

Abstract
Development of novel treatments for lymphedema has been limited by the fact that the pathophysiology of this disease is poorly understood. It remains unknown, for example, why limb swelling resulting from surgical injury resolves initially, but recurs in some cases months or years later. Finding answers for these basic questions has been hampered by the lack of adequate animal models. In the current study, we used Cre-lox mice that expressed the human diphtheria toxin receptor (DTR) driven by a lymphatic-specific promoter in order to noninvasively ablate the lymphatic system of the hind limb. Animals treated in this manner developed lymphedema that was indistinguishable from clinical lymphedema temporally, radiographically, and histologically. Using this model and clinical biopsy specimens, we show that the initial resolution of edema after injury is dependent on the formation of collateral capillary lymphatics and that this process is regulated by M2-polarized macrophages. In addition, we show that despite these initial improvements in lymphatic function, persistent accumulation of CD4+ cells inhibits lymphangiogenesis and promotes sclerosis of collecting lymphatics, resulting in late onset of edema and fibrosis. Our findings therefore provide strong evidence that inflammatory changes after lymphatic injury play a key role in the pathophysiology of lymphedema.
A transgenic mouse model recapitulates progressive lymphedema and provides insights into the cellular mechanisms governing recovery and progression.
Introduction
Improved survival for some malignancies has led to the concept of cancer as a chronic disease (1). As a result, survivorship has become a major focus of research aiming to minimize toxicity of treatments and decrease the incidence of iatrogenic complications (2). Secondary lymphedema is the most common long-term complication of surgery for solid tumors and occurs when lymphatic channels or lymph nodes are removed during cancer staging/treatment (3). Patients with lymphedema develop chronic swelling and fibrosis of the affected extremity, resulting in disfigurement, infections, and decreased quality of life (4). Lymphedema can also develop without antecedent injury due to congenital abnormalities of the lymphatic system or as a result of parasitic infections, which are most common in tropical and subtropical regions. It is estimated that nearly 5 million Americans have lymphedema, while worldwide, the disease is thought to affect as many as 200 million individuals (5, 6).
The development of effective treatment options for secondary lymphedema has been hampered by the fact that the mechanisms regulating the pathology of this disease remain poorly understood. For example, although it is clear that lymphatic injury is necessary for the development of lymphedema, it is unknown why only a subset of patients who undergo lymphadenectomy develop lymphedema (7). In addition, it is unclear why patients who do develop lymphedema do so in a delayed manner — most commonly months, or in some cases years, after surgery. In fact, in most cases temporary postsurgical edema resolves over a period of a few weeks only to recur and progressively worsen 6–12 months after surgery (3). Thus, understanding the mechanisms that regulate initial resolution of edema and delayed onset of progressive lymphatic dysfunction is a necessary step for development of novel treatment options for a disease in which little to no progress has been made in nearly 50 years.
Over the last century, a considerable effort has been expended to develop animal models of lymphedema. Although a variety of models that display some characteristics of clinical lymphedema have been developed, these models have significant limitations, including high morbidity and spontaneous resolution of swelling (8–10). The necessity for extensive surgical manipulation (i.e., full-thickness excision of large portions of skin) in most models has made it difficult to separate wound healing from lymphatic regeneration and does not accurately model human pathology, in which only lymph nodes are removed. In addition, in most models the maximal swelling that occurs is noted shortly after surgical injury and resolves spontaneously over time. In contrast, secondary lymphedema clinically develops months to years after the initial surgical injury and is a progressive disease once initiated. Thus, development of accurate models of lymphedema is a key step in understanding the pathology of this disease.
In the current study, we used inducible Cre-lox technology to create mice that express the human diphtheria toxin receptor (DTR) on their lymphatic endothelial cells (LECs) by coupling DTR with a lymphatic specific promoter (FLT4; Figure 1A). DTR activation enabled us to selectively ablate both capillary and collecting lymphatic vessels using diphtheria toxin (DT). Similar to clinical findings (3), local ablation of lymphatic vessels in the hind limb resulted in temporary edema that spontaneously improved over a period of 6–9 weeks, only to recur in a progressive manner over the course of the following year. The initial decrease in edema occurred in concert with dermal lymphatic hyperplasia and required infiltration of M2-polarized macrophages. Late development of lymphedema required infiltration of CD4+ T cells, which inhibited lymphangiogenesis, promoted collecting vessel fibrosis and lymphatic smooth muscle cell proliferation, and resulted in progressive edema and extracellular matrix (ECM) fibrosis.
Figure 1. FLT4-Cre-DTR enables selective ablation of capillary and collecting lymphatics.

(A) Schematic diagram of the inducible Cre-recombinase system. Downstream of FLT4, there is an internal ribosome entry site (IRES) gene, followed by Cre recombinase and estrogen receptor type 2 (ERT2), which induces genetic recombination to place the floxed DTR gene downstream of FLT4 in response to tamoxifen administration. (B) Immunofluorescence staining of the ear skin shows colocalization of DTR with initial lymphatics (LYVE-1+) and collecting lymphatics (CD31+ with patchy α-SMA+ coverage; scale bar: 50 μm). (C) Representative flow cytometry dot plots for LECs (CD45–PODO+CD31+) and BECs (CD45–PODO–CD31+) in inguinal (i.e., subcutaneous) lymph nodes harvested 24 hours after i.p. DT administration. PODO, podoplanin. (D) Quantification (%) of LECs and BECs from flow cytometry of inguinal lymph nodes 24 hours after i.p. DT administration (n = 4–5 animals/group; LECs, *P < 0.05 and BECs, P = NS). (E) Representative whole mount immunofluorescence staining of ear skin localizing podoplanin and α-SMA in control and DT-treated mice. Note virtually complete ablation of capillary and collecting lymphatics. (F) Top row: Gross images of popliteal lymph nodes harvested 1 week after subcutaneous DT injection in the hind limb, with resultant lymph node hemorrhage. Bottom row: Representative photomicrographs of LYVE-1 immunofluorescence localization in popliteal lymph nodes 1 week after hind limb DT injection, showing destruction of the subcapsular and medullary lymphatics (scale bar: 400 μm). (G) Top row: Representative lymphoscintigraphy heat maps and peak nodal uptake of technetium-99m (99mTc) in the popliteal lymph nodes (yellow dotted circles) of control versus DT after unilateral hind limb injection (injection site is marked by arrows). (H) Quantification of the peak nodal uptake of 99mTc in the popliteal lymph node after DT administration for control and 1, 9, and 52 weeks (n = 4/group; *P < 0.05). Note the decreased peak nodal uptake following DT administration. 2-tailed Student’s t test, ANOVA with post hoc comparison tests.
Results
DT ablates capillary and collecting lymphatics.
To confirm expression of DTR on LECs, we induced DTR expression with tamoxifen and analyzed the expression of DTR using immunofluorescence. This analysis demonstrated that LECs in initial (LYVE+) and collecting lymphatics (CD31+ with patchy α-SMA+ coverage) expressed DTR (Figure 1B). We found similar findings when we colocalized DTR with podoplanin (data not shown). In contrast, blood endothelial cells (BECs, MECA-32+) did not express DTR, confirming specific expression of the receptor (data not shown).
Intraperitoneal administration of DT resulted in an 82% depletion of LECs in subcutaneous lymph nodes 24 hours after treatment but no significant depletion of BECs (Figure 1, C and D). However, this treatment had marked systemic toxicity, leading to hemorrhagic ascites and death within 72 hours. Histological analysis of different tissues revealed that lymphatic ablation after systemic injection was variable, with minimal depletion in the trachea and diaphragm but high-level ablation in the skin, lymph nodes, and intestine (Supplemental Figure 1; supplemental material available online with this article; doi:10.1172/jci.insight.84095DS1).
Subcutaneous injection of DT ablates lymphatic vessels without systemic toxicity.
