

Abstract
Type 2 ryanodine receptor (RyR2) serves as the major intracellular Ca2+ release channel that drives heart contraction. RyR2 is activated by cytosolic Ca2+ via the process of Ca2+-induced Ca2+ release (CICR). To ensure stability of Ca2+ dynamics, the self-reinforcing CICR must be tightly controlled. Defects in this control cause sarcoplasmic reticulum (SR) Ca2+ mishandling, which manifests in a variety of cardiac pathologies that include myocardial infarction and heart failure. These pathologies are also associated with oxidative stress. Given that RyR2 contains a large number of cysteine residues, it is no surprise that RyR2 plays a key role in the cellular response to oxidative stress. RyR’s many cysteine residues pose an experimental limitation in defining a specific target or mechanism of action for oxidative stress. As a result, the current understanding of redox-mediated RyR2 dysfunction remains incomplete. Several oxidative modifications, including S-glutathionylation and S-nitrosylation, have been suggested playing an important role in the regulation of RyR2 activity. Moreover, oxidative stress can increase RyR2 activity by forming disulfide bonds between two neighboring subunits (intersubunit cross-linking). Since intersubunit interactions within the RyR2 homotetramer complex dictate the channel gating, such posttranslational modification of RyR2 would have a significant impact on RyR2 function and Ca2+ regulation. This review summarizes recent findings on oxidative modifications of RyR2 and discusses contributions of these RyR2 modifications to SR Ca2+ mishandling during cardiac pathologies.
Keywords: Ca release, Glutathione, Heart, Reactive oxygen species, Ryanodine receptor, Sarcoplasmic reticulum
1 Excitation-Contraction Coupling and SR Ca2+ Cycling
Excitation-contraction coupling (ECC) is the cellular mechanism that connects the electrical stimulus to the contraction of the heart. During the action potential (AP), a small Ca2+ influx via L-type Ca2+ channels (LTCCs) causes a massive Ca2+ release from the sarcoplasmic reticulum (SR). This global rise in cytosolic Ca2+ concentration ([Ca2+]i) triggers cell contraction with the binding of Ca2+ to the myofilament protein troponin C. This induces an allosteric change in the troponin-tropomyosin complex, allowing for myosin heads to form a cross bridge interaction with actin. Once this Ca2+-dependent interaction takes place, mechanical force is generated with the hydrolysis of ATP by the actomyosin ATPase. These cycles will persist as long as ATP and Ca2+ are present in sufficient concentration (Goldman 1987). Thus, Ca2+ transport systems involved in the movement of Ca2+ into and out of the cytosolic milieu contribute directly to the activation and relaxation of the myofilaments. In adult ventricular myocytes, Ca2+ released from the SR plays a particularly important role in cell contraction. The cardiac SR is equipped with Ca2+ handling machinery that is perfectly designed to regularly repeat the major steps of the cardiac cycle: Ca2+ release and uptake (Zima et al. 2014). SR Ca2+ release predominantly occurs via the type 2 ryanodine receptors (RyR2), whereas SR Ca2+ uptake is entirely mediated by the type 2a SR Ca-ATPase (SERCA) (Bers 2001).
1.1 Molecular Components of Ca2+ Cycling
Activated by Ca2+ influx via LTCCs, RyR2 mediates a massive Ca2+ release during systole. This mechanism is known as Ca2+-induced Ca2+ release (CICR) (Fabiato 1983). In order for membrane excitation to simultaneously activate SR Ca2+ release in ventricular myocytes, sarcolemmal invaginations called transverse tubules (T-tubules) descend deep into the myocyte (Fig. 1). The SR forms a junction with the T-tubule creating a specialized subcellular microdomain called the dyadic cleft that allows for efficient activation of CICR (Soeller and Cannell 1999). Within the dyadic cleft, RyR2s and LTCCs interact in a highly organized lattice forming the Ca2+ release unit (CRU; Fig. 1) (Cheng and Lederer 2008). It has been estimated that CRU comprises a cluster of ~100 RyRs (Franzini-Armstrong et al. 1999). However, the exact value currently remains a debated issue. Other investigators have estimated RyR2 cluster number to be smaller in size (Baddeley et al. 2009; Hayashi et al. 2009). These RyR2 clusters align with LTCCs in the dyadic cleft via junctophilins (Garbino et al. 2009). Activation of single CRU (spontaneously or by LTCC Ca2+ current) produced a local increase of [Ca2+]i called Ca2+ spark (Cheng et al. 1993). The global Ca2+ release is the result of the spatiotemporal activation of thousands of individual CRU or Ca2+ sparks (Fig. 1). Thus, the amplitude of the global Ca2+ transient during systole is the result of local subcellular recruitment of CRUs (Stern 1992).
Fig. 1.

Intracellular Ca2+ regulation in adult ventricular myocytes. (a, top panel) a representative confocal image of rabbit ventricular myocytes loaded with the voltage-sensitive fluorescent dye Di-8-ANEPPS. Di-8-ANEPPS was used to label the T-tubular system. (a, bottom panel) the diagram illustrates the main components of Ca2+ release unit (CRU) in ventricular myocytes. A significant fraction of L-type Ca2+ channels (LTCC) is localized in the T-tubule (TT), whereas the majority of ryanodine receptors (RyR2) is concentrated in the junctional SR. Ca2+-ATPase (SERCA) pumps cytosolic Ca2+ back into the SR, and the Na+-Ca2+ exchanger (NCX) removes Ca2+ from the cell. The plasmalemmal Ca2+-ATPase and mitochondria play a minor role in cardiac relaxation. (b) Confocal images of diastolic Ca2+ spark (top) and an action potential-induced Ca2+ transient (bottom). Activation of a single CRU generates a Ca2+ spark, whereas simultaneous activation of thousands of these individual release units generates a global Ca2+ transient
During diastole, there are two major Ca2+ transport systems that compete for cytosolic Ca2+: SERCAa and the sarcolemmal Na+-Ca2+ exchanger (NCX). The sarcolemmal Ca2+-ATPase and the mitochondrial Ca2+ uniporter compete as well, but are considered to be minor components (Bassani et al. 1992). Immediately after the global rise in cytosolic Ca2+, the majority of Ca2+ is sequestered back into the SR by SERCA and to a lesser extent is extruded from the cell by NCX (Fig. 1). The contribution of SERCA and NCX to decreasing [Ca2+]i during diastole is variable among animal species. It has been estimated that NCX contributes only 7% to Ca2+ removal in small rodent myocardium (Bers 2001). Whereas in rabbit, dog, and human myocardium, SERCA and NCX contribute approximately 70% and 30% to cardiac relaxation, respectively. There are two major contributors to the Ca2+ transient in the heart: LTTC-mediated Ca2+ current and SR Ca2+ release. While Ca2+ current contributes approximately 30% to the Ca2+ transient in the rabbit ventricle, the majority of Ca2+ comes from SR Ca2+ release by RyR2 (Bers 2001). At steady state, Ca2+ current and SR Ca2+ release must be balanced by Ca2+ extrusion and SR Ca2+ reuptake. Therefore, any changes in sarcolemmal Ca2+ current, SR Ca2+ release, SR Ca2+ uptake, or sarcolemmal Ca2+ extrusion can have a profound effect on Ca2+-dependent inotropy (force) and lusitropy (relaxation) (Eisner et al. 1998).
1.2 Ryanodine Receptor Complex
Predominantly expressed in cardiac muscle, the type 2 RyR is a tetrameric channel with a total molecular weight of approximately 564 kDa. RyR2 has a relatively low selectivity given its permeability to many different divalent and monovalent cations. Furthermore, the channel has a very high conductance of approximately 100 pS for divalent cations (Fill and Copello 2002). Its characteristically low selectivity for Ca2+ is suggested to be fundamental to its physiological role to produce a fast and large Ca2+ release event. While Ca2+ needs to compete with other cations for occupancy of the channel pore, it has been proposed that RyR2 has surface or vestibule charges that may enhance the permeation of Ca2+ (Gillespie 2008; Mead-Savery et al. 2009). Although cytosolic Ca2+ is the central physiological activator of RyR2, other free ions and small molecules can alter its activity including caffeine, Mg2+, H+, and ATP (Eager and Dulhunty 1998; Masumiya et al. 2001; Fill and Copello 2002). There are a number of proteins that interact with RyR2 as well, each of which can modulate the channel’s activity (Fig. 2). Proteins that interact on the cytosolic side of RyR2 include calmodulin (CaM), FK-506-binding proteins (FKBP), sorcin, and Homer-1 (for reviews, see (Bers 2004; Meissner 2004; Marx et al. 2000)). The two known kinases that are scaffolded on RyR2, protein kinase A (PKA), and calcium-/calmodulin-dependent kinase (CaMKII) have been shown to phosphorylate RyR2 at Ser-2809 and Ser-2815, respectively (Marx et al. 2000; Wehrens et al. 2004). Also, there are two known protein phosphatases that play a role in regulating RyR2 phosphorylation, including PP1 and PP2A. Spanning the SR membrane, but also associated with RyR2, are the auxiliary proteins junctin and triadin. Their function is thought to be important for RyR’s ability to sense luminal Ca2+ ([Ca2+]SR) via interactions with the SR Ca2+-binding protein calsequestrin (CASQ). All of the aforementioned proteins that make up the RyR2 complex are necessary for proper function of RyR2 channel activity.
Fig. 2.
