

Abstract
Background. Hypophosphatasia (HPP) is caused by loss-of-function mutation(s) of the gene that encodes the tissue-nonspecific isoenzyme of alkaline phosphatase (TNSALP). Consequently, cell-surface deficiency of TNSALP phosphohydrolase activity leads to extracellular accumulation of inorganic pyrophosphate, a natural substrate of TNSALP and inhibitor of mineralization. Children with HPP can manifest rickets, skeletal pain, deformity, fracture, muscle weakness, and premature deciduous tooth loss. Asfotase alfa is a recombinant, bone-targeted, human TNSALP injected s.c. to treat HPP. In 2012, we detailed the 1-year efficacy of asfotase alfa therapy for the life-threatening perinatal and infantile forms of HPP.
Methods. Here, we evaluated the efficacy and safety of asfotase alfa treatment administered to children 6–12 years of age at baseline who were substantially impaired by HPP. Two radiographic scales quantitated HPP skeletal disease, including comparisons to serial radiographs from similarly affected historical control patients.
Results. Twelve children receiving treatment were studied for 5 years. The 6-month primary endpoint was met, showing significant radiographic improvement. Additional significant improvements included patient growth, strength, motor function, agility, and quality of life, which for most patients meant achieving normal values for age- and sex-matched peers that were sustained at 5 years of treatment. For most, pain and disability resolved. Mild to moderate injection-site reactions were common and were sometimes associated with lipohypertrophy. Low anti–asfotase alfa antibody titers were noted in all patients. No evidence emerged for clinically important ectopic calcification or treatment resistance.
Conclusions. Asfotase alfa enzyme replacement therapy has substantial and sustained efficacy with a good safety profile for children suffering from HPP.
Trial Registration. ClinicalTrials.gov NCT00952484 (https://clinicaltrials.gov/ct2/show/NCT00952484) and NCT01203826 (https://clinicaltrials.gov/ct2/show/NCT01203826).
Funding. Alexion Pharmaceuticals Inc. and Shriners Hospitals for Children.
Asfotase alfa enzyme replacement therapy has substantial and sustained efficacy with a good safety profile for children suffering from hypophosphatasia.
Introduction
Hypophosphatasia is the inborn error of metabolism caused by loss-of-function mutation(s) of the gene that encodes the tissue-nonspecific isoenzyme of alkaline phosphatase (TNSALP) (1, 2). TNSALP is a cell-surface phosphohydrolase richly expressed in bone, liver, and kidney (3), and deficient TNSALP activity in hypophosphatasia leads to extracellular accumulation of its natural substrates, including inorganic pyrophosphate (PPi) (4) and pyridoxal 5′-phosphate (PLP) (5). PPi is a potent inhibitor of mineralization (6), and the excessive PPi in hypophosphatasia can block hydroxyapatite crystal formation within the skeletal matrix (7, 8), causing rickets during growth (9) or osteomalacia in adult life (10). Impaired hydrolysis of PLP in severely affected babies can lead to neurotransmitter deficiency and vitamin B6–dependent seizures (11). The clinical expressivity of hypophosphatasia is classified according to whether there are dental complications alone (odontohypophosphatasia) or to patient age when skeletal and other problems first manifest (1, 2, 12). Perinatal hypophosphatasia is almost always fatal soon after birth (13), and infantile hypophosphatasia has a mortality estimate of 50% during the first year of life (1, 2). Childhood hypophosphatasia features rickets often complicated by skeletal pain, fracturing, muscle weakness, abnormal ambulation or gait, and premature loss of deciduous teeth (1, 2, 14).
Asfotase alfa is a recombinant, bone-targeted, human TNSALP developed to treat hypophosphatasia (15). In 2012, we reported that infants and young children (≤3 years of age) with life-threatening perinatal and infantile hypophosphatasia showed substantial radiographic healing of rickets, improved respiratory status, and better physical function during 1 year of asfotase alfa treatment (9). Here, we assess the impact of 5 years of asfotase alfa treatment in a separate cohort of older children (6–12 years of age) with either infantile- or childhood-onset disease who were substantially impaired by hypophosphatasia, including effects on their skeletal manifestations, growth, physical function, and quality of life.
Results
Patient Characteristics.
Thirteen children — 11 boys and 2 girls with infantile hypophosphatasia or compromised by childhood hypophosphatasia (16) — were enrolled between September 2009 and January 2010 for treatment (Figure 1). Their case summaries are detailed in the Supplement; supplemental material available online with this article; doi:10.1172/jci.insight.85971DS1. Enrollment ages ranged from 6–12 years (Table 1). Reported skeletal abnormalities included knock knees (77%), rachitic chest (46%), bowing of leg(s) (39%), and craniosynostosis (31%) and were accompanied by muscle weakness in most patients and an unusual gait in all. Bone pain often limited activities (46%) and/or required analgesics (39%). Nearly half the patients had poor weight gain and prior difficulty feeding. Hypercalcemia had occurred in 31%. After 1 month of treatment, 1 boy withdrew for elective scoliosis surgery. Figure 2 is an example of the radiographic abnormalities that can be observed in a child with untreated hypophosphatasia.
Figure 1. Study design and patient participation.

During the 6-month initial phase involving 13 participants, 1 child withdrew for elective surgery after 1 month of treatment. The remaining 12 were assessed up to 5 years (60 months) of treatment and continue on study. Data were pooled across these 2 phases for analysis. The radiographic findings were contrasted to 2-year experience with 16 historical control patients. *ClinicalTrials.gov: NCT00952484, NCT01203826. LOCF, last observation carried forward.
