

Abstract
Liver kinase B1 (LKB1) and its downstream effector AMP‐activated protein kinase (AMPK) play critical roles in polarity establishment by regulating membrane trafficking and energy metabolism. In collagen sandwich‐cultured hepatocytes, loss of LKB1 or AMPK impaired apical ABCB11 (Bsep) trafficking and bile canalicular formation. In the present study, we used liver‐specific (albumin‐Cre) LKB1 knockout mice (LKB1−/−) to investigate the role of LKB1 in the maintenance of functional tight junction (TJ) in vivo. Transmission electron microscopy examination revealed that hepatocyte apical membrane with microvilli substantially extended into the basolateral domain of LKB1−/− livers. Immunofluorescence studies revealed that loss of LKB1 led to longer and wider canalicular structures correlating with mislocalization of the junctional protein, cingulin. To test junctional function, we used intravital microscopy to quantify the transport kinetics of 6‐carboxyfluorescein diacetate (6‐CFDA), which is processed in hepatocytes into its fluorescent derivative 6‐carboxyfluorescein (6‐CF) and secreted into the canaliculi. In LKB1−/− mice, 6‐CF remained largely in hepatocytes, canalicular secretion was delayed, and 6‐CF appeared in the blood. To test whether 6‐CF was transported through permeable TJ, we intravenously injected low molecular weight (3 kDa) dextran in combination with 6‐CFDA. In wild‐type mice, 3 kDa dextran remained in the vasculature, whereas it rapidly appeared in the abnormal bile canaliculi in LKB1−/− mice, confirming that junctional disruption resulted in paracellular exchange between the blood stream and the bile canaliculus. Conclusion: LKB1 plays a critical role in regulating the maintenance of TJ and paracellular permeability, which may explain how various drugs, chemicals, and metabolic states that inhibit the LKB1/AMPK pathway result in cholestasis. (Hepatology 2016;64:1317‐1329)
Abbreviations
- 6‐CF
6‐carboxyfluorescein
- 6‐CFDA
6‐carboxyfluorescein diacetate
- AMPK
AMP‐activated protein kinase
- IVM
intravital microscopy
- LKB1
liver kinase B1
- PBS
phosphate‐buffered saline
- TEM
transmission electron microscopy
- TJ
tight junction
- ZO‐1
zona occludens‐1
Hepatocytes have a unique polarized organization that includes up to three apical canalicular plasma membrane domains and a larger basolateral domain that faces the sinusoidal compartment of the hepatic vasculature. Segregation of these plasma membrane domains is achieved by formation of TJ between individual cells, which creates a paracellular diffusion barrier critical for proper organ function.1, 2 TJ are composed of transmembrane pore forming barrier proteins including claudins and occludin that are linked to peripheral scaffolding proteins, such as zona occludens‐1 (ZO‐1). In turn, scaffolding proteins bind to regulatory proteins, such as cingulin, and cytoskeletal proteins, including myosin, that connect the scaffold with microtubules and actin.2, 3 TJ permeability is regulated under various physiological conditions.4 For example, vasopressin, epinephrine, and angiotensin increase TJ permeability in perfused rodent liver.5 A similar effect is observed in pancreas in response to cholinergic stimulation.6 Two general mechanisms for regulation of TJ permeability have been proposed. The first involves transcriptional regulation or posttranslational modification of junctional proteins. The second involves changes in the actin cytoskeleton that modify intercellular junction contacts and permeability.4 Although it is accepted that claudins determine the size, charge, and conductance properties of the paracellular pathway,2 the mechanisms regulating flux in the intact liver and the signaling molecules involved are not known. In vivo hepatic permeability is inferred based on the differential secretion of fluid phase probes with differential size such as inulin and sucrose.7 However, direct visualization of paracellular transport in the intact liver has not been achieved.
The serine‐threonine kinase and tumor suppressor LKB1 is a well‐conserved key regulator of cell polarity, trafficking, and metabolism.8, 9, 10 In an intestinal cell line, LKB1 induces polarity in the absence of cell‐cell or cell‐matrix cues11 and is involved in establishment and maintenance of cell polarity in pancreas,12 neurons,13 bronchial epithelia,14 and cultured hepatocytes.15, 16 LKB1 activates a family of metabolic sensors called AMPK. In hepatocytes, activation of the LKB1‐AMPK pathway enhanced bile canalicular formation, whereas inhibition resulted in loss of polarity and mislocalization of apical transporters such as Bsep.15, 16, 17 In MDCK cells and cultured hepatocytes, AMPK regulates TJ assembly and disassembly in response to calcium depletion.18, 19 LKB1‐AMPK activation phosphorylates cingulin, a junctional component promoting interaction with microtubules.20 However, it is not known whether or how the LKB1‐AMPK pathway functions in the maintenance of established hepatocyte TJs in intact animals.
Due to the importance of TJ in cell polarity and the involvement of LKB1 in hepatocyte polarization, we investigated the role of LKB1 in TJ regulation and function in the intact liver. Using a combination of transmission electron microscopy (TEM), immunofluorescence, and intravital microscopy (IVM) to directly visualize biliary and paracellular transport, we determined that LKB1 plays a critical role in the regulation and maintenance of TJ structure and function in the intact liver.
Materials and Methods
REAGENTS AND ANTIBODIES
The rat monoclonal antibody against ZO‐1 clone R40.76 was used.21 Antibodies against claudin‐1 (catalog no. 71‐7800), claudin‐3 (catalog no. 34‐1700), Rab11a (catalog no. 71‐5300) and rhodamine phalloidin were obtained from Thermo Fisher Scientific (Waltham, MA). The rabbit polyclonal antibody against Par‐3 was obtained from Milipore (catalog no. 07‐330; Billerica, MA). The rabbit polyclonal antibody against Bsep was obtained from Kamiya Biomedical Company (catalog no. MC‐333; Tukwila, WA). The rabbit antibody against cingulin was a gift from Sandra Citi (University of Geneva, Geneva, Switzerland),22 and the Myosin Vb was a gift from John Hammer (NHLBI, NIH).23 Rabbit anti‐LKB1 (catalog no. CST3047), anti‐AMPK (catalog no. CST2532), and anti‐phospho‐Thr172 AMPK (catalog nos. pAMPK and CST2535) antibodies were purchased from Cell Signaling Technology (Danvers, MA). Mouse anti‐α‐tubulin (catalog no. T6199) was obtained from Sigma‐Aldrich.
