

Abstract
The physiological components that contribute to cystic fibrosis (CF) lung disease are steadily being elucidated. Gene therapy could potentially correct these defects. CFTR-null pigs provide a relevant model to test gene therapy vectors. Using an in vivo selection strategy that amplifies successful capsids by replicating their genomes with helper adenovirus coinfection, we selected an adeno-associated virus (AAV) with tropism for pig airway epithelia. The evolved capsid, termed AAV2H22, is based on AAV2 with 5 point mutations that result in a 240-fold increased infection efficiency. In contrast to AAV2, AAV2H22 binds specifically to pig airway epithelia and is less reliant on heparan sulfate for transduction. We administer AAV2H22-CFTR expressing the CF transmembrane conductance regulator (CFTR) cDNA to the airways of CF pigs. The transduced airways expressed CFTR on ciliated and nonciliated cells, induced anion transport, and improved the airway surface liquid pH and bacterial killing. Most gene therapy studies to date focus solely on Cl– transport as the primary metric of phenotypic correction. Here, we describe a gene therapy experiment where we not only correct defective anion transport, but also restore bacterial killing in CFTR-null pig airways.
An AAV capsid serotype with enhanced transduction properties is used to correct defective anion transport and restore bacterial killing in CFTR-null pig airways.
Introduction
Cystic fibrosis (CF) is an autosomal recessive disease caused by mutations in the gene encoding CF transmembrane conductance regulator (CFTR). Advanced CF lung disease is characterized by chronic bacterial airway infection, neutrophilic inflammation, adherent mucus, and progressive bronchiectasis. Newborns with CF have little or no inflammation. The mechanism by which loss of CFTR-mediated chloride and bicarbonate transport leads to chronic airway infection remains uncertain. Recent work using a CF pig model shows that the lack of bicarbonate transport results in acidic airway surface liquid (ASL), impaired antimicrobial properties, and viscous mucus in newborn CF pigs (1–3). The resulting airway infections initiate a cascade of inflammation and airway wall remodeling that progressively destroys the lung and shortens lives (4, 5).
Efficient adeno-associated virus–mediated (AAV-mediated) gene therapy can potentially correct this defect and prevent lung infections. However, while gene therapy trials in humans have shown that aerosolized AAV2 is safe, it is not efficient (6–8). A recent double-blind, placebo-controlled, phase 2b trial with a nonviral gene therapy vector (pGM169/GL67A) showed the feasibility of monthly delivery of a vector to this population and resulted in a small but significant improvement in lung function (9), indicating that enhancements in delivery could enable further progress.
AAV gene transfer efficiency can be modulated or improved by using different serotypes (10–15), site-directed modifications of the capsid (16), and directed evolution (17, 18) of viral capsids. We previously reported a directed evolution process in primary cultures of human airway epithelia. Our innovative strategy used adenovirus to amplify AAV virions that successfully infected airway epithelia. We identified a capsid, termed AAV2.5T, which efficiently transduced CF human airway epithelia. By combining beneficial properties of AAV5 and AAV2, this chimeric virus increased gene delivery efficiency 100-fold relative to AAV2 (17). Our objective herein was to investigate whether AAV-mediated CFTR gene transfer to the airway epithelia of CF pigs is sufficient to reverse the host defense defect in bacterial killing. Since AAV2.5T does not transduce pig airway epithelia (19), we employed an in vitro–in vivo directed evolution strategy to select an AAV capsid that is capable of transducing pig airway epithelia. Our findings establish that efficient AAV gene transfer of CFTR improves the early CF phenotypes of acidic ASL pH and bacterial killing defects in a relevant animal model of this human disease.
Results
Selection of AAV with tropism for porcine airway epithelia.
A pig in vitro–in vivo directed evolution-selection strategy was used to identify capsids with increased efficiency and specificity for pig airway epithelium (Figure 1A). First, to enhance the specificity of the library for pig airway, we pooled several AAV capsid libraries, including a library of shuffled AAV1, -2, -4, -5, -6, -8, and -9 cap genes (20), an error-prone AAV2 library (21), and a library in which loops of the AAV capsid were diversified (22). The pooled library was selected in pig airway epithelia cultures for 3 rounds, and in each round, the sequences of AAV-mutant capsids able to successfully infect pig epithelia were recovered, amplified, and used to generate the next pool. After the third round of in vitro selection, the AAV library was mutagenized to increase capsid diversity of the library as previously described (17). Next, the library was aerosolized into the trachea of a non-CF 6-week-old pig. After 4 days, the pig was euthanized, and epithelial cells were isolated from the trachea and bronchi and infected with WT adenovirus type 5 (Ad5) to complete the AAV replication cycle. Recovered capsids were then cloned and repackaged to create the next library. This process was repeated twice for a total of 3 rounds of in vivo selection. Progression of the in vivo selection was monitored by evaluating the abundance of recovered AAV genomes by PCR (Figure 1B). Following 3 rounds of in vivo selection, AAV DNA from the recovered clones was isolated and sequenced. A single AAV capsid variant (AAV2H22) emerged, which was identical to AAV2 except for 5 amino acid changes (E67A, S207G, Q598L, I648V, and V708I). This variant was previously found in a selection for enhanced infectivity of human glia in vitro (22). Interestingly, when selection was fully conducted in primary cultures of pig airway epithelia and not in vivo — although AAV2H22 was present — we selected primarily AAV6-based capsids (data not shown), suggesting that there could be biological differences between in vivo and in vitro approaches.
Figure 1. Selection of AAV2H22 with tropism for pig airway epithelia.