Table 1. Patient demographic and baseline characteristics.
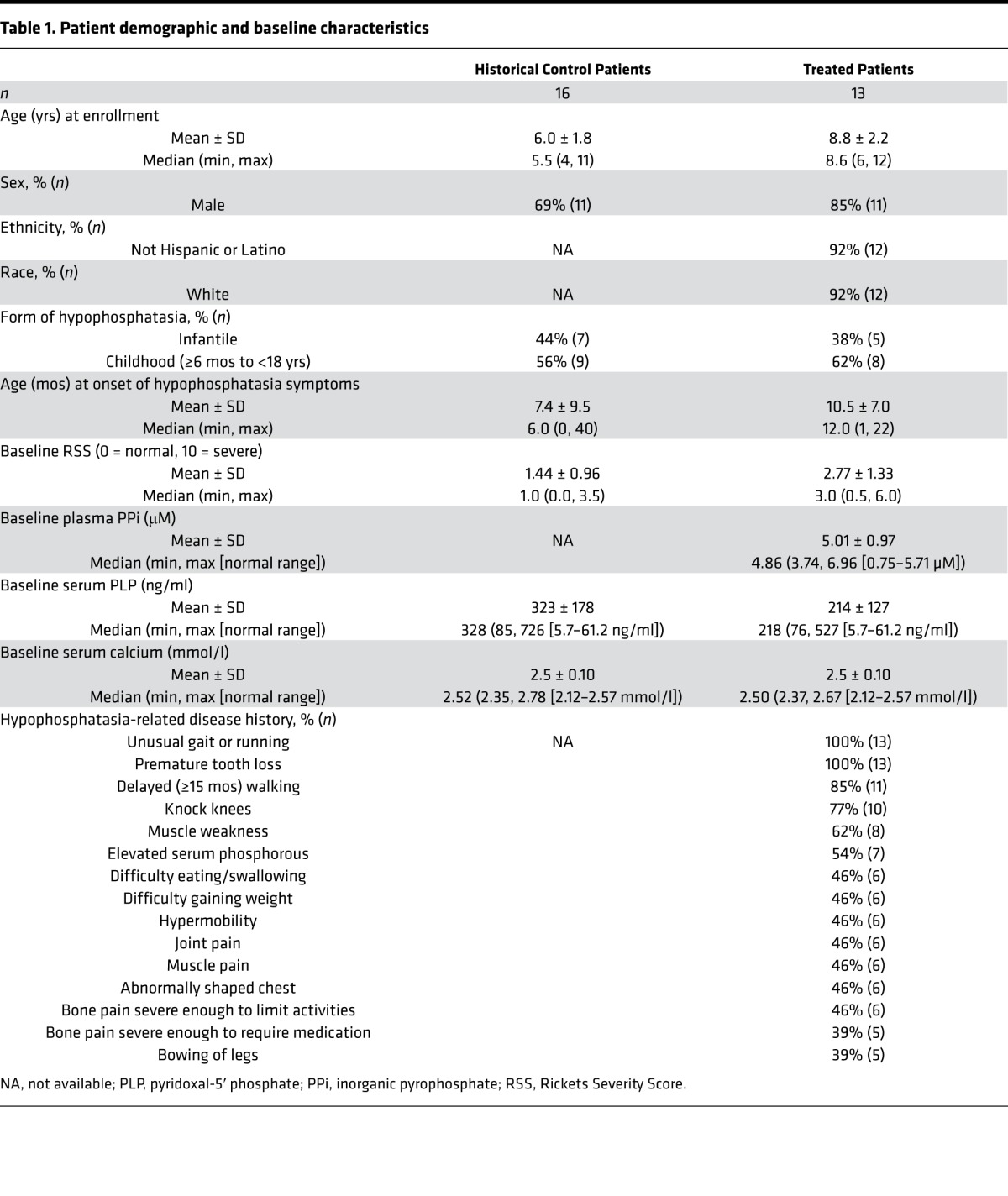
Figure 2. Skeletal features of untreated hypophosphatasia.

Untreated, the radiographic features of hypophosphatasia in children (e.g., patient 7) include, as in the wrist pictured, osteopenia, metaphyseal fraying, metaphyseal flaring, and metadiaphyseal sclerosis, and as in the knee pictured, characteristic “tongues” of radiolucency, irregularity of the provisional zone of calcification, distal metaphyseal demineralization, transverse subphyseal band of lucency, and apparent physeal widening.
The 16 historical controls — 11 boys and 5 girls with infantile or childhood hypophosphatasia — were slightly younger compared with the treatment group when their first study-eligible radiographs had been taken, and they had a lower median serum ALP level than the treatment group. All other assessed characteristics, including their medical histories and demographic findings, were similar to the treatment group (Table 1).
Primary endpoint: skeletal radiographic changes.
