

Abstract
Immune-suppressed organ transplant recipients (OTRs) can develop catastrophic squamous cell carcinoma (SCC), characterized by multiple primary tumors, extensive body surface area involvement, or metastases. There are currently no curative systemic therapies available. We previously showed that IL-22 enhances SCC proliferation. Herein, we examined links between cyclosporine (CSA), IL-22, and SCC in patients, cell lines, and mice with UV light–induced SCC. Eighteen of 114 OTRs developed catastrophic SCC, which was strongly associated with CSA treatment. We found that CSA drives T cell polarization toward IL-22–producing T22 cells, and CSA treatment increased IL-22 receptor in SCC cells. SCC tissue from OTRs showed increased expression of IL-22RA1. CSA potentiated rescue by IL-22 of serum-starved SCC cells; treatment of SCC cells with IL-22 and CSA increased both their migratory and invasive capacity. In a UV-induced model of SCC in SKH-1 immunocompetent mice, treatment with anti–IL-22 antibody reduced tumor number and tumor burden. We found that catastrophic SCC in OTRs is associated with CSA use, which may be acting by favoring T22 polarization. Since anti–IL-22 antibody administration decreased tumor number and tumor burden in vivo, blockade of the IL-22 axis may be developed as a viable therapeutic option for catastrophic SCC.
Transplant immunosuppression by cyclosporine A promotes production of IL-22 by immune cells, which in turn mediates aggressive behavior by squamous cell carcinoma.
Introduction
Cutaneous squamous cell carcinoma (SCC) is the second most common human cancer and accounts for the majority of non–melanoma-related metastases and up to 10,000 deaths in the United States each year (1, 2). Recent studies show that the number of deaths from SCC exceeds deaths from melanomas in the southern United States (2). Although most primary cutaneous SCCs (cSCCs) can be cured via surgical excision, certain subtypes may present as highly aggressive tumors, with elevated rates of proliferation and significant invasive potential (3). In particular, transplant-associated SCC (TSCC), which occurs in immune-suppressed solid organ transplant recipients (OTRs) may be considerably more aggressive than SCC in immune competent patients, with metastatic rates as high as 8% (4, 5). Additionally, the incidence of SCC in OTRs is estimated to be over 100 times greater than that of the general population (6). Furthermore, OTRs may experience catastrophic carcinomatosis defined by numerous primary cancers, extensive dysplasia involving more than 50% body surface area, or metastases (7). This can make a surgical cure difficult, or often impossible, due to an inability to achieve a clear margin or tumor-free plane (4, 7). To date, no systemic medical therapies have emerged for the treatment of TSCC, and it therefore remains a significant cause of morbidity and mortality in OTRs (4, 7). In the case of catastrophic carcinomatosis, where surgical options are no longer viable, the need for a systemic therapy is even more essential in order to offer any form of treatment with curative intent. Therefore, there exists a real need for the development of targeted medical therapy for the treatment of aggressive SCC in transplant recipients.
It is a challenge to balance medical management of catastrophic cancer in OTRs while maintaining immune suppression at appropriate levels to ensure allograft survival. The mainstay of therapy for the prevention of allograft rejection in transplant recipients has been chronic immunosuppression using calcineurin inhibitors such as cyclosporine (CSA). CSA acts by binding to intracellular cyclophilin molecules — preventing the activation of the calcium-dependent phosphatase calcineurin — and inhibits the expression of immune-related cytokines such as IL-2 and IFN-γ (8, 9). The use of these agents has been associated with an increased incidence of SCCs in particular primary cSCC, which occur proportionally to the intensity and duration of treatment (10, 11). One proposed mechanism underlying the tumor-promoting effect seen with CSA use involves decreased tumor surveillance, which may occur as a result of diminished IL-2 production and systemic immunosuppression (12). Additionally, CSA has been shown to have direct transformative effects on keratinocytes and may induce phenotypic changes that can increase cell invasiveness and unregulated tumor growth (13, 14). CSA has also been associated with the promotion and stimulation of cellular proliferation in normal human fibroblasts (15). Currently, however, the precise mechanism underlying the tumorigenic effect of CSA on SCCs — and its association with catastrophic SCC, as defined above — remains largely unknown.
Recently, we found that the TSCC immune microenvironment shows a distinct immune phenotype when compared with SCC lesions from immunocompetent patients (16). In particular, we found an increased Treg/CD8+ T cell ratio and an increased percentage of CD8+ IL-22–producing T cells (T22 cells) (16). These cells have been shown to produce IL-22 without IL-17 and are highly expressed in benign hyperproliferative conditions such as psoriasis and atopic dermatitis (17, 18). IL-22 itself has been implicated in the promotion of inflammation and the suppression of keratinocyte apoptosis via the inhibition of terminal keratinocyte differentiation (19–21). Increased expression of IL-22 and related cytokines may thus help explain the increased proliferation and subsequent poorer outcomes observed in TSCC.
We therefore investigated the role of CSA in catastrophic SCC in OTR and its relationship with the IL-22 axis in SCC in vitro. We further defined the presence of IL-22R in SCC and TSCC, as well as the mechanism through which CSA may contribute to IL-22–mediated SCC proliferation. Furthermore, we show that CSA itself may directly enhance the proliferation of cSCC cells. Finally, we demonstrate that IL-22 blockade decreases SCC number and burden in a murine model that mirrors human SCC development. Treatment with CSA may thus, in part, be responsible for the generation of a distinct and unique immune background, which promotes the development of the proliferative, invasive, and evasive SCC phenotype seen in OTR. IL-22 blockade may allow for treatment of widespread SCC without compromising CSA-mediated immune suppression.