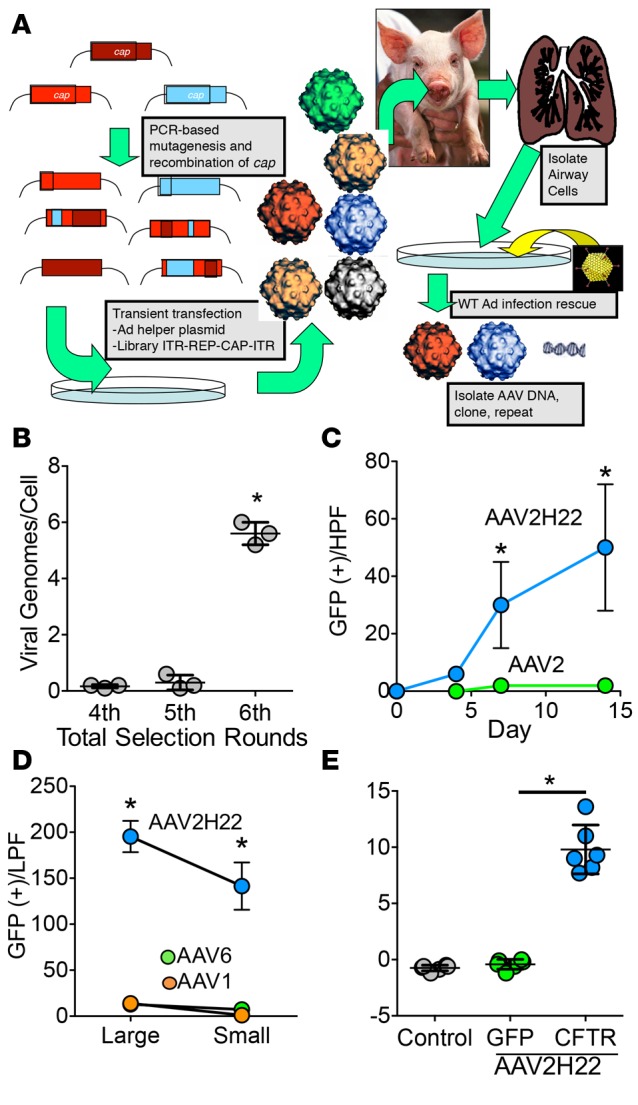
(A) After 3 rounds of in vitro selection to enrich for adeno-associated virus (AAV) with tropism for pig airway, an in vivo selection strategy was taken. Plasmids encoding an AAV viral capsid library were transfected into 293 cells, along with adenovirus helper plasmids. The viral library was aerosolized into the pig trachea, and 3 days later, the lungs were harvested and airway cells were isolated and infected with WT adenovirus (WT Ad). The capsid sequences for the successful AAVs were subcloned, and a new enriched viral library was made for each round. (B) Enrichment was monitored for in vivo rounds 4, 5, and 6 by assessing the number of viral genomes per cell after WT Ad amplification. (C) Pig airway epithelia were transduced with AAV2H22 that expressed GFP (blue line). Expression was monitored over time as GFP+ cells per high-power field (HPF, 20×). n = 6; *P < 0.01 vs. AAV2 (green line), Mann-Whitney nonparametric t test. (D) Comparison of transduction efficiency on large and small pig airway epithelia. AAV2H22 (blue) outperformed AAV1 (orange) and AAV6 (green). GFP+ cells per low power field (LPF, 10×). n = 3 epithelia from 3 different pigs, *P < 0.01 vs. AAV1 and AAV6, Mann-Whitney nonparametric t test. (E) AAV2H22-CFTRΔR–mediated correction of CF pig epithelia. CFTR-null epithelia lack CFTR activity stimulation (ΔIsccAMP) by cAMP levels elevated via forskolin (1 × 10−5 M) and IBMX (1 × 10−4 M). AAV2H22-CFTRΔR–corrected cAMP-regulated chloride current as compared with AAV2H22-GFP. n = 6; *P < 0.01, Mann-Whitney nonparametric t test.
AAV2H22 transduces pig airway epithelia with higher efficiency than AAV1, AAV2, and AAV6.
We next compared the efficacy of recombinant, GFP-encoding AAV2H22 and AAV2 to transduce the apical surface of pig airway epithelia in vitro. AAV2H22-mediated expression of GFP plateaued by 2 weeks, and it was 240 times more efficient than AAV2 (Figure 1C). Next, we asked if AAV2H22 could transduce distal small airway epithelia (<200-μm diameter) using our recently reported method to isolate, expand, and culture small airway epithelia (23). We found that AAV2H22 efficiently transduced both large and small airway epithelia (Figure 1D). We also compared AAV2H22 to AAV1, a serotype previously shown to transduce airway epithelia better than AAV2 (24), and AAV6, a predominant serotype selected from our in vitro evolution strategy. AAV1 and AAV6 showed minimal transduction of large and small airway epithelia (Figure 1D). These data demonstrate the superiority of AAV2H22 as compared with existing naturally occurring AAV serotypes and, moreover, suggest that the AAV2H22 selected in large airways is also functional in small airway epithelia.
We next asked whether AAV2H22 can correct CFTR function in organotypic airway epithelia cultures. AAV2H22-CFTRΔR vector was produced to deliver CFTR minigene of 156 bp partial deletion at R-domain (CFTRΔR) cDNA to CF pig airway epithelia. This vector genome contains a CFTR minigene (CFTRΔR contains a 156-bp partial deletion at the R-domain; refs. 17, 25) under the direction of a short 183-bp synthetic enhancer/promoter (F5Tg83) that has been shown to express efficiently in CF human airway epithelia and restore CFTR channel activity (26). Using this vector genome packaged into the AAV2H22 capsid, analysis of short-circuit current in Ussing chambers demonstrated that AAV2H22-CFTRΔR rescues cAMP-stimulated Cl– current (IsccAMP) in CF pig airway epithelia (Figure 1E).
AAV2H22 specifically binds and infects pig airway epithelia.
We hypothesized that AAV2H22 may have evolved to have higher affinity for its receptor in pig airway epithelia than AAV2. To investigate this possibility, varying concentrations of AAV2 or AAV2H22 virions were allowed to bind to the apical surface of isolated pig airway epithelia at 4°C (Figure 2A). The binding curves of AAV2H22 differed from those of AAV2 and plateaued on airway epithelia. Saturation suggests that AAV2H22 binds to a receptor(s) and binding plateaus once all receptor sites are occupied. By contrast, AAV2 binding did not saturate (Figure 2A), suggesting either a highly abundant receptor or a nonspecific interaction (27, 28), and the latter is consistent with its lower transduction efficiency. We also compared AAV2 and AAV2H22 binding to pig alveolar epithelia (29). In contrast to airway epithelia, both AAV2 and AAV2H22 demonstrated saturated binding to alveolar epithelia, and binding plateaued at a higher concentration for AAV2 (Figure 2B). Consistent with specific binding to alveolar epithelia, AAV2-GFP transduced pig alveolar epithelia 26 times better than AAV2H22-GFP (Figure 2C). Interestingly, comparison of AAV2H22 transduction between species showed that AAV2H22-GFP failed to transduce human airway epithelia (Figure 2D). Together, these data show that selectively isolating an AAV capsid that successfully transduced the large conducting airway of pigs in vivo led to an AAV with stronger tropism for pig airway epithelia compared with alveolar epithelia.