After 6 weeks of asfotase alfa administered s.c., the median Radiographic Global Impression of Change (RGI-C) score (see Methods) had improved significantly (+1.0 [0.0, +2.0; min, max]; P = 0.001). Except where noted, values are expressed as median (min, max). Further improvement occurred by 6 months (Figure 3) and persisted through 5 years (+2.2 [+1.7, +2.7]; P = 0.0005) (see Supplement). For no patient did the score worsen. However, at 5 years of treatment, the median score had not reached +3 for complete or nearly complete healing of all treated patients. In contrast, no significant change occurred in the RGI-C score of the historical controls spanning up to 2 years (Figure 3). The RGI-C responder analysis showed 69% (9/13) of treated patients achieved “responder” designation (RGI-C score ≥ +2) at 6 months of therapy (last observation carried forward [LOCF] data imputation applied) compared with 6% (1/16) of the historical controls (P = 0.007). The percentage of treated patients considered responders then further improved steadily; 75% at 2 years, 88% at 3 years, and 92% at 5 years (1 patient had withdrawn at month 1). Representative radiographs are illustrated in Figure 4. Consistent with the RGI-C findings, the Rickets Severity Score (RSS; see Methods) analysis confirmed improvement of rickets with treatment (documenting significantly decreased median RSS scores comparing the first eligible radiographs with all subsequent evaluated time points) as contrasted to the historical controls (Supplemental Table 1).
Figure 3. Skeletal improvements during treatment with asfotase alfa.

Changes in RGI-C scores during 6 months and 2 years of treatment with asfotase alfa. Individual dots illustrate the distribution of individual patient scores at each time point. Boxes represent the median RGI-C scores and represent first and third quartiles. Whisker lines above and below the boxes represent the end-range of patient scores. The RGI-C is scored on a scale ranging from –3 to +3, with 0 representing no change. Negative values represent worsening, and positive values represent improvement or healing. A score of +3 indicates nearly complete or complete healing. Median RGI-C scores for the historical controls and asfotase alfa were 0 and 2, respectively, at 6 months and 2 years. The number of patients assessed at each time point is shown below each bar. P ≤ 0.0001 by Wilcoxon signed-rank test at all time points compared with no change.
Figure 4. Representative radiographic changes.

Radiographic changes were documented in 12 treated patients and are illustrated in the Supplemental Methods. Patient 7, a 6-year-old boy at enrollment, is shown. RGI-C, Radiographic Global Impression of Change; RSS, Rickets Severity Score. Note the improvements at 6 months of treatment that persist after 5 years of therapy.
TNSALP replacement and substrates.
Twelve of the 13 treated patients had at least 1 mutant TNSALP allele identified using PCR and DNA sequencing of all coding exons and splice sites (Supplemental Table 2). The exception (patient 11) has not shown a TNSALP gene mutation despite extensive molecular investigations. The median serum ALP activity of the treatment group at baseline was 49 IU/l (min, max: 27, 68; see Supplement for age- and sex-specific normal ranges for each patient) and then rapidly increased, presumably reflecting asfotase alfa activity, with treatment. At 5 years of therapy, it remained markedly elevated at 5747 IU/l (3,039, 9,959). Baseline plasma PPi median concentration, 4.9 μM (3.7, 7.0), was near the upper limit of the age-dependent reference range (0.75–5.71 μM). Baseline plasma PLP median concentration, 218 ng/ml (76, 527), was clearly elevated (normal 5.7–61.2 ng/ml). At 6 weeks of treatment, rapid and significant reduction of plasma PPi and PLP concentrations had achieved the normal limits for most treated patients (2.0 μM [1.3, 3.1] and 10.6 ng/ml [0.9, 40.3], respectively). Then, normal levels generally persisted throughout the study (Supplemental Figure 1).
Growth.
With treatment, the first significant increase in the group’s weight Z scores (Figure 5A) was identified at 6 weeks (baseline: –1.21 [–8.2, 2.3]; 6 weeks: –0.92 [–8.0, 2.1], P = 0.0048), and the first significant increase in the group’s height Z scores was noted after 1.5 years (baseline: –1.26 [–6.6, 0.0]; 1.5 years: –0.87 [–6.6, –0.3], P = 0.0257) (Figure 5B). BMI was increased at 5 years of treatment (baseline: –0.57 [–1.4, 2.4]; 5 years: 0.05 [–1.0, 2.2], P = 0.0077) (Figure 5C).
Figure 5. Growth during treatment with asfotase alfa.

Growth was assessed by Z scores for (A) weight, (B) height, and (C) BMI. For all graphs, individual dots indicate individual patient scores at each time point. Median, min, max, and n values are given below each panel. P values at 5 years from Wilcoxon signed-rank test.
Strength.
Most treated patients became stronger for their age and sex; e.g., right hip abduction baseline median at 13.6 lbs was 7.3 and 22.1 (min, max, respectively; 50% predicted for healthy peers). At 6 months, 18.3 lbs, minimum and maximum values were 8.4 and 28.2, respectively (P = 0.0018 or 60% predicted). At 5 years, 30.7 lbs, minimum and maximum values were 19.5 and 84.4, respectively (P = 0.0079 or 91% predicted). Significant improvements were similarly documented for hip and knee extensors and knee flexors (Supplemental Figure 2).
Physical function.
Before treatment, gross motor delays and functional disability were documented in most patients; e.g., 9 of the 13 had a subnormal (<80% predicted) 6-minute walk test (6MWT). After 5 years of treatment, 7 of these 9 children achieved or surpassed 80% of the predicted distance (± 2 SD mean), indicating normal ambulation. The median score increased from 61% at baseline to 83% at 5 years (P = 0.0002; Figure 6A; Video 1; Supplemental Figure 3).
Figure 6. Functional assessment during treatment with asfotase alfa.