We next examined the effects of subcutaneous DT administration in the ear and hind limb (5 ng/d for 3 days). Using whole mount staining with podoplanin/α-SMA in ear and hind limb tissues harvested 1 week after DT injection, we found that while the blood vasculature was preserved, virtually the entire lymphatic vascular network was disrupted and fragmented dermal lymphatics were filled with CD45+ cells (Figure 1E, Supplemental Figure 2, A and B, and data not shown). DT injection in the hind limb, similar to i.p. injection, disrupted the lymphatics in the draining lymph nodes, caused profound edema of the foot, and markedly impaired uptake of technetium-99m (99mTc) radioactive tracer by the draining lymph nodes for as long as 52 weeks (Figure 1, F–H).
Injection of DT leads to the development of chronic lymphedema that models clinical lymphedema temporally, histologically, and radiographically.
Similar to the clinical scenario following lymphatic injury, hind limb lymphatic ablation in DTR mice resulted in marked swelling 1 week after injection that partially resolved over the ensuing 6–9 weeks (Figure 2, A and B). Similarly, consistent with development of clinical lymphedema, examination of animals 9, 18, and 52 weeks after injection demonstrated progressive swelling of the foot, peaking at a more than 40% increase at the 52-week time point (Figure 2, A and B). In addition, consistent with clinical histological findings, the hind limbs of DT-injected animals displayed progressive dermal fibrosis, hyperkeratosis, and subcutaneous fibroadipose tissue deposition (Figure 2, C and D, and Supplemental Figure 2, C–F). Consistent with previously published reports, analysis of lymphatic drainage using near-infrared (NIR) lymphoscintigraphy with indocyanine green (ICG) in patients with stage III lymphedema demonstrated a near complete absence of collecting lymphatics and dermal backflow (accumulation of dye in the skin) resulting from leaky initial lymphatics (Figure 2E) (11, 12). Similarly, analysis of DT-treated mice 52 weeks following lymphatic ablation demonstrated a similar radiographic appearance with loss of collecting lymphatics and dermal backflow at the site of dye injection (Figure 2E).
Figure 2. Subcutaneous injection of DT leads to development of chronic lymphedema that models clinical lymphedema temporally, histologically, and radiographically.

(A) Representative photographs of mouse hind limbs at 1, 9, and 52 weeks following DT injection as compared with normal (i.e. nontreated) mice. (B) Quantification of increasing foot diameter from baseline after DT administration over the course of the study (n = 5–6/group). (C) Quantification of fibroadipose area for histological sections from 1, 3, 6, 9, 18, and 52 weeks (*P < 0.001). (D) Quantification of scar index (collagen I:collagen III) from foot histological sections for each time point (*P < 0.001). (E) Left panels: NIR lymphangiography in human hands showing normal uptake (left) and dermal backflow with loss of lymphatics in the setting of lymphedema (right). Right panels: NIR lymphangiography of normal (left) and lymphedematous mouse hind limbs (52 weeks after lymphatic ablation; right). Note the similarity of the pattern to clinical disease. (F) Top row: Representative photomicrograph showing immunofluorescence staining of collecting lymphatic vessels (podoplanin+, green; α-SMA+, red) in normal human skin and lymphedematous skin (scale bar: 40 μm). Note the sclerosed collecting vessel in the lymphedematous tissue. Bottom row: Representative photomicrograph showing immunofluorescent staining of collecting lymphatic vessels (podoplanin+; green and α-SMA+; red) in normal mouse hind limb skin and DT treated skin (scale bar: 40 μm). Note decreased luminal area and increased wall thickness in the lymphedematous tissue. (G) Quantification of collecting lymphatic vessel area at different time points after lymphatic ablation (*P < 0.001). (H) Quantification of α-SMA thickness at different time points after lymphatic ablation (*P < 0.001). (I) Representative photomicrographs localizing CD4 and podoplanin in distal mouse hind limb tissues in age-matched normal and DT-treated animals (52 weeks after DT; scale bar: 100 μm). (J) Quantification of CD4+ cells in control and DT-treated mice 52 weeks after lymphatic ablation (*P < 0.001). (K) ELISA for TGF-β1, TNF-α, and IFN-γ in hind limb tissues harvested from control an DT-treated mice 1 and 3 weeks after DT injection (*P < 0.01). 2-tailed Student’s t test.
Previous studies have shown that clinical lymphedema results in progressive fibrosis of collecting lymphatic vessels, accumulation of α-SMA cells, and sclerosis of vessels with late-stage disease (13). Consistent with these reports, we noted progressive sclerosis and narrowing of the main hind limb collecting lymphatics as time progressed following DT administration (Figure 2, F and G). These findings were similar to clinical lymphedema biopsy specimens obtained from patients with unilateral breast cancer–related upper extremity lymphedema (Figure 2F). Collecting lymphatics harvested from mice treated with DT 52 weeks after lymphatic ablation were surrounded by a thickened layer of α-SMA positive cells and were nearly encased in fibrotic tissues (Figure 2H).
Our group has previously shown that clinical lymphedema specimens are infiltrated with CD4+ cells and that the degree of CD4+ infiltration is positively correlated with increasing lymphedema stage (14, 15). Consistent with these findings, we found that CD4+ cells accumulated in subcutaneous tissues of animals treated with DT and that the timing of this inflammatory response correlated with the observed late increase in hind limb swelling and fibrosis (Figure 2, I and J). Similar to the clinical scenario, we also noted a marked increase in protein concentration of profibrotic and inflammatory cytokines, including TGF-β1, TNF-α, and IFN-γ in hind limb tissues of animals treated with DT (Figure 2K). Taken together, our findings show that local administration of DT in the hind limb results in the development of chronic and progressive lymphedema that is temporally, histologically, and radiographically similar to clinical lymphedema.
Transient decrease in edema coincides with dermal lymphatic hyperplasia.
We used NIR lymphatic imaging with intradermal ICG injection to analyze lymphatic vessel regeneration after ablation (Figure 3A, top, and Figure 3B). One week after DT administration, we found virtually no ICG uptake, indicating dysfunction in both capillary and collecting lymphatics . However, analysis at 3, 6, and 9 weeks after injection demonstrated regeneration and hyperplasia of dermal lymphatic vessels in the distal hind limb. These LYVE-1+ vessels were tortuous and abnormal in appearance, with punctate areas of ICG pooling in the dermis (Figure 3C). By 52 weeks after DT injection, the majority of these vessels had regressed or were not functional, resulting in increasing pooling of ICG. Similar to late-stage clinical lymphedema, we never observed functional (i.e., IGC-containing) regeneration of the collecting lymphatics after DT ablation, although collecting lymphatic vessels could be seen in histological sections (see below).
Figure 3. Transient decrease in edema coincides with dermal lymphatic hyperplasia.

(A) Top row: NIR lymphatic imaging of hind limbs at various time points after lymphatic ablation (n = 5/group). Bottom: Prussian blue staining of ferritin in cross sections of popliteal lymph nodes at various times following DT administration (n = 5/group; scale bar: 200 μm). (B) Quantification of Prussian blue lymph node staining (percentage of total lymph node area) at various time points after DT administration (*P < 0.05). (C) Left: A high-power view of dermal backflow shown in NIR imaging 3 weeks after lymphatic ablation, demonstrating a dense network of small-caliber vessels with leakage of contrast at discrete points (large white dots). Right: Whole mount section of area shown in the dotted green box in the NIR image, demonstrating proliferation of LYVE-1+ vessels; original magnification, 500 μm. 2-tailed Student’s t test.
Increasing capillary lymphatic density in the early periods after DT administration also correlated with lymphatic uptake by the draining popliteal lymph node, as assessed with ferritin labeling of functional lymphatics (16) (Figure 3A, bottom, and Figure 3B). Consistent with our 99mTc lymphoscintigraphic findings, we noted either absent or low levels of ferritin staining in draining lymph nodes harvested from DT-injected animals 1, 3, or 6 weeks prior to sacrifice. At the 9-week time point, we did note low levels of uptake in the popliteal lymph node; however, this expression was virtually completely lost by 52 weeks after DT injection.
Lymphatic regeneration requires infiltration of macrophages.