LIVER‐SPECIFIC LKB1−/− MICE
The generation and maintenance of liver‐specific LKB1−/− mice (NCI mouse repository; MGI: 2387402) have been described before.17, 24 Briefly, liver‐specific LKB1 knockouts were obtained by crossing mice containing floxed LKB1 alleles with mice expressing the Cre recombinase under the control of albumin promoter, Alb‐Cre (MGI 2176228; The Jackson Laboratory, Bar Harbor, ME). Homozygous LKB1 knockouts (LKB1−/−) and their wild‐type littermates (control) were used in all experiments. Alb‐Cre LKB1−/− mice appeared to be smaller than normal and as early as 10 days after birth displayed jaundice of the paws and snout. The animals were monitored closely. Animals displaying signs indicative of distress were euthanized. The study was approved and conducted according to the animal protocols approved by the Institutional Animal Care and Use Committee, protocols 14‐738 and 15‐779, National Institute of Dental and Craniofacial Research, in compliance with the Guide for the Care and Use of Laboratory Animals (National Institutes of Health publication 86‐23, revised 1985). Male and female mice, 7 or 21‐23 days old, were used in experiments.
TISSUE SECTIONS AND IMMUNOFLUORESCENCE
Livers were excised and snap frozen in liquid nitrogen. Frozen sections were prepared by the National Heart, Lung, and Blood Institute Pathology Core. Tissue sections were fixed in 100% methanol for 20 minutes at −20°C and then rehydrated in phosphate‐buffered saline (PBS) for 30 minutes at 4°C. Sections were blocked in PBS containing 1% bovine serum albumin and 5% normal goat serum for 20 minutes at room temperature, followed by incubation with primary antibodies for 60 minutes at room temperature. Sections were washed three times for 10 minutes with the blocking solution, followed by incubation with fluorescent‐labeled secondary antibodies (Cy2, Cy3, or Cy5‐labeled; Jackson Immunoresearch, West Grove, PA) for 60 minutes at room temperature. Sections were washed and mounted with Mowiol 4‐88 (catalog no. 81381; Sigma Aldrich, St. Louis, MO) containing 1% n‐propyl gallate. For Rab11a and phalloidin labeling with ZO‐1, tissue sections were fixed in 1% paraformaldehyde in CSK buffer (10 mM 1,4‐piperazinediethanesulfonic acid, pH 6.8, 100 mM KCl, 300 mM sucrose, 2 mM MgCl2, and 2 mM ethylene glycol tetraacetic acid) for 20 minutes at 4°C, permeabilized with 0.2% Triton X‐100 (10 minutes), quenched with 50 mM NH4Cl (10 minutes), and blocked in 2% normal goat serum in PBS for 60 minutes. Primary and secondary antibody incubations were performed as above.
WESTERN BLOT ANALYSIS
Frozen liver tissue was lysed in RIPA buffer (150 mM NaCl, 0.1% Triton X‐100, 0.5% sodium deoxycholate, 0.1% sodium dodecyl sulfate, and 50 mM Tris‐HCl pH 8.0) containing protease inhibitor cocktail and was homogenized using ceramic beads with electric tissue homogenizer (Bertin Technologies) at 5000 rpm for 20 seconds, followed by centrifugation at 13,000 rpm at 4°C for 20 minutes. Then, 20 μg of protein were separated on 4%‐20% gel (Bio‐Rad, Hercules, CA) sodium dodecyl sulfate‐polyacrylamide gel electrophoresis at 100 V for 90 minutes. Proteins were transferred on ice for 1 hour at 100 V to nitrocellulose membrane, which was subsequently blocked in 5% milk‐TBS for 1 hour. Membranes were incubated with primary antibodies overnight at 4°C, followed by washing in TBST, and incubation with secondary HRP‐conjugated antibodies for 1 hour at room temperature. Membranes were washed again with TBST before protein was detected with SuperSignal West Femto Maximum Sensitivity Substrate (ThermoFischer Scientific). Films were scanned using EPSON Perfection V700 scanner and quantification of band densitometry was measured with ImageJ standard gel analysis. The mean ± SD was calculated from three individual experiments, and a Student t test was used for P value calculation.
TEM
Small liver samples (1 mm3 maximum) were fixed for 1.5 hours in 2% (wt/vol) formaldehyde and 2% (wt/vol) glutaraldehyde in 0.1 M sodium cacodylate (pH 7.2), postfixed in 1% aqueous OsO4, and stained en bloc with 2% (wt/vol) uranyl acetate. Samples were further dehydrated with a series of ethanol concentrations, penetrated with EMbed 812 (Electron Microscopy Sciences, Hatfield, PA) and placed in flat molds. The resin was subsequently polymerized at 65°C for 60 hours. Thin (70‐nm) sections were cut using a Leica EM UC7 microtome and were stained with uranyl acetate and lead citrate. The samples were examined on an FEI Tecnai 20 TEM operated at 120 kV, and images were recorded on an AMT CCD camera.
IVM OF THE LIVER
Mice at 21‐23 days after birth were anesthetized by way of an intraperitoneal injection of 100 mg/kg ketamine (Fort Dodge Animal Health, Fort Dodge, IA) and 20 mg/kg xylazine (Akorn Inc., Decatur, IL) for control mice (approximately 8 g) and 25 mg/kg ketamine and 5 mg/kg xylazine for LKB1−/− mice (approximately 4 g). Body temperature was monitored using a rectal probe. A total of 20 U heparin was injected intraperitoneally to avoid ischemia. The tip of the left lateral lobe of the liver was exposed by way of a 2‐3 cm incision made along the ribcage. After cutting of the skin, the muscle was cut using a cauterizer to avoid bleeding. The incision was cooled with wet gauze. Qtracker 655 Vascular Label (ThermoFischer Scientific) was injected retro‐orbitally, 20 μL per mouse, to visualize sinusoids and blood flow. For positioning of the mouse on the microscope stage, a piece of cardboard with a rectangle window large enough to surround the exposed lobe was taped to the microscope stage, which reduced motion artifacts due to heartbeat and respiration. The liver lobe was separated with a narrow gauze strip followed by application of water‐based 0.3% carbomer 940 gel (Snowdrift Farm, Tucson, AZ [also available from Lubrizon, Wickliffe, OH]) to prevent tissue dehydration during imaging. Additional fluorescent probes were administered by way of retro‐orbital injection including 0.2 μg/g 6‐carboxyfluorescein diacetate (6‐CFDA; Sigma‐Aldrich) or 3 kDa Texas Red dextran (ThermoFischer Scientific), 30 μL in control and 20 μL in LKB1−/−, as noted. Imaging was performed as described earlier using an IX81 inverted confocal microscope equipped with a Fluoview 1000 scanning head (OlympusAmerica, Center Valley, PA).25 6‐Carboxyfluorescein (6‐CF) and Alexa 488 were excited with a 488‐nm laser, Texas Red‐dextran and Alexa 594 were excited with a 561‐nm laser, and Qtracker 655 was excited with a 633‐nm laser. For the time‐lapse imaging, 20‐μm stacks with 1‐μm step size were acquired every 2 minutes over the course of up to 1 hour. Time‐lapse series were acquired using a UPLSAPO 40XS, 40X N.A. 1.25 silicon oil objective (Olympus America).