(A) Percent predicted 6MWT distance median achieves the normal range after 6 months of treatment and is sustained at 5 years of therapy. P ≤ 0.0005 by paired t test for the mean difference between each time point and baseline. (B) Gross motor function, assessed using the BOT-2 Running Speed and Agility subtest (17), shows median scores reaching the normal range (gray area) by 1 year of treatment that were sustained. Median, min, max, and n values are given within the figure. (C) Disability assessment using the CHAQ (24) shows patient improvement. *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001 mean difference for each time point compared with baseline by paired t test. Median, min, max, and n values are given below each figure. For all graphs, individual dots indicate individual patient scores at each time point. Gray area represents the normal range.
The median value for the Bruininks-Oseretsky Test of Motor Proficiency, Second Edition (BOT-2) Strength and Agility Composite Score at baseline (28.0 [20.0, 37.0]; percentile rank = 1.0) was greater than 2 SD below the median score of 50 for healthy, age-matched peers (17). During treatment, it improved, reaching the normal range after 1 year, and was significantly better after 5 years (46.0 [33.0, 64.0]; P ≤ 0.0001; percentile rank = 35.0) (Figure 6B; Video 2; Supplemental Figure 4). The magnitude of change in the standard scores indicated gross motor function within the normal range (± 1 SD mean).
Pain and disability assessments.
Improvements in the above effort-dependent functional tests were substantiated by the parent-reported Child Health Assessment Questionnaire (CHAQ) and Pediatric Outcomes Data Collection Instrument (PODCI) results. These clinically meaningful advances persisted through 5 years. The elevated CHAQ pain (20.0 [0.0, 72.0]) and disability (1.0 [0.0, 2.3]) medians at baseline were 0 at 5 years of treatment, indicating no pain or disability for most patients (0.0 [0.0, 60.0], P = 0.1125; 0.0 [0.0, 1.0], P = 0.0002, respectively) (Figure 6C). The PODCI global function median score, which was abnormal at baseline (27.0 [–2.0, 55.0]), improved to slightly above normal at the 5-year assessment (52.0 [28.0, 57.0], P = 0.0002).
Safety.
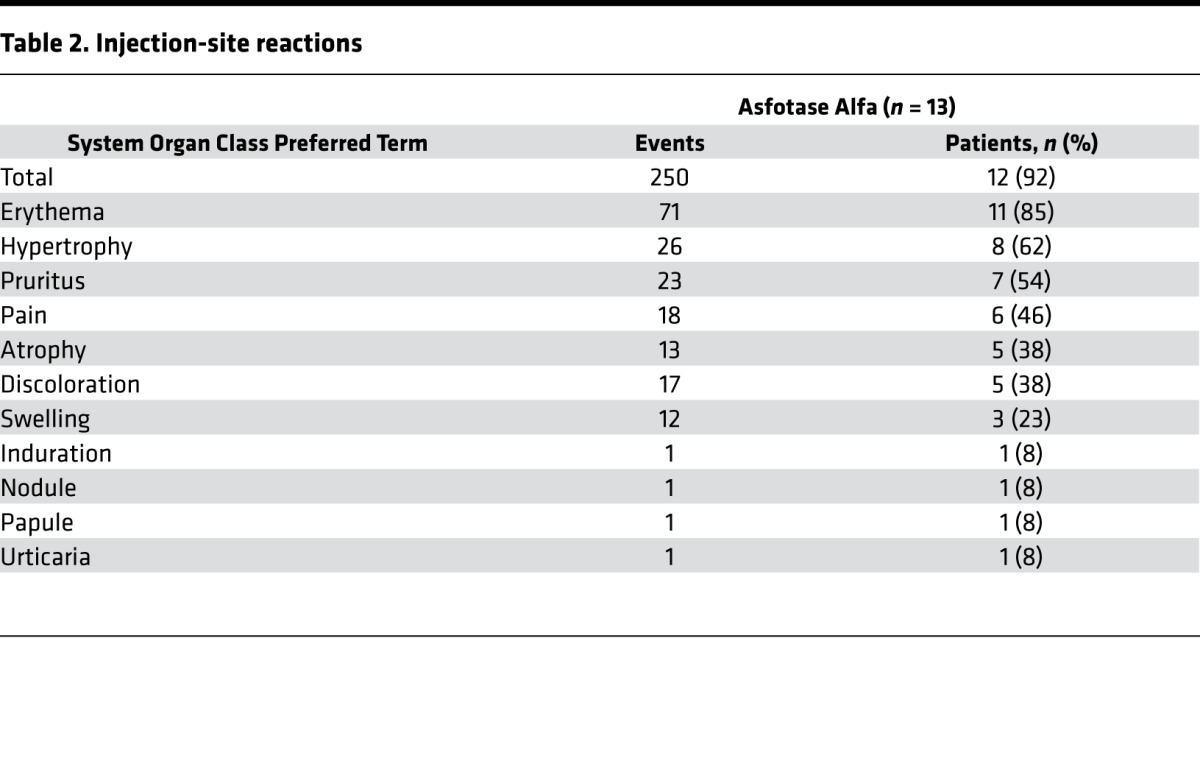
There were no deaths, serious adverse events (AEs), or withdrawals due to AEs. One patient receiving asfotase alfa 9 mg/kg/week s.c. underwent dose reduction for a low plasma PLP level. Four patients had injection-associated reactions (IARs) (10 events) during the first month of treatment. All were injection-site reactions (ISRs), with the exception of nausea and peripheral edema in 1 patient, and all were assessed by the investigators as mild. Each patient experienced at least 1 treatment-emergent AE; 86% mild and 14% moderate in severity. Most AEs (53%) were considered by the investigators to be unrelated to the treatment. When we excluded the ISRs, 2% of AEs were deemed treatment related. The most common therapy-related AEs were ISRs that were typically mild (Table 2 and Supplemental Figure 5A). For 8 patients, 26 events were considered focal lipohypertrophy, of which 8 were moderate in severity (Supplemental Figure 5B); the others were mild at data cutoff, and no dose adjustments were made. The lipohypertrophy occurred for 6 patients receiving 6 injections weekly and 2 patients receiving 3 injections weekly, all receiving the same weekly dose (mg/kg) of asfotase alfa. At 3 years of treatment and thereafter, localized lipohypertrophy persisted in 6 of the 8 patients. The dose of asfotase alfa was reduced for 1 patient with mild ISRs (multiple macules). To minimize ISRs, injection-site rotation was emphasized for all patients.