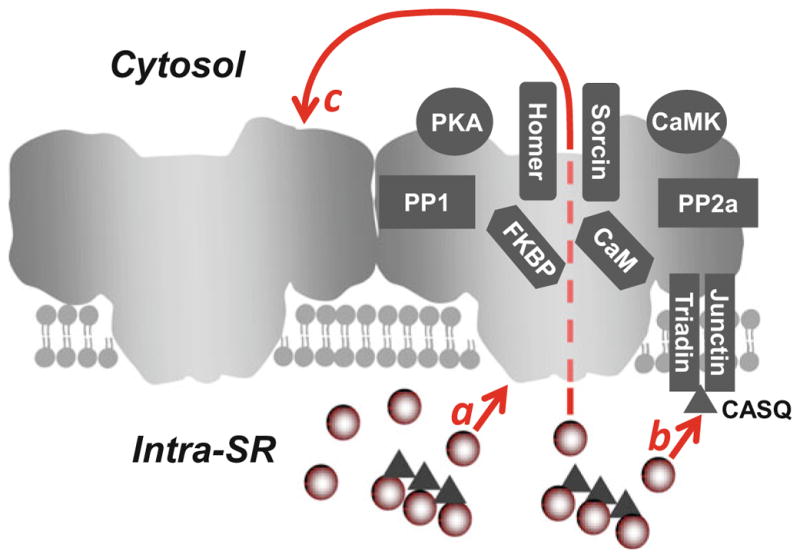
Regulation of cardiac ryanodine receptor. On the cytosolic side, RyR2 interacts with calmodulin (CaM), FK-506-binding proteins (FKBP), homer, sorcin, two major protein kinases (PKA and CaMKII), and two phosphatases (PP1 and PP2A). Luminal Ca2+ regulates RyR2 activity by directly binding to the luminal side of the channel (a). Moreover, triadin and junction form the luminal Ca2+ sensor via interactions with the SR Ca2+-buffering protein calsequestrin (CASQ; b). Luminal Ca2+ can also indirectly regulate RyR2 by acting on the cytosolic Ca2+ activation site of neighboring channels by a “feed-through” mechanism (c)
1.3 Ca2+-Induced Ca2+ Release
Unlike ECC in skeletal muscle, local Ca2+ entry from LTCC is an absolute requirement for SR Ca2+ release in cardiac muscle. Fabiato and Fabiato were the first to characterize cardiac CICR (Fabiato and Fabiato 1975). They showed that SR Ca2+ release by CICR was graded, having a dependence on both time and trigger. Moreover, it was shown that the introduction of high [Ca2+] immediately after the Ca2+ pulse trigger would cause a decrease in SR Ca2+ release. It was concluded that this characteristic was due to a Ca2+-dependent inactivation site on the cytosolic side of RyR2 (Fabiato 1985). While the activation characteristics of RyR2 have since been confirmed, inactivation of RyR2 or termination of CICR still remains controversial (Stern and Cheng 2004). A more recent study was unable to show that high [Ca2+]i promotes inactivation of CICR (Nabauer and Morad 1990; Stevens et al. 2009). Their results suggest the existence of other CICR termination mechanisms. In vitro and in vivo studies have shown that RyR2 channel activity and CICR termination are indeed dependent on [Ca2+]SR (Sitsapesan and Williams 1994; Terentyev et al. 2002; Gyorke et al. 2004; Qin et al. 2008). Furthermore, it has been demonstrated that SR Ca2+ release terminates at a critical level of [Ca2+]SR, which is dependent on RyR2 channel gating (Zima et al. 2008). For example, RyR2 sensitization to cytosolic Ca2+ by the presence of caffeine results in a lower SR termination level, increased SR Ca2+ fractional release, and increased cytosolic Ca2+ transient (Domeier et al. 2009). These characteristics of increased RyR2 activity are suggested to have pathological significance in the event of arrhythmogenic spontaneous Ca2+ waves and promoting abnormally low SR Ca2+ content as seen in the failing heart (Zima et al. 2014).
1.4 Luminal Ca2+ and Inter-RyR2 Regulation of CICR
While Fabiato was the first to suggest an inactivation mechanism for RyR2 ([Ca2+]i-dependent), a number of other mechanisms have since been proposed that implicate luminal Ca2+ regulation ([Ca2+]SR-dependent) of RyR2 in the termination of CICR. Without changes in RyR2 channel activity, SR Ca2+ release terminates at a critical level that remains constant on a beat-to-beat basis (Domeier et al. 2009). Moreover, the termination level observed from a single RyR cluster (during Ca2+ spark events) also remains constant (Zima et al. 2008). This intrinsic property of RyR2 implies that there is the potential for a Ca2+ sensor to exist on the luminal side of the channel (Fig. 2a). While few argue the existence of a luminal Ca2+ binding site on RyR2 that acts as the primary [Ca2+]SR sensor (Chen et al. 2014), multiple studies claim that the luminal Ca2+ sensing mechanism is solely regulated by a complex made up of junctin, triadin, and calsequestrin (Gyorke et al. 2004; Qin et al. 2008) (Fig. 2b). Furthermore, evidence linking the naturally occurring mutations in calsequestrin and triadin to catecholaminergic polymorphic ventricular tachycardia (CPTV) provide further support for the latter mechanism (Liu et al. 2006; Roux-Buisson et al. 2012).
While the dependence of SR Ca2+ release on SR Ca2+ load is widely accepted as fact, the theory that RyR2 activity is solely regulated by a luminal Ca2+ mechanism remains highly debatable. Other mechanisms have been proposed that explain termination of SR Ca2+ release as a RyR2 current-dependent process (Guo et al. 2012; Cannell et al. 2013; Bovo et al. 2015b). Recently proposed mechanism called induction decay (Cannell et al. 2013) or pernicious attrition (Guo et al. 2012) explains the local CICR dependence on SR Ca2+ load by the magnitude of the trans-SR Ca2+ driving force. This simply means that as RyR2 Ca2+ current increases with increasing SR Ca2+ load, inter-RyR2 CICR within a cluster will facilitate Ca2+ release (Fig. 2c). Intuitively, as SR Ca2+ load falls, local Ca2+ release will fail to sustain inter-RyR2 CICR leading to the termination of SR Ca2+ release. While RyR2 gating is stochastic in nature, the decreased local [Ca2+]i will no longer be sufficient to activate channels that have spontaneously closed. Therefore, any modifications that promote the open conformation of RyR2 or enhance the channel’s sensitivity to [Ca2+]i would likely hinder this termination process and, therefore, allow for a greater amount of SR Ca2+ release (Domeier et al. 2009; Bovo et al. 2015b).
1.5 SR Ca2+ Leak
The RyR2, being the main Ca2+ release channel in the SR, is responsible for triggered Ca2+ release during systole as well as non-triggered Ca2+ release during diastole. Non-triggered Ca2+ release events are referred to as SR Ca2+ leak. Ca2+ leak mediated by RyR2 can occur as spontaneous Ca2+ sparks as well as non-spark-mediated Ca2+ leak (Zima et al. 2010). Ca2+ sparks are observed as spatially restricted rises in [Ca2+]i that predominantly occur at junctional SR, which lie adjacent to the z-lines of sarcomeres. Cheng and colleagues were the first to visualize Ca2+ sparks in 1993 (Cheng et al. 1993). Soon after, a growing body of work described the average Ca2+ spark to increase local [Ca2+]i by approximately 300–500 nM and have a spatial width of about 2 μm (Lopez-Lopez et al. 1994; Cannell et al. 1995; Satoh et al. 1997). While Ca2+ sparks occur at a very low frequency in a healthy myocyte, a significantly increased Ca2+ sparks propensity during diastole observed during sympathetic stimulation with β-adrenergic receptor (β-AR) agonists (Bovo et al. 2012; Santiago et al. 2013) or in failing heart (Kubalova et al. 2005; Domeier et al. 2009). As the frequency of Ca2+ sparks during diastole increases over time, Ca2+ sparks can summate, causing local [Ca2+]i to rise to a level at which neighboring clusters become activated. Consequently, the subsequent activation of several RyR2 clusters within a small region can propagate an asynchronous global SR Ca2+ release event known as a spontaneous Ca2+ wave. Ca2+ waves are a form of SR Ca2+ leak that is sufficient to induce spontaneous APs and is therefore considered an important trigger for cardiac arrhythmias (Schlotthauer and Bers 2000; Xie and Weiss 2009; Shiferaw et al. 2012). Although the majority of SR Ca2+ leak arises via RyR2 clusters that are responsible for the spark generation, sparks are very rare events during rest in healthy ventricular myocytes (Bovo et al. 2014). The absence of Ca2+ sparks can be explained if SR Ca2+ leak is mainly composed of unsynchronized openings of individual RyRs in a release cluster (non-spark-mediated Ca2+ leak) (Zima et al. 2010; Santiago et al. 2010).
2 SR Ca2+ Cycling in Heart Disease
In the healthy myocardium, SR Ca2+ handling is robust and dynamic in nature, allowing the molecular machinery of the sarcomere to respond in an appropriate fashion. In fact, the intrinsic ability of the myocardium to modify amplitude and duration of each Ca2+ transient is fundamental property of the heart so that it can match cardiac output with the demand of the body (Lakatta 2004). In both ischemic and non-ischemic etiologies of heart disease, the heart undergoes a large number of changes that contribute to the disease phenotype as well as to the progression into heart failure (HF). In these pathophysiological states, both the contractile function and the electrical properties of the myocardium become dysfunctional. Abnormal Ca2+ handling is considered to be a major downstream effect that ultimately promotes the cardiac disease phenotypes. The inability of the failing heart to maintain an adequate SR Ca2+ load is an important contributor to the hearts’ lack of capacity to meet cardiac demand (Houser et al. 2000; Bers 2001; Zima et al. 2014). Another consequence of impaired SR Ca2+ handling is the increased risk for cardiac arrhythmias. For instance, the abrupt increase in [Ca2+]i as a result of stress (e.g., β-AR activation) can cause the formation of pro-arrhythmic Ca2+ waves (Bers 2001; Janse 2004; Pogwizd and Bers 2004; Eisner et al. 2009).
2.1 Ischemia/Reperfusion
In the event that a coronary artery becomes obstructed as a result of atherosclerotic plaques, the downstream blood flow slows or completely ceases, creating a hypoxic environment for the non-perfused myocardium. In the ischemic environment, metabolites build up within the interstitium and intracellularly due to the energy consumption of the working myocardium and the lack of blood perfusion. Commonly associated with ischemia/reperfusion (I/R) injury are complex cellular metabolic changes including a decrease in [ATP] and a subsequent increase in free [Mg2+], [ADP], and inorganic phosphate ([Pi]) as well as a drop in intracellular pH (Opie and Clusin 1990; Opie 1993). Furthermore, increased reactive oxygen species (ROS) generation is prominent as a result of I/R injury (Misra et al. 2009). All the aforementioned factors are known to modulate the activity of the proteins required for intracellular Ca2+ cycling, particularly RyR2. Consequently, the enhanced RyR2-mediated Ca2+ leak results in the occurrence of pro-arrhythmic Ca2+ waves (Belevych et al. 2009, 2012). As a result, a major complication associated with reperfusing blood to the ischemic region is the increased risk of arrhythmogenesis (Yavuz 2008). These bouts of arrhythmias have the potential of initiating reentrant tachyarrhythmias, which can devolve into fibrillation and ultimately sudden cardiac death (Bunch et al. 2007).
2.2 Heart Failure
Congestive HF can be simply defined as the inability for cardiac output to meet the metabolic demand from the body. Today, HF remains the leading cause of hospitalization for ages 65 and older in the United States and Europe. Researchers have documented different cellular changes of the myocardium that prove to be dependent on the etiology of HF (Sen et al. 2000). Cardiovascular disease risk factors can promote a variety of cellular changes in the myocardium, which ultimately affect the development of HF. Myocardial infarction (MI), due to coronary artery disease, is the leading cause of ischemic HF, while increased cardiac afterload (e.g., hypertension) is a common cause of non-ischemic HF (Cowburn et al. 1998). Due to progressive cardiac remodeling, patients who suffer from HF have a poor prognosis as well as a low quality of life. In patients suffering from end-stage HF, death either results from cardiac pump failure or arrhythmias (Lane et al. 2005).