IMAGE ANALYSIS AND QUANTIFICATION
Original OIF files were opened in ImageJ and corrected for three‐dimensional drift as necessary.
Quantification of canaliculi width and length was performed on maximal projections of 5 μm z‐stacks using ImageJ. At least three stacks from two independent control or LKB1−/− mice were used, and 30‐45 cells were analyzed. The shortest distance between two opposite ZO‐1‐positive junctions (for canalicular width) or the maximal continuous ZO‐1 labeling (for canalicular length) were measured. For canalicular width/length per cell, values obtained were summed and divided by the number of cells.
To measure 6‐CF transport, sum projections were generated using ImageJ, background was subtracted and 6‐CF mean fluorescence was measured. Fluorescence over time was plotted as the mean ± SEM (control, n = 4; LKB1−/−, n = 3). To address the relationship between 6‐CF and Qtracker 655 vascular label, stacks of images collected over time were analyzed in four dimensions with Imaris software v7.6.3 (Bitplane, Belfast, UK). Quantification of colocalization was assessed in three dimensions, and pixel codistribution was calculated for green and red fluorescent patterns. The degree of colocalization of 6‐CF (green) and Qtracker (red) was quantified and expressed as a Pearson colocalization coefficient (1, perfect correlation or perfect colocalization; 0, no correlation; ‐1, perfect inverse correlation). For measurements of paracellular transport, 20 squares measuring 10 × 10 μm were drawn using ImageJ in the canalicular area based on 6‐CF fluorescence. The regions were transferred to the 3‐kDa dextran channel and mean intensity was measured. Mean and standard deviation over time of normalized mean fluorescence were calculated and presented in the graph. Similar results were obtained from three independent experiments.
Results
LOSS OF LKB1 LEADS TO ABNORMAL BILE CANALICULAR MORPHOLOGY
Liver‐specific LKB1 knockout mice (LKB1−/−) exhibit a severe phenotype, including weight loss, dehydration, and jaundice, and die approximately 28 days after birth.17, 24 Therefore, mice at 21‐23 days after birth were used in this study. Despite their clinical condition, hematoxylin and eosin staining of LKB1−/− liver sections showed no sign of inflammation or necrosis compared with controls24 (Fig. 1A), suggesting that the severe phenotype results from depletion of LKB1 in hepatocytes and is not secondary to hepatocellular necrosis or inflammation.
Figure 1.
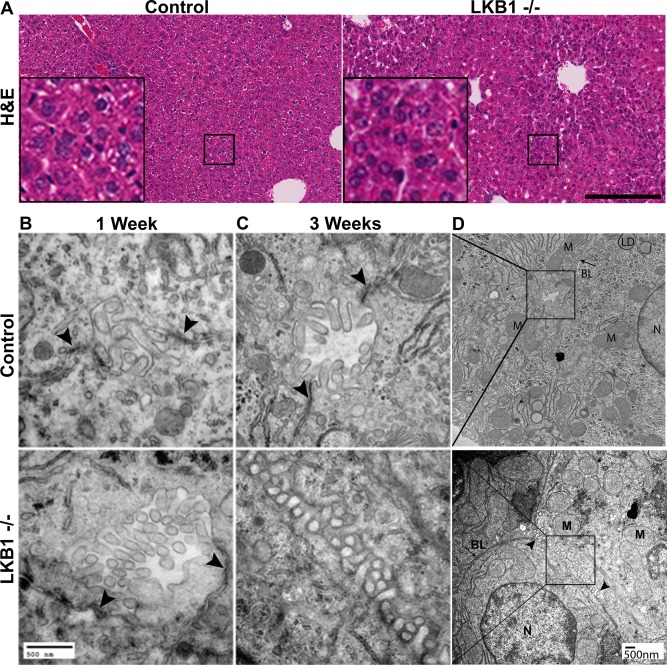
Hepatocytes and tissue architecture in control and LKB1−/− mice. (A) Livers from 3‐week‐old control and LKB1−/− mice were stained with hematoxylin and eosin. Scale bar: 200 μm. (B, C) TEM of liver sections from 1‐week‐old (B) and 3‐week‐old (C) control and LKB1−/− mice. Arrowheads indicate TJs. Bile canaliculi morphology was altered in the LKB1−/− mouse (C). Scale bar: 500 nm. (D) Lower magnification of the insets shown in panel C. Scale bar: 500 nm. Abbreviations: BL, basolateral; LD, lipid droplet; M, mitochondria; N, nucleus. Scale bar: 500 nm.
TEM studies were performed for qualitative evaluation of canalicular morphology. In 1‐week‐old control and LKB1−/− mice canalicular channels, well‐defined canaliculi, microvilli, and TJs were observed (Fig. 1B), which is consistent with previous reports that LKB1 protein depletion is initially detected 10‐14 days after birth.17, 24 Similarly, liver sections from 3‐week‐old control mice displayed normal morphology, including clearly identified TJs (Fig. 1C, upper panel). However, in LKB1−/− liver sections, TJs were reduced in number and were difficult to recognize (Fig. 1C, lower panel). The canalicular spaces extended into the lateral membrane domain of the hepatocytes (Fig. 1D). Bile canaliculi with normal appearance were occasionally observed in LKB1−/− mice due to regional heterogeneity.
LKB1 DEPLETION RESULTS IN DISRUPTED TJ STRUCTURE AND MISLOCALIZATION OF CINGULIN
To investigate the role of LKB1 in TJ maintenance in hepatocytes, we initially evaluated the distribution of ZO‐1, which links junctional barrier proteins, such as claudins, to the actin cytoskeleton2 (Fig. 2). Liver tissue sections from control and LKB1‐deficient mice were processed for immunofluorescence as described in the Materials and Methods section. ZO‐1 staining labeled the borders of the canaliculi in control mice with the typical linear morphology. However, in LKB1−/− mice, ZO‐1 staining was tortuous and disorganized. Quantification of canalicular morphology based on ZO‐1 labeling revealed that LKB1−/− mice had significantly longer and wider canaliculi compared with control mice (Fig. 2).
Figure 2.
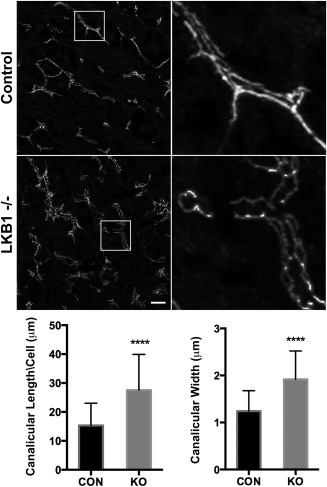
Loss of LKB1 leads to changes in canalicular morphology. Liver sections were stained with ZO‐1 antibody in control and LKB1−/− mice left top and bottom panels, respectively. Insets on the right depict a higher magnification of the boxed area. Scale bar: 10 μm. ZO‐1 labeling was used for indirect evaluation of canalicular width and length. For canalicular length (bottom left), a mean of 15.30 ± 1.17 (n = 43) was measured in control mice (black bar), whereas a mean of 27.53 ± 2.12 (n = 34) was measured in LKB1−/− mice (gray bar). ****P < 0.0001. For canalicular width (bottom right), a mean of 1.24 ± 0.08 (n = 29) was measured in control (black bar) while 1.91 ± 0.1 (n = 36) was measured in LKB1−/− (gray bar). ****P < 0.0001. Similar results were obtained from three independent experiments. Abbreviations: CON, control; KO, knockout.