Results
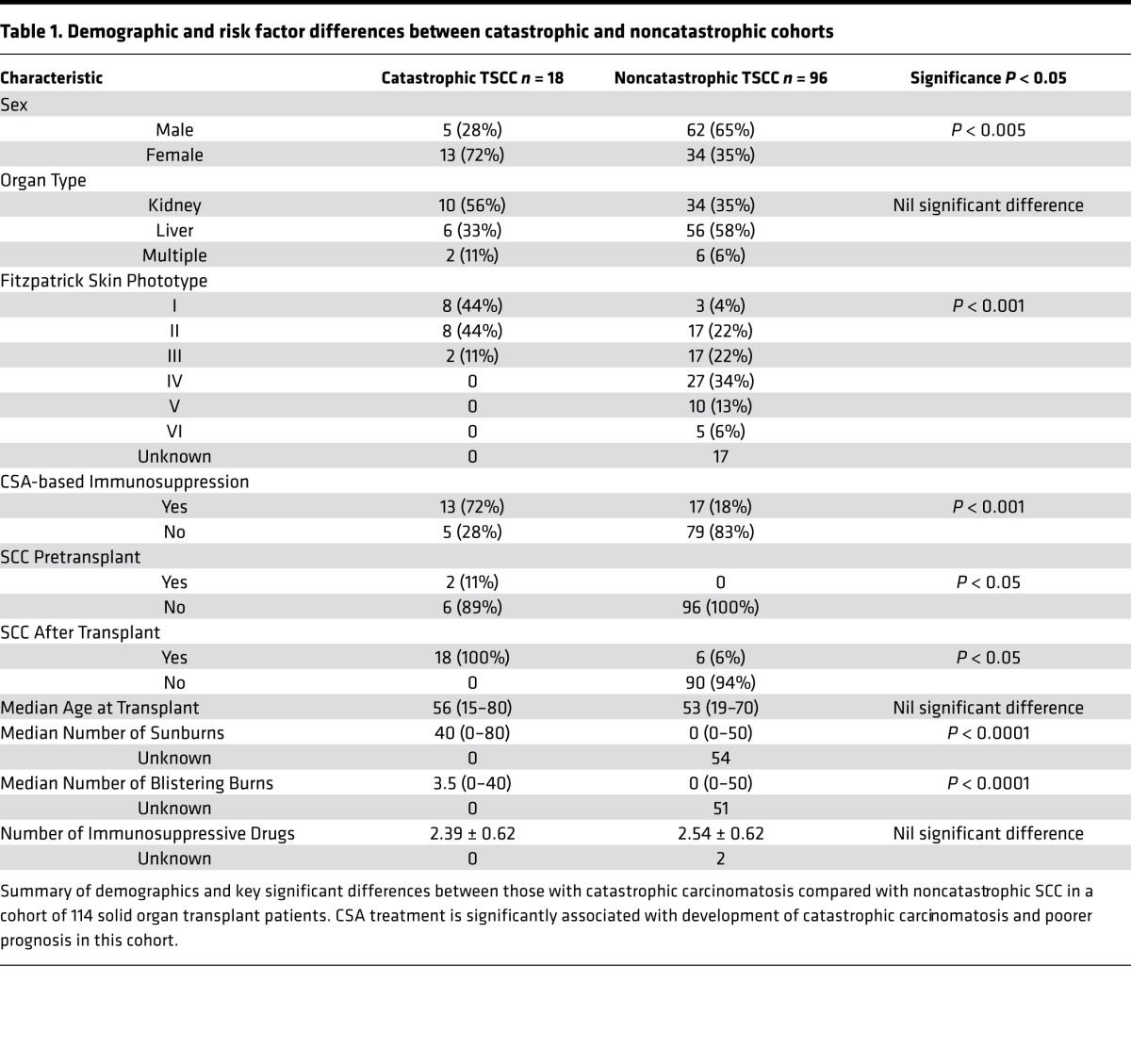
Cyclosporine A treatment is associated with increased risk for catastrophic SCC.
We studied 114 OTRs followed in the High Risk Skin Cancer Clinic and Section of Mohs and Dermatologic Surgery in the Department of Dermatology at NYU Langone Medical Center and looked for factors associated with catastrophic cutaneous carcinomatosis, defined by one or more of the following: (i) development of 10 or more primary SCCs over 1 year; (ii) in-transit, regional, or distant metastases; and (iii) severe field disease, defined as greater than 50% body surface area involvement of dysplastic skin. Eighteen of 114 OTRs met this definition of catastrophic carcinomatosis, representing 16% of all OTRs in our population (Table 1). There were no significant differences in the type of organ transplant between those with catastrophic carcinomatosis and those without, with 56% of the catastrophic cohort receiving a kidney transplant, 33% receiving a liver transplant, and 11% receiving multiple organ transplants (Table 1). An immune suppressive regimen including CSA was associated with significant risk for catastrophic SCC (P < 0.005, Table 1) on univariate analysis. Catastrophic carcinomatosis was also associated with skin types I–III (fair skin with tendency to burn more easily than tan) (P < 0.001), female gender (P < 0.004), number of sunburns (P < 0.001), number of blistering sunburns (P < 0.001), and history of SCC prior to transplant (P < 0.05). OTRs with catastrophic SCC were more likely to develop SCC sooner after transplant than OTRs who did not develop catastrophic disease. We found that approximately 80% of OTRs with catastrophic disease developed their first posttransplant SCC within 10 months of receiving their allograft. Based on our clinical findings of rapid SCC development and morbid course in CSA-treated OTRs in our cohort, along with our prior work showing increased T22 polarization in TSCC (16), we set out to explore the effects of CSA on IL-22–mediated SCC proliferation, T cell polarization, and IL-22 receptor expression.
Table 1. Demographic and risk factor differences between catastrophic and noncatastrophic cohorts.
Cyclosporine A augments IL-22–mediated rescue from serum starvation and potentiates the effects of IL-22 in driving accelerated SCC growth.
To further explore the relationship between CSA and IL-22–mediated tumor growth, as well as the effect of CSA itself on SCC proliferation, we cultured A431 cells in serum starvation conditions and investigated the influence of CSA and IL-22 treatment, both individually and in combination, on cell proliferation. We found that, similar to our previous results (16), IL-22 (40 or 100 ng/ml) rescued A431 SCC cells from serum starvation in a dose-dependent fashion (Figure 1A). Cells cultured with IL-22 were proliferative, with large colony formation and a significant increase in cell numbers, as compared with those grown in serum starvation alone (IL-22 40 ng/ml: 1.75 × 105 cells; IL-22 100 ng/ml: 3.2 × 105 cells; 0.1% FBS alone: 1.0 × 105 cells; P < 0.05; Figure 1A). Furthermore, treatment with CSA (50 ng/ml) enhanced A431 cell proliferation for all conditions and further potentiated the hyperproliferation induced by IL-22, with dramatically enhanced colony formation (CSA 50 ng/ml: 1.7 × 105 cells, Figure 1A) and elevated cell numbers seen when treated in combination (CSA + IL-22 40 ng/ml: 2.9 × 105 cells vs. IL-22 alone: 1.7 × 105, P < 0.05, Figure 1A). This suggests that CSA may be augmenting IL-22–driven SCC proliferation in OTRs.
Figure 1. CSA favors IL-22 response and effects in SCC.

(A) Treatment of A431 squamous cell carcinoma (SCC) cells with IL-22 results in significantly increased proliferation, shown at ×4 magnification; proliferation is further amplified with cyclosporine A (CSA) and IL-22 in combination. Data represent mean of 3 experiments ± SEM. *P < 0.05, **P < 0.01, ****P < 0.0001 determined by one-way ANOVA with Dunnett’s multiple comparisons test. (B) Representative dot plot analysis of IFN-γ, IL-4, IL-17, and IL-22 expression in CD4+ and CD8+ T cells with and without CSA. Numbers indicate percent gated cells. Percentage change in cytokine expression with CSA treatment is shown below the plots. Data represent mean of 3 experiments ± SEM. (C) IL10RB and IL22RA1 receptor expression in patient tissue samples from tumor (SCC) and adjacent non–tumor-bearing skin (S Adj), as well as transplant patient tumor samples with catastrophic outcomes (TSCC) and adjacent non–tumor-bearing skin (T Adj) as detected by RT-PCR and normalized to Keratin14 to account for variations in epithelial tissue between the samples. Data represent mean of 5 experiments ± SEM. *P < 0.05 and **P < 0.01 determined by one-way ANOVA with Tukey’s multiple comparisons test. (D) CSA induces IL-22 receptor subunit expression in A431 SCC cells in vitro. Data represent mean of 3 experiments ± SEM. *P < 0.05 determined by one-way ANOVA with Dunnett’s multiple comparisons test.