Table 2. Injection-site reactions.
Surveillance of the patients’ radiographs showed no evidence of ectopic calcification. On ophthalmoscopy, the retinal examinations were consistently unremarkable. However, 46% (6 of 13) of the patients showed small refractile deposits in the conjunctiva or cornea presumed to contain calcium. For 5 patients, these were considered possibly or probably related to the treatment. No deposit was deemed clinically apparent or significant. No patient developed nephrocalcinosis, although after approximately 2 years of treatment, renal sonography revealed “focal echogenicity” consistent with a small renal stone present for 6 months in 1 kidney of 1 patient.
All 12 patients tested would show anti–asfotase alfa antibodies. Five of the 6 patients who began treatment in the 3 mg/kg dose group were antibody positive by Week 6, and all were antibody positive at Week 12 of treatment. In those starting in the 2 mg/kg dose group, 1 patient was antibody positive by Week 6, and 5 patients were positive by Week 48. In 5 of the 12 patients, the antibodies were neutralizing in vitro at 1 or more time points, but with no apparent compromise of therapeutic efficacy.
Discussion
Hypophosphatasia features the greatest range of severity of all skeletal diseases (1) and represents the last rickets or osteomalacia to have a medical treatment (2, 18). Asfotase alfa is a first-in-class, bone-targeted, enzyme replacement therapy for hypophosphatasia (2).
In this study, asfotase alfa was administered for the first time to older children (6–12 years old) impaired by the infantile and childhood forms of hypophosphatasia, all of whom had skeletal involvement, muscle weakness, and/or difficulties with ambulation. Due to the rarity of hypophosphatasia (1), a concurrent control group was not possible, but evaluation of the primary outcome objective (12) concerning the radiographic changes of this disorder was improved using radiographs from a pediatric historical control group, also with infantile or childhood hypophosphatasia, assessed concomitantly by the same radiologists.
Skeletal improvement with asfotase alfa therapy was verified using 2 scales to study (9) the sequential radiographs of the 12 treated patients and the 16 age- and sex-matched historical control patients. Both scales documented no change in the controls but instead documented substantial healing of the skeletal disease after 6 months of treatment that persisted through the 5 years of therapy. Skeletal healing did not become complete for all patients.
As the weakness improved and the skeleton mended, there was substantially better physical function, including walking distance, and most patients showed improved growth. Gross motor function, strength for all proximal muscles tested, agility, and stamina typically achieved their normal ranges and together were the basis for independent and age-appropriate performance of daily living activities. For most patients, their pain and disability resolved. Consistent with the radiographic findings, the treated patients demonstrated better skeletal mineralization assessed as bone mineral density quantitated by dual-energy x-ray absorptiometry and histomorphometry of transiliac crest bone biopsies (data not shown). Low titers of anti–asfotase alfa antibodies were found in all patients tested, but there was no evidence of resistance to this biologic.
Although the pathogenesis of the muscle weakness of hypophosphatasia is not understood (1, 2), it can be an important complication. Notably, the muscle weakness, hyperphosphatemia, and rickets or osteomalacia characteristic of hypophosphatasia (1, 2) are hallmarks of toxicity from the first-generation bisphosphonate, etidronate, a synthetic analog of PPi (19). Perhaps the high extracellular levels of PPi in hypophosphatasia somehow compromise muscle function and are corrected by asfotase alfa treatment.
No deaths or safety-related discontinuation of therapy occurred during this study. Asfotase alfa treatment was generally well tolerated and safe. Although it markedly increased circulating ALP activity, no clinically important ectopic mineralization occurred. Possibly, avoidance of ectopic calcification in our treated patients reflected reduction of extracellular PPi levels into the normal range but not overcorrection (i.e., suppression) (19). The most common AEs were mild to moderate ISRs. Lipohypertrophy appeared in some children, but its pathogenesis is unknown.
In conclusion, asfotase alfa treatment has substantial efficacy for children impaired by hypophosphatasia. Better stature and improved strength and agility accompanied significant healing of the skeletal manifestations and persisted through 5 years of treatment. The treatment was well tolerated and seemed safe, with a favorable benefit-risk profile. In 2015, asfotase alfa became the only approved medical therapy for hypophosphatasia in Japan and the only approved medical therapy for pediatric-onset hypophosphatasia in Canada, the European Union, and the United States.
Methods
Study design.