SR Ca2+ mishandling is one of the hallmark changes that take place in HF. This SR dysfunction is an important contributor to the heart’s depressed contractile function and the increased incidence of arrhythmias, implicating RyR2 and SERCA dysfunction as the primary cause. In a majority of studies, HF is associated with increased SR Ca2+ leak and decreased SR Ca2+ reuptake mediated by RyR2 and SERCA, respectively. However, there is conflicting evidence with respect to the contribution of RyR2 to impaired SR Ca2+ cycling. Furthermore, pathophysiological differences have been observed between ischemic and idiopathic HF with regard to SR Ca2+ cycling dysfunction. A study by Sen et al. found that impaired SERCA activity was the primary impairment in ischemic HF, whereas SERCA activity in idiopathic HF was not significantly different when compared to healthy myocardium (Sen et al. 2000). Furthermore, impaired SR Ca2+ release was only observed in idiopathic HF. These results suggest that the underlying mechanisms responsible for SR Ca2+ cycling dysfunction may depend on the etiology of HF.
Moreover, HF is commonly defined as a condition of chronic oxidative stress due to compromised energetics. The impaired cardiac metabolism is considered to play an important role in the progression of disease (Mak and Newton 2001; Ventura-Clapier et al. 2004; Santos et al. 2011). As the disease progresses, oxidative stress worsens due to the increasing energy demand and workload of the failing heart, thus perpetuating a deleterious cycle (Seddon et al. 2007). Although HF is associated with a large number of complex changes, the focus of this review is directed at understanding the role oxidative stress on SR Ca2+ regulation and RyR2 function. To date, RyR2 has been characterized in HF as having an increased phosphorylation level, an increased oxidation level, and a decreased association of auxiliary proteins. All of the aforementioned have been associated with increased channel activity. While functionally important phosphorylation sites on RyR2 have been characterized (Marx et al. 2000; Wehrens et al. 2004; Xiao et al. 2006), the specific mechanisms of oxidative modifications of RyR2 and their contribution to defective SR Ca2+ cycling remain incomplete.
3 Oxidative Stress
Oxidative stress is a prominent feature in the onset and progression of a number of disease states, including cardiovascular disease. Although the generation of ROS has been shown to play an important role in normal cell signaling, during periods of oxidative stress, excessive ROS production can have detrimental effects on normal protein function and cell viability. Furthermore, increased ROS production can induce lipid peroxidation and DNA damage that can compromise the structural and genetic integrity of the cell.
In order to counteract any ROS produced, the cell has an intrinsic antioxidant system that allows for the breakdown of ROS into nontoxic molecules. Some of the major components of the cellular antioxidant system include superoxide dismutase (SOD), catalase, and glutathione peroxidase (Turrens 2003; Yamawaki et al. 2003; Slodzinski et al. 2008), which act as selective scavenging enzymes. The nonspecific antioxidants include glutathione and thioredoxin systems. Reduced glutathione (GSH), a highly abundant low-molecular-weight thiol, is considered the largest of antioxidant pools and is arguably the most important antioxidant system in the myocardium. GSH is considered the main line of defense against ROS, because it is ubiquitous throughout all cellular compartments.
Thus, oxidative stress can be simply defined as increased ROS production that overwhelms the cellular antioxidant defense (Giordano 2005). Depending on the etiology of disease, oxidative stress can manifest at different time points and from different sources. The following will briefly review acute oxidative stress in I/R as well as chronic oxidative stress in HF.
3.1 Acute Oxidative Stress in Ischemia/Reperfusion Injury
It is has been well characterized that restoring blood flow to the ischemic region drastically increases ROS production, which further increases oxidative stress. The generation of ROS, due to an increased supply of oxygen to ischemic myocardium, has been implicated as the underlying factor that promotes I/R injury (Vanden Hoek et al. 1996; Zweier et al. 1987). In this condition, the electron transport chain (ETC) in the mitochondria becomes uncoupled, producing superoxide anion (O2·−) (Turrens 1997). This sudden burst of ROS overwhelms the intrinsic antioxidant system. The GSH/GSSG ratio can decrease considerably during I/R (Ceconi et al. 1988; Werns et al. 1992), which can contribute to mitochondrial ROS spill over (Aon et al. 2007, 2010; Brown et al. 2010). Other sources of ROS, including NADPH oxidase (NOX), uncoupled nitric oxide synthase (NOS), and xanthine oxidase (XO), are also believed to play a role in I/R injury (Becker 2004; Zweier and Talukder 2006; Angelos et al. 2006). A recent study implicated Ca2+-dependent delayed after depolarizations (DADs) as the major mechanism of arrhythmogenesis in a dog model of acute MI (Belevych et al. 2012). It has been shown that the occurrence of DADs could be prevented with intravenous perfusion of the ROS scavenger Tempol (Xing et al. 2009). The results from these studies implicate oxidative stress as a major factor in the generation of cardiac arrhythmias after MI.
During MI, excessive β-AR stimulation manifests in the ischemic region due to elevated concentrations of catecholamines (Lameris et al. 2000). Both ex vivo and in vivo I/R studies have shown that the main source of endogenous catecholamines is in fact from nonexocytotic release at sympathetic nerve endings that innervate the myocardium (Lameris et al. 2000; Kurz et al. 1995). β-AR stimulation is considered to be an important contributor in I/R injury. Increased β-AR stimulation can further increase energy demand and intracellular ROS production (Christensen and Videbaek 1974; Bovo et al. 2015a). Studies that block PKA activation via beta blockers or direct inhibition of PKA has proven to be effective in reducing infarct size (Makaula et al. 2005; Spear et al. 2007). A recent study done by Nagasaka et al. showed that mitochondrial ROS production was significantly increased in the presence of PKA catalytic subunit in permeabilized myocytes (Nagasaka et al. 2007).
3.2 Chronic Oxidative Stress in Heart Failure
The mechanisms that are responsible for the progression of heart failure are very complex and have been under intensive investigation for many years. However, one of the common features that have been implicated to play an important role in the pathophysiology of HF is chronic oxidative stress (Belch et al. 1991; Hill and Singal 1997). Both experimental and clinical studies have measured an increase in ROS production in HF (Mak and Newton 2001; Ventura-Clapier et al. 2004; Giordano 2005; Santos et al. 2011). HF is commonly associated with morphological and functional abnormalities in mitochondria (Schaper et al. 1991). While compromised mitochondrial function is considered a significant cause of oxidative stress (Balaban et al. 2005; Turrens 2003; Giordano 2005), the molecular mechanisms of this defect in HF are not fully understood. In a mouse model of MI-induced HF, an increase in ROS production and lipid oxidation were associated with impaired mitochondrial function (Ide et al. 2001). Furthermore, in a canine model of HF, the mitochondrial ETC was significantly more prone to uncoupling and subsequent ROS production (Ide et al. 1999). These results also provide evidence for a positive correlation between depressed contractility and the level of ROS production.
Moreover, it has been shown that antioxidant activity progressively deteriorates in HF (Hill and Singal 1997). During HF progression, myocardium switches energy substrate from fatty acids to glucose. These adaptive changes in cellular metabolism are associated with decreased expression of mitochondrial transcription factors and proteins (Ventura-Clapier et al. 2004; Santos et al. 2011). In our recent studies, we found that the mitochondrial ROS defense is substantially reduced in HF, especially at the mitochondrial type 2 SOD (SOD-2) level (unpublished results). While the impaired SOD-2 function has been implicated in numerous diseases (including Parkinson, cancer, diabetes) (Turrens 2003; Miao and St Clair 2009), its role in HF has never been explored. We suggest that the SOD-2 decline is the critical step in a chain of events that lead to oxidative stress and HF progression. Foremost, SOD-2 is the only defense line against mitochondrial O2·−, whereas H2O2 can be neutralized by several enzymes (including peroxidase, peroxiredoxin, and catalase). Thus, SOD-2 downregulation would have more significant impact on ROS level than downregulation of any other ROS-scavenging enzyme. Second, O2·− reacts extremely rapidly with nitric oxide (NO) forming highly reactive peroxynitrite (ONOO·−) (Ferdinandy and Schulz 2003). Thus, the downregulation of SOD-2 in HF would have the significant impact on nitroso-redox balance: an increase of reactive ONOO·− production and a decrease of cardioprotective NO. In support of this hypothesis, Sod2−/− knockout mice are characterized by a maladaptive cardiac hypertrophy and cardiomyopathy (Makino et al. 2011; Lebovitz et al. 1996). Thus, restoring SOD-2 defense can be an effective strategy to improve cardiac function and delay HF progression.
4 Oxidative Posttranslational Modifications of RyR2
Recent emphasis has been placed on the study of oxidative posttranslational modifications (PTMs) and their important role in the regulation of heart function. Among all cardiac ion transporters and channels, RyR2 appears to be the most sensitive to redox modification (Zima and Blatter 2006; Hool and Corry 2007), thus linking oxidative stress to Ca2+ regulation. RyR2 has approximately 360 cysteine residues per tetrameric channel, with an estimated 84 of those are in a reduced free thiol state (Xu et al. 1998). Each free thiol residue can serve as a target for a number of oxidative modifications including S-nitrosylation, S-glutathionylation, or disulfide cross bridge formation. To date, a number of in vitro studies have shown that both ROS and other free radicals can induce changes in RyR2 channel activity. Bilayer studies have shown that RyR2 channel activity is increased in the presence of ROS, whereas reducing agents decrease the RyR2 activity (for review, see Zima and Blatter (2006)). Elevated ROS production, which is associated with increased cardiac demand, has been suggested to play a role in the augmentation of SR Ca2+ release (Heinzel et al. 2006). Therefore, oxidative PTMs of RyR2 may function as a mechanism for positive inotropy in the healthy heart. However, in the case of MI or HF, abnormally elevated ROS level can cause irregular Ca2+ cycling and, therefore, contractile dysfunction and arrhythmias. It has been shown that abnormal SR Ca2+ release in myocytes from infarcted (Belevych et al. 2009) and failing (Terentyev et al. 2008) heart was associated with an increase in RyR2 oxidation.
4.1 S-Glutathionylation
In the presence of oxidative stress, free thiols of cysteine residues are the first subjected to oxidation. Depending on the degree of oxidative stress, protein free thiols can be oxidized by ROS to form sulfenic (R-SOH), sulfinic (R-SO2H), or sulfonic (R-SO3H) acid products (Giles and Jacob 2002). GSH attenuates ROS production during oxidative stress either by directly scavenging free radicals or acting as a substrate for the major antioxidant enzyme glutathione peroxidase. Also, GSH can readily react with protein sulfenic acids forming the reversible S-glutathionylation of RyR2. The reversible reduction of S-glutathionylation is carried out mainly by the enzyme glutaredoxin. The formation of sulfinic and sulfonic acids, however, are considered biologically irreversible. Thus, the formation of the protein-glutathione-mixed disulfide is thought to have a protective role during changes in cellular redox state (Townsend 2007). However, it has been proposed that S-glutathionylation may play a role in promoting protein disulfide formation of both intra- and intermolecular species (Bass et al. 2004; Cumming et al. 2004; Brennan et al. 2004).