Figure 2. AAV2H22 binds specifically to the apical surfaces of pig airway epithelia.
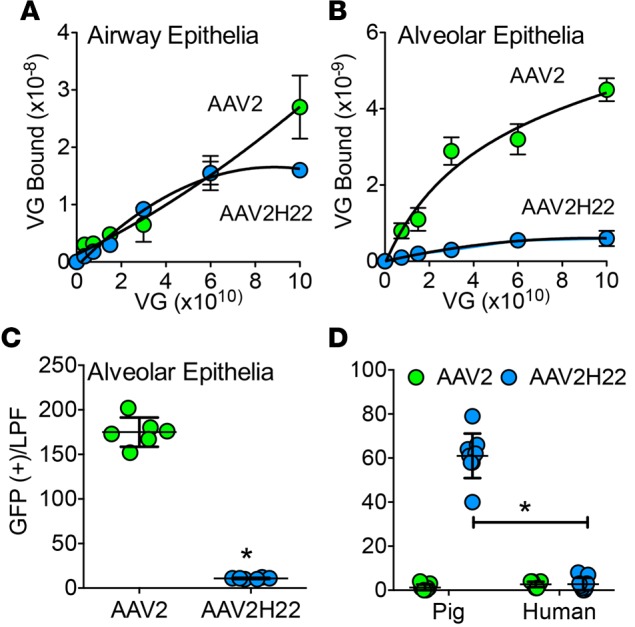
(A and B) AAV2H22 or AAV2 virions were allowed to bind the apical surfaces of pig airway (A) or alveolar (B) epithelia at the indicated doses for 1 hour at 4°C. In airway epithelia, AAV2H22 (blue) plateaued at 6 × 1010 VG, whereas AAV2 (green) did not reach saturation. In alveolar epithelia, both AAV2 and AAV2H22 saturated, but AAV2 had more apparent binding sites. n = 6–12. (C) Consistent with B, AAV2-GFP transduced pig alveolar epithelia more efficiently than AAV2H22-GFP. n = 6; *P < 0.01, Mann-Whitney nonparametric t test. (D) In contrast to pig, AAV2H22-GFP did not infect human airway epithelia. n = 3 from different donors in 4 different experiments; *P < 0.01, Mann-Whitney nonparametric t test.
AAV2H22 does not require heparan sulfate proteoglycans for transduction of HeLa or CHO cells.
Due to the differences in the binding of AAV2 and AAV2H22 in pig airway and alveolar epithelia, we hypothesized that AAV2H22 also utilizes a different primary attachment receptor from AAV2 in cell lines whose primary receptor is heparan sulfate proteoglycan (HSPG) (27, 28). PgsD-677 cells (30) are unable to produce HSPG due to mutations in N-acetylglucosaminyltransferase and glucuronosyltransferase, enzymes required for the polymerization of heparan sulfate chains. We found that AAV2-GFP transduction dropped by approximately 83% in pgsD-677 cells compared with parental CHO-K1 cells, but AAV2H22-GFP was only partially inhibited (Figure 3A). As a control, transduction of AAV5-GFP, which uses sialic acid as a receptor (28), was not inhibited in pgsD-677 cells. This result indicates that AAV2H22-GFP transduces pgsD-677 cells via a receptor other than HSPG.
Figure 3. AAV2H22 is less reliant than AAV2 on heparan sulfate proteoglycan (HSPG) as a receptor in CHO and HeLa cells.
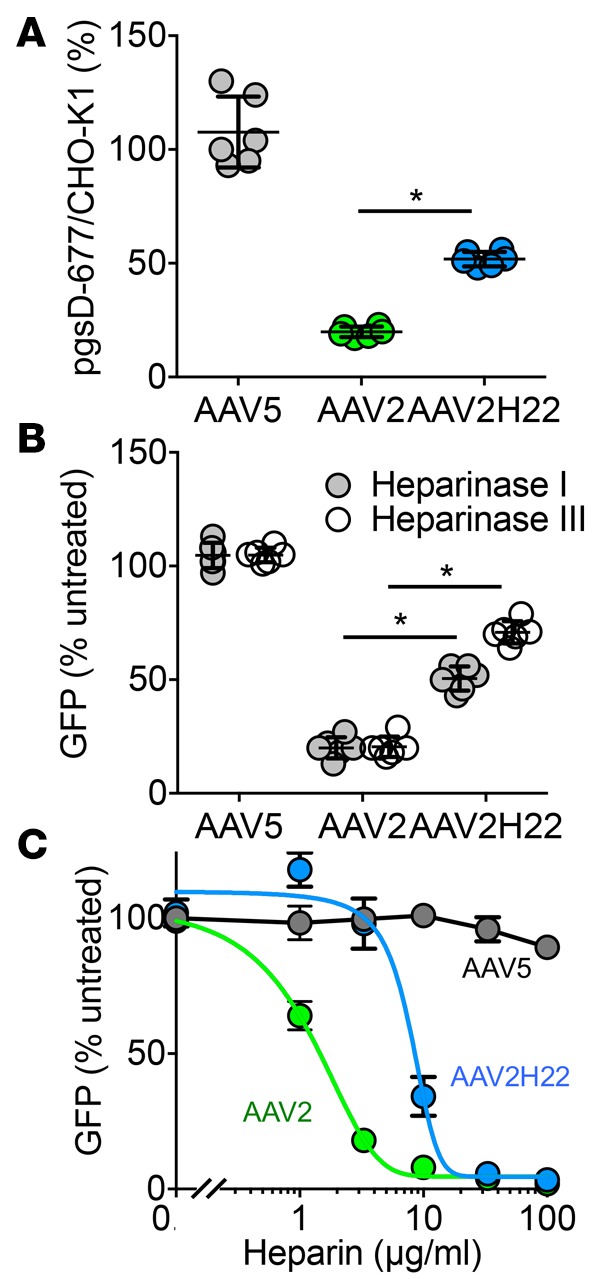
(A) The parental cell line CHO-K1, which expresses HSPG, and mutant cell line pgsD-677, which does not express HSPG, were transduced with AAV2-GFP, AAV5-GFP, or AAV2H22-GFP, and the number of GFP+ cells was determined. Data were quantified as a percentage of GFP+ CHO-K1 cells. n = 6; *P < 0.01 vs. AAV5 *P < 0.01, Mann-Whitney nonparametric t test. (B) The effect of heparinase was determined in HeLa cells treated with heparinase I or heparinase III and inoculated with AAV5, AAV2H22, or AAV2. n = 6; *P < 0.01 vs. untreated *P < 0.01, Mann-Whitney nonparametric t test. (C) AAV2, AAV5, or AAV2H22 were incubated with increasing concentrations of heparin for 30 minutes at 37°C, followed by transduction on HeLa cells. The number of GFP+ cells was determined and quantified as the % change relative to untreated. n = 6; *P < 0.01 vs. untreated *P < 0.01, Mann-Whitney nonparametric t test.