Cyclosporine A favors polarization of CD4+ and CD8+ T cells toward T22.
We have previously shown that TSCC is associated with a distinct immune phenotype when compared with SCC lesions from immune competent patients, with a significantly decreased percentage of CD4+IFN-γ producing cells, and significantly increased percentage of CD8+IL-22–producing cells (16). We found that CSA-treated transplant patients were likely to have more frequent and more aggressive SCC (Table 1), and we were interested in determining what effect CSA might have in driving T cell phenotype. We stimulated PBMC-derived T cells taken from healthy volunteers and cultured with or without CSA (1,000 ng/ml) for 1 hour. Following incubation with CSA, there was a 90% decrease in the percentage of CD4+ IFN-γ–producing cells (90.3% ± 1.7%, P < 0.001, Figure 1B). Similarly, IL-17–producing T cells (T-17) induction was also dramatically suppressed, with an approximate 97% decrease in the percentage of CD4+ IL-17–producing cells (96.2% ± 1.6%, P < 0.001, Figure 1B). In contrast, treatment with CSA had no effect on numbers of IL-4– or IL-22–producing cells, and the percentage of CD4+ IL-22–producing cells remained relatively constant (15% ± 23%, Figure 1B). This resulted in an elevated proportion of Th22 cells within the total CD4+ T cell population. We evaluated the effect of CSA on the CD8+ T cell population and again found a dramatic suppression of IFN-γ– and IL-17–producing cells with an 85% and 49% reduction in CD8+ IFN-γ– and IL-17–producing cells, respectively (84.7% ± 1.2%, 49.28% ± 11%, P < 0.001). CSA treatment had no effect on numbers of IL-4– or IL-22–producing cells, with the percentage of IL-22–secreting cells remaining relatively unchanged (146.7% ± 26.7%, Figure 1B). The net effect of CSA treatment is an increased proportion of T22 cells as compared with other T cell subtypes.
IL-22 receptor is most highly expressed in TSCC and is induced by cyclosporine A.
We next studied IL-22 receptor expression in TSCC and wondered whether it might be affected by CSA treatment. We performed quantitative PCR (qPCR) analysis on mRNA extracted from human SCC, from TSCC lesions from patients with catastrophic outcomes, from their adjacent nontumor bearing skin, and from normal skin taken from healthy volunteers. We found a significant upregulation of the IL-22 receptor subunit IL10RB in the peritumoral region of SCC, as compared with normal skin, the peritumoral region of TSCC (P < 0.05), and SCC tumor (P < 0.01). Furthermore, we found that TSCC lesions contained an approximately 3-fold increase in mean mRNA expression for the IL-22 receptor subunit IL22RA1 as compared with SCC lesions from immunocompetent patients (P < 0.05, Figure 1C). These findings suggest that the IL-22 receptor is indeed highly expressed in TSCC lesions and the human SCC microenvironment in vivo. We were interested in evaluating the effect of CSA on the expression of the IL-22 receptor in SCC cells. We therefore cultured A431 cells with CSA (25, 50, 100, and 1,000 ng/ml) for 24 hours and performed reverse transcription PCR (RT-PCR) to determine IL-22 expression. We found that mean mRNA expression for the IL-22 receptor subunit IL10RB was increased approximately 2-fold following treatment with CSA. A 4-fold increase was observed following culture with 50 ng/ml CSA (P < 0.05, Figure 1D). Similarly, mean mRNA expression for the IL-22 receptor subunit IL22RA1 was increased 2-fold following treatment with CSA, with a 4-fold increase following culture with 50 ng/ml CSA (Figure 1D). Incubation with CSA therefore results in the effective upregulation of mRNA expression for both subunits of the IL-22 receptor on SCC cells. This indicates that CSA treatment might render SCC more susceptible to IL-22 stimulation.
CSA and IL-22 enhance migration and invasion by A431 SCC cells in vitro.
Since we found that IL-22 enhanced proliferation of SCC and that this was further enhanced by CSA, we wondered whether IL-22 or CSA might affect migration or invasion by SCC. We performed scratch assays to assess migration of A431 SCC cells in vitro. Scratches were followed at time points of 8, 24, 32, 48, 56, and 72 hours. We found that A431 cells treated with IL-22 (20 ng/ml) had a greater rate of closure at all time points (Figure 2A) with IL-22–treated cells showing significantly greater closure than control at 48 hours (Figure 2A, 47.7% ± 6.5% vs. 28.4% ± 6.5%, P = 0.05). Using the same technique, A431 cells were treated with CSA (50 ng/ml), and the CSA-treated cells showed accelerated migratory capacity with significantly greater percentage of closure seen at both 24 hours (11.2% ± 2.4% vs. 27.3% ± 5.7%, P < 0.05, Figure 2A) and 48 hours (27.4% ± 1.7% vs. 49% ± 9.1%, P < 0.05, Figure 2A). This shows that CSA or IL-22 can enhance migratory capacity of SCC cells and indicates the potential for enhanced invasive capacity. To further determine the influence of CSA and IL-22 on migration of A431 cells through extracellular matrices, we treated A431 cells with IL-22 (40 ng/ml) or CSA (25 ng/ml), both individually and in combination in an invasion assay. We found that treatment with CSA alone, IL-22 alone, and IL-22 and CSA in combination all significantly increased the invasive capacity of treated cells compared with untreated cells (fold changes of 1.3, P < 0.01; 1.9, P < 0.05; and 1.7, P < 0.001, respectively, Figure 2B). Combination treatment did not show increased effect over CSA or IL-22 alone. Thus, a microenvironment rich in both CSA and IL-22, as is found in TSCC, might favor SCC invasiveness.
Figure 2. CSA and IL-22 enhance migration and invasion of A431 cutaneous SCC cells in vitro.

(A) Scratch assays performed on A431 squamous cell carcinoma (SCC) cells demonstrate significantly increased migration at 48 hours when treated with IL-22 and at 24 and 48 hours when treated with cyclosporine A (CSA). Images shown at ×4 magnification. Data represent mean of 3 experiments ± SEM. *P < 0.05 determined by t test. (B) Invasion of SCC cells through a membrane is significantly increased compared with control when treated with CSA and IL-22, both alone and in combination. Images at ×10 magnification. Scale bars: 400 μm. Data represent mean of 5 experiments ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001 determined by t test.
Treatment with anti–IL-22 antibody decreases SCC tumor number and tumor burden.