In all tissues, the ratio between oxidized and reduced GSH (GSSG/GSH) is an important indicator of the redox state. Changes in cellular redox environment potentially affects the activity of many proteins, including RyR2 (Zima and Blatter 2006). As a result, oxidative stress can potentially promote abnormally elevated [Ca2+]i in the myocardium during diastole (Kourie 1998). In cardiomyocytes, cytosolic glutathione is mainly reduced under normal physiological conditions. During oxidative stress, however, the GSSG/GSH ratio can increase significantly (Ceconi et al. 1988; Werns et al. 1992) as well as total protein-glutathione-mixed disulfides (Tang et al. 2011). The increased formation of glutathione-mixed disulfides is a common feature of oxidative stress due to the abundance of glutathione and the ready conversion of reactive thiols. Recent studies have been implicated glutathione mixed disulfides as a critical signaling mechanism that plays a causative role, rather than a protective role, in cardiovascular disease. With respect to SR Ca2+ cycling, however, it is unclear if increased glutathione-mixed disulfide is beneficial or detrimental. S-glutathionylation of RyR2 is also thought to play a role in myocardial preconditioning before an ischemic insult. For example, tachycardia-induced preconditioning was proven to reduce the infarct size after ischemia (Domenech et al. 1998). It was later identified that tachycardia stimulated NADPH oxidase-dependent S-glutathionylation of RyR2, increasing RyR2 Ca2+ release and decreasing SR Ca2+ leak in SR microsomal preparations (Sanchez et al. 2005). It still remains controversial whether or not increased single-channel activity or increased Ca2+ release from SR microsomes can also coincide with decreased SR Ca2+ leak within a cellular milieu.
4.2 S-Nitrosylation
The vast body of research studying ischemic preconditioning has yielded many different molecular mechanisms (Zaugg et al. 2003). Given its complex nature, the crucial downstream targets that give a tissue the ability to resist ischemic injury make up a sizeable list that has steadily grown over the recent years. NO signaling, an important regulator in many physiologic processes, and subsequent protein S-nitrosylation is commonly identified as an important molecular intermediate allowing for ischemic preconditioning. Several cardioprotection studies defined many downstream targets of NO, having identified the cardioprotective effect as the result of covalently linked NO with reactive protein thiols (S-nitrosylation). These downstream targets include proteins that are involved in mitochondrial metabolism, apoptosis, ROS defense, protein trafficking, myofilament contraction, and Ca2+ handling. Overall, increased S-nitrosylation in the myocardium can be antiapoptotic and anti-inflammatory (Sun and Murphy 2010; Lima et al. 2010). As mentioned previously, the reperfusion of blood or reintroduction of O2 to the ischemic tissue stimulates oxidative phosphorylation in impaired mitochondria, which results in a burst of ROS production. Recent studies have found that S-nitrosylation of mitochondrial protein complexes (I and IV) of the ETC inhibits their activity, which limits oxidative phosphorylation (Zhang et al. 2005; Sun et al. 2007; Rassaf et al. 2014). Furthermore, S-nitrosylation of myofilament proteins decreases their sensitivity to Ca2+, decreasing myofilament cross bridge formation, which subsequently reduces ATP consumption (Nogueira et al. 2009). By promoting energy conservation in the myocardium, S-nitrosylation limits ROS production from uncoupled mitochondria during I/R.
The cardioprotective effects of S-nitrosylation on Ca2+ machinery, although independent, complements the effect seen in the mitochondria. In both I/R and HF, impaired Ca2+ cycling commonly leads to an increase in diastolic [Ca2+]i as well as depleted [Ca2+]SR. In a state of [Ca2+]i overload, the diastolic function of the heart is impaired, and the likelihood of arrhythmogenesis is increased. Evidence of S-nitrosylation-dependent cardioprotection has been documented for the major components of Ca2+ cycling, preventing [Ca2+]i overload (Loyer et al. 2008). For LTCC, S-nitrosylation of the channel has been shown to reduce the channel activity. Also, SERCA activity has been reported to increase in response to S-nitrosylation. Paradoxically, RyR2 activity has been shown to increase in response to S-nitrosylation (Zima and Blatter 2006; Gonzalez et al. 2009). Other studies, however, showed that hyponitrosylation of RyR2 caused the channel to be more susceptible to oxidation by ROS, leading to increased SR Ca2+ leak and arrhythmogenesis (Gonzalez et al. 2010). Moreover, it has been suggested that S-nitrosylation can potentially prevent irreversible oxidation of cysteine residues (Sun and Murphy 2010). Thus, S-nitrosylation of RyR2 may act as a protective PTM against oxidative stress and detrimental SR Ca2+ leak. By maintaining SR Ca2+ load and preventing [Ca2+]i overload, S-nitrosylation plays a very important role in cardiac function during periods of oxidative stress. Recent work from Gonzalez et al. identified that enhanced xanthine oxidase superoxide production caused a decrease in cardiac RyR2 S-nitrosylation with an overall decrease in free thiols, promoting SR Ca2+ leak in heart failure rats (Gonzalez et al. 2010).
4.3 Intersubunit Cross-Linking
The protein-protein interaction between RyR2 subunits has been implicated in channel gating (Abramson and Salama 1989; Kimlicka et al. 2013; Strauss and Wagenknecht 2013) and, therefore, likely plays an important role in regulating SR Ca2+ release. In the past decade, there has been a great amount of progress in defining the quaternary structure of RyR (Serysheva et al. 2008; Cornea et al. 2009; Tung et al. 2010; Zalk et al. 2015), particularly for the skeletal type 1 isoform (RyR1). Although only a small portion of the cytosolic domain has been crystallized to date, high-resolution cryo-EM studies have provided insight into conformational changes that occur as a result of channel activation. By superimposing the 3-D crystal structure of the N-terminal domain (1–532 amino acids) of RyR within the 3-D matrix created using images from cryo-EM, it was determined that the intersubunit gap between N-terminal domains becomes widened by ~7 Å in the open conformation. These results suggest that any protein-protein interactions that are taking place in the closed conformation are likely disrupted as the result of channel opening (van Petegem 2015). Although the N-terminal domain is only responsible for a small portion of the intersubunit interaction, it is of particular interest because a large number of disease mutations have been found to localize within it. In fact, a majority of these mutations were found facing the intersubunit boundary. Thus, it is highly plausible that these mutations affect the channel function by disrupting normal interdomain interactions. Moreover, the mutations were all associated with a gain-of-function phenotype (increased RyR channel activity) (Kimlicka et al. 2013). These functional results are consistent with the structural evidence, supporting the claim that RyR channel activity is indeed affected by changes in the intersubunit interactions.
Recent work by Han et al. demonstrated that in the presence of an oxidant, RyR1 undergoes covalent disulfide cross-linking between neighboring subunits (intersubunit cross-linking) that is reversible with the reducing agent dithiothreitol. In parallel, cryo-EM images showed that RyR1, which normally has a cytosolic structure that resembles a pinwheel (Fig. 3), undergoes major morphological changes as a result of H2O2 treatment. These morphological changes, however, are reversible with the treatment of dithiothreitol (Han et al. 2006). A different cryo-EM study used a nonselective cross-linking agent (glutaraldehyde) to induce intersubunit cross-linking. In these conditions, RyR1 adopted a conformation that resembled that of the open state. In both these studies, the authors suggest that intersubunit cross-linking leads to activation of RyR1 as a result of structural changes that directly affect gating of the channel (Aghdasi et al. 1997; Strauss and Wagenknecht 2013).
Fig. 3.

Oxidative stress can increase diastolic SR Ca2+ leak by inducing intersubunit cross-linking within the ryanodine receptor complex. The RyR2 tetramer is shown as viewed from the cytoplasmic face
Abramson and Salama were the first to suggest that intersubunit cross-linking is involved in the gating of RyR1 (Abramson and Salama 1989). They argue that thiol oxidation is a necessary requirement for RyR1 channel opening. In order for this hypothesis to be correct, the transition from conducting to nonconducting states would have to coincide with the reduction of the principal disulfides regulating gating. Furthermore, because the cytosolic environment is maintained at a highly reduced state, this proposed gating mechanism assumes that dynamic disulfide formation is present without oxidative stress. Recently, a study done by Zissimopoulos et al. provides some biochemical evidence to support this hypothesis for both RyR1 and RyR2. The major limitation to this study, however, was that full-length RyR was not used in the experimental approach (Zissimopoulos et al. 2013). Their work shows that N-terminal fragments of RyR2 self-assemble into oligomers similar to that of RyR1. Interestingly, unlike RyR1, N-terminal fragments of RyR2 were covalently linked by endogenous disulfide bonds in ambient conditions (absence of exogenous ROS treatment). Even though both RyR1 and RyR2 N-terminal fragments formed disulfide-linked oligomers with H2O2 in a dose-dependent manner, the authors suggest that a difference in sequence homology between isoforms may explain the disparity in disulfide bond formation. Alternatively, if the requisite cysteine residues are in fact conserved between isoforms, other oxidative PTMs (e.g., glutathionylation, nitrosylation) that resist disulfide formation may be unique to RyR1.
In light of recent work, the susceptibility for intersubunit cross-linking appears to be increased for the cardiac isoform of RyR2 (Zissimopoulos et al. 2013). Although some work has been done to examine the effects of intersubunit cross-linking on RyR2 function, no studies have examined its role in SR Ca2+ cycling within the cellular environment. We have recently discovered that the redox-mediated RyR2 cross-linking has a significant impact on the channel activity and SR Ca2+ release (Mazurek et al. 2014). We found that the RyR2 cross-linking increased the open probability of RyR2 measured in lipid bilayers and RyR2-mediated Ca2+ leak in isolated ventricular myocytes. Lastly, we found a positive correlation between the cross-linking level and SR Ca2+ leak. When the cross-linking reached the maximum level, further oxidation of RyR2 did not enhance SR Ca2+ leak. These results clearly demonstrate that the intersubunit cross-linking is a strong regulator of cardiac RyR2 function in vivo and in vitro (Fig. 3).
5 Conclusion
Ca2+ released through RyR2 is essential for initiating a robust myocardial contraction. Consequently, defects in RyR2 regulation contribute to contractile dysfunction in a variety of cardiac pathologies (Janssen and de Tombe 1997; Marks 2000; Gyorke and Carnes 2008; Yano 2008; Zima and Terentyev 2013). In particular, abnormal RyR2 activity due to cysteine oxidation (Zima and Blatter 2006; Hool and Corry 2007) causes SR Ca2+ mishandling, arrhythmias, and contractile dysfunction in infarcted (Belevych et al. 2009) and failing hearts (Mochizuki et al. 2007; Terentyev et al. 2008; Domeier et al. 2009; Belevych et al. 2011). Since RyR2 cysteine oxidation is implicated in the progression of cardiac disease, these cysteine residues are promising targets for therapeutic intervention. RyR2 contains as many as 90 cysteine residues per monomer, and the redox status of these residues can affect RyR2 function (Zima and Blatter 2006). However, the functionally important redox-sensing sites on RyR2 have yet to be characterized. As a result, RyR2 oxidation has always been treated as a nonselective PTM. In contrast, three functionally important phosphorylation sites on RyR2 have been characterized (Marx et al. 2000; Wehrens et al. 2004; Xiao et al. 2006), and clinical studies targeting these sites are currently underway (Lehnart 2007; Lompre et al. 2010; van Oort et al. 2010). Thus, identifying the functionally important RyR2 cysteines is essential for understanding the molecular mechanisms of RyR2 regulation and SR Ca2+ mishandling during oxidative stress.