To evaluate further TJ structure, we examined the distribution of filamentous actin using phalloidin labeling. As expected, the actin cytoskeleton was associated with TJ primarily in control liver sections and colocalized with ZO‐1 staining (Fig. 3A,B). In the absence of LKB1, actin colocalization with ZO‐1 was unchanged; however, similar to ZO‐1 staining, it revealed the disrupted morphology and distribution of the junctions (Fig. 3B). Next, we labeled the transmembrane TJ protein, claudin‐1. In control mouse sections, claudin‐1 had a linear pattern similar to (but not overlapping with) ZO‐1, which is consistent with the fact that the former is a transmembrane apical protein and the latter is cytosolic. In LKB1−/− mice, claudin‐1 showed disorganized distribution highlighting the significantly wider canaliculi and was also observed in internal compartments (Fig. 3C). Similar distribution was observed for claudin‐3 (Supporting Fig. 1A). The regulatory protein cingulin links TJ with the cytoskeleton, regulates signal transduction,26 and has been identified as an AMPK effector.20, 27 In control liver sections, cingulin localized in close proximity with ZO‐1 at TJ. However, in LKB1−/− sections, cingulin was infrequently associated with TJ and was predominantly cytosolic (Fig. 3D). Western blot analysis of phosphorylated AMPK levels revealed a significant reduction in phosphorylated AMPK levels in the absence of LKB1, which may explain the altered distribution of cingulin (Fig. 3E).
Figure 3.
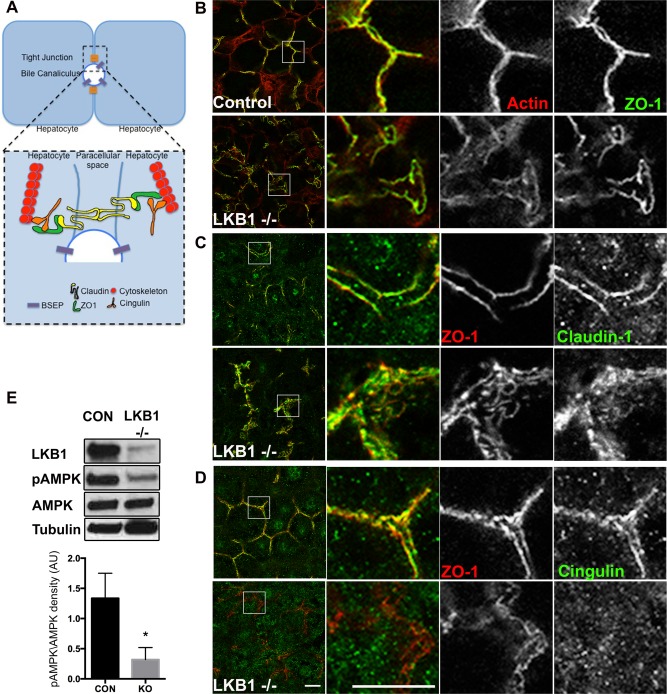
Loss of LKB1 leads to disruption of TJ components and reduced AMPK activation. (A) Simplified schematic representation of TJ organization in hepatocytes. TJs are at the interface of lateral and apical membranes and are composed of transmembrane proteins (claudin; yellow), scaffold proteins (ZO‐1; green), regulatory proteins (cingulin; orange) and cytoskeleton (actin or microtubules; red). (B‐D) Liver sections from control (upper panels) or LKB1−/− (lower panels) were stained with phalloidin, claudin‐1, or cingulin and ZO‐1 antibodies. Note the redistribution of cingulin to the cytosol in LKB1−/− mice compared with control mice. Scale bar: 10 μm. (E) Western blot analysis of LKB1, AMPK, and pAMPK. Total cell lysates of livers from control and LKB1−/− mice were immunoblotted to determine levels of LKB1, AMPK, and phospho‐Thr172 AMPK (pAMPK). Representative western blots are shown. Quantification of pAMPK/AMPK from three independent experiments was plotted as the mean ± SEM (control 1.33 ± 0.23; LKB1−/− 0.31 ± 0.11; P < 0.005).
Previous reports noted the delayed trafficking of the apical transporter Bsep in the absence of LKB1.17, 24 In control liver sections, Bsep localized to apical membranes, whereas ZO‐1 highlighted the adjacent TJ (Supporting Fig. 1B). In the absence of LKB1, Bsep was observed in internal compartments and only partially localized to apical membranes (Supporting Fig. 1B). Additionally, residual Bsep that was properly transported to the apical membrane highlighted the much‐widened apical domain (Supporting Fig. 1B). β‐Catenin, a major component of adherens junctions, had comparable distribution in control and LKB1−/− mice (Supporting Fig. 1C).
LKB1 regulates polarity in part through the regulation of membrane trafficking.17, 28 Therefore, we determined the distribution of Rab11a and Myosin Vb (recycling endosomes) and Par‐3 (polarity regulating protein). Under control conditions, Rab11a and Myosin Vb are localized in apical recycling endosomes adjacent to apical membranes, whereas Par3 localized to TJ with ZO‐1. Loss of LKB1 revealed only minor changes in their distribution (Supporting Fig. 1D‐1F).
Our findings show that the TJ proteins claudin‐1, claudin‐3, and actin remained associated with the abnormal junctions in the absence of LKB1, whereas cingulin lost association with TJ and became mostly cytosolic.
DELAYED CANALICULAR SECRETION IN LKB1‐DEFICIENT MICE
To test junctional barrier function, we adopted a previously described method to evaluate efflux transporter activity using 6‐CFDA and IVM.29, 30 6‐CFDA is a nonfluorescent, membrane‐permeant probe. Deacetylation of 6‐CFDA in the cytosol of hepatocytes yields a fluorescent, charged molecule, 6‐CF, that is visible and membrane‐impermeant. Consequently, 6‐CF is secreted into the bile canaliculi by ABCC2 and fluorescence can be used to quantitatively measure transporter activity.