Because macrophages are a known source of lymphangiogenic cytokines in a variety of settings (17), we next sought to determine whether macrophage infiltration correlated with lymphatic regeneration after DT administration. Analysis of hind limb tissues harvested after DT ablation showed a massive influx of F4/80+ cells in the early periods after injury, peaking at 1 week and decreasing progressively thereafter (Figure 4, A and B). Macrophage infiltration preceded lymphatic vessel regeneration, and the number of macrophages was inversely related to the dermal lymphatic vessel density (LVD, Figure 4C). The majority of macrophages present at the 3- and 6-week time points also expressed CD206, a marker of M2 polarization (Figure 4, A and D). In addition, consistent with M2 polarization, we noted that some of these macrophages also stained positively for LYVE-1 (Figure 4A and Supplemental Figure 3, A–C). These LYVE-1+ macrophages cells tended to aggregate around and were closely associated with regenerating lymphatic vessels, but did not become incorporated into newly formed vessels (Figure 5).
Figure 4. Lymphatic regeneration after DT administration is associated with macrophage infiltration and M2 differentiation.

(A) Representative triple immunofluorescence localization of F4/80 (top), CD206 (middle), and LYVE-1 (bottom) in the hind limbs of control (normal) and DT-treated mice 1 and 3 weeks after DT administration (scale bar: 100 μm). (B) Quantification of macrophage (F4/80+) infiltration at various time points following lymphatic ablation with DT. (C) Quantification of lymphatic vessel density over time in control and DT-treated mice. (D) Quantification of F4/80+ and CD206+ dual positive cells in hind limb tissues over time. (E) Real-time quantitative PCR for VEGF-A, VEGF-C, and LYVE-1 in cell-sorted CD11b+ cells harvested from hind limb tissues before (control) and 1 or 3 weeks after DT administration (*P < 0.01). (F) ELISA for VEGF-A, VEGF-C, IL-4, and IL-13 in hind limb tissues harvested from control and DT-treated animals 1 and 3 weeks following DT administration (*P < 0.05). 2-tailed Student’s t test.
Figure 5. LYVE-1+ macrophages are intimately associated with but do not incorporate into regenerated lymphatics.
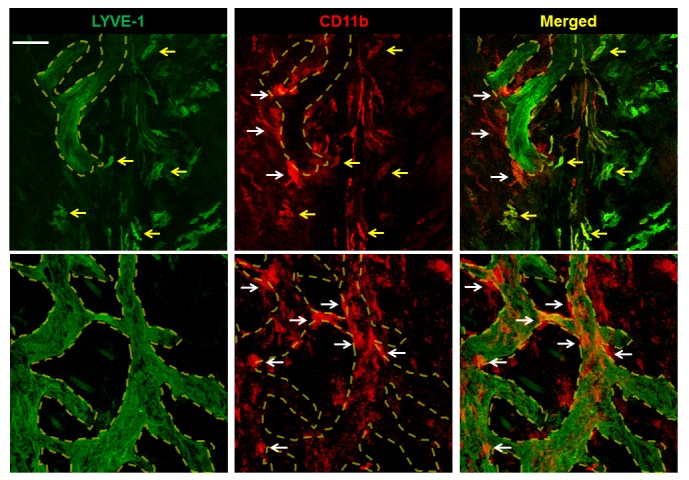
Representative dual immunofluorescence localization of LYVE-1 and CD11b in the distal hind limb skin 4 weeks after DT administration. Regenerating lymphatic vessels are outlined in dotted yellow lines. LYVE-1+CD11b+ macrophages (yellow arrows) are near the outer surface of lymphatic vessels, while LYVE-1–CD11b+ macrophages (white arrows) are intimately associated with them (scale bar: 100 μm).
The temporal relationship between macrophage infiltration and lymphatic vessel regeneration suggested that these cells are required for the early lymphangiogenic response. This hypothesis was supported by quantitative PCR (qPCR) experiments performed on cell-sorted CD11b+ macrophages harvested from the distal hind limb of DT-treated and control mice demonstrating increased VEGF-A (1.5- to 2-fold; 1 and 3 weeks after DT) and VEGF-C (2-fold increase 3 weeks after DT) mRNA expression (Figure 4E). In addition, consistent with our histological observations, we noted that expression of LYVE-1 by macrophages increased dramatically at both 1 and 3 weeks after DT administration. These findings were confirmed with protein analysis demonstrating elevated concentrations of VEGF-A and VEGF-C in hind limb tissues harvested 1 and 3 weeks after DT treatment (Figure 4F). In addition, we noted that the expression of Th2 cytokines (IL-4 and IL-13) was also significantly increased in tissues harvested from DT-treated mice, suggesting that the expression of both pro-lymphangiogenic (VEGF-A and VEGF-C) and anti-lymphangiogenic (IL-4, IL-13, TGF-β1, and IFN-γ) cytokines is increased after lymphatic ablation.
To test the hypothesis that macrophage infiltration is necessary for capillary lymphatic regeneration and partial resolution of lymphedema after lymphatic ablation, we ablated lymphatic vessels and then depleted macrophages with clodronate liposomes (CLs) administered i.p. (110 mg/kg 3 times weekly). Control animals were similarly treated but given PBS liposomes (PLs). As expected, this treatment was highly effective in depleting macrophages systemically (Supplemental Figure 4, A and B). More importantly, macrophage depletion completely prevented the temporary resolution of edema, resulting in markedly increased hind limb edema and fibroadipose deposition (Figure 6, A–D). Consistent with this observation, we found virtually no evidence of regenerating functional capillary lymphatic vessels using NIR imaging (Figure 6E). Immunofluorescent colocalization of macrophages (CD11b+) and lymphatics (LYVE-1+) in distal hind limb tissues of DT-treated mice demonstrated a marked decrease in lymphangiogenesis (2-fold decrease lymphatic vessel density) and very effective depletion of macrophages as compared with controls (Figure 6, E–G). This decrease in macrophages corresponded with a marked decrease in VEGF-A, as well as in VEGF-C protein expression in hind limb tissues (Figure 6, H and I). Lymphatic vessels present in macrophage-depleted mice were severely hypoplastic, with few branch points. Decreased lymphatic regeneration in macrophage-depleted animals resulted in increased CD4+ cell infiltration, increased accumulation of surrounding α-SMA+ cells around collecting lymphatics, and accelerated collecting lymphatic vessel sclerosis (Supplemental Figure 4, C–G). Taken together, these findings indicate that the initial resolution of edema after lymphatic injury is dependent on proliferation of capillary lymphatic vessels, which is regulated by an influx of macrophages with M2 differentiation, and that inhibition of this response accelerates and exacerbates chronic lymphedema.
Figure 6. Lymphatic regeneration requires infiltration of macrophages.

(A) Representative images of hind limbs following DT administration in control (PBS liposomes) and macrophage-depleted (clodronate liposome) animals (n = 5/group). (B) Quantification of increase in foot diameter from baseline in control and macrophage-depleted DT treated animals over time (*P < 0.001). (C) Representative H&E-stained cross sections of the distal hind limb harvested 4 weeks after DT administration in control and macrophage-depleted animals. Fibroadipose tissue of the hypodermis is outlined with dotted black lines and marked with an asterisk (scale bar: 1 mm). (D) Quantification of fibroadipose deposition as a percentage of the total cross-sectional area of the distal hind limb (*P < 0.001). (E) Left: Representative NIR imaging of control (top) and macrophage-depleted (bottom) animals 4 weeks after DT administration (n = 5/group). Middle and right: Whole mount dual immunofluorescence localization of LYVE-1 (middle) and LYVE-1/CD11b (right), demonstrating lymphatic vessels in the distal hind limbs of control (top) but not macrophage-treated (bottom) animals. Note the diminutive and hypoplastic appearance of lymphatics in macrophage-depleted mice (scale bar: 200 μm). (F) Quantification of dermal lymphatic vessels in hind limb tissue sections in control and macrophage-depleted animals 4 weeks after DT treatment (*P < 0.001). (G) Quantification of F4/80+ cells in hind limb tissue sections in control and macrophage-depleted animals 4 weeks after DT treatment (*P < 0.001). (H) ELISA for VEGF-A in hind limb protein from control and depleted mice at 3 weeks (*P < 0.01). (I) ELISA for VEGF-C in hind limb protein from each group of mice (*P < 0.01). 2-tailed Student’s t test.