We found several lines of evidence linking IL-22 with enhanced proliferation and invasion by SCC. We next investigated the effect of IL-22 blockade on SCC progression in mice. Exposure of immune competent SKH-1 mice to UV light (UVB) models the effects of UVB on SCC development in humans. SKH-1 mice were exposed to UVB irradiation for 15 weeks to induce tumor development. Mice were then divided into 2 groups; one group (treatment arm) received the anti–IL-22 antibody (0.3 mg/ml, i.p.) 3 times weekly until week 22. The other group (control arm) was treated with vehicle alone. This same antibody was used in previous murine studies that led to the development of fezakinumab, which was used in human trails for rheumatoid arthritis and psoriasis, and is currently being investigated for use in atopic dermatitis (NCT01941537).Treatment with the anti–IL-22 antibody resulted in decreased number and overall tumor burden between weeks 18 and 22 (Figure 3, A and B, P < 0.05). All treated mice tolerated the anti–IL-22 antibody without any adverse effects. Thus, we were able to decrease number and burden of UVB-induced tumors via systemic treatment with anti–IL-22 antibody.
Figure 3. Anti–IL-22 treatment reduces tumor number and burden in SKH-1 mice.

(A) Tumors with a diameter of >1 mm were measured weekly. SKH-1 mice treated with anti–IL-22 for 9 weeks following 15 weeks of UVB exposure exhibited significantly decreased tumor number and overall tumor burden. Data represent mean values with n = 9 and n = 10 mice per data point for anti–IL-22 and vehicle-treatment groups, respectively; **P < 0.05 at week 18 determined by one-way ANOVA and Wilcoxon-Mann Whitney test to account multiple comparisons. (B) Skin tumor formation on representative mice from each treatment group at week 18, following 1 week of anti–IL-22 treatment.
Discussion
Over the last several decades, there have been considerable advances in the field of organ transplantation, which have led to a significant increase in the number of surgeries performed each year and the overall posttransplant survival rate of OTRs (22, 23). As OTRs now live longer, complications of chronic immunosuppressive therapy have emerged as major causes of non–transplant-related morbidity and mortality. In particular, there is a dramatic increase in the incidence of aggressive SCC. TSCC lesions are furthermore associated with greatly elevated rates of metastases and recurrence, leading to a 3-year mortality, which can reach as high as 46% (7, 24, 25). In the present study, we focused on CSA, a mainstay of immunosuppressive therapy in OTR. We found that CSA treatment of OTRs is positively correlated with catastrophic SCC. Furthermore, CSA potentiated IL-22–mediated effects on proliferation of SCC in vitro and induced IL-22 receptor expression on SCC cells. This recapitulated our finding of increased IL-22R in TSCC samples from OTRs. In addition, treatment with anti–IL-22 antibody resulted in decreased tumor burden and tumor number in a murine model of UVB induced SCC. Taken together, these results identify the IL-22 pathway as a potential therapeutic target in treating OTR with TSCC and catastrophic carcinomatosis, a condition with no currently available medical treatment.
Catastrophic SCC was defined by Berg and Otley as (i) development of 10 or more primary SCCs over 1 year; (ii) in-transit, regional, or distant metastases; and (iii) severe field disease (>50% body surface area involvement of dysplastic skin) (7). Using a cohort of our OTRs, we showed that CSA use was associated with significant risk for catastrophic SCC. This is the first report to link CSA therapy directly to catastrophic disease, and it is consistent with prior reports linking CSA, a calcineurin inhibitor (CNI), with overall SCC risk. It is important to distinguish between catastrophic and noncatastrophic SCC, since noncatastrophic skin cancer is usually easily managed by excision while catastrophic skin cancer brings morbidity and may shorten life (7). Other factors associated with catastrophic SCC in OTRs include skin types I–III, female gender, number of sunburns, number of blistering sunburns, diagnosis of SCC prior to transplant, and development of first posttransplant SCC within 10 months of allograft. These criteria may be useful in determining which OTRs require most stringent screening efforts. This also may be useful in determining appropriate risk-to-benefit ratio when deciding whether to begin or continue CNI therapy. Since the incidence of SCC in OTRs appears to be directly proportional to the duration and intensity of immunosuppression therapy (11), one might consider an alternative in a fair-skinned allograft recipient with SCCs prior to transplant.
Much has been written about CSA and overall SCC risk, but there is little data on mechanisms responsible for CSA-mediated acceleration of carcinogenesis. Previous studies have suggested mechanisms for the association between CSA and SCC. These include decreased tumor surveillance, which may occur as a result of diminished IL-2 production, and systemic immunosuppression (12). Additionally, CSA has been shown to have direct transformative effects on keratinocytes and may induce phenotypic changes, which can increase cell invasiveness and unregulated tumor growth (13, 14). CSA has also been associated with the promotion of fibroblast proliferation (15). Past studies in SCID mice have supported a role of TGF-β in immune evasion by SCC; our early work supports a role for TGF-β in SCC development (26, 27). The current study sheds light on potential mechanisms for CSA-induced SCC in transplant patients utilizing tissue from catastrophic patients in our cohort.
We have previously shown the TSCC tumor environment to contain a unique T cell profile when compared with SCC from immunocompetent patients (16). Specifically, we found that TSCC showed a significantly decreased percentage of CD4+ IFN-γ–producing cells and a significantly increased number of CD8+ IL-22–producing cells (16). Our current findings suggest that CSA may actually be responsible for driving this disparate T cell phenotype through the selective inhibition of IFN-γ and IL-17. IFN-γ–producing cells are key mediators of the innate and adaptive immune systems and are critical for the generation of antitumor immunity (28).
IL-22 is involved in multiple inflammatory processes and has also been shown to enhance progression of gastric, hepatocellular, and skin carcinoma (29–34). It mediates its effects via a heterodimeric receptor that is highly expressed in epithelial cells of the skin and other organs. Binding of IL-22 to its receptor results in the activation of signaling cascades, which ultimately lead to the promotion of cell cycle progression and the inhibition of apoptosis. This fits in well with our in vitro findings where CSA augments IL-22 rescue of SCC cells from serum starvation. In addition, CSA increased IL-22 receptor expression in SCC cells; this appears to recapitulate our finding of increased IL-22R expression in TSCC samples from OTR. In addition to being able to proliferate, skin cancer cells need to invade the dermis, access lymphatics, and evade immune detection in order to grow and spread. Our findings demonstrated that both IL-22 and CSA increased SCC cell proliferation and invasion in vitro. It has been shown that TSCC lesions are associated with greatly elevated rates of metastases and recurrence, leading to a 3-year mortality, which can reach as high as 46% (7, 24, 25). CSA treatment of OTR may be driving SCC proliferation, migration, and invasion via the IL-22 axis.
Sun exposure is a known contributor to the development of SCC (35–37). Oberyszyn and colleagues have developed a model of UVB-induced SCC in immune-competent hairless SKH-1 mice (38). Over a period of 15 weeks of UV exposure, mice develop new cutaneous tumors that demonstrate enhanced growth and progression when treated with CSA following UV irradiation compared with UV exposure alone (38). We found that CSA induced IL-22 receptor expression in SCC and enhanced IL-22–mediated SCC proliferation. This indicates a role for CSA in driving aggressive behavior by SCC through the IL-22 axis. We saw a reduction in tumor burden and number when we blocked this axis by treating UV -ed mice with anti–IL-22.