For IVM, control and LKB1−/− mice were anesthetized and the left lobe of the liver was exposed as described in the Materials and Methods section. Prior to imaging, the vascular label Qtracker 655 was injected retro‐orbitally to evaluate blood flow and identify regions for imaging. For time‐lapse imaging, 20‐μm stacks were acquired every 2 minutes for up to an hour. After acquisition of the first stack, 0.2 mg/kg 6‐CFDA was retro‐orbitally injected. Figure 4A shows single frames from control or LKB1−/− representative time lapses. For quantification of 6‐CF fluorescence over time, sum projections were generated using ImageJ, as described in the Materials and Methods section. Mean fluorescence of 6‐CF from entire frames over time was plotted as the mean ± SEM (control, n = 4; LKB1−/−, n = 3 [Fig. 3B]). In control mice, 6‐CF fluorescence increased sharply in the first 2 minutes and peaked at 4‐6 minutes (Fig. 4B). Fluorescence originated from both hepatocytes cytosol and bile canaliculi (Fig. 4A). Subsequently, a gradual decrease in fluorescence was observed representing transport of 6‐CF from the cytosol into the canaliculus followed by excretion and loss of 6‐CF signal by 50 minutes after injection (data not shown). In LKB1−/− mice, 6‐CF largely remained in hepatocytes (compare 6‐CF fluorescence at 34 minutes after injection in control and LKB1−/− mice); its secretion was greatly delayed likely due to mislocalization of the transporter. The lower fluorescence intensity observed in LKB1−/− mice could be due to changes in pH. Surprisingly, although 6‐CF excretion was delayed, green fluorescence appeared in the blood and accumulated over time (Fig. 4A, inset). This was confirmed in colocalization analysis of 6‐CF with respect to the vasculature marker (Qtracker 655). Following the analysis, Pearson's coefficient was calculated (1, perfect correlation or perfect colocalization; 0, no correlation; ‐1, perfect inverse correlation). Figure 4C presents the Pearson's coefficient values from one control mouse and one LKB1−/− mouse over the course of the respective experiments. In the control, Pearson's coefficient values ranged from −0.0858 to −0.0039, indicating no correlation between the two channels. In the LKB1−/− mouse, the range was 0.2243‐0.4097. Notably, LKB1−/− mice had relatively high basal Pearson's coefficient values due to elevated serum bilirubin levels, resulting in background fluorescence.17, 24 However, Pearson's coefficient increased over time, demonstrating increased colocalization of 6‐CF labeling with Qtracker. This overall trend was observed in four control mice and three LKB1−/− mice, as shown in Fig. 4D.
Figure 4.
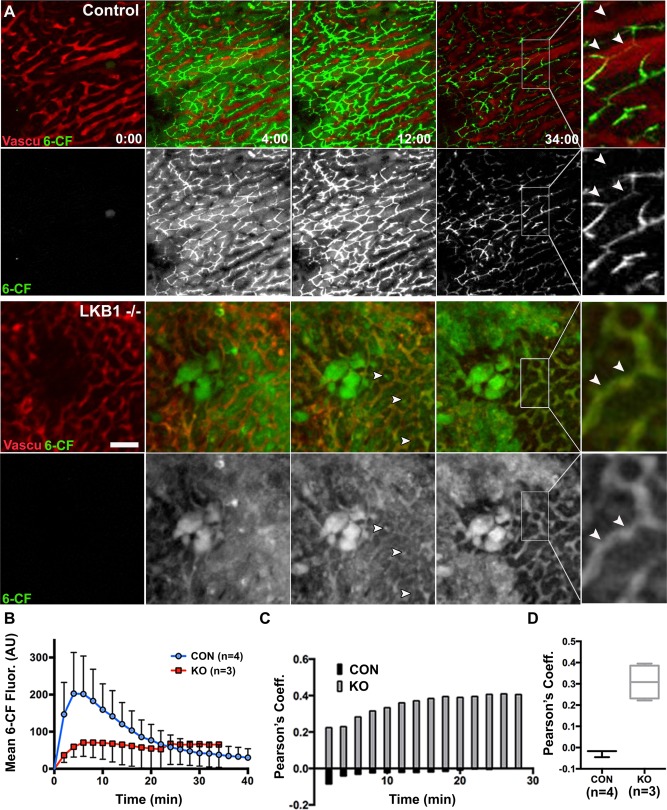
IVM of biliary excretion in the intact liver. (A) Mice were anesthetized and positioned on the microscope stage for IVM of the liver as described in the Materials and Methods section. Single frames from time‐lapse imaging of control (upper panels) or LKB1−/− (lower panels) are presented. Blood flow and sinusoid space were visualized with Qtracker 655 probe (red) that was injected systemically before imaging. To measure biliary excretion, 6‐CFDA was injected after acquisition of the first z‐stack. Note that there were fewer canalicular structures in LKB1−/− mice (arrowheads) and 6‐CF signal from hepatocytes was observed throughout, suggesting delayed excretion compared with control mice. Inset: Qtracker and 6‐CF label the vasculature and canaliculi, respectively, in control mice; in LKB1−/− mice, they overlapped in blood vessels (white arrowheads). Scale bar: 50 μm. (B) Measurement of 6‐CF fluorescence in control mice (blue; n = 4) and LKB1−/− mice (red; n = 3) are plotted as the mean + SEM. (C) The degree of colocalization of Qtracker (red) and 6‐CF (green) was quantified and expressed as a Pearson coefficient (1, perfect correlation; 0, no correlation; and −1, inverse correlation). Representative Pearson's coefficient values are shown for control mice (black bars) and LKB1−/− mice (gray bars) over time (in minutes). (D) Pearson's coefficient values at t = 34 minutes from control mice (black; n = 4) or LKB1−/− mice (gray; n = 3). Abbreviations: CON, control; KO, knockout.
Theoretically, 6‐CF may be transported from hepatocytes directly back to the blood or diffuse from the canalicular space through permeable TJ. We showed previously that expression of the basolateral ABCC3 and ABCC4, which can transport organic anions from liver into the circulation, was not induced.17 Therefore, we postulated that 6‐CF might be returned to the circulation through permeable TJs by way of the paracellular pathway.
LKB1 REGULATES PARACELLULAR PERMEABILITY IN THE LIVER
To test the possibility that permeability of TJs increase in the absence of LKB1, we modified the previously described IVM approach by adding a low molecular weight Texas red‐dextran [3 kDa; Stokes radius 2.6 nm31]. If TJs are permeable and solutes can diffuse from the bile canaliculi into the blood, then the 3 kDa dextran injected systemically would have access into the canalicular space.
To explore this possibility, control and LKB1−/− mice livers were imaged in vivo. Blood flow was visualized using Qtracker 655 followed by injection of 6‐CFDA to identify canaliculi and 3 kDa dextran to evaluate TJ permeability.32 In control mice, 3 kDa dextran remained in the vasculature throughout the experiment and was never observed to coincide with 6‐CF in the canalicular space (Fig. 5 and Supporting Movie S3). However, in LKB1 −/− liver, 3kDa dextran rapidly (2 minutes after injection) appeared in the canalicular space and accumulated over time (Fig. 5 and Supporting Movie S4). Quantification of 3 kDa dextran fluorescence in the canalicular area was measured based on 6‐CF accumulation, and intensity over time revealed that whereas 3 kDa dextran does not accumulate in the canalicular space in control mice, it increases rapidly in LKB1−/− mice. These results suggest that LKB1 may be involved in the regulation of paracellular permeability.