To further investigate the requirement for HSPG, HeLa cells were treated with heparinase I or heparinase III to cleave HSPG at the linkage between hexosamines and O-sulfated iduronic acids or the 1–4 linkages between glucosamine and glucuronic acid, respectively. Both heparinase I and III inhibited AAV2-GFP gene transfer by more than 80%, whereas AAV2H22-GFP was inhibited by less than 50% (Figure 3B), further suggesting that AAV2H22 may utilize a non-HSPG receptor. By contrast, AAV5 was unaffected.
Finally, we tested whether AAV2H22 transduction could be inhibited by soluble heparin sodium salt from porcine intestinal mucosa, which should function as a soluble decoy receptor that competes for binding to the AAV2 capsid. When we incubated AAV2-GFP, AAV5-GFP, or AAV2H22-GFP with increasing concentrations of heparin, the concentration of heparin that reduced transduction to 50% (IC50) of AAV2-GFP transduction was ~2 μg/ml of heparin, whereas AAV2H22-GFP transduction did not reach an IC50 until ~9 μg/ml heparin (Figure 3C).
We had previously shown that HSPG are not present on the surface of airway epithelia (14). Moreover, treating the surface of airway epithelia with heparinase I or heparinase III had no effect on AAV2H22 transduction (data not shown), suggesting that the difference seen in HeLa cells is not responsible for the increased efficiency on airway epithelia.
Together, these data suggest that AAV2H22 retains some ability to bind and utilize HSPGs for infection, but also has evolved to bind an additional receptor that is abundant in the pig airway epithelium.
Modeling AAV2H22 mutations in major capsid protein.
AAV2H22 is identical to AAV2 except for 5 amino acid changes (E67A, S207G, Q598L, I648V, and V708I); some but not all of the 2H22 mutations match the amino acid identity of other serotypes (AAV1, -2, -6, -7, -8, and -9). We modeled the major capsid protein of AAV2H22, superimposed on its parent AAV2 (31), and found that the 2 mutations Q598L and V708I were predicted to lie on the capsid surface. The Q598L amino acid replacement is located in subloop 4 in the surface of the AAV2 capsid, one of the regions implicated in binding to the HSPG receptor (32, 33). Shen et al. found subloop 4 to be required for transduction and likely important for the interaction with other capsid regions (34). Position 708 is located in a plateau that interacts with the monoclonal antibody A20 (35). Substitution at 708 prevents binding of AAV2 to A20 without interfering with HSPG binding or transduction efficiency (36). Three of the mutations in the evolved AAV2H22 virus were not predicted to reside on the surface of the virus (E67A, S207G, and I648V). Importantly, E67A is located in the capsid protein VP1, which has been shown to be important for endocytosis, endosomal escape, and trafficking through the endocytic pathway to the nucleus (37). Thus, the amino acid changes in the evolved AAV2H22 may contribute to the increased tropism for pig airway epithelia.
We asked whether surface-exposed mutations in AAV2H22 are necessary or sufficient for transduction in pig airway epithelia. Individual mutations that occurred in AAV2H22 were introduced into AAV2 by site-directed mutagenesis (Figure 4A). A Q598L or V708I mutation in AAV2-GFP was sufficient to enhance gene transfer to pig airway epithelia (Figure 4B); however, reverting these mutations back to WT within the evolved AAV2H22-GFP demonstrated that neither mutation was required for transduction of pig airway epithelia (Figure 4C). Interestingly, AAV2-GFP with the V708I substitution and AAV2H22-GFP with L598 reverted back to Q598 both outperformed AAV2H22 (Figure 4, B and C). It is possible that some mutations were selected for functions other than their ability to infect pig airway epithelia (e.g., replication, packaging in 293 cells, or interaction with adenovirus). Moreover, the V708I mutation may have additional advantages in vivo, such as minimizing antibody neutralization as previously described (36). Next, we examined whether the non–surface-exposed mutations in AAV2H22 (E67A, S207G, and I648V) are required or sufficient to enhance gene transfer to pig airway epithelia using the same site-directed mutagenesis strategy. We found that E67A enhanced AAV2-mediated gene transfer of GFP but is not required (Figure 4, D and E). This residue is close to the PLA2 catalytic domain (37) and, thus, may affect the ability of AAV2 to escape the endosome. Analysis of S207G demonstrates that this mutation was not sufficient for gene transfer, and more importantly, reverting the mutation back to 207S resulted in enhanced gene transfer by AAV2H22 (Figure 4, D and E). Thus, this mutation may have indirect effects that complement other mutations, or it may have evolved under a different selective pressure (e.g., replication, packaging in 293 cells, or interaction with adenovirus). Finally, I648V enhanced gene transfer of AAV2, and the reversion mutant impaired gene transfer by AAV2H22. This mutation is in an interloop region of unknown function. In summary, these mutations may offer a comprehensive selective advantage in packaging, in vivo infection, and/or in vivo evasion of immunity. Future work may investigate the receptor used by this virus at the apical surface of airway epithelia.
Figure 4. Surface-exposed mutations affect pig airway epithelial transduction.