CD4+ cell depletion prevents lymphatic vessel sclerosis and improves lymphatic function.
We have previously shown that depletion or genetic absence of CD4+ cells (but not macrophages, CD8+, or CD25+ cells) prevents development of lymphedema in a mouse tail model (15). Although these results were exciting, it is possible that some of the observed changes were related to improvements in wound healing rather than decreased lymphedema. Therefore, to test the hypothesis that CD4+ cells contribute to progressive onset of lymphedema after lymphatic injury, we injected transgenic mice with DT in the hind limbs and allowed them to recover for 9 weeks. At this point, animals were divided into 2 groups such that experimental animals underwent systemic CD4+ cell depletion using weekly injections of monoclonal anti-CD4 neutralizing antibodies (CD4 mAb), while controls were treated with nonspecific isotype control monoclonal antibodies for an additional 9 weeks.
Mice treated with CD4 mAb beginning 9 weeks after lymphatic ablation had significantly decreased hind limb swelling and fibroadipose deposition as compared with controls 18 weeks after DT injection (Figure 7, A–D). CD4+ depletion also decreased accumulation of α-SMA+ cells around collecting lymphatics and inhibited sclerosis/obstruction of these structures (Figure 7, E–G). ICG lymphangiography demonstrated that animals treated with isotype control antibodies had virtually no identifiable functional capillary or collecting vessels in the distal hind limb (Figure 7H). In contrast, CD4 mAb–treated animals consistently demonstrated the presence of functional, albeit abnormal-appearing lymphatic vessels. These observations were corroborated by histological analysis demonstrating increased dermal lymphatic vessel density in CD4 mAb–treated animals as compared with controls (Figure 7, I and J). In addition, we saw a marked reduction in infiltrating CD4+ cells (>80% decrease) in the hind limb tissues of mice treated with CD4 mAb as analyzed by histological staining and flow cytometry (Figure 7, I and K, and Supplemental Figure 5). Interestingly, CD4+ cells tended to cluster around the lymphatic vessels of control mice, and this response was lost in CD4 mAb–treated animals. Finally, analysis of popliteal lymph nodes after distal hind limb ferritin injection demonstrated a nearly 3-fold increase in subcortical ferritin staining in CD4-depleted animals (Figure 7, L and M). Taken together, these findings suggest that progressive accumulation of CD4+ cells after lymphatic injury regulates late fibroadipose tissue deposition, lymphatic vessel sclerosis, and lymphatic dysfunction. In addition, CD4+ cell accumulation in the late stages of disease impairs lymphatic function by inhibiting lymphangiogenesis.
Figure 7. CD4+ cell depletion prevents lymphatic vessel sclerosis and improves lymphatic function.

(A) Representative images of hind limbs 18 weeks after DT administration and treatment with isotype control or CD4 mAb (beginning 9 weeks after DT injection). (B) Quantification of increase in foot diameter from baseline in isotype and CD4 mAb–treated mice over time. Red arrow indicates the time at which weekly isotype or CD4 mAb administration was initiated (n = 5/ group; *P < 0.001). (C) Representative H&E image of distal hind limb cross sections harvested 18 weeks after DT administration in isotype and CD4 mAb–treated mice. Fibroadipose tissue of the hypodermis is outlined with dotted black lines and marked with an asterisk (scale bar: 1 mm). (D) Quantification of fibroadipose tissue area in the distal hind limb of control and CD4 mAb–treated animals 18 weeks after DT treatment (*P < 0.001). (E) Representative high-power photomicrographs of collecting lymphatics (podoplanin, green; α-SMA, red) from the distal hind limb of control and CD4 mAb–treated mice 18 weeks following DT administration. Note the luminal narrowing and increase in perilymphatic smooth muscle cell deposition in isotype control–treated mice (scale bar: 20 μm). (F) Quantification of smooth muscle wall thickness surrounding collecting lymphatic vessels in the distal hind limbs of isotype control– and CD4 mAb–treated mice harvested 18 weeks after DT administration (*P = 0.004). (G) Quantification of collecting lymphatic vessel area in distal hind limbs of isotype and CD4 mAb–treated mice (*P = 0.005) 18 weeks after DT administration. (H) Representative NIR lymphangiography of control and CD4 mAb–treated mice demonstrating ICG-containing lymphatic vessels only in CD4-depleted mice (white arrows) (n = 5/group). (I) Representative colocalization of LYVE-1 and CD4+ cells in distal hind limb tissues of isotype and CD4 mAb–treated mice 18 weeks after DT administration (scale bar: 50 μm). (J) Quantification of lymphatic vessel density (LVD) in distal hind limbs of isotype or CD4 mAb–treated animals 18 weeks after DT administration (*P < 0.001). (K) Quantification of CD4+ cells in the tissues of the distal hind limbs of mice treated with isotype or CD4 mAbs 18 weeks after DT administration (*P < 0.001). (L) Representative low- and high-power photomicrographs of Prussian blue–stained popliteal lymph nodes harvested 18 weeks after DT administration from isotype- and CD4 mAb–treated mice (scale bar: 200 μm). (M) Quantification of Prussian blue (ferritin) staining area of popliteal lymph nodes in isotype- and CD4 mAb–treated mice (*P = 0.007). 2-tailed Student’s t test.
Discussion
A large number of clinical reports have analyzed the histological, radiological, and epidemiological aspects of lymphedema (18–21). More recently, a few studies have analyzed genetic changes in an effort to identify predisposing factors for lymphedema (22–24). Although these studies have been enormously helpful in identifying risk factors for lymphedema, clinical studies are inherently limited in identifying pathologic pathways, since this approach precludes time course analysis, provides relatively small amounts of tissues in biopsies, is confounded by patient factors, and most importantly, does not readily allow targeted interventions.
The limitations of clinical studies have led to the description of a large number of animal models of lymphedema. Surgical models of lymphedema usually require extensive surgical intervention with high animal morbidity resulting in variable swelling and, only in rare cases, chronic lymphedema. As a result, there is currently no surgical model that accurately mimics the clinical disease temporally or physiologically. In the current study, we describe a technique to ablate both capillary and collecting lymphatic vessels using local injections of DT. This approach, in contrast to previous studies reporting the use of LYVE-1/DTR mice, (25) resulted in the development of chronic lymphedema in the hind limb that mirrors the clinical disease temporally, radiographically, and histologically. Thus, similar to clinical lymphedema, treatment of transgenic mice hind limbs with DT resulted in initial swelling that improved over a short period of time only to return in a progressive fashion with increasing CD4+ cell accumulation (14, 26), worsening fibroadipose tissue deposition (20, 27), and collecting vessel sclerosis (13, 28).
Other studies have reported on nonsurgical methods of lymphatic ablation. For example, Jang et al., as in our study, reported on the use of DT-mediated ablation of lymphatics using LYVE-1/DTR mice (25). However, their report focused on systemic toxicity of this treatment, as subcutaneous injection of DT into the ears of these animals did not cause lymphedema. This difference from our findings likely stems from the fact that LYVE-1 is not expressed at high levels in the collecting lymphatic vessels of adult mice, thereby sparing the deep system of DT-mediated injury (29). In contrast, the use of the FLT4 promoter in our study targeted both capillary and collecting lymphatics, thereby enabling us to induce long-term lymphedema. In addition, because LYVE-1 is expressed by a subset of macrophages, the use of LYVE-1/DTR models complicates modeling of lymphedema, since our study indicates that these cells play a key role in the pathology of the disease. Other studies have reported on nonsurgical ablation of lymphatic vessels using lymphatic-specific photodynamic therapy (30). However, because lymphatic destruction is limited to small, superficial regions of the skin, this approach does not enable modeling of lymphedema.