Figure 5.
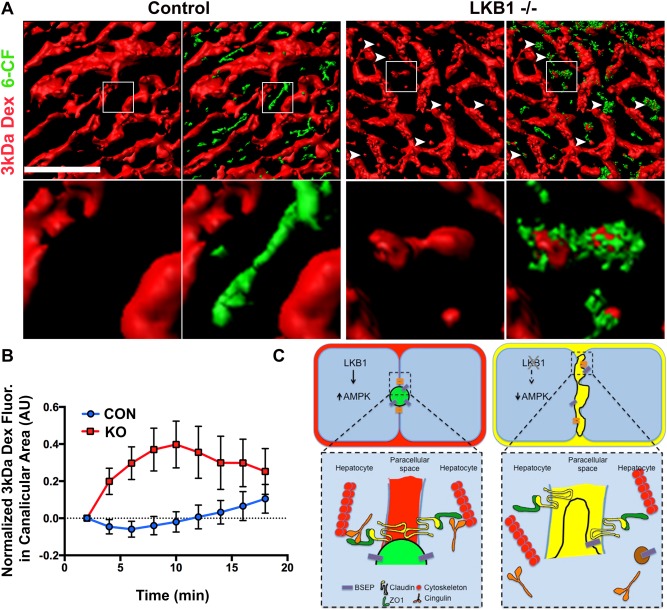
IVM of TJs permeability and the paracellular pathway. (A) Mice were anesthetized and positioned on the microscope stage for IVM of the liver as described in the Materials and Methods section. Blood flow and sinusoid space were visualized with a Qtracker 655 probe (not shown) that was injected systemically prior to imaging. 3 kDa dextran and 6‐CFDA were injected after acquisition of the first z‐stack. Volume rendering of the different probes was generated; a single frame of 3 kDa dextran (red) or an overlay of 3 kDa and 6‐CF (green) are shown. The enlarged inset in the lower panel corresponds to the boxed area in the upper panel. Note that in LKB1−/−, but not control, 3 kDa dextran is present in the canaliculi area also labeled by 6‐CF (white arrowheads). Scale bar: 50 μm. (B) Measurement of 3 kDa dextran in the bile canaliculi area was plotted as the mean ± SD of 20 regions for representative control mice (blue) or LKB1−/− mice (red). Similar results were obtained in three independent experiments. (C) Proposed model summarizing the experimental observations. LKB1‐deficient mice had reduced levels of phosphorylated AMPK levels, which may be the cause of the observed abnormal canaliculi morphology and distribution of TJ proteins. Notably, the apical pole extended into the lateral domain of hepatocytes and the junctional regulatory protein, cingulin (orange), localized primarily in the cytosol. Mislocalization of additional TJ proteins, and membrane trafficking regulators may explain the delayed biliary excretion. Mixing of biliary (green in control mice) and blood (red in control mice) content (represented as yellow) was observed in LKB1‐deficient mice.
Discussion
AMPK is a key substrate of LKB1. Following phosphorylation of threonine 172, active AMPK regulates multiple metabolic effects, including polarity and mitochondrial dynamics.33 AMPK activates a large number of downstream targets, including cingulin,20, 27 Par‐1,34 and CLIP170, a plus‐end microtubular protein.35 In addition, AMPK indirectly regulates ATP synthesis by mitochondria by increasing cellular glucose uptake and glycolysis10 and up‐regulating mitochondrial biogenesis36 and directly by regulating mitochondrial fusion to enhance ATP synthesis by oxidative phosphorylation.37, 38, 39 Notably, AMPK phosphorylation was reduced significantly in LKB1−/− mouse liver (Fig. 3E). Therefore, although several mechanisms may be responsible for the multiple cellular effects observed after LKB1 depletion, it is likely that AMPK is responsible for the observed effects on TJ structure and function.
TJ structure and regulation are complex, as reviewed elsewhere (2) and schematized in Fig. 3A. Notably, TJ create the functional barrier, which restricts paracellular movement of water and solutes between blood and bile.2, 4, 40 Transmembrane proteins seal TJ by interacting with partners on adjacent cells and also bind directly to the scaffolding protein ZO‐1 at the TJ. In turn, ZO‐1 binds directly to peri‐junctional F‐actin and other components of the cytoskeleton and is proposed to coordinate barrier function with cytoskeletal changes.41 Dynamic coupling between the barrier and the cytoskeleton is presumably required since the TJ must maintain an Ångström‐level paracellular seal in the face of continuous intercellular movements, apoptosis, and epithelial restitution.42 Although the physiological cues that lead to LKB1 activation are not known, it is possible that signaling pathways downstream of LKB1 regulate paracellular flux via changes in actin organization, actomyosin contractility, or occluding.43, 44
Extensive TEM study of liver specimens revealed a substantial paucity of bile canaliculi and TJ in LKB1‐deficient mice. Remarkably, this was associated with extension of bile canaliculi with abundant microvilli into the normal lateral plasma membrane domain at one or both borders of the canalicular membrane. While major disruptions in canalicular morphology were observed, the TJ components investigated show only modestly diminished fidelity in their subcellular localization with the exception of cingulin. Cingulin, which is an AMPK target27 and associates with microtubules in response to AMPK phosphorylation,20 loses TJ localization and is widely distributed throughout the cytoplasm (Fig. 3D). In contrast, ZO‐1, claudin‐1, claudin‐3, and actin remain mostly associated with the residual canalicular structures and show only minor redistribution to other areas of the cell (Fig. 2, Fig. 3B,C, and Supporting Fig. 1A). Detailed mechanistic study of the role of LKB1 in TJ function will require substantial study beyond the scope of the present report.
The phenotype observed in LKB1−/− mice is primarily functional and is not associated with hepatocellular necrosis or inflammation24 (Fig. 1A). The major effects are metabolic, probably involving the multiple roles of AMPK,36 and are manifested by weight loss and death. Jaundice also largely results from canalicular and junctional defects.45 Various experimental and clinical cholestatic disorders are associated with increased expression of ABCC3 and C4, which localize to the sinusoidal plasma membrane domain of hepatocytes and facilitate transport of retained conjugated bilirubin from liver back into the circulation. In LKB1−/− mouse liver, expression of these normally quiescent transporters was not increased.17 An alternative or additional mechanism by which conjugated bilirubin can be transferred from bile into the circulation is through permeable TJs by way of the paracellular pathway. Interestingly, Schossleitner et al.46 demonstrated recently that cingulin regulates endothelial barrier function and that loss of cingulin from TJ results in increased junctional permeability.