(A) Diagram highlighting capsid mutations found in AAV2 or AAV2H22. Site-directed mutagenesis was used to introduce mutations to the AAV2 capsid (top) or to revert AAV2H22 to WT (AAV2) capsid sequence (bottom). (B and C) Effect of surface-exposed mutations on GFP transduction efficiency. Residues L598 and I708 were sufficient to augment AAV2 transduction efficiency (B) but were not required for AAV2H22 transduction (C). (D and E) Effect of non–surface-exposed mutations on GFP transduction efficiency. Residues A67 and V648, but not G207, in AAV2 were sufficient to augment GFP transduction (D). However, none were required for AAV2H22 transduction (E). n = 12; *P < 0.01 vs. AAV2 (B and D) or AAV2H22 (C and E). *P < 0.01, Mann-Whitney nonparametric t test.
AAV2H22 restores CFTR expression and activity in pig airway in vivo.
In a proof-of-concept study, four 1-week-old CF pigs received 1.96 × 1012 viral genomes (VG) of AAV2H22-CFTRΔR into the lungs via aerosol delivery and 6 age-matched CF pigs served as a control. Due to low in vitro transduction efficiency in pig airway epithelia cells, AAV2 was not examined in pigs in vivo. Two weeks later, the pigs were euthanized, and CFTR expression in tracheal airway was assessed. IHC on airway tissue sections revealed CFTR protein expression at the apical surface of ciliated cells in non-CF pigs (Figure 5A), and by contrast, no CFTR expression was detected in the control CF pig (Figure 5B). Importantly, the CF pigs that received AAV2H22-CFTRΔR exhibited CFTR expression and localization similar to non-CF pigs (Figure 5, C and D).
Figure 5. AAV2H22-CFTRΔR transduces pig airway epithelia in vivo.

(A–G) Gut-corrected CFTR-null pigs (1 week old) were transduced with 1.96 × 1012 VG of AAV2H22-CFTRΔR. Two weeks later, pigs were studied and compared with age-matched WT and nontreated CFTR-null pigs. (A–D) Confocal images of CFTR expression. (A) WT. (B) CFTR-null control. (C) CFTR-null transduced with AAV2H22-CFTRΔR. (D) Confocal image from C superimposed on differential interference contrast (DIC) image to show cilia. Red, CFTR; green, ZO-1 in A and cadherin in B–D; blue, nuclei (DAPI). (E) Correction of cAMP-stimulated Isc in the trachea of CFTR-null pigs transduced with AAV2H22-CFTRΔR relative to untreated controls. (F) Short-circuit currents of nasal epithelia in a CFTR-null pig transduced with AAV2H22-CFTRΔR in the right nostril. Left nostril was used as control. Inhibition of Na+ current with amiloride (Amil.) (1 × 10−4 M) hyperpolarized apical membrane voltage and increased the driving force for Cl− secretion. Inhibition of non-CFTR Cl− channels with 4,4’-diisothiocyano-2,2’-stilbenedisulfonic acid (DIDS) (1 × 10−4 M). CFTR activity stimulation (ΔIsccAMP) by cAMP levels elevated via forskolin (1 × 10−5 M) and IBMX (1 × 10−4 M). (G) Airway surface liquid (ASL) pH was measured in vivo using pH-sensitive planar optical probe placed on tracheal surface. (H) S. aureus–coated grids were placed on tracheal surface of WT and CFTR-null pigs transduced with AAV2H22-CFTRΔR or control. Data are percentage dead bacteria. For each pig, 2–3 grids were used for 1 minute, 5–16 fields were counted per grid, each field contained ~100–1,000 bacteria, and data from each field were averaged. For A–H, n = 3 CFTR-null pigs transduced with AAV2H22; n = 6 untreated CFTR-null pigs; n = 6 non-CF pigs. *P < 0.05 vs. untreated CFTR-null *P < 0.01, Mann-Whitney nonparametric t test. For F, n = 1 nostril with contralateral nostril as a control. The non-CF control pigs (E, G, and H) were shared with a companion manuscript by Cooney et al. (58).
Additionally, short-circuit current from the excised AAV2H22-CFTRΔR transduced trachea was measured in Ussing chambers, and we observed cAMP-stimulated Cl– current to be about half the current of non-CF pigs of similar age (Figure 5E). We also aerosolized, 1 × 1011 VG of AAV2H22-CFTRΔR in the right nostril of 1-week-old CF pigs, with the left nostril receiving vehicle control. Two weeks later, the freshly excised nasal septum was studied in Ussing chambers. Whereas the control left turbinate epithelia remained CF-like (38), the paired right turbinate epithelia exhibited a pronounced change in short-circuit current (Isc) after stimulation with 3-isobutyl-1-methylxanthine (IBMX) and forskolin that is consistent with reconstituted rescued CFTR expression (Figure 5F).
We have previously reported in newborn CF pigs (in vivo) that loss of CFTR-mediated HCO3– secretion results in decreased ASL pH and impaired bacterial killing (1). We found that, in contrast to untreated CF pigs, AAVH22-mediated CFTRΔR expression restored the tracheal ASL pH alkalinity to levels observed in non-CF pigs of similar age (Figure 5G). Finally, to investigate the effect of AAV2H22-mediated CFTRΔR expression on host defense, we examined bacterial killing using bacteria-coated grids (1). S. aureus chemically linked to gold grids were placed on the surface of tracheal explants from AAV2H22-CFTRΔR–treated pigs or control pigs for 60 seconds. A fluorescent live/dead stain showed that 3-week-old CF pigs have impaired bacterial killing, whereas expression of CFTRΔR with AAV2H22 resulted in a partial rescue of bacterial killing (Figure 5H). These data demonstrate that CFTR gene therapy with AAV2H22 can restore ASL pH to WT levels and partially correct the bacterial killing defect in CF pig airway epithelia. The partial correction of bacterial killing could indicate regional differences in ASL pH restoration or additional defects in host defense.
Discussion
We developed a combined in vitro–in vivo strategy to select an AAV capsid with enhanced tropism and transduction efficiency for pig airway epithelia to test the hypothesis that gene transfer can restore CFTR expression and correct the functional defects observed in CF. Our in vivo selection approach involved AAV library administration to the live pig airway and rescue of successful infectious variants by subsequent ex vivo coinfection with a WT adenovirus. Our selection yielded an AAV capsid that is based on AAV2 with 5 individual mutations. This viral capsid, termed AAV2H22, efficiently and selectively transduces pig airway epithelia. In addition, AAV2H22-delivered CFTRΔR partially corrects transepithelial anion transport, ASL pH, and bacterial killing defects in a relevant, large-animal CF model. In short, for the first time to our knowledge, we show that AAV-mediated gene transfer to the conducting airway epithelium in vivo can complement CFTR activity and enhance host defense in CF.