Patients who undergo lymphadenectomy have temporary swelling of the extremities that generally resolves over the ensuing 4–6 weeks. The results of our study suggest that this initial resolution is dependent on capillary lymphatic regeneration rather than restoration of the collecting lymphatic vessels. We found that this response is regulated by macrophage infiltration, M2 differentiation, elaboration of lymphangiogenic cytokines, and possibly by direct interaction of macrophages with newly formed lymphatic vessels. These findings are supported by previous studies demonstrating a major role for macrophages in regulating lymphangiogenesis in a variety of settings, including wound healing, inflammation, and tumor metastasis (31, 32). In addition, in an interesting recent article, Ogata and colleagues used a mouse model of lymphatic injury to show that macrophages play a key role in the early regulation of lymphangiogenesis and that this process may be regulated by interactions with Th1 and Th17 cells (33). Our findings are also supported by lymphangiographic studies in a canine model of axillary lymphadenectomy demonstrating resolution of edema concurrent with development of hyperplastic collateral lymphatics that bypassed the zone of lymphatic injury (34). In the current study, in contrast to that by Ogata and colleagues, we found that macrophage depletion tended to accelerate development of progressive lymphedema, suggesting that the degree of lymphatic stasis is a major regulator of chronic inflammation and pathology. This discrepancy likely reflects the different models and the timing of analysis, with Ogata et al. utilizing a lymphatic injury/lymphadenectomy model with resultant mild edema that resolved over a 28-day period; and our group using a more extensive model of lymphatic injury that resulted in chronic and progressive changes that persisted for a year. Therefore, additional work is required to delineate the cellular mechanisms by which macrophages and T cells interact to regulate initial swelling. However, consistent with previous reports utilizing lymphangiogenic cytokines to promote lymphatic regeneration after lymphatic injury (35, 36), our findings suggest that early therapeutic interventions that improve formation of collateral lymphatics after lymphatic injury may prevent progression to late-stage disease by providing functional bypass channels around the zone of injury.
The findings of our current study taken together with previous reports suggest that macrophages play a dual role in the pathology of lymphedema by regulating lymphangiogenesis and fibrosis. Our findings with macrophage depletion suggest that these cells are necessary for lymphatic regeneration after lymphatic ablation and that loss of this response markedly increases the fibrotic response. This effect maybe due to increased lymphatic fluid stasis resulting from lack of lymphatic regeneration, thereby resulting in increased CD4+ cell infiltration and expression of profibrotic cytokines. Macrophage infiltration in the later stages (i.e., 52 weeks) is markedly decreased as compared with the early time periods after lymphatic injury. This is important because macrophages have also been shown to play a role in regulation of fibrosis (37). Thus, early macrophage infiltration is important for lymphatic regeneration, while late cellular infiltration may modulate fibrotic responses.
An interesting observation of our study was that many infiltrating macrophages expressed LYVE-1, a marker of lymphatic differentiation. However, unlike some prior studies (38, 39), but in agreement with others (40), we did not note incorporation of macrophages into newly formed lymphatic capillaries. Instead, macrophages tended to surround newly formed vessels, suggesting that these cells regulate lymphangiogenesis by a paracrine process (41, 42). This hypothesis is supported by previous studies demonstrating that macrophages promote lymphangiogenesis by producing lymphangiogenic growth factors and expressing matrix metalloproteinases that degrade the ECM (43). The latter effect may be a necessary step for lymphangiogenesis in the setting of fibrous tissue deposition after lymphatic injury. This concept is supported by previous studies demonstrating increased tail swelling in MMP-9–knockout mice in a mouse tail model of lymphedema (44).
Using both animal models and clinical biopsy specimens, our group has previously shown that CD4+ cells plays a key role in the development of lymphedema (14). Our current study further supports this hypothesis and provides substantial evidence that CD4+ cells mediate late lymphatic dysfunction by a number of physiologic changes, including ECM fibrosis, sclerosis of collecting lymphatics, and inhibition of lymphangiogenesis. Thus, lymphatic injury initiates a CD4+ cell inflammatory response resulting in a vicious cycle of ongoing fibrosis, impaired lymphangiogenesis and worsening lymphatic function. In this sense lymphedema may represent a form of fibrotic organ failure of the lymphatic system. This hypothesis is supported by the fact that CD4+ cells are strongly implicated in the regulation of fibrosis in a number of organ systems, including idiopathic pulmonary fibrosis, liver fibrosis, and scleroderma (45). This hypothesis is also supported by previous studies demonstrating that CD4+ cells are key regulators of smooth muscle cell proliferation and atherosclerosis (46–48), as well as studies demonstrating that ECM composition is a key regulator of interstitial fluid transport (49). Progressive lymphatic and ECM fibrosis also provides a rationale for known risk factors of lymphedema, such as radiation and obesity, as these factors independently increase fibrosis (50–52). Indeed, previous studies have shown that radiation-induced fibrosis markedly decreases interstitial fluid transport and that inhibition of fibrosis effectively preserves lymphatic function independent of LEC survival in this setting (53). Finally, a number of recent studies have shown that T cells are potently antilymphangiogenic and that this effect is mediated, at least in part, by the expression of inflammatory cytokines such as IFN-γ, IL-4, and IL-13 (54–56). The findings of the current study are consistent with these studies, since we found that depletion of CD4+ cells increased LVD histologically and radiographically. It is important to note that recent studies by Ogata and colleagues suggest that subsets of T cells may promote lymphangiogenesis by interacting with macrophages in the early periods following lymphatic injury, emphasizing that the immune responses to lymphatic injury are complex, temporally regulated, and require additional study.
In summary, the findings of the current study suggest that the pathology of lymphedema can be conceptualized as a 2-phase process (Supplemental Figure 6). In the early periods following lymphatic injury, lymphangiogenesis is initiated by infiltration of macrophages with M2 differentiation that secrete lymphangiogenic cytokines and interact with LECs to promote formation of hyperplastic capillary lymphatics that bypass the zone of injury. The late stages of disease are mediated by progressive infiltration of CD4+ cells, which promote ECM fibrosis, collecting lymphatic sclerosis, and inhibit lymphangiogenesis. The findings of this study are important, since a better understanding of the pathophysiology of lymphedema will enable development of novel preventive and therapeutic options for this disabling disease.
Methods
Generation of FLT4-DTR mice.
FLT4-CreERT2 mice (a gift from Sagrario Ortega, CNIO) (57, 58) were crossed with human DTR floxed C57BL/6 mice (C57BL/6-Gt(ROSA)26Sortm1(HBEGF)Awai/J; The Jackson Laboratory). Tamoxifen administration to double mutant mice activates the CreERT2 recombinase under the transcriptional control of the FLT4 regulatory elements, resulting in efficient induction of DTR expression in LECs of adult mice. Gene expression of both transgenes was confirmed by genotyping (Transnetyx), and double homozygous mice were backcrossed for 6 generations to ensure consistency. Cre expression was induced in adult male mice by using tamoxifen (300 mg/kg/d subcutaneously for 5 days) followed by either systemic (100 ng i.p.) or local (5 ng subcutaneously daily ×3 doses) DT delivery (58).
Analysis of lymphatic function.
Foot diameter measurements were performed in standardized locations using a digital caliper, and changes were confirmed using histological cross sections obtained from the same region to quantify fibroadipose tissue area and dermal thickness using Panoramic Viewer (3DHISTECH). Lymph node uptake of interstitial fluid was determined by measuring the uptake of high-molecular-weight Fe-III equine splenic ferritin (Sigma-Aldrich) in the popliteal lymph node after injection into the hind limb, performed as previously described (59). Standardized sections of the lymph nodes were prepared, and ferritin uptake was assessed as a percentage of the total cross-sectional area (MetaMorph software; Molecular Devices). Lymphatic function was also assessed using lymphoscintigraphy as previously described using 99mTc sulfur colloid injected intradermally in the distal hind limb (60).