Demonstration and quantification of the paracellular pathway in liver is rendered difficult due to instability of hepatocyte cultures. In perfused liver, the paracellular pathway is examined by calculating plasma to bile ratio of fluid phase probes varying in size.7 To date, no direct visualization and measurement of hepatic TJ permeability have been demonstrated in vivo. The present study demonstrates direct dynamic transfer of 3 kDa dextran from the blood to the canalicular space in LKB1−/− liver but not in control mouse liver (Fig. 5). These observations reveal paracellular permeability and are consistent with appearance of 6‐CF in sinusoidal blood following its uptake and secretion by hepatocytes in LKB1−/− mouse liver (Fig. 4A [inset] and Fig. 4C). The Stokes radius of 3 kDa dextran is 2.6 nm,31 indicating that the TJ opening in LKB1−/− mouse liver exceeds this dimension. It may be possible to quantify TJ permeability change using dextrans of different sizes.
IVM of the liver was initially described in the early 1950s by Hanzon47 but was subsequently neglected. Technical advances in microscopy, deep tissue imaging, fluorescent probes, transgenic animal models, and image analysis have made in situ studies of subcellular, cellular, and tissue processes within a living organism possible.30, 48 Importantly, concepts derived from cell cultures and other species can be evaluated at the organ level under varied developmental, experimental, and genetic conditions. In the present study, we used transgenic LKB1−/− mice; however, virtually any transgenic mouse can be studied similarly under varied experimental conditions. Finally, IVM offers an opportunity to bridge the knowledge gap between the cell, organ, and organism to better understand physiological and pathological processes.
The present study reveals that LKB1 is essential for regulating TJ and paracellular permeability. These findings are relevant to the pathogenesis of cholestasis induced by drugs, chemicals, viruses, and metabolic states, which inhibit the LKB1 pathway.
Author names in bold designate shared co‐first authorship.
Supporting information
Additional Supporting Information may be found at onlinelibrary.wiley.com/doi/10.1002/hep.28724/suppinfo.
Supporting Information
Supporting Information Movie 1
Supporting Information Movie 2
Supporting Information Movie 3
Supporting Information Movie 4
Acknowledgment
We thank Julie Donaldson for critical reading of the manuscript and Daniela Malide for help with colocalization analysis. The liver IVM protocol was developed and optimized with the help of Kirstin Meyer, Max Planck Institute, Dresden.
Potential conflict of interest: Nothing to report.
This research was supported by the Intramural Research Program of the National Institutes of Health, National Institute of Dental and Craniofacial Research.
REFERENCES
- 1. Treyer A, Musch A. Hepatocyte Polarity. Comprehensive Physiology 2013;3:243–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Van Itallie CM, Anderson JM. Architecture of tight junctions and principles of molecular composition. Semin Cell Dev Biol 2014;36:157–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Furuse M. Molecular basis of the core structure of tight junctions. Cold Spring Harb Perspect Biol 2010;2:a002907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Turner JR, Buschmann MM, Romero‐Calvo I, Sailer A, Shen L. The role of molecular remodeling in differential regulation of tight junction permeability. Semin Cell Dev Biol 2014;36:204–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Lowe PJ, Miyai K, Steinbach JH, Hardison WG. Hormonal regulation of hepatocyte tight junctional permeability. Am J Physiol 1988;255:G454–G461. [DOI] [PubMed] [Google Scholar]
- 6. Kuijpers GA, Van Nooy IG, Vossen ME, Stadhouders AM, Van Uyen A, De Pont JJ, et al. Tight junctional permeability of the resting and carbachol stimulated exocrine rabbit pancreas. Histochemistry 1985;83:257–264. [DOI] [PubMed] [Google Scholar]
- 7. Lake JR, Licko V, Van Dyke RW, Scharschmidt BF. Biliary secretion of fluid‐phase markers by the isolated perfused rat liver. Role of transcellular vesicular transport. J Clin Invest 1985;76:676–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Jansen M, ten Klooster JP, Offerhaus GJ, Clevers H. LKB1 and AMPK family signaling: the intimate link between cell polarity and energy metabolism. Physiol Rev 2009;89:777–798. [DOI] [PubMed] [Google Scholar]
- 9. Baas AF, Smit L, Clevers H. LKB1 tumor suppressor protein: PARtaker in cell polarity. Trends Cell Biol 2004;14:312–319. [DOI] [PubMed] [Google Scholar]
- 10. Shaw RJ, Lamia KA, Vasquez D, Koo SH, Bardeesy N, Depinho RA, et al. The kinase LKB1 mediates glucose homeostasis in liver and therapeutic effects of metformin. Science 2005;310:1642–1646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Baas AF, Kuipers J, van der Wel NN, Batlle E, Koerten HK, Peters PJ, et al. Complete polarization of single intestinal epithelial cells upon activation of LKB1 by STRAD. Cell 2004;116:457–466. [DOI] [PubMed] [Google Scholar]
- 12. Hezel AF, Gurumurthy S, Granot Z, Swisa A, Chu GC, Bailey G, et al. Pancreatic LKB1 deletion leads to acinar polarity defects and cystic neoplasms. Mol Cell Biol 2008;28:2414–2425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Shelly M, Cancedda L, Heilshorn S, Sumbre G, Poo MM. LKB1/STRAD promotes axon initiation during neuronal polarization. Cell 2007;129:565–577. [DOI] [PubMed] [Google Scholar]
- 14. Xu X, Jin D, Durgan J, Hall A. LKB1 controls human bronchial epithelial morphogenesis through p114RhoGEF‐dependent RhoA activation. Mol Cell Biol 2013;33:2671–2682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Fu D, Wakabayashi Y, Ido Y, Lippincott‐Schwartz J, Arias IM. Regulation of bile canalicular network formation and maintenance by AMP‐activated protein kinase and LKB1. J Cell Sci 2010;123:3294–3302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Fu D, Wakabayashi Y, Lippincott‐Schwartz J, Arias IM. Bile acid stimulates hepatocyte polarization through a cAMP‐Epac‐MEK‐LKB1‐AMPK pathway. Proc Natl Acad Sci U S A 2011;108:1403–1408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Homolya L, Fu D, Sengupta P, Jarnik M, Gillet JP, Vitale‐Cross L, et al. LKB1/AMPK and PKA control ABCB11 Trafficking and polarization in hepatocytes. PLoS One 2014;9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Zheng B, Cantley LC. Regulation of epithelial tight junction assembly and disassembly by AMP‐activated protein kinase. Proc Natl Acad Sci U S A 2007;104:819–822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Zhang L, Li J, Young LH, Caplan MJ. AMP‐activated protein kinase regulates the assembly of epithelial tight junctions. Proc Natl Acad Sci U S A 2006;103:17272–17277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Yano T, Matsui T, Tamura A, Uji M, Tsukita S. The association of microtubules with tight junctions is promoted by cingulin phosphorylation by AMPK. J Cell Biol 2013;203:605–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Anderson JM, Stevenson BR, Jesaitis LA, Goodenough DA, Mooseker MS. Characterization of ZO‐1, a protein component of the tight junction from mouse liver and Madin‐Darby canine kidney cells. J Cell Biol 1988;106:1141–1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Citi S, Sabanay H, Jakes R, Geiger B, Kendrick‐Jones J. Cingulin, a new peripheral component of tight junctions. Nature 1988;333:272–276. [DOI] [PubMed] [Google Scholar]