Evolution and viruses.
For successful infection, viruses must both locate bona fide receptors and avoid decoys. We previously showed that AAVs need to bind a receptor that is internalized in order to initiate airway gene transfer (17, 19). At the same time, they must avoid being bound by abundant low-affinity receptors present on the mucus glycocalyx (39). Interestingly, we found that AAV2H22 is less dependent on heparan sulfate as a receptor and that higher doses of soluble heparin are required to inhibit its transduction. This result suggests that heparan sulfate binding in ASL may contribute to the decreased AAV2 gene transfer efficiency as compared with AAV2H22. The identity of the receptor or receptors for AAV2H22 remains unknown. Of note, one of the mutations in the AAV2H22 capsid (L598Q) resides in subloop 4 and is reportedly important for AAV2 binding to the laminin-A receptor (40). Both heparan sulfate and laminin-A are expressed primarily on the basolateral surface of airway epithelia, which is normally inaccessible for virus binding. Thus, our AAV2H22 avoids being bound to soluble heparan sulfate and sialic acids in mucins and is able to reach receptors on the apical surface that efficiently mediate gene transfer. A recent publication identified a previously uncharacterized type I transmembrane protein, KIAA0319L as the AAV receptor (AAVR) (41). AAVR was shown to be sufficient and required for different AAV serotypes. However, we do not know if AAVR is expressed in pig airway epithelia.
Vectors for pig airways are different than for human airways.
Translational progression for gene therapy traditionally begins with assessing a vector and transgene in vitro, is followed by efficacy analysis in a murine model of the target disease, progresses to safety studies in nonhuman primates, and finally investigates safety and efficacy in humans. Not surprisingly, some vectors that are promising in mice subsequently mediate low gene transfer efficacy in humans (6, 42, 43). Interestingly, when we selected for viruses that transduce human or pig airway epithelia, these viruses were efficient only on cells from the species from which they were selected. These results indicate that the selection pressures applied on viruses to transduce human and pig cells yield viruses with different receptor tropism, which can prevent transduction in other species. This is not surprising, based on the extensive literature for swine influenza viruses and how these viruses rarely evolve to infect humans (44).
A more subtle but surprising finding is the difference in binding and transduction efficacy between the large, small, and alveolar airway epithelia. Many viruses exhibit analogous regional tropism, but the underlying mechanisms remain unclear. Fortunately, advances in tissue-specific organotypic modeling may allow this to be addressed directly in the future. Moreover, these techniques may allow the use of targeted evolution in human lungs. It remains unclear which of these epithelia is the correct therapeutic target to alleviate CF pathology or, alternatively, whether submucosal glands represent the most physiologically important site for CFTR correction, as submucosal glands from CF pigs and humans secrete mucus that is more viscous and acidic — and at lower volumes — than normal (45–47). We recently reported that, following methacholine stimulation, mucus in CF pigs is adherent to the opening of the submucosal glands and impairs mucociliary transport (48, 49). As a result, aerosolized AAV is unlikely to target the submucosal glands, given such complex chemical, biological, and structural barriers. However, future directed evolution studies will consider isolating regional epithelia and submucosal glands to identify whether AAV capsid variants with tropism for these tissues can be engineered and whether there is a difference between normal and CF animals. These future studies may offer tools that allow us to directly address the importance of regional versus broad vector targeting in our preclinical model for CF gene therapy.
There are major challenges with developing biological therapies in animal models. First, if a virus is efficient in an animal model, it may not be efficient in humans. For example, AAV2 efficiently corrected hemophilia B in mice and dogs (50). However, it proved to be less efficient in humans (51). Moreover, similar to the pig specificity observed here, previous directed evolution on human airway epithelia yielded an AAV capsid that was efficient in human but not mouse or pig airway epithelia (17). Second, a biologic that is safe in animal models may not be safe in humans. For example, an immunomodulatory humanized agonistic anti-CD28 monoclonal antibody that was developed for multiple sclerosis, rheumatoid arthritis, and certain cancers was deemed to be safe in animal models (52). However, when tested in humans, the anti-CD28 antibody reacted with Tregs and produced adverse events (53). These examples illustrate that we need to change the vector design and assessment paradigm, where safety and efficacy of animal-specific biologics need to be tested in relevant animal models; however, we also need to recognize that the biologics may be different than the ones that will work in humans. Translational researchers will need to develop creative clinical experiments that minimize risk and allow for testing efficacy and safety in humans.
AAV strengths and limitations.
A major strength of AAV is its impressive safety record in humans with CF. AAV2 that expresses CFTR has been aerosolized to more than 125 people with CF without any major adverse events (6, 7, 42, 54, 55). AAVs form an icosahedral capsid assembled from 60 capsid viral protein monomers (VP monomers). The capsid is composed of 3 VPs — VP1, VP2, and VP3 — with a stoichiometry of 1:1:10. AAV contains a single-stranded DNA that, after nuclear uncoating, is converted into a transcriptionally competent double-stranded genome that persists episomally and integrates into the host genome at low efficiency (56). We now report an efficient AAV capsid with specificity for pig airway epithelia in vitro and after airway administration in vivo, and a key question for future investigation will be the duration of subsequent gene expression in the lung following AAV delivery. Persistent CFTR expression will be desirable for treating CF lung disease, which starts at birth and progresses throughout the lifetime of people with CF. rAAV-mediated gene expression can, in general, be long lasting, and we and others have reported that AAV transduction of airway epithelia in vitro achieves transgene expression for months. The encouraging transduction efficiency of AAV2H22 and the level of CFTRΔR expression in CF pigs demonstrated in these proof-of-concept studies motivate investigation of the hypothesis that AAV2H22-CFTRΔR could persist for months in the airways and prevent early lung disease. However, it is conceivable that repeated administration or permanent modification of airway progenitor cells may be needed for long-term CFTR maintenance (57). Lentiviruses stably integrate into the genome; however, they are more difficult to produce to high titer than AAV, have unclear safety profiles in the lungs, and — similar to AAV — could benefit from progenitor cell delivery to maintain expression. A companion study by McCray and colleagues investigates the application of lentiviral vectors to deliver CFTR to newborn CF pig airways (58). Together, these approaches are expected to become part of a toolkit that will provide critically needed long-term solutions for people with CF.