Immunosuppressive therapy is a major component of OTR treatment to maintain transplanted organs. The majority of OTRs are managed on multiple immunosuppressive agents, which generally include one calcineurin inhibitor (CSA or tacrolimus), along with an antimetabolite (azathioprine or mycophenolate) and prednisolone (39). Tacrolimus has become increasingly popular in transplant patients over recent years, largely due to studies suggesting it has a lower risk of acute rejection in comparison to CSA; however, despite this popularity, there have been no clear benefits seen in long-term rejection rates (40, 41). Tacrolimus, while also acting via calcineurin inhibition, has different toxicities and a differing side effect profile; because of this, it is not the best suited CNI for every patient (39, 42). In recent years, there has also been a shift away from CNI-based therapy after transplant due to the availability of newer agents such as sirolimus. However, a recent study of over 3,000 transplant patients shows no reduction in SCC incidence with sirolimus (43). In addition, recent reviews suggest that CSA may have a favorable safety profile compared with tacrolimus and favorable cardiac profile compared with sirolimus (44). Since the differing toxicities of each of these classes of immunosuppressant appear to be dose related, combinations of agents including CSA are often used in order to minimize toxicities while maintaining efficacy in preventing allograft rejection (12, 45). Thus, CSA will likely continue to be a significant component of immunosuppressive therapy in OTR, either in higher doses for appropriate patient subsets or in combination with other immunosuppressive agents. The ultimate goal of this research is to define the mechanism(s) by which CSA increases SCC and to tailor therapy to targets that will lessen CSA’s tumorigenic side effects, without compromising the level of immunosuppression required to maintain the transplant. CSA causes immunosuppression via dual mechanisms — both via calcineurin inhibition and via an independent inhibition of the JNK and P38 signaling pathways (46). By targeting the IL-22 pathway downstream of these effects, it is hypothesized that the specific effects of the IL-22 pathway can be dissected out, allowing targeted therapy of SCC while permitting concurrent immunosuppression via alternate pathways. The present study focusing on CSA’s effects on IL-22 may lead to such a targeted therapy. Such therapy might someday allow OTRs to maintain their immune suppressive regimens without the risk of catastrophic skin cancers. Reference to our newly described risk factors for catastrophic SCC might provide criteria for consideration for such targeted therapy.
We found that CSA increases the risk for catastrophic SCC in our cohort. We have identified a link between CSA and the IL-22 axis that can influence proliferation, invasion, and immune evasion by SCC. We showed that IL-22 blockade can decrease SCC burden and size in vivo using a murine system that mirrors development of UVB-induced SCC development in humans. Further clarification of this relationship can facilitate identification of novel points for intervention that may allow reduction of morbidity and mortality associated with catastrophic carcinomatosis in this population, while allowing adequate immune suppression to proceed concurrently. Successful targeting of this pathway might allow patients to remain on immune suppression that optimizes allograft survival while decreasing probability of catastrophic SCC.
Methods
Participants.
Participants selected for cohort analysis were chosen from patient volunteers who had previously received an organ transplant — either kidney, liver, or pancreas — and now presented for dermatologic evaluation. A total of 114 patients volunteered to participate. IRB approval at NYU Langone Medical Center was obtained prior to enrolling patients to participate in the study. Data including demographics, risk factors, and associations were collected, and univariate analysis with SPSS was performed to determine any significant associations.
Cell culture for proliferation studies and cell counts.
A cSCC cell line A431 (obtained from ATCC) was cultured in DMEM supplemented with 10% or 0.1% FBS (serum starvation medium) at 37°C for 48 hours to synchronize cellular proliferation at G0 phase. Subsequently, cells were divided into 2 groups, with the one group receiving treatment with 50 ng/ml CSA (Sigma-Aldrich) and the second group receiving a vehicle control. Both groups were then grown in one of the following 4 conditions: (i) 0.1% FBS, (ii) 0.1% FBS + 40 ng/ml IL-22 (R&D Systems), (iii) 0.1% FBS +100 ng/ml IL-22, and (iv) 10% FBS (full growth media). Both CSA and IL-22 were reconstituted in PBS prior to administration according to the manufacturer’s protocol. After 72 hours, the cells were photographed under light microscopy, harvested by trypsinization, and counted using an automated cell counter (Countess Automated Cell Counter, Invitrogen).
T cell isolation and flow cytometry.
Whole blood samples were obtained from healthy volunteers (n = 3), following which PBMC-derived T cells were isolated and cultured with or without the addition of CSA (1,000 ng/ml) for 24 hours. T cells were then activated for 4 hours using anti-CD3/CD28 beads, in the presence of 10 μg/ml brefeldin A (Sigma-Aldrich). EDTA (2 mM; Fisher Scientific) was added for 10 minutes on ice to stop activation. Cells were then incubated in aqua marina live/dead dye (Invitrogen) on ice for 30 minutes for dead cell discrimination and subsequently fixed with 4% paraformaldehyde (BD Biosciences) on ice for 20 minutes. Cells were permeabilized in FACSPerm (BD Biosciences), blocked in 1:50 mouse serum (BD Biosciences), and incubated for 30 minutes on ice with the following anti-human, mouse monoclonal antibodies: CD3-Pacific Blue (eBioscience, clone 500A2), CD4-Phycoerythrin-Cy7 (eBioscience, clone RPA-T4), CD8–PerCp-Cy5.5 (BD Pharmingen, clone RPA-T7), IFN-γ–Alexa Fluor 700 (BD Pharmingen, clone L243), IL-4–Phycoerythrin (BD Pharmingen, clone 8D4-8), IL-17–Alexa Fluor 488 (eBioscience, clone eBio17B7), and IL-22–Allophycocyanin (R&D Systems, clone 142928). Samples were acquired using an LSR-II flow cytometer (BD Biosciences) and analyzed with FlowJo software (Tree Star Inc.). Live CD3+CD4+ and CD3+CD8+ cells were first gated, following which the frequencies of the cells producing indicated cytokines were analyzed.
RNA isolation.
Samples were obtained from surgical discard in Mohs micrographic surgery, from normal skin, tumor, and adjacent nontumor bearing skin, and tumors were obtained from transplant patients with catastrophic carcinomatosis and adjacent nontumor bearing skin. Total RNA isolation was carried out as described previously (26, 47). Briefly, SCC (n = 9) and TSCC (n = 7) tumor samples were removed at Mohs micrographic surgery, and patient-matched, site-matched peritumoral skin was obtained at the time of repair after clear margins were achieved. Normal skin was obtained from normal volunteers (n = 9). All samples were snap-frozen and stored in liquid nitrogen. Individual frozen samples were placed in 1 ml of room temperature RLT Lysis buffer with 1% β-mercaptoethanol (QIAGEN) and immediately homogenized at full power for 30 seconds using a PowerGen 1000 homogenizer (Fisher Scientific). Homogenates were sonicated on ice for 20 seconds at full power. DNA was removed with on-column DNase digestion using an RNase-free DNase Set (QIAGEN). RNA was isolated using the RNeasy Mini Kit (QIAGEN) according to the manufacturer’s recommendations. Total RNA concentration and purity was evaluated using an Ultraspec 2100 prospectrophotometer (Amersham Biosciences, GE Healthcare).