ICG lymphangiography was performed using a modification of previously published techniques (61). Briefly, 15 μl of 0.15 mg/ml ICG (Sigma-Aldrich) was injected intradermally in the dorsal aspect of the hind limb and visualized using a custom-made EVOS EMCCD camera (Life Technologies) and a LED light source (CoolLED). Video and static images were obtained using a Zeiss V12 SteREO Lumar microscope (Caliper Life Sciences).
Immunostaining.
Immunostaining was performed using paraffin and frozen sections as previously described (14). Primary antibodies used for immunohistochemical stains included LYVE-1, CD45, and CD4 (catalog AF2125, MAB114, and AF554, respectively; R&D Systems), α-SMA (catalog C6198; Sigma-Aldrich), CD11b (catalog 557395; BD Biosciences), and F4/80, podoplanin, DTR, CD206, and collagen I (catalog ab16911, ab11936, AF259-NA, ab64693, ab34710, respectively; Abcam). Immunofluorescence and whole mount staining were performed using Alexa Fluor–conjugated secondary antibodies (Life Technologies) (60). Images were scanned using Mirax imaging software (Carl Zeiss) or confocal microscopy (Leica Microsystems), processed with Imaris (Bitplane), and analyzed using MetaMorph software. For analysis of collecting lymphatic vessels, 2 major collecting vessels flanking the dorsal caudal femoral artery were identified at a point located 2 mm proximal to the tarsal joint. The cross-sectional luminal area of these 2 large collectors was measured using Panoramic Viewer software, and the thickness of the smooth muscle lining was assessed by measuring the thickest point of α-SMA+ cells in each cross section.
Cell counts of inflammatory cells and lymphatic vessels were performed by 2 reviewers blinded to the experimental protocol in 4 randomly-selected, 40× high-power fields in 4–6 animals per group. Collagen I deposition in the dermis and subcutaneous tissues was quantified from stained tissue sections using MetaMorph software and confirmed using picrosirius red staining and polarized light microscopy as previously reported (14).
Human lymphedema samples.
Six patients with postsurgical upper extremity lymphedema were identified at the MD Anderson Cancer Center, and full-thickness 5-mm punch biopsies were obtained from identical locations of the lymphedematous and contralateral control limbs. Dual immunofluorescence staining for podoplanin and α-SMA+ was performed to allow identification and analysis of collecting lymphatic vessels. The study was approved by the IRBs of both the MD Anderson Cancer Center and MSKCC.
Macrophage depletion.
Macrophages were depleted beginning the same day as DT ablation of hind limb lymphatics using CLs (110 mg/kg i.p. 3 times weekly; Foundation Clodronate Liposomes); control mice were treated with an equivalent volume of PBS-containing liposomes (62, 63).
CD4+ cell depletion.
We used well-described monoclonal antibodies against mouse CD4 (clone GK1.5; 5 μg/g/dose; Bio X Cell) or isotype control antibodies (Bio X Cell) administered i.p. (14, 15). CD4 mAb and isotype control administration was started 9 weeks after DT administration and was continued for 9 weeks.
Flow cytometry.
Flow cytometry was performed on isolated lymph nodes, skin, or spleens as previously described (15). Briefly, tissues were digested in dispase (0.8 mg/ml), collagenase (0.2 mg/ml), and DNAse I (0.1 mg/ml) at 37°C for 30 minutes. Digested tissues were filtered and resuspended in 2% FCS/sodium azide solution in PBS; stained with fluorophore-conjugated antibodies for CD45, CD31, podoplanin, and TCR-β (catalog 103107, 102509, 127407, and 109223; BioLegend), LYVE-1 and F4/80 (catalog 50-0443-80 and 11-4801-82; eBioscience); and analyzed with a FACSCalibur flow cytometer (BD Biosciences) using FlowJo software (Tree Star).
qPCR.
To isolate hind limb macrophages, hind limb skin was digested as previously described (15, 64), and CD11b+ macrophages were isolated using magnetic beads (MACS, Miltenyi Biotec) according to the manufacturer’s recommendations. RNA extraction was performed on these macrophages using TRIzol (Invitrogen, Life Technologies) according the manufacturer’s recommendations. RNA quality and quantity were assessed using an Agilent Bioanalyzer (Agilent Technologies). The isolated RNA was converted to cDNA using a TaqMan reverse transcriptase kit (Roche), and relative expression of gene expression between groups was performed using delta-delta CT PCR analysis and normalizing gene expression using GAPDH RNA amplification as previously described (14). Relative expression was calculated using the formula: 2[–(Ct gene of interest – Ct endogenous control) sample A – (Ct gene of interest – Ct endogenous control) sample B)]. All samples were performed in triplicate. The primers used for the PCR targets of interest were for VEGF-A, VEGF-C, and LYVE-1 (Mm01281449_m1, Mm00437310_m1, and Mm00475056_m1 respectively; Applied Biosystems, Life Technologies).
Statistics.
Values are presented as mean ± SD, with P ≤ 0.05 considered significant. Differences between 2 groups were analyzed using the unpaired, 2-tailed Student’s t test, while multiple groups were assessed using ANOVA with post hoc tests to compare between groups. All measurements were performed by 2 reviewers blinded to the experimental protocol, and a minimum of 4–6 animals were used for each group.
Study approval.
All human tissues were obtained with written informed consent and studies approved by the IRB committees of both the MSKCC and the MD Anderson Cancer Center. All mouse studies and protocols were approved by the IACUC at Memorial Sloan Kettering Cancer Center.
Author contributions
JCG, RPK, and BJM conceived of the concept and designed the experiments. JCG, ILS, RPK, and SO developed methods. IMC and SO designed, generated, and characterized the FLT4-CreERT2 mouse. DC provided human biopsy samples for analysis. JJD provided clinical ICG images. JCG, GEH, RPK, ILS, JST, GDGN, and JZ performed the experiments. JCG, GEH, RPK, ILS, JST, GDGN, JZ, and BJM analyzed the data. JCG, GEH, RPK, SO and BJM prepared and edited the manuscript.
Supplementary Material
Acknowledgments
We thank Mesruh Turkekul for expert technical and histological assistance. We also thank the Molecular Cytology Core at MSKCC for assistance with both histology and tissue imaging. This work was supported by an R01 grant from the NIH (no. HL111130-01) awarded to BJM; a Pilot Grant from the Plastic Surgery Foundation (no. 312436) awarded to JCG; and a grant from the STARR Foundation awarded to BJM. Salary support for JCG was provided by a Research Fellowship Grant from the Plastic Surgery Educational Foundation (no. 312436). Salary support for GDGN was provided by a T32 grant from the NIH (CA009685). SO received a grant from the Ministry of Science and Innovation of Spain (BIO2009-09488). This work was partially supported by NIH/NCI Cancer Center Support Grant P30 CA008748. Sources of unding: NIH R01 HL111130-01 and STARR foundation grant awarded to BJM; Plastic Surgery Education Foundation Research Fellowship Grant 312436 awarded to JCG; NIH T32 CA009685 to GDGN; Ministry of Science and Innovation of Spain Grant BIO2009-09488 to SO; NIH/NCI Cancer Center Support Grant P30 CA008748.
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Reference information:JCI Insight. 2016;1(15):e84095. doi:10.1172/jci.insight.84095.
Contributor Information
David Chang, Email: dchang@surgery.bsd.uchicago.edu.
Jamie Zampell, Email: zampellj@gmail.com.
Sagrario Ortega, Email: Sortega@cnio.es.
