in Msh2LoxP/LoxPVilCre mice
Msh2LoxP/LoxPVilCre mice display a strong predisposition to intestinal cancers at 10 months of age (30) making them a good model to study the effects of baicalein treatment on the growth of Msh2-deficient tumors in the colon (Supplementary Fig. S10A and S10B). Azoxymethane (AOM) and dextran sulfate sodium (DSS) can be used to accelerate and enhance onset of colon tumors in Msh2 knockout mice (31). To study the effect of baicalein on the chronic-AOM-DSS-induced colon tumors, male and female Msh2LoxP/LoxPVilCre mice were fed a control or baicalein-supplemented diet for four weeks and then treated with AOM and DSS as shown in Supplementary Fig. S10C. Both male and female mice in the baicalein-supplemented diet group gained slightly more weight than mice in the control diet group (Supplementary Fig. S10D and S10E). In male mice, the number of tumors in the baicalein-supplemented diet group was significantly less than that in control diet group (Fig. 7A and 7C and Supplementary Fig. S10F). The female mice also showed a similar trend toward reduced tumor formation when fed baicalein-containing diet (Fig. 7B and 6C and Supplementary Fig. S10F). To study the role of baicalein on the acute-AOM-DSS-induced colon tumors, female Msh2LoxP/LoxPVilCre mice were fed a control or baicalein-supplemented diet for four weeks and then treated with AOM and DSS as shown in Supplementary Fig. S11A. The mice in the baicalein-supplemented diet group gained slightly more weight than mice in the control diet group (Supplementary Fig. S11B). The colon condition of both diet groups is shown in Supplementary Fig. S11C. The mice showed a trend toward reduced tumor formation when fed a baicalein-containing diet, compared to the control diet group (Supplementary Fig. S11D and S11E). Male Msh2LoxP/LoxPVilCre mice were fed a control or baicalein-supplemented diet for four weeks and then treated with AOM and DSS as shown in Supplementary Fig. S11F. The mice in the baicalein-supplemented diet group gained slightly more weight than mice in control diet group (Supplementary Fig. S11G). The colon condition of both diet groups is shown in Supplementary Fig. S11H. The number of tumors in the baicalein-supplemented diet group was significantly less than that in control diet group (Supplementary Fig. S11I and S11J).
Figure 7. Baicalein inhibits the growth of AOM-DSS-induced colon tumors in MSH2LoxP/LoxPVilCre mice, and the model for baicalein activity in MutSα-proficient and MutSα-deficient cells.
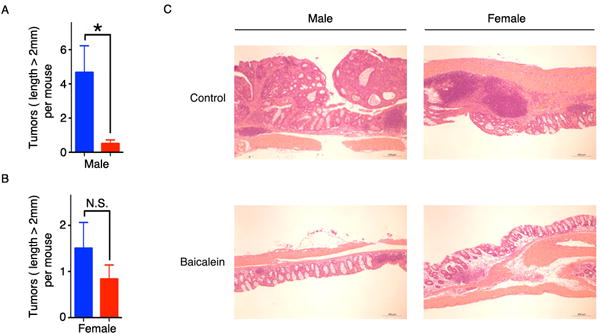
A, B, AOM-DSS was used to induce colon tumors in MSH2LoxP/LoxPVilCre mice fed a control or baicalein-supplemented diet. The number of tumors (length > 2mm) in the colons of male mice (A) or female mice (B) was counted. C, Representative images of hematoxylin-eosin (H&E) staining of colon sections. The scale bar indicates 200 μm. Data in all panels are shown as mean ± S.E. *p < 0.05, versus control diet by two-tailed Student’s t-test.
To further evaluate the effect of baicalein on the AOM-DSS-induced colon tumors, WT male mice were fed a control or baicalein-supplemented diet for four weeks and then treated with AOM and DSS as shown in Supplementary Fig. S12A. The mice in baicalein-supplemented diet group gained slightly more weight than mice in control diet group (Supplementary Fig. S12B). The number of severe colon tumors in the WT mice was lower compared to Msh2LoxP/LoxPVilCre mice as indicated by the lack of tumors more than 2 mm in WT mice. In the WT mice, there was not much difference in the number of tumors between the two groups (Supplementary Fig. S12C, S12D and S12E).
Discussion
In this manuscript, we provide a molecular mechanism for the activation of the MutSα-dependent damage checkpoint that allows for cell survival in the presence of baicalein, as well as an alternative pathway explaining the baicalein-induced cell death observed in the absence of MutSα both in vitro and in vivo. Baicalein simultaneously binds directly to mismatched DNA and to MutSα. The interaction between baicalein and MutSα dissociates MutSα from CHK2, resulting in the activation of CHK2 by ATM. The activated checkpoint allows the DNA repair machinery to remove damage, resulting in cell survival. In the absence of MutSα, baicalein-bound mismatched DNA is converted to DSBs by XPF more frequently and without activation of CHK2, resulting in cell death (Supplementary Fig. 13). Although MutSα has previously been implicated in damage signaling following DNA methylation and exposure to ionizing radiation(5,32), our data provide the first direct evidence that MutSα plays an important role in signaling DSBs to the checkpoint machinery.