This open-label study (NCT00952484 and NCT01203826) sponsored by Alexion Pharmaceuticals Inc. evaluated the efficacy, safety, tolerability, and pharmacokinetics of asfotase alfa given to children compromised by hypophosphatasia. The study began as a 6-month assessment of 6- to 12-year-old, Tanner stage ≤2 survivors of infantile hypophosphatasia or patients with childhood hypophosphatasia (16). Those who completed this initial study entered an extension phase (Figure 1). Our report summarizes the findings during their first 5 years of treatment. The data cutoff was November 5, 2014; the study is ongoing.
Inclusion criteria required radiographic skeletal features of hypophosphatasia-related rickets, serum ALP activity below age-adjusted lower limits of normal, and plasma PLP levels at least twice the upper limit of normal. Serum 25-hydroxyvitamin D had to be greater than or equal to 20 ng/ml (50 nM). Exclusion criteria included treatable rickets, hypocalcemia, hypophosphatemia, or bisphosphonate exposure (see Supplemental Methods). Radiographic skeletal changes during the first 2 years of asfotase alfa treatment were compared with similar but untreated pediatric historical controls with hypophosphatasia who had been studied at Shriners Hospital for Children but were too old for the treatment study or declined participation. Their bilateral wrist and/or knee radiographs, previously obtained at intervals spanning 6 months to 2 years between 5 and 12 years of age, were evaluated as close to 6 months or 1–2 years apart for comparison with the treatment group.
Asfotase alfa was provided by the sponsor, Alexion Pharmaceuticals Inc. Patients to receive s.c. injections of asfotase alfa (40 or 100 mg/ml concentration) were initially randomized to 2 or 3 mg/kg thrice weekly (i.e., 6 or 9 mg/kg/week) for 6 months (Figure 1). In the extension study, patients initially received 3 mg/kg/week either as 0.5 mg/kg 6 times weekly or 1 mg/kg thrice weekly until preliminary analysis led to a protocol amendment that doubled the dose to 6 mg/kg/week to maintain therapeutic efficacy. Doses could be adjusted by the investigator together with the sponsor for safety concerns (any time) or lack of efficacy (extension phase only). Since a separately identified historical control patient group was used for comparison, there was no randomization between the drug-treated and control patients.
Outcome assessments.
The primary endpoint objective, evaluation of changes in the skeletal manifestations of hypophosphatasia (see Supplemental Methods), was obtained using the RGI-C (9) and RSS (20). The RGI-C, a 7-point ordinal scale, assesses hypophosphatasia skeletal disease in wrists and knees, including physeal widening, irregularity of the provisional zone of calcification, metaphyseal flaring, radiolucencies, and patchy osteosclerosis, as well as recent fractures (see Supplemental Methods) (9). The changes compared with baseline were quantified independently by 3 pediatric radiologists blinded to treatment time points and group assignments. For the RGI-C, +3 signifies complete or nearly complete healing, whereas –3 represents severe worsening. The scores per patient were averaged across radiologists at each time point. An RGI-C “responder” had a score greater than or equal to +2 (i.e., substantial or near/complete healing) (9). The RSS, which was developed to assess nutritional rickets, was scored by the same individual rater to: (i) evaluate the treated patients’ radiographs throughout the study and (ii) evaluate the historical control radiographs representing 6-month, 1-year, and 2-year intervals. Knee and wrist growth-plate abnormalities and metaphyseal fraying and cupping were rated according to a 10-point scale (0 = no rickets; 10 = severe rickets), with scores for each radiograph determined in random order (Supplemental Table 3) (20).
Secondary endpoint assessments included changes in TNSALP substrate levels (i.e., plasma PPi and PLP); growth (i.e., height, weight, and BMI Z scores); strength of muscle groups that move the hips and knees, measured using a hand-held dynamometer (21); walking ability and endurance/speed, using a 6MWT (22); physical function and strength by BOT-2 (17); global function, including pain and interference with normal activities, by the PODCI (23); and disability by the CHAQ (24) (Supplemental Methods). Normative PODCI scores for a healthy population have a mean of 50 with standard deviation of 10 and range from –77 (worst) to +58 (best). The CHAQ disability scores range from 0–3, with higher scores indicating greater disability.
Safety and tolerability primary endpoint surveillance included evaluation of AEs, deaths, vital signs, physical examination, laboratory testing (standard biochemistry, hematology, and urinalysis; Covance Inc.), ophthalmologic examination for ectopic calcification (during the extension phase), renal ultrasound for nephrocalcinosis, and anti–asfotase alfa antibody formation.
Statistics.
Data management and statistical analyses (SAS Version 9.2, SAS Institute Inc.) were performed by the sponsor. Except where noted, values are expressed as median (min, max). The RGI-C median scores were compared using Wilcoxon rank-sum test. Missing values were imputed with LOCF, and patients with no postbaseline assessment were assigned “no change” (e.g., RGI-C = 0). Observed median and mean within-group changes were also assessed for difference from 0 using the Wilcoxon signed-rank test and paired t test. All analyses were 2-sided, with significance defined as P less than 0.05. Otherwise, descriptive statistics are reported.
Study approval.
Safety and efficacy surveillance included an independent monitoring board. The study was designed by the authors and sponsor. The protocol, its amendments, and informed consent/assent documents were approved by the governing IRBs for Shriners Hospital for Children and for the University of Manitoba. Written informed consent was obtained from the parent(s) or legal guardian(s) and assent from the study patients. All authors affirm data completeness and accuracy and protocol adherence.