- 23. Nedvetsky PI, Stefan E, Frische S, Santamaria K, Wiesner B, Valenti G, et al. A role of myosin Vb and Rab11‐FIP2 in the aquaporin‐2 shuttle. Traffic 2007;8:110–123. [DOI] [PubMed] [Google Scholar]
- 24. Woods A, Heslegrave AJ, Muckett PJ, Levene AP, Clements M, Mobberley M, et al. LKB1 is required for hepatic bile acid transport and canalicular membrane integrity in mice. Biochem J 2011;434:49–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Masedunskas A, Sramkova M, Parente L, Sales KU, Amornphimoltham P, Bugge TH, et al. Role for the actomyosin complex in regulated exocytosis revealed by intravital microscopy. Proc Natl Acad Sci U S A 2011;108:13552–13557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Aijaz S, D'Atri F, Citi S, Balda MS, Matter K. Binding of GEF‐H1 to the tight junction‐associated adaptor cingulin results in inhibition of Rho signaling and G1/S phase transition. Dev Cell 2005;8:777–786. [DOI] [PubMed] [Google Scholar]
- 27. Ducommun S, Deak M, Sumpton D, Ford RJ, Nunez Galindo A, Kussmann M, et al. Motif affinity and mass spectrometry proteomic approach for the discovery of cellular AMPK targets: identification of mitochondrial fission factor as a new AMPK substrate. Cell Signal 2015;27:978–988. [DOI] [PubMed] [Google Scholar]
- 28. Gissen P, Arias IM. Structural and functional hepatocyte polarity and liver disease. J Hepatol 2015;63:1023–1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ryan JC, Ghabril M, Decker B, Dunn K. Quantitative intravital microscopy of liver transport. Mol Biol Cell 2012;23. [Google Scholar]
- 30. Weigert R, Porat‐Shliom N, Amornphimoltham P. Imaging cell biology in live animals: ready for prime time. J Cell Biol 2013;201:969–979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Larina O, Bhat P, Pickett JA, Launikonis BS, Shah A, Kruger WA, Edwardson JM, et al. Dynamic regulation of the large exocytotic fusion pore in pancreatic acinar cells. Mol Biol Cell 2007;18:3502–3511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Xiang RL, Mei M, Cong X, Li J, Zhang Y, Ding C, et al. Claudin‐4 is required for AMPK‐modulated paracellular permeability in submandibular gland cells. J Mol Cell Biol 2014;6:486–497. [DOI] [PubMed] [Google Scholar]
- 33. Toyama EQ, Herzig S, Courchet J, Lewis TL Jr, Loson OC, Hellberg K, et al. Metabolism. AMP‐activated protein kinase mediates mitochondrial fission in response to energy stress. Science 2016;351:275–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Lizcano JM, Goransson O, Toth R, Deak M, Morrice NA, Boudeau J, et al. LKB1 is a master kinase that activates 13 kinases of the AMPK subfamily, including MARK/PAR‐1. EMBO J 2004;23:833–843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Nakano A, Kato H, Watanabe T, Min KD, Yamazaki S, Asano Y, et al. AMPK controls the speed of microtubule polymerization and directional cell migration through CLIP‐170 phosphorylation. Nat Cell Biol 2010;12:583–590. [DOI] [PubMed] [Google Scholar]
- 36. Hardie DG. AMP‐activated protein kinase‐an energy sensor that regulates all aspects of cell function. Genes Dev 2011;25:1895–1908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Nunnari J, Suomalainen A. Mitochondria: in sickness and in health. Cell 2012;148:1145–1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Mihaylova MM, Shaw RJ. The AMPK signalling pathway coordinates cell growth, autophagy and metabolism. Nat Cell Biol 2011;13:1016–1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Fu D, Mitra K, Sengupta P, Jarnik M, Lippincott‐Schwartz J, Arias IM. Coordinated elevation of mitochondrial oxidative phosphorylation and autophagy help drive hepatocyte polarization. Proc Natl Acad Sci U S A 2013;110:7288–7293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Derangeon M, Spray DC, Bourmeyster N, Sarrouilhe D, Herve JC. Reciprocal influence of connexins and apical junction proteins on their expressions and functions. Biochim Biophys Acta 2009;1788:768–778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Fanning AS, Anderson JM. Zonula occludens‐1 and‐2 are cytosolic scaffolds that regulate the assembly of cellular junctions. Ann N Y Acad Sci 2009;1165:113–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Guillot C, Lecuit T. Mechanics of epithelial tissue homeostasis and morphogenesis. Science 2013;340:1185–1189. [DOI] [PubMed] [Google Scholar]
- 43. Buschmann MM, Shen L, Rajapakse H, Raleigh DR, Wang Y, Wang Y, et al. Occludin OCEL‐domain interactions are required for maintenance and regulation of the tight junction barrier to macromolecular flux. Mol Biol Cell 2013;24:3056–3068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Clayburgh DR, Barrett TA, Tang Y, Meddings JB, Van Eldik LJ, Watterson DM, et al. Epithelial myosin light chain kinase‐dependent barrier dysfunction mediates T cell activation‐induced diarrhea in vivo. J Clin Invest 2005;115:2702–2715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Anderson JM. Leaky junctions and cholestasis: a tight correlation. Gastroenterology 1996;110:1662–1665. [DOI] [PubMed] [Google Scholar]
- 46. Schossleitner K, Rauscher S, Groger M, Friedl HP, Finsterwalder R, Habertheuer A, et al. Evidence that cingulin regulates endothelial barrier function in vitro and in vivo. Arterioscler Thromb Vasc Biol 2016;36:647–654. [DOI] [PubMed] [Google Scholar]
- 47. Hanzon V. Liver cell secretion under normal and pathologic conditions studied by fluorescence microscopy on living rats. Acta Physiol Scand Suppl 1952;28:1–268. [PubMed] [Google Scholar]
- 48. Porat‐Shliom N, Chen Y, Tora M, Shitara A, Masedunskas A, Weigert R. In vivo tissue‐wide synchronization of mitochondrial metabolic oscillations. Cell Rep 2014;9:514–521. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional Supporting Information may be found at onlinelibrary.wiley.com/doi/10.1002/hep.28724/suppinfo.
Supporting Information
Supporting Information Movie 1
Supporting Information Movie 2
Supporting Information Movie 3
Supporting Information Movie 4