Methods
Isolation, expansion, and culture of primary epithelial cells
Generation of gut-corrected CFTR−/− pigs has been previously reported (59). All human lung tissues were obtained from the University of Iowa Cell Culture Core Facility. Primary human and pig airway epithelia from non-CF human donor lungs, CF pigs, and non-CF pigs (Exemplar Genetics) were isolated from trachea and bronchi of donor lungs as previously described (60). Cells were seeded onto collagen-coated, semipermeable membranes (Millipore) and grown at the air-liquid interface as previously described (60, 61). Isolation, expansion, and culture of pig large proximal (including trachea and bronchi) and small distal (diameter ~200 μm) airway epithelia were performed as previously described (23, 29). Briefly, large and small airway cells were expanded by culturing in F media in the presence of 10 μM Y-27632, a ROCK inhibitor, and irradiated fibroblast feeder cells NIH-3T3-J2. After 2 passages of amplification, expanded cells were then seeded onto collagen-coated, semipermeable membranes in the absence of feeder cells and ROCK inhibitor and maintained in Small Airway Growth Media (Lonza) supplemented with 10 ng/ml keratinocyte growth factor (KGF) for 1 week, after which cells were maintained in USG media. Experiments were performed on matched large and small airway epithelia isolated from the same animal and cultured under identical conditions. Pig alveolar epithelia were isolated and cultured as previously described (29, 62, 63). Cell lines were obtained from the ATCC except AAV293 cells (Stratagene California) and cultured as follows: HeLa, CHO-K1, and CHO pgsD-677 were cultured in IMDM (Mediatech), and AAV293 cells were cultured in DMEM (Gibco). All cells were maintained at 37°C and 5% CO2.
AAV library production
DNA shuffling and mutagenesis were used as previously described (17, 64–68) to create a viral library from the following AAV serotypes: AAV1, AAV2, AAV4, AAV5, AAV6, AAV8, and AAV9. Additionally, a library was constructed from random peptide insertions into AAV2 surface loops, as previously described, to provide further diversity to the library (66).
Selection of evolved AAV specific for pig airway epithelia
A pig in vitro–in vivo evolution-selection strategy was used to identify capsids with improved efficiency and specificity for airway epithelium (Figure 1A).
In vitro selection in organotypic primary pig airway epithelia.
The AAV library was inoculated on the apical surface of pig airway epithelial cultures at an MOI of 100 VG/cell diluted in EMEM (Invitrogen) at a total volume of 35 μl per epithelium. Three epithelia were used from 3 different non-CF pigs for each round of selection. The cultures were incubated for 4 hours at 37°C in the presence of virus, followed by removal of the virus by aspiration and 2 washes with 250 μl of phosphate buffered saline. Three days after adding the AAV library to the cells, 100 pfu/cell of WT Ad5, diluted in EMEM for a final volume of 20 μl, was adsorbed to the basolateral surface of the airway epithelia. Forty hours after Ad5 infection, epithelia were freeze-thawed 3 times, followed by heat inactivation at 95°C for 10 minutes in a solution of 150 mM NaCl and 50 mM Tris (pH 8.0). Successful variants were cloned into pSub2 as previously described (69), repackaged in new libraries, and subjected to further rounds of selection for a total of 3 rounds in pig airway epithelial cultures (17).
In vivo selection in pigs.
Although we initially attempted selection directly in vivo, no AAV capsids were recovered after round 3. Therefore, we took a preenrichment approach. After preselecting the virus library for 3 rounds in vitro in cultured pig airway epithelia, we subjected the resulting recovered variants to further shuffling and mutagenesis. For each round of selection, 4- to 8-week-old non-CF pigs were sedated using ketamine (20 mg/kg, i.m. injection; Akorn, Inc), xylazine (2 mg/kg, i.m. injection; Biomeda-MTC), and propofol (2 mg/kg, i.v. injection; Fresenius Kabi USA, LLC), and then approximately 1 × 1011 viral genomes were aerosolized into the airways using a LABneb catheter control unit (Trudell Medical International) and an AeroProbe Intracorporeal Nebulizing Catheter, laboratory version (Trudell Medical International). Three days after infection, the pig was sedated and then euthanized by intracardiac injection of Euthasol (Virbac Corporation). The lungs were removed, and airway epithelial cells were isolated from the trachea, carina, and primary bronchi and plated as previously described (60, 61). Four to 5 days after plating, WT Ad5 was added at a concentration of ~100 pfu/cell to allow the AAV variants that successfully infected the cells to replicate. Cells were harvested 36 hours later, DNA was isolated (Qiagen DNA Blood Kit), and PCR was performed to detect AAV DNA. Successful variants were cloned as previously described, repackaged in new libraries, and subjected to further rounds of selection (17). A total of 3 rounds of in vivo selection were performed.
AAV transduction assays
Self-complementary AAV1 (scAAV1), scAAV2, scAAV6, or scAAV2H22 vectors harboring enhanced GFP were diluted in EMEM and added to the apical surface of pig or human airway epithelia (10,000 VG/cell) or added to CHO-K1 and CHO pgsD-677 (10,000 VG/cell). Samples were incubated for 4 hours at 37°C, followed by 2 washes with PBS. GFP+ cells were visualized with an Olympus IX71 fluorescence microscope (10× magnification, low-power field [LPF]; 20× magnification, high-power field [HPF]), and data were calculated as the number of GFP+ cells per field.
Viral binding assays
Virions of AAV2-CMV-eGFP or AAV2H22-CMV-eGFP were allowed to bind to the apical surface of pig airway or alveolar epithelia for 1 hour on ice followed by 2 washes with 250 μl of ice cold phosphate buffered saline. Total DNA was extracted, and quantitative PCR (qPCR) was performed using SYBR Green Extract-N-Amp (Sigma Aldrich) according to the manufacturer’s instructions. qPCR primers were designed for the CMV promoter of the virus (5′-AAATCAACGGGACTTTCCAA-3′ and 5′-GGTTCACTAAACGAGCTC-3′) and compared with known amounts of viral DNA standards to calculate the number of bound viral genomes. Electron microscopy of viral preparations established that less than 10% of the capsids were empty, meaning they do not contain DNA.