References
- 1.Ferlay J, et al. Cancer incidence and mortality patterns in Europe: estimates for 40 countries in 2012. Eur J Cancer. 2013;49(6):1374–1403. doi: 10.1016/j.ejca.2012.12.027. [DOI] [PubMed] [Google Scholar]
- 2.Lerman R, Jarski R, Rea H, Gellish R, Vicini F. Improving symptoms and quality of life of female cancer survivors: a randomized controlled study. Ann Surg Oncol. 2012;19(2):373–378. doi: 10.1245/s10434-011-2051-2. [DOI] [PubMed] [Google Scholar]
- 3.Cormier JN, Askew RL, Mungovan KS, Xing Y, Ross MI, Armer JM. Lymphedema beyond breast cancer: a systematic review and meta-analysis of cancer-related secondary lymphedema. Cancer. 2010;116(22):5138–5149. doi: 10.1002/cncr.25458. [DOI] [PubMed] [Google Scholar]
- 4.Warren AG, Brorson H, Borud LJ, Slavin SA. Lymphedema: a comprehensive review. Ann Plast Surg. 2007;59(4):464–472. doi: 10.1097/01.sap.0000257149.42922.7e. [DOI] [PubMed] [Google Scholar]
- 5.Moffatt CJ, et al. Lymphoedema: an underestimated health problem. QJM. 2003;96(10):731–738. doi: 10.1093/qjmed/hcg126. [DOI] [PubMed] [Google Scholar]
- 6.Rockson SG, Rivera KK. Estimating the population burden of lymphedema. Ann N Y Acad Sci. 2008;1131:147–154. doi: 10.1196/annals.1413.014. [DOI] [PubMed] [Google Scholar]
- 7.Petrek JA, Heelan MC. Incidence of breast carcinoma-related lymphedema. Cancer. 1998;83(12 Suppl American):2776–2781. doi: 10.1002/(sici)1097-0142(19981215)83:12b+<2776::aid-cncr25>3.0.co;2-v. [DOI] [PubMed] [Google Scholar]
- 8.Yang CY, et al. Developing a lower limb lymphedema animal model with combined lymphadenectomy and low-dose radiation. Plast Reconstr Surg Glob Open. 2014;2(3):e84095. doi: 10.1097/GOX.0000000000000064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tabibiazar R, et al. Inflammatory manifestations of experimental lymphatic insufficiency. PLoS Med. 2006;3(7):e84095. doi: 10.1371/journal.pmed.0030254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hadamitzky C, Pabst R. Acquired lymphedema: an urgent need for adequate animal models. Cancer Res. 2008;68(2):343–345. doi: 10.1158/0008-5472.CAN-07-2454. [DOI] [PubMed] [Google Scholar]
- 11.Rasmussen JC, et al. Human lymphatic architecture and dynamic transport imaged using near-infrared fluorescence. Transl Oncol. 2010;3(6):362–372. doi: 10.1593/tlo.10190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hara H, Mihara M, Seki Y, Todokoro T, Iida T, Koshima I. Comparison of indocyanine green lymphographic findings with the conditions of collecting lymphatic vessels of limbs in patients with lymphedema. Plast Reconstr Surg. 2013;132(6):1612–1618. doi: 10.1097/PRS.0b013e3182a97edc. [DOI] [PubMed] [Google Scholar]
- 13.Mihara M, et al. Pathological steps of cancer-related lymphedema: histological changes in the collecting lymphatic vessels after lymphadenectomy. PLoS One. 2012;7(7):e84095. doi: 10.1371/journal.pone.0041126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Avraham T, et al. Th2 differentiation is necessary for soft tissue fibrosis and lymphatic dysfunction resulting from lymphedema. FASEB J. 2013;27(3):1114–1126. doi: 10.1096/fj.12-222695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zampell JC, Yan A, Elhadad S, Avraham T, Weitman E, Mehrara BJ. CD4(+) cells regulate fibrosis and lymphangiogenesis in response to lymphatic fluid stasis. PLoS One. 2012;7(11):e84095. doi: 10.1371/journal.pone.0049940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Leu AJ, Berk DA, Lymboussaki A, Alitalo K, Jain RK. Absence of functional lymphatics within a murine sarcoma: a molecular and functional evaluation. Cancer Res. 2000;60(16):4324–4327. [PubMed] [Google Scholar]
- 17.Kataru RP, et al. Critical role of CD11b+ macrophages and VEGF in inflammatory lymphangiogenesis, antigen clearance, and inflammation resolution. Blood. 2009;113(22):5650–5659. doi: 10.1182/blood-2008-09-176776. [DOI] [PubMed] [Google Scholar]
- 18.Rockson SG. Lymphedema. Am J Med. 2001;110(4):288–295. doi: 10.1016/S0002-9343(00)00727-0. [DOI] [PubMed] [Google Scholar]
- 19.Mortimer PS, Rockson SG. New developments in clinical aspects of lymphatic disease. J Clin Invest. 2014;124(3):915–921. doi: 10.1172/JCI71608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Olszewski WL, Jamal S, Manokaran G, Lukomska B, Kubicka U. Skin changes in filarial and non-filarial lymphoedema of the lower extremities. Trop Med Parasitol. 1993;44(1):40–44. [PubMed] [Google Scholar]
- 21.Szuba A, Rockson SG. Lymphedema: classification, diagnosis and therapy. Vasc Med. 1998;3(2):145–156. doi: 10.1177/1358836X9800300209. [DOI] [PubMed] [Google Scholar]
- 22.Newman B, et al. Possible genetic predisposition to lymphedema after breast cancer. Lymphat Res Biol. 2012;10(1):2–13. doi: 10.1089/lrb.2011.0024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Finegold DN, et al. Connexin 47 mutations increase risk for secondary lymphedema following breast cancer treatment. Clin Cancer Res. 2012;18(8):2382–2390. doi: 10.1158/1078-0432.CCR-11-2303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Finegold DN, et al. HGF and MET mutations in primary and secondary lymphedema. Lymphat Res Biol. 2008;6(2):65–68. doi: 10.1089/lrb.2008.1524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jang JY, et al. Conditional ablation of LYVE-1+ cells unveils defensive roles of lymphatic vessels in intestine and lymph nodes. Blood. 2013;122(13):2151–2161. doi: 10.1182/blood-2013-01-478941. [DOI] [PubMed] [Google Scholar]
- 26.Olszewski WL, Engeset A, Romaniuk A, Grzelak I, Ziolkowska A. Immune cells in peripheral lymph and skin of patients with obstructive lymphedema. Lymphology. 1990;23(1):23–33. [PubMed] [Google Scholar]
- 27.Tassenoy A, et al. Demonstration of tissue alterations by ultrasonography, magnetic resonance imaging and spectroscopy, and histology in breast cancer patients without lymphedema after axillary node dissection. Lymphology. 2006;39(3):118–126. [PubMed] [Google Scholar]
- 28.Suami H, Pan WR, Taylor GI. Changes in the lymph structure of the upper limb after axillary dissection: radiographic and anatomical study in a human cadaver. Plast Reconstr Surg. 2007;120(4):982–991. doi: 10.1097/01.prs.0000277995.25009.3e. [DOI] [PubMed] [Google Scholar]
- 29.Mäkinen T, et al. PDZ interaction site in ephrinB2 is required for the remodeling of lymphatic vasculature. Genes Dev. 2005;19(3):397–410. doi: 10.1101/gad.330105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kilarski WW, et al. Optimization and regeneration kinetics of lymphatic-specific photodynamic therapy in the mouse dermis. Angiogenesis. 2014;17(2):347–357. doi: 10.1007/s10456-013-9365-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ran S, Montgomery KE. Macrophage-mediated lymphangiogenesis: the emerging role of macrophages as lymphatic endothelial progenitors. Cancers (Basel) 2012;4(3):618–657. doi: 10.3390/cancers4030618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Riabov V, Gudima A, Wang N, Mickley A, Orekhov A, Kzhyshkowska J. Role of tumor associated macrophages in tumor angiogenesis and lymphangiogenesis. Front Physiol. 2014;5:e84095. doi: 10.3389/fphys.2014.00075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ogata F, et al. Excess lymphangiogenesis cooperatively induced by macrophages and CD4(+) T cells drives the pathogenesis of lymphedema. J Invest Dermatol. 2016;136(3):706–714. doi: 10.1016/j.jid.2015.12.001. [DOI] [PubMed] [Google Scholar]