Author contributions:
MPW conceived and helped design the study. MPW, KLM, CRG, AY, KEM, DP, SM, KPF, DDT, KK, ALR, and KH acquired the data. MPW, KLM, CRG, TO, WHM, DP, SM, KPF, DDT, and ALR analyzed and interpreted the data. All the authors made substantial contributions to drafting or revising the article critically for important intellectual content and approved the final version to be published; each author takes public responsibility for it.
Supplementary Material
Acknowledgments
We thank the parents and patients for making this study possible. Identifiable patients in Videos 1 and 2 have provided a signed statement of informed consent to publish their image (in print and online). We are grateful to Thomas E. Herman, Rebecca Hulett, Geetika Khanna, and Thomas Thacher for radiographic evaluations; Jill Mayhew for performing the physical therapy assessments; Beth Leiro for central scoring of source documents; and Catherine Siener and Elizabeth Malkus for performing the assessments for the Shriners Hospital for Children site. Ian Clark, (Winnipeg), James Hoekel, and Mark Rallo (St. Louis) provided the ophthalmologic assessments. Deborah Wenkert, Hal Landy, and Alison Skrinar helped design and initiate the study. Agustin Melian helped analyze and interpret the data and provided critical comments for the draft manuscript. These data were presented in part at the following meetings: 92nd Annual Meeting of the Endocrine Society 2010, San Diego, California, USA; 33rd Annual Meeting of the American Society for Bone and Mineral Research 2011, San Diego, California, USA; 53rd Annual Meeting of the European Society for Paediatric Endocrinology 2014, Dublin, Ireland; 35th Annual Meeting of the American Society for Bone and Mineral Research 2014, Houston, Texas, USA; and 97th Annual Meeting of the Endocrine Society 2015, San Diego, California, USA. Eileen Sawyer, and Stephanie Rock of Alexion Pharmaceuticals Inc. helped create, edit, and finalize the manuscript. SVS Writing Group (Chicago, Illinois, USA) and PharmaWrite LLC (Princeton, New Jersey, USA), funded by Alexion Pharmaceuticals Inc., worked to prepare the report.
Footnotes
Tatjana Odrljin’s present affiliation is Shire, Lexington, Massachusetts, USA
Role of funding source: Alexion Pharmaceuticals Inc. was involved in the trial design and data interpretation and funded the data collection and analyses. Editorial and writing support was funded by Alexion Pharmaceuticals Inc. and Shriners Hospitals for Children, which also funded the acquisition of the historical control radiographs.
Conflict of interest: M.P. Whyte is a study investigator for Alexion Pharmaceuticals Inc. and has received research grants, honoraria, and travel support from Alexion Pharmaceuticals Inc. K.L. Madson received honoraria, research grants, and travel support from Alexion Pharmaceuticals Inc. D. Phillips is a consultant for Alexion Pharmaceuticals Inc., which sponsored the study. K. Hamilton has received honorarium and travel support from Alexion Pharmaceuticals Inc. K.P. Fujita, D.D. Thompson, and S. Moseley are employees with stock options at Alexion Pharmaceuticals Inc. T. Odrljin is a former employee with stock options of Alexion Pharmaceuticals Inc. and has a patent pending for asfotase alfa. C. Rockman-Greenberg is study investigator for Alexion Pharmaceuticals Inc. and has received honoraria and travel support from Alexion Pharmaceuticals Inc. The institution for M.P. Whyte, K.L. Madson, A.L. Reeves, and K.E. Mack (Shriners Hospitals for Children) has received research grants from Alexion Pharmaceuticals Inc. in support for the submitted work.
Reference information:JCI Insight. 2016;1(9):e85971. doi:10.1172/jci.insight.85971.
Contributor Information
Michael P. Whyte, Email: mwhyte@shrinenet.org.
Katherine L. Madson, Email: kmadson@shrinenet.org.
Dawn Phillips, Email: dawnphillipspt@gmail.com.
Amy Yakimoski, Email: ayakimoski@chrim.ca.
Kim Hamilton, Email: kimberly_erin@hotmail.com.
Kori Kagan, Email: kkagan@exchange.hsc.mb.ca.
Kenji P. Fujita, Email: fujitak@alxn.com.
David D. Thompson, Email: thompson.david.d@gmail.com.
Scott Moseley, Email: MoseleyS@alxn.com.
Tatjana Odrljin, Email: odrljit@yahoo.com.