Baicalein has been reported to have some anti-inflammatory(33) and anti-proliferative(16) effects. Given these properties, it is no surprise that baicalein previously has been reported to kill several different cancer cell types, including colon cancer. Many mechanisms of action have been proposed to explain these effects, including reduction of reactive oxygen species (ROS), attenuation of NFκB signaling, suppression of COX-2 gene expression, upregulation of death receptor 5, and inhibition of cell cycle checkpoints (18,19,21). Although the current study also provides strong evidence that the regulation of cell cycle progression is involved in the baicalein-induced death of MutSα-deficient cells, our study differs from previous reports describing the anti-tumor effects of baicalein in that it implicates CHK2/ATM-mediated G2/M phase arrest. Additional studies in our lab have shown a decrease in ROS following treatment with the concentrations of baicalein used in this manuscript (data not shown). However, further studies are needed to investigate the contribution of baicalein’s antioxidant activity, as well as the potential contributions of NFκB, COX-2, and death receptor 5, in the baicalein-induced death of MutSα-deficient cells.
Although baicalein is not the only small molecule that has been reported to kill MutSα-deficient cancer cells in vitro, baicalein offers several advantages over other MSH2-mutant sensitizing drugs such as methotrexate(34) and psoralen(35). A common critique of therapeutic strategies that target DNA repair deficiencies is that drugs that cause DNA damage often increase malignancy in the long term by inducing mutation rates. Indeed, methotrexate tested positive in many of the standard genotoxicity assays (Chemical Carcinogenesis Research Information System http://toxnet.nlm.nih.gov/cgi-bin/sis/htmlgen?CCRIS), and the ultraviolet radiation required to activate psoralen is also a mutagen. Baicalein, on the other hand, does not increase mutagenesis in HEK293T due to the ability of the DNA repair polymerase Polη to bypass baicalein-induced lesions in an error-free manner(15). However, due to MutSα interaction of baicalein, it is possible that baicalein would affect MMR pathway for reducing mutagenesis. In addition, whereas the majority of chemotherapeutic agents have no clear targets, here we showed that baicalein targets both MutSα and mismatched DNA, providing the molecular mechanisms by which baicalein would target the MMR-deficient tumor population and serve as a personalized cancer therapy. Although the IC50 values of baicalein in MutSα-deficient cancer cells are within the range of those reported for other types of cancers (10–264 μM)(21), they far exceed the nanomolar values required for the successful use of baicalein as a human therapeutic. In addition, high Kd of baicalein for mismatched DNA also raises the possibility of side and off target effects. Thus, future studies will involve collaborations with structural biologists and synthetic chemists to optimize the structure of this lead compound and improve its potency.
Supplementary Material
Acknowledgments
We thank P. Hsieh for providing MutSα expression baculovirus constructs; S. Anderson (NHGRI) for flow cytometry; Dr. D. Bodine (NHGRI), P. Hsieh (NIDDK), W. Yang (NIDDK), G. Li (Univ. Kentucky) and members in the Myung laboratory for helpful discussions and comments on the manuscript. K. Myung thanks E. Cho.
Financial Support: This research was supported in part by the intramural research program of the National Center for Advancing Translational Sciences to R. Huang, M. Xia, D. Maloney, the National Human Genome research institute and NIH grant (MH092164-01) to K. Myung. This work was also supported by the Institute for Basic Science (IBS-R022-D1-2015) to K.Myung.
Footnotes
There is no conflict to disclose.