RT-PCR.
All primers and probes used for the evaluation of target genes IL22RA1 and IL10RB were predesigned by Applied Biosystems and proven to have maximal coverage. Gene expression levels were normalized to housekeeping genes keratin 14 (KRT14, Applied Biosystems) and human acidic ribosomal protein (RPLP0, GenBank accession number NM-001002, Applied Biosystems). The RT-PCR reaction was carried out using 10 ng total RNA and EZ PCR Core Reagents (Applied Biosystems) according to the manufacturer’s directions. The samples were amplified and quantified on an Applied Biosystems PRISM 7900 HT using the following thermal cycler conditions: 2 minutes at 50°C, 30 minutes at 60°C, 5 minutes 95°C; and 40 cycles of 15 seconds at 95°C followed by 60 seconds at 60°C.
Cell culture with CSA for RT-PCR and IL-22 receptor expression.
Six-well plates were seeded at a concentration of 1 × 105 cells per well with A431 cells and cultured in full growth media using 3 ml DMEM supplemented with 10% FBS at 37°C for 48 hours. Subsequently, CSA was added to the wells using several different concentrations (25, 50, 100, and 1,000 ng/ml). CSA was reconstituted in EtOH prior to administration according to the manufacturer’s protocol. A positive control and negative control were included, consisting of cells grown in full growth media alone, as well as those treated with an EtOH vehicle respectively. After 24 hours of incubation with CSA, cells were washed and collected using RLT lysis buffer with 1% β-mercaptoethanol in preparation for subsequent RNA extraction and RT-PCR analysis.
Scratch assay.
A431 cells were cultured in 10% FBS DMEM media to 90% confluence and seeded at a density of 500,000 cells per well. Following incubation at 37°C in 5% CO2 for 24 hours, a scratch was made using a 1-ml pipette tip, and cells were treated with IL-22 (20 ng/ml) or CSA (50 ng/ml). Scratch closure was photographed at 0, 8, 24, 32, 48, 56, and 72 hours. Rate of closure and percentage closure were subsequently calculated using ImageJ software (NIH), and data analyses were performed using GraphPad Prism 6 software.
Invasion assay.
To investigate the effects of CSA and IL-22 on the invasive properties of SCC, cell invasion assays were carried out using both BioCoat (Corning Inc.) and CultureCoat (Trevigen) invasion assay systems as per manufacturers’ protocols. A431 cells (ATCC) were seeded at appropriate densities in the top chamber of the assay with 5% FBS used as a chemoattractant in the bottom chamber. Both top and bottom chambers were dosed with CSA (25 ng/ml) or IL-22 (40 ng/ml), or a combination of both CSA and IL-22, at aforementioned concentrations. Epidermal growth factor (EGF) (100 ng/ml) was used as positive control. Plates were incubated for 24 hours at 37°C in 5% CO2, and the assays were quantified using the Spectramax M3 plate reader (Molecular Devices) at fluorescent wavelengths suggested by the manufacturer. Data were analyzed using GraphPad Prism 6 software, and images were acquired using an EVOS fluorescence microscope (NYU Langone Medical Center Small Instrument Fleet CORE).
Murine study.
Murine studies were performed in collaboration with the Oberyszyn Lab at The Ohio State University using the SKH-1 hairless mice system as previously described (38, 48). Female SKH-1 outbred hairless mice (6–8 weeks old; Charles River Laboratories) were housed in the vivarium at The Ohio State University. Mice were dorsally exposed to 2240 J/m2 UVB, previously determined to be one minimal erythema dose, 3 times weekly on nonconsecutive days for 15 weeks. UVB dose was calculated using UVX radiometer and UVB sensor (UVP) and emitted by Phillips FS40 UV bulbs (American Ultraviolet Company). After 15 weeks, UVB treatments were terminated and mice were assigned to one of 2 treatment groups: anti–IL-22 (0.3 mg/ml, n = 9) or vehicle control (n = 10) so that each group contained the same total number of tumors. Rat anti–mouse IL-22 antibody was obtained as part of a Material Transfer Agreement from Pfizer, and injections were dissolved in phosphate buffered saline. Antibody or vehicle treatment was administered i.p. 3 times a week for 9 weeks. The area (length × width) of individual tumors on each mouse was measured weekly to quantify tumor number, size, and total tumor area. Only tumors greater than 1 mm in any direction were included in the data set.
Provision of materials.
Rat anti–mouse IL-22 antibody was obtained as part of a Material Transfer Agreement from Pfizer.
Statistics.
Unless otherwise specified, statistical analyses were performed using GraphPad Prism software. Comparisons of cell counts were performed using a 2-tailed, Student’s t test, with P < 0.05 considered to be significant. One-way ANOVA tests were used for the statistical comparison of flow cytometry data, supplemented with the Bonferroni post-hoc analysis.
Wilcoxon-Mann Whitney test was used as appropriate to account for multiple comparisons, with P < 0.05 considered to be significant. Averaged results of multiple experiments are presented as the arithmetic mean ± SEM.
Study approval.
All human studies were reviewed and approved by the IRB at NYU Langone Medical Center. Human subjects were provided with written informed consent prior to involvement in the study. Written informed consent was obtained for all patients before their participation, and the study was performed with strict adherence to the Declaration of Helsinki Principles. The animal study described was reviewed and approved by the Institutional Animal Care and Use Committee at The Ohio State University and according to the requirements established by the American Association for Accreditation of Laboratory Animal Care. All procedures were approved by the Institutional Animal Care and Use Committee before the initiation of any studies.
Author contributions
HM, VY, NR, CO, TB, TMO, KLT, JG, JGK, DF, and JAC conceived and designed the experiments. HM, VY, NR, CO, TB, TMO, KLT, and JG performed the experiments. MA, HM, VY, NR, CO, TB, TMO, KLT, JG, JGK, DF, and JAC analyzed the data. TMO, JGK, DF, and JAC contributed reagents/materials/analysis tools. MA, HM, VY, NR, CO, TB, DF, and JAC wrote the paper.
Acknowledgments
Funding was provided by the Cancer Research Institute’s Clinic and Laboratory Integration Program grant.
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Reference information:JCI Insight. 2016;1(8):e86434. doi:10.1172/jci.insight.86434.
Contributor Information
Melody Abikhair, Email: melodyabikhair@gmail.com.
Hiroshi Mitsui, Email: hmitsui@yamanashi.ac.jp.
Valerie Yanofsky, Email: vyanofsky@chpnet.org.
Nazanin Roudiani, Email: Nazanin.Roudiani@nyumc.org.
Channa Ovits, Email: cgo1024@gmail.com.
Teddy Bryan, Email: teddy.bryan@gmail.com.
Juana Gonzalez, Email: jgonzalez@rockefeller.edu.
Diane Felsen, Email: dfelsen@med.cornell.edu.