Acknowledgments
This work was supported by the NIH Grant (R01HL130231), the Research Career Development Award from the Schweppe Foundation, and the RFC grant from Loyola University Chicago to A.V.Z.
References
- Abramson JJ, Salama G. Critical sulfhydryls regulate calcium release from sarcoplasmic reticulum. J Bioenerg Biomembr. 1989;21:283–294. doi: 10.1007/BF00812073. [DOI] [PubMed] [Google Scholar]
- Aghdasi B, Zhang JZ, Wu Y, Reid MB, Hamilton SL. Multiple classes of sulfhydryls modulate the skeletal muscle Ca2+ release channel. J Biol Chem. 1997;272:3739–3748. doi: 10.1074/jbc.272.6.3739. [DOI] [PubMed] [Google Scholar]
- Angelos MG, Kutala VK, Torres CA, He G, Stoner JD, Mohammad M, Kuppusamy P. Hypoxic reperfusion of the ischemic heart and oxygen radical generation. Am J Physiol Heart Circ Physiol. 2006;290:H341–H347. doi: 10.1152/ajpheart.00223.2005. [DOI] [PubMed] [Google Scholar]
- Aon MA, Cortassa S, Maack C, O’Rourke B. Sequential opening of mitochondrial ion channels as a function of glutathione redox thiol status. J Biol Chem. 2007;282:21889–21900. doi: 10.1074/jbc.M702841200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aon MA, Cortassa S, O’Rourke B. Redox-optimized ROS balance: a unifying hypothesis. Biochim Biophys Acta. 2010;1797:865–877. doi: 10.1016/j.bbabio.2010.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baddeley D, Jayasinghe ID, Lam L, Rossberger S, Cannell MB, Soeller C. Optical single-channel resolution imaging of the ryanodine receptor distribution in rat cardiac myocytes. Proc Natl Acad Sci U S A. 2009;106:22275–22280. doi: 10.1073/pnas.0908971106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balaban RS, Nemoto S, Finkel T. Mitochondria, oxidants, and aging. Cell. 2005;120:483–495. doi: 10.1016/j.cell.2005.02.001. [DOI] [PubMed] [Google Scholar]
- Bass R, Ruddock LW, Klappa P, Freedman RB. A major fraction of endoplasmic reticulum-located glutathione is present as mixed disulfides with protein. J Biol Chem. 2004;279:5257–5262. doi: 10.1074/jbc.M304951200. [DOI] [PubMed] [Google Scholar]
- Bassani RA, Bassani JW, Bers DM. Mitochondrial and sarcolemmal Ca2+ transport reduce [Ca2+]i during caffeine contractures in rabbit cardiac myocytes. J Physiol. 1992;453:591–608. doi: 10.1113/jphysiol.1992.sp019246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker LB. New concepts in reactive oxygen species and cardiovascular reperfusion physiology. Cardiovasc Res. 2004;61:461–470. doi: 10.1016/j.cardiores.2003.10.025. [DOI] [PubMed] [Google Scholar]
- Belch JJ, Bridges AB, Scott N, Chopra M. Oxygen free radicals and congestive heart failure. Br Heart J. 1991;65:245–248. doi: 10.1136/hrt.65.5.245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belevych AE, Terentyev D, Viatchenko-Karpinski S, Terentyeva R, Sridhar A, Nishijima Y, Wilson LD, Cardounel AJ, Laurita KR, Carnes CA, Billman GE, Gyorke S. Redox modification of ryanodine receptors underlies calcium alternans in a canine model of sudden cardiac death. Cardiovasc Res. 2009;84:387–395. doi: 10.1093/cvr/cvp246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belevych AE, Terentyev D, Terentyeva R, Nishijima Y, Sridhar A, Hamlin RL, Carnes CA, Gyorke S. The relationship between arrhythmogenesis and impaired contractility in heart failure: role of altered ryanodine receptor function. Cardiovasc Res. 2011;90:493–502. doi: 10.1093/cvr/cvr025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belevych AE, Terentyev D, Terentyeva R, Ho HT, Gyorke I, Bonilla IM, Carnes CA, Billman GE, Gyorke S. Shortened Ca2+ signaling refractoriness underlies cellular arrhythmogenesis in a postinfarction model of sudden cardiac death. Circ Res. 2012;110:569–577. doi: 10.1161/CIRCRESAHA.111.260455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bers DM. Excitation-contraction coupling and cardiac contractile force. Kluwer Academic Publishers; Dordrecht: 2001. [Google Scholar]
- Bers DM. Macromolecular complexes regulating cardiac ryanodine receptor function. J Mol Cell Cardiol. 2004;37:417–429. doi: 10.1016/j.yjmcc.2004.05.026. [DOI] [PubMed] [Google Scholar]
- Bovo E, Lipsius SL, Zima AV. Reactive oxygen species contribute to the development of arrhythmogenic Ca2+ waves during beta-adrenergic receptor stimulation in rabbit cardiomyocytes. J Physiol. 2012;590:3291–3304. doi: 10.1113/jphysiol.2012.230748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bovo E, de Tombe PP, Zima AV. The role of dyadic organization in regulation of sarcoplasmic reticulum Ca(2+) handling during rest in rabbit ventricular myocytes. Biophys J. 2014;106:1902–1909. doi: 10.1016/j.bpj.2014.03.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bovo E, Mazurek SR, de Tombe PP, Zima AV. Increased Energy Demand during Adrenergic Receptor Stimulation Contributes to Ca(2+) Wave Generation. Biophys J. 2015a;109:1583–1591. doi: 10.1016/j.bpj.2015.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bovo E, Mazurek SR, Fill M, Zima AV. Cytosolic Ca(2)(+) buffering determines the intra-SR Ca(2)(+) concentration at which cardiac Ca(2)(+) sparks terminate. Cell Calcium. 2015b;58:246–253. doi: 10.1016/j.ceca.2015.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brennan JP, Wait R, Begum S, Bell JR, Dunn MJ, Eaton P. Detection and mapping of widespread intermolecular protein disulfide formation during cardiac oxidative stress using proteomics with diagonal electrophoresis. J Biol Chem. 2004;279:41352–41360. doi: 10.1074/jbc.M403827200. [DOI] [PubMed] [Google Scholar]
- Brown DA, Aon MA, Frasier CR, Sloan RC, Maloney AH, Anderson EJ, O’Rourke B. Cardiac arrhythmias induced by glutathione oxidation can be inhibited by preventing mitochondrial depolarization. J Mol Cell Cardiol. 2010;48:673–679. doi: 10.1016/j.yjmcc.2009.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bunch TJ, Hohnloser SH, Gersh BJ. Mechanisms of sudden cardiac death in myocardial infarction survivors: insights from the randomized trials of implantable cardioverter-defibrillators. Circulation. 2007;115:2451–2457. doi: 10.1161/CIRCULATIONAHA.106.683235. [DOI] [PubMed] [Google Scholar]
- Cannell MB, Cheng H, Lederer WJ. The control of calcium release in heart muscle. Science. 1995;268:1045–1049. doi: 10.1126/science.7754384. [DOI] [PubMed] [Google Scholar]
- Cannell MB, Kong CH, Imtiaz MS, Laver DR. Control of sarcoplasmic reticulum Ca2+ release by stochastic RyR gating within a 3D model of the cardiac dyad and importance of induction decay for CICR termination. Biophys J. 2013;104:2149–2159. doi: 10.1016/j.bpj.2013.03.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ceconi C, Curello S, Cargnoni A, Ferrari R, Albertini A, Visioli O. The role of glutathione status in the protection against ischaemic and reperfusion damage: effects of N-acetyl cysteine. J Mol Cell Cardiol. 1988;20:5–13. doi: 10.1016/s0022-2828(88)80174-3. [DOI] [PubMed] [Google Scholar]
- Chen W, Wang R, Chen B, Zhong X, Kong H, Bai Y, Zhou Q, Xie C, Zhang J, Guo A, Tian X, Jones PP, O’Mara ML, Liu Y, Mi T, Zhang L, Bolstad J, Semeniuk L, Cheng H, Zhang J, Chen J, Tieleman DP, Gillis AM, Duff HJ, Fill M, Song LS, Chen SR. The ryanodine receptor store-sensing gate controls Ca2+ waves and Ca2+–triggered arrhythmias. Nat Med. 2014;20:184–192. doi: 10.1038/nm.3440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng H, Lederer WJ. Calcium sparks. Physiol Rev. 2008;88:1491–1545. doi: 10.1152/physrev.00030.2007. [DOI] [PubMed] [Google Scholar]
- Cheng H, Lederer WJ, Cannell MB. Calcium sparks: elementary events underlying excitation-contraction coupling in heart muscle. Science. 1993;262:740–744. doi: 10.1126/science.8235594. [DOI] [PubMed] [Google Scholar]
- Christensen NJ, Videbaek J. Plasma catecholamines and carbohydrate metabolism in patients with acute myocardial infarction. J Clin Invest. 1974;54:278–286. doi: 10.1172/JCI107763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cornea RL, Nitu F, Gruber S, Kohler K, Satzer M, Thomas DD, Fruen BR. FRET-based mapping of calmodulin bound to the RyR1 Ca2+ release channel. Proc Natl Acad Sci U S A. 2009;106:6128–6133. doi: 10.1073/pnas.0813010106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowburn PJ, Cleland JG, Coats AJ, Komajda M. Risk stratification in chronic heart failure. Eur Heart J. 1998;19:696–710. doi: 10.1053/euhj.1997.0820. [DOI] [PubMed] [Google Scholar]
- Cumming RC, Andon NL, Haynes PA, Park M, Fischer WH, Schubert D. Protein disulfide bond formation in the cytoplasm during oxidative stress. J Biol Chem. 2004;279:21749–21758. doi: 10.1074/jbc.M312267200. [DOI] [PubMed] [Google Scholar]
- Domeier TL, Blatter LA, Zima AV. Alteration of sarcoplasmic reticulum Ca2+ release termination by ryanodine receptor sensitization and in heart failure. J Physiol. 2009;587:5197–5209. doi: 10.1113/jphysiol.2009.177576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Domenech RJ, Macho P, Velez D, Sanchez G, Liu X, Dhalla N. Tachycardia preconditions infarct size in dogs: role of adenosine and protein kinase C. Circulation. 1998;97:786–794. doi: 10.1161/01.cir.97.8.786. [DOI] [PubMed] [Google Scholar]
- Eager KR, Dulhunty AF. Activation of the cardiac ryanodine receptor by sulfhydryl oxidation is modified by Mg2+ and ATP. J Membr Biol. 1998;163:9–18. doi: 10.1007/s002329900365. [DOI] [PubMed] [Google Scholar]