References
- 1. Whyte MP. Hypophosphatasia. In: Thakker RV, Whyte MP, Eisman JA, Igarashi T, eds. Genetics of Bone Biology and Skeletal Disease. New York, New York, USA: Academic Press; 2013:337–360. [Google Scholar]
- 2.Whyte MP. Hypophosphatasia - aetiology, nosology, pathogenesis, diagnosis and treatment. Nat Rev Endocrinol. 2016;12(4):233–246. doi: 10.1038/nrendo.2016.14. [DOI] [PubMed] [Google Scholar]
- 3. Millán JL. Mammalian Alkaline Phosphatases. From Biology to Applications in Medicine and Biotechnology. Weinheim, Germany: Wiley-VCH Verlag GmbH & Co.; 2006. [Google Scholar]
- 4.Russell RG. Excretion of inorganic pyrophosphate in hypophosphatasia. Lancet. 1965;2(7410):461–464. doi: 10.1016/s0140-6736(65)91422-4. [DOI] [PubMed] [Google Scholar]
- 5.Whyte MP, Mahuren JD, Vrabel LA, Coburn SP. Markedly increased circulating pyridoxal-5′-phosphate levels in hypophosphatasia. Alkaline phosphatase acts in vitamin B6 metabolism. J Clin Invest. 1985;76(2):752–756. doi: 10.1172/JCI112031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fleisch H, Russell RG, Straumann F. Effect of pyrophosphate on hydroxyapatite and its implications in calcium homeostasis. Nature. 1966;212(5065):901–903. doi: 10.1038/212901a0. [DOI] [PubMed] [Google Scholar]
- 7.Anderson HC, Hsu HH, Morris DC, Fedde KN, Whyte MP. Matrix vesicles in osteomalacic hypophosphatasia bone contain apatite-like mineral crystals. Am J Pathol. 1997;151(6):1555–1561. [PMC free article] [PubMed] [Google Scholar]
- 8. Whyte MP. Hypophosphatasia: nature’s window on alkaline phosphatase function in man. In: Bilezikian JP, Raisz LG, Rodan GA, eds. Principles of Bone Biology. 3rd ed. San Diego: Academic Press; 2008:1573-1598. [Google Scholar]
- 9.Whyte MP, et al. Enzyme-replacement therapy in life-threatening hypophosphatasia. N Engl J Med. 2012;366(10):904–913. doi: 10.1056/NEJMoa1106173. [DOI] [PubMed] [Google Scholar]
- 10.Whyte MP, Mumm S, Deal C. Adult hypophosphatasia treated with teriparatide. J Clin Endocrinol Metab. 2007;92(4):1203–1208. doi: 10.1210/jc.2006-1902. [DOI] [PubMed] [Google Scholar]
- 11.Baumgartner-Sigl S, et al. Pyridoxine-responsive seizures as the first symptom of infantile hypophosphatasia caused by two novel missense mutations (c.677T>C, p.M226T; c.1112C>T, p.T371I) of the tissue-nonspecific alkaline phosphatase gene. Bone. 2007;40(6):1655–1661. doi: 10.1016/j.bone.2007.01.020. [DOI] [PubMed] [Google Scholar]
- 12.Whyte MP, et al. Asfotase alfa treatment improves survival for perinatal and infantile hypophosphatasia. J Clin Endocrinol Metab. 2016;101(1):334–342. doi: 10.1210/jc.2015-3462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Leung EC, Mhanni AA, Reed M, Whyte MP, Landy H, Greenberg CR. Outcome of perinatal hypophosphatasia in manitoba mennonites: a retrospective cohort analysis. JIMD Rep. 2013;11:73–78. doi: 10.1007/8904_2013_224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Seshia SS, Derbyshire G, Haworth JC, Hoogstraten J. Myopathy with hypophosphatasia. Arch Dis Child. 1990;65(1):130–131. doi: 10.1136/adc.65.1.130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Millán JL, et al. Enzyme replacement therapy for murine hypophosphatasia. J Bone Miner Res. 2008;23(6):777–787. doi: 10.1359/JBMR.071213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Whyte MP, et al. Hypophosphatasia: validation and expansion of the clinical nosology for children from 25 years experience with 173 pediatric patients. Bone. 2015;75:229–239. doi: 10.1016/j.bone.2015.02.022. [DOI] [PubMed] [Google Scholar]
- 17.Deitz JC, Kartin D, Kopp K. Review of the Bruininks-Oseretsky Test of Motor Proficiency, Second Edition (BOT-2) Phys Occup Ther Pediatr. 2007;27(4):87–102. doi: 10.1080/J006v27n04_06. [DOI] [PubMed] [Google Scholar]
- 18. Whyte MP. Hypophosphatasia. In: Glorieux FH, Jeuppner H, Pettifor JM, eds. Pediatric Bone: Biology & Diseases. 3rd ed. San Diego: Elsevier (Academic Press); 2012:771–794. [Google Scholar]
- 19.Otero JE, et al. Severe skeletal toxicity from protracted etidronate therapy for generalized arterial calcification of infancy. J Bone Miner Res. 2013;28(2):419–430. doi: 10.1002/jbmr.1752. [DOI] [PubMed] [Google Scholar]
- 20.Thacher TD, Fischer PR, Pettifor JM, Lawson JO, Manaster BJ, Reading JC. Radiographic scoring method for the assessment of the severity of nutritional rickets. J Trop Pediatr. 2000;46(3):132–139. doi: 10.1093/tropej/46.3.132. [DOI] [PubMed] [Google Scholar]
- 21.Brinkmann JR. Comparison of a hand-held and fixed dynamometer in measuring strength of patients with neuromuscular disease. J Orthop Sports Phys Ther. 1994;19(2):100–104. doi: 10.2519/jospt.1994.19.2.100. [DOI] [PubMed] [Google Scholar]
- 22.Henricson E, et al. Percent-predicted 6-minute walk distance in duchenne muscular dystrophy to account for maturational influences. PLoS Curr. 2012;4:e85971. doi: 10.1371/currents.RRN1297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Daltroy LH, Liang MH, Fossel AH, Goldberg MJ. The POSNA pediatric musculoskeletal functional health questionnaire: report on reliability, validity, and sensitivity to change. Pediatric Outcomes Instrument Development Group. Pediatric Orthopaedic Society of North America. J Pediatr Orthop. 1998;18(5):561–571. doi: 10.1097/00004694-199809000-00001. [DOI] [PubMed] [Google Scholar]
- 24.Singh G, Athreya BH, Fries JF, Goldsmith DP. Measurement of health status in children with juvenile rheumatoid arthritis. Arthritis Rheum. 1994;37(12):1761–1769. doi: 10.1002/art.1780371209. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.