References
- 1.Housman TS, et al. Skin cancer is among the most costly of all cancers to treat for the Medicare population. J Am Acad Dermatol. 2003;48(3):425–429. doi: 10.1067/mjd.2003.186. [DOI] [PubMed] [Google Scholar]
- 2.Karia PS, Han J, Schmults CD. Cutaneous squamous cell carcinoma: estimated incidence of disease, nodal metastasis, and deaths from disease in the United States, 2012. J Am Acad Dermatol. 2013;68(6):957–966. doi: 10.1016/j.jaad.2012.11.037. [DOI] [PubMed] [Google Scholar]
- 3.Lohmann CM, Solomon AR. Clinicopathologic variants of cutaneous squamous cell carcinoma. Adv Anat Pathol. 2001;8(1):27–36. doi: 10.1097/00125480-200101000-00005. [DOI] [PubMed] [Google Scholar]
- 4.Carucci JA. Cutaneous oncology in organ transplant recipients: meeting the challenge of squamous cell carcinoma. J Invest Dermatol. 2004;123(5):809–816. doi: 10.1111/j.1523-1747.2004.23440.x. [DOI] [PubMed] [Google Scholar]
- 5.Ong CS, Keogh AM, Kossard S, Macdonald PS, Spratt PM. Skin cancer in Australian heart transplant recipients. J Am Acad Dermatol. 1999;40(1):27–34. doi: 10.1016/S0190-9622(99)70525-6. [DOI] [PubMed] [Google Scholar]
- 6.Lindelof B, Sigurgeirsson B, Gabel H, Stern RS. Incidence of skin cancer in 5356 patients following organ transplantation. Br J Dermatol. 2000;143(3):513–519. [PubMed] [Google Scholar]
- 7.Berg D, Otley CC. Skin cancer in organ transplant recipients: Epidemiology, pathogenesis, and management. J Am Acad Dermatol. 2002;47(1):1–17. doi: 10.1067/mjd.2002.125579. quiz 18–20. [DOI] [PubMed] [Google Scholar]
- 8.Tsuda K, et al. Calcineurin inhibitors suppress cytokine production from memory T cells and differentiation of naive T cells into cytokine-producing mature T cells. PLoS One. 2012;7(2):e86434. doi: 10.1371/journal.pone.0031465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Grinyo JM, et al. Low-dose cyclosporine with mycophenolate mofetil induces similar calcineurin activity and cytokine inhibition as does standard-dose cyclosporine in stable renal allografts. Transplantation. 2004;78(9):1400–1403. doi: 10.1097/01.TP.0000141227.63639.63. [DOI] [PubMed] [Google Scholar]
- 10.Jonas S, et al. De novo malignancies after liver transplantation using tacrolimus-based protocols or cyclosporine-based quadruple immunosuppression with an interleukin-2 receptor antibody or antithymocyte globulin. Cancer. 1997;80(6):1141–1150. doi: 10.1002/(SICI)1097-0142(19970915)80:6<1141::AID-CNCR18>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- 11.Christenson LJ, Cherikh WS, Otley CC, Salasche SJ, Kauffman HM. Allograft and overall survival of patients with posttransplant skin cancer. Dermatol Surg. 2011;37(2):183–191. doi: 10.1111/j.1524-4725.2010.01848.x. [DOI] [PubMed] [Google Scholar]
- 12.Leblanc KG, Jr, Hughes MP, Sheehan DJ. The role of sirolimus in the prevention of cutaneous squamous cell carcinoma in organ transplant recipients. Dermatol Surg. 2011;37(6):744–749. doi: 10.1111/j.1524-4725.2011.01973..x. [DOI] [PubMed] [Google Scholar]
- 13.Hojo M, et al. Cyclosporine induces cancer progression by a cell-autonomous mechanism. Nature. 1999;397(6719):530–534. doi: 10.1038/17401. [DOI] [PubMed] [Google Scholar]
- 14.Wu X, et al. Opposing roles for calcineurin and ATF3 in squamous skin cancer. Nature. 2010;465(7296):368–372. doi: 10.1038/nature08996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cotrim P, Martelli-Junior H, Graner E, Sauk JJ, Coletta RD. Cyclosporin A induces proliferation in human gingival fibroblasts via induction of transforming growth factor-beta1. J Periodontol. 2003;74(11):1625–1633. doi: 10.1902/jop.2003.74.11.1625. [DOI] [PubMed] [Google Scholar]
- 16.Zhang S, et al. Increased Tc22 and Treg/CD8 ratio contribute to aggressive growth of transplant associated squamous cell carcinoma. PLoS One. 2013;8(5):e86434. doi: 10.1371/journal.pone.0062154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nograles KE, et al. IL-22-producing “T22” T cells account for upregulated IL-22 in atopic dermatitis despite reduced IL-17-producing TH17 T cells. J Allergy Clin Immunol. 2009;123(6):1244–1252 e1242. doi: 10.1016/j.jaci.2009.03.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Res PC, et al. Overrepresentation of IL-17A and IL-22 producing CD8 T cells in lesional skin suggests their involvement in the pathogenesis of psoriasis. PLoS One. 2010;5(11):e86434. doi: 10.1371/journal.pone.0014108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wolk K, et al. IL-22 regulates the expression of genes responsible for antimicrobial defense, cellular differentiation, and mobility in keratinocytes: a potential role in psoriasis. Eur J Immunol. 2006;36(5):1309–1323. doi: 10.1002/eji.200535503. [DOI] [PubMed] [Google Scholar]
- 20.Boniface K, et al. A role for T cell-derived interleukin 22 in psoriatic skin inflammation. Clin Exp Immunol. 2007;150(3):407–415. doi: 10.1111/j.1365-2249.2007.03511.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zheng Y, et al. Interleukin-22, a T(H)17 cytokine, mediates IL-23-induced dermal inflammation and acanthosis. Nature. 2007;445(7128):648–651. doi: 10.1038/nature05505. [DOI] [PubMed] [Google Scholar]
- 22.United Network for Organ Sharing Transplant trends. [April 18, 2016];UNOS Web site. https://www.unos.org/data/transplant-trends/#transplants_by_donor_type
- 23.Wu JJ, Orengo IF. Squamous cell carcinoma in solid-organ transplantation. Dermatol Online J. 2002;8(2):e86434. [PubMed] [Google Scholar]
- 24.Carroll RP, Ramsay HM, Fryer AA, Hawley CM, Nicol DL, Harden PN. Incidence and prediction of nonmelanoma skin cancer post-renal transplantation: a prospective study in Queensland, Australia. Am J Kidney Dis. 2003;41(3):676–683. doi: 10.1053/ajkd.2003.50130. [DOI] [PubMed] [Google Scholar]