- Eisner DA, Trafford AW, Diaz ME, Overend CL, O’Neill SC. The control of Ca release from the cardiac sarcoplasmic reticulum: regulation versus autoregulation. Cardiovasc Res. 1998;38:589–604. doi: 10.1016/s0008-6363(98)00062-5. [DOI] [PubMed] [Google Scholar]
- Eisner DA, Kashimura T, Venetucci LA, Trafford AW. From the ryanodine receptor to cardiac arrhythmias. Circ J. 2009;73:1561–1567. doi: 10.1253/circj.cj-09-0478. [DOI] [PubMed] [Google Scholar]
- Fabiato A. Calcium-induced release of calcium from the cardiac sarcoplasmic reticulum. Am J Physiol. 1983;245:C1–C14. doi: 10.1152/ajpcell.1983.245.1.C1. [DOI] [PubMed] [Google Scholar]
- Fabiato A. Time and calcium dependence of activation and inactivation of calcium-induced release of calcium from the sarcoplasmic reticulum of a skinned canine cardiac Purkinje cell. J Gen Physiol. 1985;85:247–289. doi: 10.1085/jgp.85.2.247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabiato A, Fabiato F. Contractions induced by a calcium-triggered release of calcium from the sarcoplasmic reticulum of single skinned cardiac cells. J Physiol. 1975;249:469–495. doi: 10.1113/jphysiol.1975.sp011026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferdinandy P, Schulz R. Nitric oxide, superoxide, and peroxynitrite in myocardial ischaemia-reperfusion injury and preconditioning. Br J Pharmacol. 2003;138:532–543. doi: 10.1038/sj.bjp.0705080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fill M, Copello JA. Ryanodine receptor calcium release channels. Physiol Rev. 2002;82:893–922. doi: 10.1152/physrev.00013.2002. [DOI] [PubMed] [Google Scholar]
- Franzini-Armstrong C, Protasi F, Ramesh V. Shape, size, and distribution of Ca(2+) release units and couplons in skeletal and cardiac muscles. Biophys J. 1999;77:1528–1539. doi: 10.1016/S0006-3495(99)77000-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garbino A, van Oort RJ, Dixit SS, Landstrom AP, Ackerman MJ, Wehrens XH. Molecular evolution of the junctophilin gene family. Physiol Genomics. 2009;37:175–186. doi: 10.1152/physiolgenomics.00017.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giles GI, Jacob C. Reactive sulfur species: an emerging concept in oxidative stress. Biol Chem. 2002;383:375–388. doi: 10.1515/BC.2002.042. [DOI] [PubMed] [Google Scholar]
- Gillespie D. Energetics of divalent selectivity in a calcium channel: the ryanodine receptor case study. Biophys J. 2008;94:1169–1184. doi: 10.1529/biophysj.107.116798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giordano FJ. Oxygen, oxidative stress, hypoxia, and heart failure. J Clin Invest. 2005;115:500–508. doi: 10.1172/JCI200524408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldman YE. Kinetics of the actomyosin ATPase in muscle fibers. Annu Rev Physiol. 1987;49:637–654. doi: 10.1146/annurev.ph.49.030187.003225. [DOI] [PubMed] [Google Scholar]
- Gonzalez DR, Treuer A, Sun QA, Stamler JS, Hare JM. S-Nitrosylation of cardiac ion channels. J Cardiovasc Pharmacol. 2009;54:188–195. doi: 10.1097/FJC.0b013e3181b72c9f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez DR, Treuer AV, Castellanos J, Dulce RA, Hare JM. Impaired S-nitrosylation of the ryanodine receptor caused by xanthine oxidase activity contributes to calcium leak in heart failure. J Biol Chem. 2010;285:28938–28945. doi: 10.1074/jbc.M110.154948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo T, Gillespie D, Fill M. Ryanodine receptor current amplitude controls Ca2+ sparks in cardiac muscle. Circ Res. 2012;111:28–36. doi: 10.1161/CIRCRESAHA.112.265652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gyorke S, Carnes C. Dysregulated sarcoplasmic reticulum calcium release: potential pharmacological target in cardiac disease. Pharmacol Ther. 2008;119:340–354. doi: 10.1016/j.pharmthera.2008.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gyorke I, Hester N, Jones LR, Gyorke S. The role of calsequestrin, triadin, and junctin in conferring cardiac ryanodine receptor responsiveness to luminal calcium. Biophys J. 2004;86:2121–2128. doi: 10.1016/S0006-3495(04)74271-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han HM, Wei RS, Lai FA, Yin CC. Molecular nature of sulfhydryl modification by hydrogen peroxide on type 1 ryanodine receptor. Acta Pharmacol Sin. 2006;27:888–894. doi: 10.1111/j.1745-7254.2006.00386.x. [DOI] [PubMed] [Google Scholar]
- Hayashi T, Martone ME, Yu Z, Thor A, Doi M, Holst MJ, Ellisman MH, Hoshijima M. Three-dimensional electron microscopy reveals new details of membrane systems for Ca2+ signaling in the heart. J Cell Sci. 2009;122:1005–1013. doi: 10.1242/jcs.028175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinzel FR, Luo Y, Dodoni G, Boengler K, Petrat F, Di LF, de GH, Schulz R, Heusch G. Formation of reactive oxygen species at increased contraction frequency in rat cardiomyocytes. Cardiovasc Res. 2006;71:374–382. doi: 10.1016/j.cardiores.2006.05.014. [DOI] [PubMed] [Google Scholar]
- Hill MF, Singal PK. Right and left myocardial antioxidant responses during heart failure subsequent to myocardial infarction. Circulation. 1997;96:2414–2420. doi: 10.1161/01.cir.96.7.2414. [DOI] [PubMed] [Google Scholar]
- Hool LC, Corry B. Redox control of calcium channels: from mechanisms to therapeutic opportunities. Antioxid Redox Signal. 2007;9:409–435. doi: 10.1089/ars.2006.1446. [DOI] [PubMed] [Google Scholar]
- Houser SR, Piacentino V, III, Weisser J. Abnormalities of calcium cycling in the hypertrophied and failing heart. J Mol Cell Cardiol. 2000;32:1595–1607. doi: 10.1006/jmcc.2000.1206. [DOI] [PubMed] [Google Scholar]
- Ide T, Tsutsui H, Kinugawa S, Utsumi H, Kang D, Hattori N, Uchida K, Arimura K, Egashira K, Takeshita A. Mitochondrial electron transport complex I is a potential source of oxygen free radicals in the failing myocardium. Circ Res. 1999;85:357–363. doi: 10.1161/01.res.85.4.357. [DOI] [PubMed] [Google Scholar]
- Ide T, Tsutsui H, Hayashidani S, Kang D, Suematsu N, Nakamura K, Utsumi H, Hamasaki N, Takeshita A. Mitochondrial DNA damage and dysfunction associated with oxidative stress in failing hearts after myocardial infarction. Circ Res. 2001;88:529–535. doi: 10.1161/01.res.88.5.529. [DOI] [PubMed] [Google Scholar]
- Janse MJ. Electrophysiological changes in heart failure and their relationship to arrhythmogenesis. Cardiovasc Res. 2004;61:208–217. doi: 10.1016/j.cardiores.2003.11.018. [DOI] [PubMed] [Google Scholar]
- Janssen PM, de Tombe PP. Uncontrolled sarcomere shortening increases intracellular Ca2+ transient in rat cardiac trabeculae. Am J Physiol. 1997;272:H1892–H1897. doi: 10.1152/ajpheart.1997.272.4.H1892. [DOI] [PubMed] [Google Scholar]
- Kimlicka L, Lau K, Tung CC, Van PF. Disease mutations in the ryanodine receptor N-terminal region couple to a mobile intersubunit interface. Nat Commun. 2013;4:1506. doi: 10.1038/ncomms2501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kourie JI. Interaction of reactive oxygen species with ion transport mechanisms. Am J Physiol. 1998;275:C1–C24. doi: 10.1152/ajpcell.1998.275.1.C1. [DOI] [PubMed] [Google Scholar]
- Kubalova Z, Terentyev D, Viatchenko-Karpinski S, Nishijima Y, Gyorke I, Terentyeva R, da Cunha DN, Sridhar A, Feldman DS, Hamlin RL, Carnes CA, Gyorke S. Abnormal intrastore calcium signaling in chronic heart failure. Proc Natl Acad Sci U S A. 2005;102:14104–14109. doi: 10.1073/pnas.0504298102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurz T, Offner B, Schreieck J, Richardt G, Tolg R, Schomig A. Nonexocytotic noradrenaline release and ventricular fibrillation in ischemic rat hearts. Naunyn Schmiedebergs Arch Pharmacol. 1995;352:491–496. doi: 10.1007/BF00169382. [DOI] [PubMed] [Google Scholar]
- Lakatta EG. Beyond Bowditch: the convergence of cardiac chronotropy and inotropy. Cell Calcium. 2004;35:629–642. doi: 10.1016/j.ceca.2004.01.017. [DOI] [PubMed] [Google Scholar]
- Lameris TW, de ZS, Alberts G, Boomsma F, Duncker DJ, Verdouw PD, Veld AJ, van den Meiracker AH. Time course and mechanism of myocardial catecholamine release during transient ischemia in vivo. Circulation. 2000;101:2645–2650. doi: 10.1161/01.cir.101.22.2645. [DOI] [PubMed] [Google Scholar]
- Lane RE, Cowie MR, Chow AW. Prediction and prevention of sudden cardiac death in heart failure. Heart. 2005;91:674–680. doi: 10.1136/hrt.2003.025254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lebovitz RM, Zhang H, Vogel H, Cartwright J, Jr, Dionne L, Lu N, Huang S, Matzuk MM. Neurodegeneration, myocardial injury, and perinatal death in mitochondrial superoxide dismutase-deficient mice. Proc Natl Acad Sci U S A. 1996;93:9782–9787. doi: 10.1073/pnas.93.18.9782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehnart SE. Novel targets for treating heart and muscle disease: stabilizing ryanodine receptors and preventing intracellular calcium leak. Curr Opin Pharmacol. 2007;7:225–232. doi: 10.1016/j.coph.2006.09.010. [DOI] [PubMed] [Google Scholar]
- Lima B, Forrester MT, Hess DT, Stamler JS. S-nitrosylation in cardiovascular signaling. Circ Res. 2010;106:633–646. doi: 10.1161/CIRCRESAHA.109.207381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu N, Colombi B, Memmi M, Zissimopoulos S, Rizzi N, Negri S, Imbriani M, Napolitano C, Lai FA, Priori SG. Arrhythmogenesis in catecholaminergic polymorphic ventricular tachycardia: insights from a RyR2 R4496C knock-in mouse model. Circ Res. 2006;99:292–298. doi: 10.1161/01.RES.0000235869.50747.e1. [DOI] [PubMed] [Google Scholar]