- 34.Suami H, Yamashita S, Soto-Miranda MA, Chang DW. Lymphatic territories (lymphosomes) in a canine: an animal model for investigation of postoperative lymphatic alterations. PLoS One. 2013;8(7):e84095. doi: 10.1371/journal.pone.0069222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rockson SG. Preclinical models of lymphatic disease: the potential for growth factor and gene therapy. Ann N Y Acad Sci. 2002;979:e84095. doi: 10.1111/j.1749-6632.2002.tb04868.x. [DOI] [PubMed] [Google Scholar]
- 36.Tammela T, et al. Therapeutic differentiation and maturation of lymphatic vessels after lymph node dissection and transplantation. Nat Med. 2007;13(12):1458–1466. doi: 10.1038/nm1689. [DOI] [PubMed] [Google Scholar]
- 37.Duffield JS, et al. Selective depletion of macrophages reveals distinct, opposing roles during liver injury and repair. J Clin Invest. 2005;115(1):56–65. doi: 10.1172/JCI22675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Maruyama K, et al. Inflammation-induced lymphangiogenesis in the cornea arises from CD11b-positive macrophages. J Clin Invest. 2005;115(9):2363–2372. doi: 10.1172/JCI23874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lee JY, et al. Podoplanin-expressing cells derived from bone marrow play a crucial role in postnatal lymphatic neovascularization. Circulation. 2010;122(14):1413–1425. doi: 10.1161/CIRCULATIONAHA.110.941468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gordon EJ, Rao S, Pollard JW, Nutt SL, Lang RA, Harvey NL. Macrophages define dermal lymphatic vessel calibre during development by regulating lymphatic endothelial cell proliferation. Development. 2010;137(22):3899–3910. doi: 10.1242/dev.050021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jeon BH, et al. Profound but dysfunctional lymphangiogenesis via vascular endothelial growth factor ligands from CD11b+ macrophages in advanced ovarian cancer. Cancer Res. 2008;68(4):1100–1109. doi: 10.1158/0008-5472.CAN-07-2572. [DOI] [PubMed] [Google Scholar]
- 42.Conrad C, et al. Multipotent mesenchymal stem cells acquire a lymphendothelial phenotype and enhance lymphatic regeneration in vivo. Circulation. 2009;119(2):281–289. doi: 10.1161/CIRCULATIONAHA.108.793208. [DOI] [PubMed] [Google Scholar]
- 43.Cross MJ, Dixelius J, Matsumoto T, Claesson-Welsh L. VEGF-receptor signal transduction. Trends Biochem Sci. 2003;28(9):488–494. doi: 10.1016/S0968-0004(03)00193-2. [DOI] [PubMed] [Google Scholar]
- 44.Rutkowski JM, Moya M, Johannes J, Goldman J, Swartz MA. Secondary lymphedema in the mouse tail: lymphatic hyperplasia, VEGF-C upregulation, and the protective role of MMP-9. Microvasc Res. 2006;72(3):161–171. doi: 10.1016/j.mvr.2006.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wynn TA, Ramalingam TR. Mechanisms of fibrosis: therapeutic translation for fibrotic disease. Nat Med. 2012;18(7):1028–1040. doi: 10.1038/nm.2807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hancock WW, Adams DH, Wyner LR, Sayegh MH, Karnovsky MJ. CD4+ mononuclear cells induce cytokine expression, vascular smooth muscle cell proliferation, and arterial occlusion after endothelial injury. Am J Pathol. 1994;145(5):1008–1014. [PMC free article] [PubMed] [Google Scholar]
- 47.Emeson EE, Shen ML, Bell CG, Qureshi A. Inhibition of atherosclerosis in CD4 T-cell-ablated and nude (nu/nu) C57BL/6 hyperlipidemic mice. Am J Pathol. 1996;149(2):675–685. [PMC free article] [PubMed] [Google Scholar]
- 48.Zhou X, Robertson AK, Hjerpe C, Hansson GK. Adoptive transfer of CD4+ T cells reactive to modified low-density lipoprotein aggravates atherosclerosis. Arterioscler Thromb Vasc Biol. 2006;26(4):864–870. doi: 10.1161/01.ATV.0000206122.61591.ff. [DOI] [PubMed] [Google Scholar]
- 49.Rutkowski JM, Markhus CE, Gyenge CC, Alitalo K, Wiig H, Swartz MA. Dermal collagen and lipid deposition correlate with tissue swelling and hydraulic conductivity in murine primary lymphedema. Am J Pathol. 2010;176(3):1122–1129. doi: 10.2353/ajpath.2010.090733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ugur S, et al. Risk factors of breast cancer-related lymphedema. Lymphat Res Biol. 2013;11(2):72–75. doi: 10.1089/lrb.2013.0004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sakorafas GH, Peros G, Cataliotti L, Vlastos G. Lymphedema following axillary lymph node dissection for breast cancer. Surg Oncol. 2006;15(3):153–165. doi: 10.1016/j.suronc.2006.11.003. [DOI] [PubMed] [Google Scholar]
- 52.Savetsky IL, et al. Obesity increases inflammation and impairs lymphatic function in a mouse model of lymphedema. Am J Physiol Heart Circ Physiol. 2014;307(2):H165–H172. doi: 10.1152/ajpheart.00244.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Avraham T, et al. Radiation therapy causes loss of dermal lymphatic vessels and interferes with lymphatic function by TGF-beta1-mediated tissue fibrosis. Am J Physiol, Cell Physiol. 2010;299(3):C589–C605. doi: 10.1152/ajpcell.00535.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Shin K, et al. TH2 cells and their cytokines regulate formation and function of lymphatic vessels. Nat Commun. 2015;6:e84095. doi: 10.1038/ncomms7196. [DOI] [PubMed] [Google Scholar]
- 55.Savetsky IL, et al. Th2 cytokines inhibit lymphangiogenesis. PLoS One. 2015;10(6):e84095. doi: 10.1371/journal.pone.0126908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kataru RP, et al. T lymphocytes negatively regulate lymph node lymphatic vessel formation. Immunity. 2011;34(1):96–107. doi: 10.1016/j.immuni.2010.12.016. [DOI] [PubMed] [Google Scholar]
- 57.Martinez-Corral I, et al. Vegfr3-CreER (T2) mouse, a new genetic tool for targeting the lymphatic system. Angiogenesis. 2016;19(3):433–445. doi: 10.1007/s10456-016-9505-x. [DOI] [PubMed] [Google Scholar]
- 58.Aschen SZ, et al. Lymph node transplantation results in spontaneous lymphatic reconnection and restoration of lymphatic flow. Plast Reconstr Surg. 2014;133(2):301–310. doi: 10.1097/01.prs.0000436840.69752.7e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Isaka N, Padera TP, Hagendoorn J, Fukumura D, Jain RK. Peritumor lymphatics induced by vascular endothelial growth factor-C exhibit abnormal function. Cancer Res. 2004;64(13):4400–4404. doi: 10.1158/0008-5472.CAN-04-0752. [DOI] [PubMed] [Google Scholar]
- 60.Weitman ES, et al. Obesity impairs lymphatic fluid transport and dendritic cell migration to lymph nodes. PLoS One. 2013;8(8):e84095. doi: 10.1371/journal.pone.0070703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zhou Q, Wood R, Schwarz EM, Wang YJ, Xing L. Near-infrared lymphatic imaging demonstrates the dynamics of lymph flow and lymphangiogenesis during the acute versus chronic phases of arthritis in mice. Arthritis Rheum. 2010;62(7):1881–1889. doi: 10.1002/art.27464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bu L, Gao M, Qu S, Liu D. Intraperitoneal injection of clodronate liposomes eliminates visceral adipose macrophages and blocks high-fat diet-induced weight gain and development of insulin resistance. AAPS J. 2013;15(4):1001–1011. doi: 10.1208/s12248-013-9501-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.van Rooijen N, van Kesteren-Hendrikx E. Clodronate liposomes: perspectives in research and therapeutics. J Liposome Res. 2002;12(1-2):81–94. doi: 10.1081/LPR-120004780. [DOI] [PubMed] [Google Scholar]
- 64.Ghanta S, et al. Regulation of inflammation and fibrosis by macrophages in lymphedema. Am J Physiol Heart Circ Physiol. 2015;308(9):H1065–H1077. doi: 10.1152/ajpheart.00598.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.