- 25.Gogia R, Binstock M, Hirose R, Boscardin WJ, Chren MM, Arron ST. Fitzpatrick skin phototype is an independent predictor of squamous cell carcinoma risk after solid organ transplantation. J Am Acad Dermatol. 2013;68(4):585–591. doi: 10.1016/j.jaad.2012.09.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bluth MJ, et al. Myeloid dendritic cells from human cutaneous squamous cell carcinoma are poor stimulators of T-cell proliferation. J Invest Dermatol. 2009;129(10):2451–2462. doi: 10.1038/jid.2009.96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Haider AS, et al. Genomic analysis defines a cancer-specific gene expression signature for human squamous cell carcinoma and distinguishes malignant hyperproliferation from benign hyperplasia. J Invest Dermatol. 2006;126(4):869–881. doi: 10.1038/sj.jid.5700157. [DOI] [PubMed] [Google Scholar]
- 28.Ikeda H, Old LJ, Schreiber RD. The roles of IFN gamma in protection against tumor development and cancer immunoediting. Cytokine Growth Factor Rev. 2002;13(2):95–109. doi: 10.1016/S1359-6101(01)00038-7. [DOI] [PubMed] [Google Scholar]
- 29.Kim MJ, et al. Change in inflammatory cytokine profiles after transarterial chemotherapy in patients with hepatocellular carcinoma. Cytokine. 2013;64(2):516–522. doi: 10.1016/j.cyto.2013.07.021. [DOI] [PubMed] [Google Scholar]
- 30.Qin S, Ma S, Huang X, Lu D, Zhou Y, Jiang H. Th22 cells are associated with hepatocellular carcinoma development and progression. Chin J Cancer Res. 2014;26(2):135–141. doi: 10.3978/j.issn.1000-9604.2014.02.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Souza JM, Matias BF, Rodrigues CM, Murta EF, Michelin MA. IL-17 and IL-22 serum cytokine levels in patients with squamous intraepithelial lesion and invasive cervical carcinoma. Eur J Gynaecol Oncol. 2013;34(5):466–468. [PubMed] [Google Scholar]
- 32.Wen Z, et al. High expression of interleukin-22 and its receptor predicts poor prognosis in pancreatic ductal adenocarcinoma. Ann Surg Oncol. 2014;21(1):125–132. doi: 10.1245/s10434-013-3322-x. [DOI] [PubMed] [Google Scholar]
- 33.Zhang W, et al. Antiapoptotic activity of autocrine interleukin-22 and therapeutic effects of interleukin-22-small interfering RNA on human lung cancer xenografts. Clin Cancer Res. 2008;14(20):6432–6439. doi: 10.1158/1078-0432.CCR-07-4401. [DOI] [PubMed] [Google Scholar]
- 34.Ziesche E, Bachmann M, Kleinert H, Pfeilschifter J, Muhl H. The interleukin-22/STAT3 pathway potentiates expression of inducible nitric-oxide synthase in human colon carcinoma cells. J Biol Chem. 2007;282(22):16006–16015. doi: 10.1074/jbc.M611040200. [DOI] [PubMed] [Google Scholar]
- 35.de Gruijl FR, van Kranen HJ, Mullenders LH. UV-induced DNA damage, repair, mutations and oncogenic pathways in skin cancer. J Photochem Photobiol B. 2001;63(1–3):19–27. doi: 10.1016/s1011-1344(01)00199-3. [DOI] [PubMed] [Google Scholar]
- 36.Brash DE, et al. A role for sunlight in skin cancer: UV-induced p53 mutations in squamous cell carcinoma. Proc Natl Acad Sci U S A. 1991;88(22):10124–10128. doi: 10.1073/pnas.88.22.10124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nakazawa H, et al. UV and skin cancer: specific p53 gene mutation in normal skin as a biologically relevant exposure measurement. Proc Natl Acad Sci U S A. 1994;91(1):360–364. doi: 10.1073/pnas.91.1.360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wulff BC, Kusewitt DF, VanBuskirk AM, Thomas-Ahner JM, Jason DF, Oberyszyn TM. Sirolimus reduces the incidence and progression of UVB-induced skin cancer in SKH mice even with co-administration of cyclosporine A. J Clin Investig Dermatol. 2008;128(10):2467–2473. doi: 10.1038/jid.2008.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Meier-Kriesche HU, et al. Immunosuppression: evolution in practice and trends, 1994–2004. Am J Transplant. 2006;6(5p2):1111–1131. doi: 10.1111/j.1600-6143.2006.01270.x. [DOI] [PubMed] [Google Scholar]
- 40.Woodward RS, Kutinova A, Schnitzler MA, Brennan DC. Renal graft survival and calcineurin inhibitor. Transplantation. 2005;80(5):629–633. doi: 10.1097/01.tp.0000184842.01686.CA. [DOI] [PubMed] [Google Scholar]
- 41.Wiesner RH. A long-term comparison of tacrolimus (FK506) versus cyclosporine in liver transplantation: a report of the United States FK506 Study Group. Transplantation. 1998;66(4):493–499. doi: 10.1097/00007890-199808270-00014. [DOI] [PubMed] [Google Scholar]
- 42.Webster AC, Woodroffe RC, Taylor RS, Chapman JR, Craig JC. Tacrolimus versus ciclosporin as primary immunosuppression for kidney transplant recipients: meta-analysis and meta-regression of randomised trial data. BMJ. 2005;331(7520):e86434. doi: 10.1136/bmj.38569.471007.AE. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Asgari MM, Arron ST, Warton EM, Quesenberry CP, Jr, Weisshaar D. Sirolimus use and risk of cutaneous squamous cell carcinoma (SCC) in solid organ transplant recipients (SOTRs) J Am Acad Dermatol. 2015;73(3):444–450. doi: 10.1016/j.jaad.2015.05.029. [DOI] [PubMed] [Google Scholar]
- 44.Euvrard S, et al. Sirolimus and secondary skin-cancer prevention in kidney transplantation. N Engl J Med. 2012;367(4):329–339. doi: 10.1056/NEJMoa1204166. [DOI] [PubMed] [Google Scholar]
- 45.Colegio OR, Hanlon A, Olasz EB, Carucci JA. Sirolimus reduces cutaneous squamous cell carcinomas in transplantation recipients. J Clin Oncol. 2013;31(26):3297–3298. doi: 10.1200/JCO.2013.50.6840. [DOI] [PubMed] [Google Scholar]
- 46.Reynolds NJ, Al-Daraji WI. Calcineurin inhibitors and sirolimus: mechanisms of action and applications in dermatology. Clin Exp Dermatol. 2002;27(7):555–561. doi: 10.1046/j.1365-2230.2002.01148.x. [DOI] [PubMed] [Google Scholar]
- 47.Kaporis HG, et al. Human basal cell carcinoma is associated with Foxp3+ T cells in a Th2 dominant microenvironment. J Invest Dermatol. 2007;127(10):2391–2398. doi: 10.1038/sj.jid.5700884. [DOI] [PubMed] [Google Scholar]
- 48.Thomas-Ahner JM, Wulff BC, Tober KL, Kusewitt DF, Riggenbach JA, Oberyszyn TM. Gender differences in UVB-induced skin carcinogenesis, inflammation, and DNA damage. Cancer Res. 2007;67(7):3468–3474. doi: 10.1158/0008-5472.CAN-06-3798. [DOI] [PubMed] [Google Scholar]