- Lompre AM, Hajjar RJ, Harding SE, Kranias EG, Lohse MJ, Marks AR. Ca2+ cycling and new therapeutic approaches for heart failure. Circulation. 2010;121:822–830. doi: 10.1161/CIRCULATIONAHA.109.890954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Lopez JR, Shacklock PS, Balke CW, Wier WG. Local, stochastic release of Ca2+ in voltage-clamped rat heart cells: visualization with confocal microscopy. J Physiol. 1994;480(Pt 1):21–29. doi: 10.1113/jphysiol.1994.sp020337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loyer X, Gomez AM, Milliez P, Fernandez-Velasco M, Vangheluwe P, Vinet L, Charue D, Vaudin E, Zhang W, Sainte-Marie Y, Robidel E, Marty I, Mayer B, Jaisser F, Mercadier JJ, Richard S, Shah AM, Benitah JP, Samuel JL, Heymes C. Cardiomyocyte overexpression of neuronal nitric oxide synthase delays transition toward heart failure in response to pressure overload by preserving calcium cycling. Circulation. 2008;117:3187–3198. doi: 10.1161/CIRCULATIONAHA.107.741702. [DOI] [PubMed] [Google Scholar]
- Mak S, Newton GE. The oxidative stress hypothesis of congestive heart failure: radical thoughts. Chest. 2001;120:2035–2046. doi: 10.1378/chest.120.6.2035. [DOI] [PubMed] [Google Scholar]
- Makaula S, Lochner A, Genade S, Sack MN, Awan MM, Opie LH. H-89, a non-specific inhibitor of protein kinase A, promotes post-ischemic cardiac contractile recovery and reduces infarct size. J Cardiovasc Pharmacol. 2005;45:341–347. doi: 10.1097/01.fjc.0000156825.80951.14. [DOI] [PubMed] [Google Scholar]
- Makino N, Maeda T, Oyama J, Sasaki M, Higuchi Y, Mimori K, Shimizu T. Antioxidant therapy attenuates myocardial telomerase activity reduction in superoxide dismutase-deficient mice. J Mol Cell Cardiol. 2011;50:670–677. doi: 10.1016/j.yjmcc.2010.12.014. [DOI] [PubMed] [Google Scholar]
- Marks AR. Cardiac intracellular calcium release channels: role in heart failure. Circ Res. 2000;87:8–11. doi: 10.1161/01.res.87.1.8. [DOI] [PubMed] [Google Scholar]
- Marx SO, Reiken S, Hisamatsu Y, Jayaraman T, Burkhoff D, Rosemblit N, Marks AR. PKA phosphorylation dissociates FKBP12.6 from the calcium release channel (ryanodine receptor): defective regulation in failing hearts. Cell. 2000;101:365–376. doi: 10.1016/s0092-8674(00)80847-8. [DOI] [PubMed] [Google Scholar]
- Masumiya H, Li P, Zhang L, Chen SR. Ryanodine sensitizes the Ca(2+) release channel (ryanodine receptor) to Ca(2+) activation. J Biol Chem. 2001;276:39727–39735. doi: 10.1074/jbc.M106557200. [DOI] [PubMed] [Google Scholar]
- Mazurek SR, Bovo E, Zima AV. Regulation of sarcoplasmic reticulum Ca(2+) release by cytosolic glutathione in rabbit ventricular myocytes. Free Radic Biol Med. 2014;68:159–167. doi: 10.1016/j.freeradbiomed.2013.12.003. [DOI] [PubMed] [Google Scholar]
- Mead-Savery FC, Wang R, Tanna-Topan B, Chen SR, Welch W, Williams AJ. Changes in negative charge at the luminal mouth of the pore alter ion handling and gating in the cardiac ryanodine-receptor. Biophys J. 2009;96:1374–1387. doi: 10.1016/j.bpj.2008.10.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meissner G. Molecular regulation of cardiac ryanodine receptor ion channel. Cell Calcium. 2004;35:621–628. doi: 10.1016/j.ceca.2004.01.015. [DOI] [PubMed] [Google Scholar]
- Miao L, St Clair DK. Regulation of superoxide dismutase genes: implications in disease. Free Radic Biol Med. 2009;47:344–356. doi: 10.1016/j.freeradbiomed.2009.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misra MK, Sarwat M, Bhakuni P, Tuteja R, Tuteja N. Oxidative stress and ischemic myocardial syndromes. Med Sci Monit. 2009;15:RA209–RA219. [PubMed] [Google Scholar]
- Mochizuki M, Yano M, Oda T, Tateishi H, Kobayashi S, Yamamoto T, Ikeda Y, Ohkusa T, Ikemoto N, Matsuzaki M. Scavenging free radicals by low-dose carvedilol prevents redox-dependent Ca2+ leak via stabilization of ryanodine receptor in heart failure. J Am Coll Cardiol. 2007;49:1722–1732. doi: 10.1016/j.jacc.2007.01.064. [DOI] [PubMed] [Google Scholar]
- Nabauer M, Morad M. Ca2(+)-induced Ca2+ release as examined by photolysis of caged Ca 2+ in single ventricular myocytes. Am J Physiol. 1990;258:C189–C193. doi: 10.1152/ajpcell.1990.258.1.C189. [DOI] [PubMed] [Google Scholar]
- Nagasaka S, Katoh H, Niu CF, Matsui S, Urushida T, Satoh H, Watanabe Y, Hayashi H. Protein kinase A catalytic subunit alters cardiac mitochondrial redox state and membrane potential via the formation of reactive oxygen species. Circ J. 2007;71:429–436. doi: 10.1253/circj.71.429. [DOI] [PubMed] [Google Scholar]
- Nogueira L, Figueiredo-Freitas C, Casimiro-Lopes G, Magdesian MH, Assreuy J, Sorenson MM. Myosin is reversibly inhibited by S-nitrosylation. Biochem J. 2009;424:221–231. doi: 10.1042/BJ20091144. [DOI] [PubMed] [Google Scholar]
- Opie LH. The mechanism of myocyte death in ischaemia. Eur Heart J. 1993;14(Suppl G):31–33. doi: 10.1093/eurheartj/14.suppl_g.31. [DOI] [PubMed] [Google Scholar]
- Opie LH, Clusin WT. Cellular mechanism for ischemic ventricular arrhythmias. Annu Rev Med. 1990;41:231–238. doi: 10.1146/annurev.me.41.020190.001311. [DOI] [PubMed] [Google Scholar]
- Pogwizd SM, Bers DM. Cellular basis of triggered arrhythmias in heart failure. Trends Cardiovasc Med. 2004;14:61–66. doi: 10.1016/j.tcm.2003.12.002. [DOI] [PubMed] [Google Scholar]
- Qin J, Valle G, Nani A, Nori A, Rizzi N, Priori SG, Volpe P, Fill M. Luminal Ca2+ regulation of single cardiac ryanodine receptors: insights provided by calsequestrin and its mutants. J Gen Physiol. 2008;131:325–334. doi: 10.1085/jgp.200709907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rassaf T, Totzeck M, Hendgen-Cotta UB, Shiva S, Heusch G, Kelm M. Circulating nitrite contributes to cardioprotection by remote ischemic preconditioning. Circ Res. 2014;114:1601–1610. doi: 10.1161/CIRCRESAHA.114.303822. [DOI] [PubMed] [Google Scholar]
- Roux-Buisson N, Cacheux M, Fourest-Lieuvin A, Fauconnier J, Brocard J, Denjoy I, Durand P, Guicheney P, Kyndt F, Leenhardt A, Le MH, Lucet V, Mabo P, Probst V, Monnier N, Ray PF, Santoni E, Tremeaux P, Lacampagne A, Faure J, Lunardi J, Marty I. Absence of triadin, a protein of the calcium release complex, is responsible for cardiac arrhythmia with sudden death in human. Hum Mol Genet. 2012;21:2759–2767. doi: 10.1093/hmg/dds104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez G, Pedrozo Z, Domenech RJ, Hidalgo C, Donoso P. Tachycardia increases NADPH oxidase activity and RyR2 S-glutathionylation in ventricular muscle. J Mol Cell Cardiol. 2005;39:982–991. doi: 10.1016/j.yjmcc.2005.08.010. [DOI] [PubMed] [Google Scholar]
- Santiago DJ, Curran JW, Bers DM, Lederer WJ, Stern MD, Rios E, Shannon TR. Ca sparks do not explain all ryanodine receptor-mediated SR Ca leak in mouse ventricular myocytes. Biophys J. 2010;98:2111–2120. doi: 10.1016/j.bpj.2010.01.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santiago DJ, Rios E, Shannon TR. Isoproterenol increases the fraction of spark-dependent RyR-mediated leak in ventricular myocytes. Biophys J. 2013;104:976–985. doi: 10.1016/j.bpj.2013.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santos CX, Anilkumar N, Zhang M, Brewer AC, Shah AM. Redox signaling in cardiac myocytes. Free Radic Biol Med. 2011;50:777–793. doi: 10.1016/j.freeradbiomed.2011.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satoh H, Blatter LA, Bers DM. Effects of [Ca2+]i, SR Ca2+ load, and rest on Ca2+ spark frequency in ventricular myocytes. Am J Physiol. 1997;272:H657–H668. doi: 10.1152/ajpheart.1997.272.2.H657. [DOI] [PubMed] [Google Scholar]
- Schaper J, Froede R, Hein S, Buck A, Hashizume H, Speiser B, Friedl A, Bleese N. Impairment of the myocardial ultrastructure and changes of the cytoskeleton in dilated cardiomyopathy. Circulation. 1991;83:504–514. doi: 10.1161/01.cir.83.2.504. [DOI] [PubMed] [Google Scholar]
- Schlotthauer K, Bers DM. Sarcoplasmic reticulum Ca(2+) release causes myocyte depolarization. Underlying mechanism and threshold for triggered action potentials. Circ Res. 2000;87:774–780. doi: 10.1161/01.res.87.9.774. [DOI] [PubMed] [Google Scholar]
- Seddon M, Looi YH, Shah AM. Oxidative stress and redox signalling in cardiac hypertrophy and heart failure. Heart. 2007;93:903–907. doi: 10.1136/hrt.2005.068270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sen L, Cui G, Fonarow GC, Laks H. Differences in mechanisms of SR dysfunction in ischemic vs. idiopathic dilated cardiomyopathy. Am J Physiol Heart Circ Physiol. 2000;279:H709–H718. doi: 10.1152/ajpheart.2000.279.2.H709. [DOI] [PubMed] [Google Scholar]
- Serysheva II, Ludtke SJ, Baker ML, Cong Y, Topf M, Eramian D, Sali A, Hamilton SL, Chiu W. Subnanometer-resolution electron cryomicroscopy-based domain models for the cytoplasmic region of skeletal muscle RyR channel. Proc Natl Acad Sci U S A. 2008;105:9610–9615. doi: 10.1073/pnas.0803189105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiferaw Y, Aistrup GL, Wasserstrom JA. Intracellular Ca2+ waves, afterdepolarizations, and triggered arrhythmias. Cardiovasc Res. 2012;95:265–268. doi: 10.1093/cvr/cvs155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sitsapesan R, Williams AJ. Regulation of the gating of the sheep cardiac sarcoplasmic reticulum Ca(2+)-release channel by luminal Ca2+ J Membr Biol. 1994;137:215–226. doi: 10.1007/BF00232590. [DOI] [PubMed] [Google Scholar]
- Slodzinski MK, Aon MA, O’Rourke B. Glutathione oxidation as a trigger of mitochondrial depolarization and oscillation in intact hearts. J Mol Cell Cardiol. 2008;45:650–660. doi: 10.1016/j.yjmcc.2008.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soeller C, Cannell MB. Examination of the transverse tubular system in living cardiac rat myocytes by 2-photon microscopy and digital image-processing techniques. Circ Res. 1999;84:266–275. doi: 10.1161/01.res.84.3.266. [DOI] [PubMed] [Google Scholar]