References
- 1.Li GM. Mechanisms and functions of DNA mismatch repair. Cell research. 2008;18(1):85–98. doi: 10.1038/cr.2007.115. [DOI] [PubMed] [Google Scholar]
- 2.Kolodner RD, Alani E. Mismatch repair and cancer susceptibility. Current opinion in biotechnology. 1994;5(6):585–94. doi: 10.1016/0958-1669(94)90079-5. [DOI] [PubMed] [Google Scholar]
- 3.Fishel R. Signaling mismatch repair in cancer. Nature medicine. 1999;5(11):1239–41. doi: 10.1038/15191. [DOI] [PubMed] [Google Scholar]
- 4.Jiricny J. The multifaceted mismatch-repair system. Nature reviews Molecular cell biology. 2006;7(5):335–46. doi: 10.1038/nrm1907. [DOI] [PubMed] [Google Scholar]
- 5.Brown KD, Rathi A, Kamath R, Beardsley DI, Zhan Q, Mannino JL, et al. The mismatch repair system is required for S-phase checkpoint activation. Nature genetics. 2003;33(1):80–4. doi: 10.1038/ng1052. [DOI] [PubMed] [Google Scholar]
- 6.Palomaki GE, McClain MR, Melillo S, Hampel HL, Thibodeau SN. EGAPP supplementary evidence review: DNA testing strategies aimed at reducing morbidity and mortality from Lynch syndrome. Genetics in medicine: official journal of the American College of Medical Genetics. 2009;11(1):42–65. doi: 10.1097/GIM.0b013e31818fa2db. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.de la Chapelle A. Genetic predisposition to colorectal cancer. Nature reviews Cancer. 2004;4(10):769–80. doi: 10.1038/nrc1453. [DOI] [PubMed] [Google Scholar]
- 8.Heinen CD, Schmutte C, Fishel R. DNA repair and tumorigenesis: lessons from hereditary cancer syndromes. Cancer biology & therapy. 2002;1(5):477–85. doi: 10.4161/cbt.1.5.160. [DOI] [PubMed] [Google Scholar]
- 9.Lynch HT, Lynch JF, Lynch PM, Attard T. Hereditary colorectal cancer syndromes: molecular genetics, genetic counseling, diagnosis and management. Familial cancer. 2008;7(1):27–39. doi: 10.1007/s10689-007-9165-5. [DOI] [PubMed] [Google Scholar]
- 10.Mueller J, Gazzoli I, Bandipalliam P, Garber JE, Syngal S, Kolodner RD. Comprehensive molecular analysis of mismatch repair gene defects in suspected Lynch syndrome (hereditary nonpolyposis colorectal cancer) cases. Cancer Res. 2009;69(17):7053–61. doi: 10.1158/0008-5472.CAN-09-0358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Le DT, Uram JN, Wang H, Bartlett BR, Kemberling H, Eyring AD, et al. PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. N Engl J Med. 2015;372(26):2509–20. doi: 10.1056/NEJMoa1500596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.van Geel RM, Beijnen JH, Bernards R, Schellens JH. Treatment Individualization in Colorectal Cancer. Curr Colorectal Cancer Rep. 2015;11(6):335–44. doi: 10.1007/s11888-015-0288-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Devaud N, Gallinger S. Chemotherapy of MMR-deficient colorectal cancer. Fam Cancer. 2013;12(2):301–6. doi: 10.1007/s10689-013-9633-z. [DOI] [PubMed] [Google Scholar]
- 14.Hewish M, Lord CJ, Martin SA, Cunningham D, Ashworth A. Mismatch repair deficient colorectal cancer in the era of personalized treatment. Nature reviews Clinical oncology. 2010;7(4):197–208. doi: 10.1038/nrclinonc.2010.18. [DOI] [PubMed] [Google Scholar]
- 15.Fox JT, Sakamuru S, Huang R, Teneva N, Simmons SO, Xia M, et al. High-throughput genotoxicity assay identifies antioxidants as inducers of DNA damage response and cell death. Proc Natl Acad Sci U S A. 2012;109(14):5423–8. doi: 10.1073/pnas.1114278109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chen CH, Huang LL, Huang CC, Lin CC, Lee Y, Lu FJ. Baicalein, a novel apoptotic agent for hepatoma cell lines: a potential medicine for hepatoma. Nutrition and cancer. 2000;38(2):287–95. doi: 10.1207/S15327914NC382_19. [DOI] [PubMed] [Google Scholar]
- 17.Fu Y, Luo J, Jia Z, Zhen W, Zhou K, Gilbert E, et al. Baicalein Protects against Type 2 Diabetes via Promoting Islet beta-Cell Function in Obese Diabetic Mice. Int J Endocrinol. 2014;2014:846742. doi: 10.1155/2014/846742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kim DH, Hossain MA, Kang YJ, Jang JY, Lee YJ, Im E, et al. Baicalein, an active component of Scutellaria baicalensis Georgi, induces apoptosis in human colon cancer cells and prevents AOM/DSS-induced colon cancer in mice. Int J Oncol. 2013;43(5):1652–8. doi: 10.3892/ijo.2013.2086. [DOI] [PubMed] [Google Scholar]
- 19.Taniguchi H, Yoshida T, Horinaka M, Yasuda T, Goda AE, Konishi M, et al. Baicalein overcomes tumor necrosis factor-related apoptosis-inducing ligand resistance via two different cell-specific pathways in cancer cells but not in normal cells. Cancer Res. 2008;68(21):8918–27. doi: 10.1158/0008-5472.CAN-08-1120. [DOI] [PubMed] [Google Scholar]