- Spear JF, Prabu SK, Galati D, Raza H, Anandatheerthavarada HK, Avadhani NG. beta1-Adrenoreceptor activation contributes to ischemia-reperfusion damage as well as playing a role in ischemic preconditioning. Am J Physiol Heart Circ Physiol. 2007;292:H2459–H2466. doi: 10.1152/ajpheart.00459.2006. [DOI] [PubMed] [Google Scholar]
- Stern MD. Theory of excitation-contraction coupling in cardiac muscle. Biophys J. 1992;63:497–517. doi: 10.1016/S0006-3495(92)81615-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stern MD, Cheng H. Putting out the fire: what terminates calcium-induced calcium release in cardiac muscle? Cell Calcium. 2004;35:591–601. doi: 10.1016/j.ceca.2004.01.013. [DOI] [PubMed] [Google Scholar]
- Stevens SC, Terentyev D, Kalyanasundaram A, Periasamy M, Gyorke S. Intra-sarcoplasmic reticulum Ca2+ oscillations are driven by dynamic regulation of ryanodine receptor function by luminal Ca2+ in cardiomyocytes. J Physiol. 2009;587:4863–4872. doi: 10.1113/jphysiol.2009.175547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strauss JD, Wagenknecht T. Structure of glutaraldehyde cross-linked ryanodine receptor. J Struct Biol. 2013;181:300–306. doi: 10.1016/j.jsb.2013.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun J, Murphy E. Protein S-nitrosylation and cardioprotection. Circ Res. 2010;106:285–296. doi: 10.1161/CIRCRESAHA.109.209452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun J, Morgan M, Shen RF, Steenbergen C, Murphy E. Preconditioning results in S-nitrosylation of proteins involved in regulation of mitochondrial energetics and calcium transport. Circ Res. 2007;101:1155–1163. doi: 10.1161/CIRCRESAHA.107.155879. [DOI] [PubMed] [Google Scholar]
- Tang H, Viola HM, Filipovska A, Hool LC. Ca(v)1.2 calcium channel is glutathionylated during oxidative stress in guinea pig and ischemic human heart. Free Radic Biol Med. 2011;51:1501–1511. doi: 10.1016/j.freeradbiomed.2011.07.005. [DOI] [PubMed] [Google Scholar]
- Terentyev D, Viatchenko-Karpinski S, Valdivia HH, Escobar AL, Gyorke S. Luminal Ca2+ controls termination and refractory behavior of Ca2+–induced Ca2+ release in cardiac myocytes. Circ Res. 2002;91:414–420. doi: 10.1161/01.res.0000032490.04207.bd. [DOI] [PubMed] [Google Scholar]
- Terentyev D, Gyorke I, Belevych AE, Terentyeva R, Sridhar A, Nishijima Y, de Blanco EC, Khanna S, Sen CK, Cardounel AJ, Carnes CA, Gyorke S. Redox modification of ryanodine receptors contributes to sarcoplasmic reticulum Ca2+ leak in chronic heart failure. Circ Res. 2008;103:1466–1472. doi: 10.1161/CIRCRESAHA.108.184457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Townsend DM. S-glutathionylation: indicator of cell stress and regulator of the unfolded protein response. Mol Interv. 2007;7:313–324. doi: 10.1124/mi.7.6.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tung CC, Lobo PA, Kimlicka L, Van PF. The amino-terminal disease hotspot of ryanodine receptors forms a cytoplasmic vestibule. Nature. 2010;468:585–588. doi: 10.1038/nature09471. [DOI] [PubMed] [Google Scholar]
- Turrens JF. Superoxide production by the mitochondrial respiratory chain. Biosci Rep. 1997;17:3–8. doi: 10.1023/a:1027374931887. [DOI] [PubMed] [Google Scholar]
- Turrens JF. Mitochondrial formation of reactive oxygen species. J Physiol. 2003;552:335–344. doi: 10.1113/jphysiol.2003.049478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Petegem Ryanodine receptors: allosteric ion channel giants. J Mol Biol. 2015;427:31–53. doi: 10.1016/j.jmb.2014.08.004. [DOI] [PubMed] [Google Scholar]
- van Oort RJ, McCauley MD, Dixit SS, Pereira L, Yang Y, Respress JL, Wang Q, De Almeida AC, Skapura DG, Anderson ME, Bers DM, Wehrens XH. Ryanodine receptor phosphorylation by calcium/calmodulin-dependent protein kinase II promotes life-threatening ventricular arrhythmias in mice with heart failure. Circulation. 2010;122:2669–2679. doi: 10.1161/CIRCULATIONAHA.110.982298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanden Hoek TL, Shao Z, Li C, Zak R, Schumacker PT, Becker LB. Reperfusion injury on cardiac myocytes after simulated ischemia. Am J Physiol. 1996;270:H1334–H1341. doi: 10.1152/ajpheart.1996.270.4.H1334. [DOI] [PubMed] [Google Scholar]
- Ventura-Clapier R, Garnier A, Veksler V. Energy metabolism in heart failure. J Physiol. 2004;555:1–13. doi: 10.1113/jphysiol.2003.055095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wehrens XH, Lehnart SE, Reiken SR, Marks AR. Ca2+/calmodulin-dependent protein kinase II phosphorylation regulates the cardiac ryanodine receptor. Circ Res. 2004;94:e61–e70. doi: 10.1161/01.RES.0000125626.33738.E2. [DOI] [PubMed] [Google Scholar]
- Werns SW, Fantone JC, Ventura A, Lucchesi BR. Myocardial glutathione depletion impairs recovery of isolated blood-perfused hearts after global ischaemia. J Mol Cell Cardiol. 1992;24:1215–1220. doi: 10.1016/0022-2828(92)93088-2. [DOI] [PubMed] [Google Scholar]
- Xiao B, Zhong G, Obayashi M, Yang D, Chen K, Walsh MP, Shimoni Y, Cheng H, Ter KH, Chen SR. Ser-2030, but not Ser-2808, is the major phosphorylation site in cardiac ryanodine receptors responding to protein kinase A activation upon beta-adrenergic stimulation in normal and failing hearts. Biochem J. 2006;396:7–16. doi: 10.1042/BJ20060116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie LH, Weiss JN. Arrhythmogenic consequences of intracellular calcium waves. Am J Physiol Heart Circ Physiol. 2009;297:H997–H1002. doi: 10.1152/ajpheart.00390.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xing D, Chaudhary AK, Miller FJ, Jr, Martins JB. Free radical scavenger specifically prevents ischemic focal ventricular tachycardia. Heart Rhythm. 2009;6:530–536. doi: 10.1016/j.hrthm.2008.12.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu L, Eu JP, Meissner G, Stamler JS. Activation of the cardiac calcium release channel (ryanodine receptor) by poly-S-nitrosylation. Science. 1998;279:234–237. doi: 10.1126/science.279.5348.234. [DOI] [PubMed] [Google Scholar]
- Yamawaki H, Haendeler J, Berk BC. Thioredoxin: a key regulator of cardiovascular homeostasis. Circ Res. 2003;93:1029–1033. doi: 10.1161/01.RES.0000102869.39150.23. [DOI] [PubMed] [Google Scholar]
- Yano M. Ryanodine receptor as a new therapeutic target of heart failure and lethal arrhythmia. Circ J. 2008;72:509–514. doi: 10.1253/circj.72.509. [DOI] [PubMed] [Google Scholar]
- Yavuz S. Surgery as early revascularization after acute myocardial infarction. Anadolu Kardiyol Derg. 2008;8(Suppl 2):84–92. [PubMed] [Google Scholar]
- Zalk R, Clarke OB, des GA, Grassucci RA, Reiken S, Mancia F, Hendrickson WA, Frank J, Marks AR. Structure of a mammalian ryanodine receptor. Nature. 2015;517:44–49. doi: 10.1038/nature13950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaugg M, Lucchinetti E, Uecker M, Pasch T, Schaub MC. Anaesthetics and cardiac preconditioning. Part I. Signalling and cytoprotective mechanisms. Br J Anaesth. 2003;91:551–565. doi: 10.1093/bja/aeg205. [DOI] [PubMed] [Google Scholar]
- Zhang J, Jin B, Li L, Block ER, Patel JM. Nitric oxide-induced persistent inhibition and nitrosylation of active site cysteine residues of mitochondrial cytochrome-c oxidase in lung endothelial cells. Am J Physiol Cell Physiol. 2005;288:C840–C849. doi: 10.1152/ajpcell.00325.2004. [DOI] [PubMed] [Google Scholar]
- Zima AV, Blatter LA. Redox regulation of cardiac calcium channels and transporters. Cardiovasc Res. 2006;71:310–321. doi: 10.1016/j.cardiores.2006.02.019. [DOI] [PubMed] [Google Scholar]
- Zima AV, Terentyev D. The biophysics of the failing heart. Springer; 2013. Sarcoplasmic reticulum Ca homeostasis and heart failure; pp. 5–31. [Google Scholar]
- Zima AV, Picht E, Bers DM, Blatter LA. Termination of cardiac Ca2+ sparks: role of intra-SR [Ca2+], release flux, and intra-SR Ca2+ diffusion. Circ Res. 2008;103:e105–e115. doi: 10.1161/CIRCRESAHA.107.183236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zima AV, Bovo E, Bers DM, Blatter LA. Ca2+ spark-dependent and -independent sarcoplasmic reticulum Ca2+ leak in normal and failing rabbit ventricular myocytes. J Physiol. 2010;588:4743–4757. doi: 10.1113/jphysiol.2010.197913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zima AV, Bovo E, Mazurek SR, Rochira JA, Li W, Terentyev D. Ca handling during excitation-contraction coupling in heart failure. Pflugers Arch. 2014;466:1129–1137. doi: 10.1007/s00424-014-1469-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zissimopoulos S, Viero C, Seidel M, Cumbes B, White J, Cheung I, Stewart R, Jeyakumar LH, Fleischer S, Mukherjee S, Thomas NL, Williams AJ, Lai FA. N-terminus oligomerization regulates the function of cardiac ryanodine receptors. J Cell Sci. 2013;126:5042–5051. doi: 10.1242/jcs.133538. [DOI] [PubMed] [Google Scholar]
- Zweier JL, Talukder MA. The role of oxidants and free radicals in reperfusion injury. Cardiovasc Res. 2006;70:181–190. doi: 10.1016/j.cardiores.2006.02.025. [DOI] [PubMed] [Google Scholar]
- Zweier JL, Flaherty JT, Weisfeldt ML. Direct measurement of free radical generation following reperfusion of ischemic myocardium. Proc Natl Acad Sci U S A. 1987;84:1404–1407. doi: 10.1073/pnas.84.5.1404. [DOI] [PMC free article] [PubMed] [Google Scholar]