- 20.Wang Y, Han E, Xing Q, Yan J, Arrington A, Wang C, et al. Baicalein upregulates DDIT4 expression which mediates mTOR inhibition and growth inhibition in cancer cells. Cancer Lett. 2015;358(2):170–9. doi: 10.1016/j.canlet.2014.12.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li-Weber M. New therapeutic aspects of flavones: the anticancer properties of Scutellaria and its main active constituents Wogonin, Baicalein and Baicalin. Cancer Treat Rev. 2009;35(1):57–68. doi: 10.1016/j.ctrv.2008.09.005. [DOI] [PubMed] [Google Scholar]
- 22.Geng H, Du C, Chen S, Salerno V, Manfredi C, Hsieh P. In vitro studies of DNA mismatch repair proteins. Analytical biochemistry. 2011;413(2):179–84. doi: 10.1016/j.ab.2011.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nehme A, Baskaran R, Nebel S, Fink D, Howell SB, Wang JY, et al. Induction of JNK and c-Abl signalling by cisplatin and oxaliplatin in mismatch repair-proficient and -deficient cells. British journal of cancer. 1999;79(7–8):1104–10. doi: 10.1038/sj.bjc.6690176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cenni B, Kim HK, Bubley GJ, Aebi S, Fink D, Teicher BA, et al. Loss of DNA mismatch repair facilitates reactivation of a reporter plasmid damaged by cisplatin. British journal of cancer. 1999;80(5–6):699–704. doi: 10.1038/sj.bjc.6690412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Koi M, Umar A, Chauhan DP, Cherian SP, Carethers JM, Kunkel TA, et al. Human chromosome 3 corrects mismatch repair deficiency and microsatellite instability and reduces N-methyl-N′-nitro-N-nitrosoguanidine tolerance in colon tumor cells with homozygous hMLH1 mutation. Cancer Res. 1994;54(16):4308–12. [PubMed] [Google Scholar]
- 26.Wang Y, Cortez D, Yazdi P, Neff N, Elledge SJ, Qin J. BASC, a super complex of BRCA1-associated proteins involved in the recognition and repair of aberrant DNA structures. Genes & development. 2000;14(8):927–39. [PMC free article] [PubMed] [Google Scholar]
- 27.Park C, Marqusee S. Pulse proteolysis: a simple method for quantitative determination of protein stability and ligand binding. Nature methods. 2005;2(3):207–12. doi: 10.1038/nmeth740. [DOI] [PubMed] [Google Scholar]
- 28.Lomenick B, Hao R, Jonai N, Chin RM, Aghajan M, Warburton S, et al. Target identification using drug affinity responsive target stability (DARTS) Proc Natl Acad Sci U S A. 2009;106(51):21984–9. doi: 10.1073/pnas.0910040106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rossi M, Meyer R, Constantinou P, Caruso F, Castelbuono D, O’Brien M, et al. Molecular structure and activity toward DNA of baicalein, a flavone constituent of the Asian herbal medicine “Sho-saiko-to”. Journal of natural products. 2001;64(1):26–31. doi: 10.1021/np000068s. [DOI] [PubMed] [Google Scholar]
- 30.Kucherlapati MH, Lee K, Nguyen AA, Clark AB, Hou H, Jr, Rosulek A, et al. An Msh2 conditional knockout mouse for studying intestinal cancer and testing anticancer agents. Gastroenterology. 2010;138(3):993–1002 e1. doi: 10.1053/j.gastro.2009.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kohonen-Corish MR, Daniel JJ, te Riele H, Buffinton GD, Dahlstrom JE. Susceptibility of Msh2-deficient mice to inflammation-associated colorectal tumors. Cancer Res. 2002;62(7):2092–7. [PubMed] [Google Scholar]
- 32.Wang Y, Qin J. MSH2 and ATR form a signaling module and regulate two branches of the damage response to DNA methylation. Proc Natl Acad Sci U S A. 2003;100(26):15387–92. doi: 10.1073/pnas.2536810100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shen YC, Chiou WF, Chou YC, Chen CF. Mechanisms in mediating the anti-inflammatory effects of baicalin and baicalein in human leukocytes. European journal of pharmacology. 2003;465(1–2):171–81. doi: 10.1016/s0014-2999(03)01378-5. [DOI] [PubMed] [Google Scholar]
- 34.Martin SA, McCarthy A, Barber LJ, Burgess DJ, Parry S, Lord CJ, et al. Methotrexate induces oxidative DNA damage and is selectively lethal to tumour cells with defects in the DNA mismatch repair gene MSH2. EMBO molecular medicine. 2009;1(6–7):323–37. doi: 10.1002/emmm.200900040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wu Q, Christensen LA, Legerski RJ, Vasquez KM. Mismatch repair participates in error-free processing of DNA interstrand crosslinks in human cells. EMBO reports. 2005;6(6):551–7. doi: 10.1038/sj.embor.7400418. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.