Heparinase treatment
Heparinase I and heparinase III (H2519 and H8891, respectively; Sigma Aldrich) were diluted in phosphate buffered saline (with calcium and magnesium). HeLa cells were treated with heparinase I (0.01 milliIU/mL) or heparinase III (0.001 milliIU) for 2 hours at 37ºC, followed by removal of the enzyme mixture and inoculation with 7.5 × 104 VG/cell of GFP-expressing AAV2, AAV5, or AAV2H22 diluted in EMEM for 1 hour at 37°C. Cells were washed twice with phosphate buffered saline to remove unbound virions. After culture in fresh medium for 24 hours, the number of GFP+ cells was determined by flow cytometry.
Heparin competition
Heparin sodium salt from porcine intestinal mucosa (H3149-250KU, Sigma Aldrich) was diluted in EMEM at concentrations of 0, 1, 3, 10, 30, or 100 μg/ml, and mixed with GFP-expressing AAV2, AAV5, or AAV2H22. The heparin and virus mixtures were incubated at 37ºC for 1 hour, followed by inoculation of the HeLa cells with the mixtures at a dose of 7.5 × 104 VG/cell for 1 hour at 37°C. After removal of the heparin and virus mixture, cells were cultured for 24 hours, and the number of GFP+ cells were determined by flow cytometry.
Three-dimensional modeling of the AAV capsid
Three-dimensional modeling of the AAV2 capsid was performed using PyMol (http://www.pymol.org/) and the published AAV2 VP3 X-ray crystallography structure (Protein Databank accession no. 1LP3; http://www.rcsb.org/) in order to identify surface-exposed amino acids (31).
Site-directed mutagenesis in AAV2 and evolved AAV2H22
Single nucleotide changes were engineered into AAV2 or AAV2H22 by site-directed mutagenesis using the QuikChange Lightning site-directed mutagenesis kit (Agilent Technologies) per the manufacturer’s recommendation.
AAV-CFTR expression vector
A 4.88 kb recombinant AAV2 (rAAV2) genome, which expresses CFTRΔR under the control of a short synthetic enhancer/promoter F5tg83 and synthetic polyadenylation signal vector (26), was packaged into the AAV2H22 capsid to produce the AAV2H22-CFTRΔR. The replication-competent AAV (rcAAV) library and rAAV vectors were packaged and purified via iodixanol gradient centrifugation as previously described. Viral vectors were harvested and titered via qPCR to obtain DNase-resistant genomic titers (16).
Electrophysiological measurements in freshly excised and cultured epithelia
Cultured pig airway epithelia.
AAV2H22-CFTRΔR (5,000 VG/cell) was diluted in EMEM and added to the apical surface of pig airway epithelia. Two weeks after AAV transduction, cells were mounted in Ussing chambers and studied as previously described (70–72).
In vivo experiments.
AAV2H22-CFTRΔR (1.96 × 1012 VG in 100 μl) was aerosolized into the lungs of 1-week-old CF pigs (n = 3), and 1 × 1011 VG was aerosolized into one nare (n = 3). To improve the time course of gene expression, virus was administered in the presence of 200 μM doxorubicin (Calbiochem, ref. 73). Doxorubicin has been shown to effectively increase viral capsid ubiquitination and transduction of recombinant AAV type 2. Two weeks after infection, animals were euthanized and epithelial tissues were immediately excised from the trachea and the nasal turbinate/septum of the pigs. Tissues were studied in Ussing chambers as previously described (1, 48, 74, 75). Apical and basolateral chambers contained the same bathing solution containing (mM): 135 NaCl, 2.4 K2HPO4, 0.6 KH2PO4, 1.2 CaCl2, 1.2 MgCl2, 10 dextrose, 5 HEPES (pH = 7.4) at 37°C, and gassed with compressed air. CFTR-mediated Cl– current was measured using a previously described protocol (72). Cellular levels of cAMP were increased with forskolin (10μM) and IBMX (100 μM), and the cAMP-stimulated current was measured.
Immunofluorescence of pig airways
Two weeks after viral inoculation, the pigs were sedated with ketamine and xylazine and euthanized by intracardiac injection of Euthasol (Virbac). Tissues from the tracheal lobe were fixed with 4% paraformaldehyde for 1 hour at room temperature, followed by 30% sucrose for 24 hours at 4oC. Excess sucrose was removed, and the tissues were embedded in optimal cutting temperature (OCT) compound (Tissue Tek by Sakura Finetek) and flash frozen in liquid nitrogen. Tissues were then sectioned using a cryostat into 7-μm sections and mounted on Superfrost Plus microscope slides (Thermo Fisher Scientific). Cryosectioned tissues were permeabilized in 0.2% Triton X-100 (Thermo Fisher Scientific) in PBS, and blocked in Super-Block (Thermo Fisher Scientific) with 5% normal goat serum (Jackson ImmunoResearch Laboratories). Tissue sections were incubated for 2 hours at 37°C in anti-CFTR antibody 596 (CF Folding Consortium) and polyclonal antibody to ZO-1 or Cadherin (Invitrogen and Zymed, respectively) (all at 1:100 dilution), followed by secondary antibodies (goat anti–mouse Alexa Fluor 488 and goat anti–rabbit Alexa Fluor 568 [Molecular Probes; Invitrogen] at 1:1,000 dilution). Sections were mounted with Vectashield containing DAPI (Vector Laboratories) to visualize nuclei. Images were acquired using sequential scanning on an Olympus Fluoview FV1000 confocal microscope with a UPLSAPO ×60 oil lens.
Bacteria-killing assay
The S. aureus grid assay was performed as previously described (1). Briefly, gold grids (TEM grids, 200 mesh, Ted Pella Inc.) were immersed in 1 mM 11-mercaptoundecanoic acid (HS(CH2)10COOH, MUA, Sigma Aldrich) solution for 1 hour at room temperature, and then exposed to 1-ethyl-3-(3-diethylaminopropyl)carbodiimide (EDC) and N-hydroxysuccinimide (NHS) (molar ratio 1:1) for 30 minutes to activate the carboxyl groups of MUA. Grids were then placed in PBS, pH 7.4, containing 10 μg ml−1 streptavidin (Sigma Aldrich) for 30 minutes at 37°C and rinsed with PBS. S. aureus isolate 43SA was cultured to log-phase growth and 1 × 108 bacteria were mixed with 0.1 mg ml−1 N-hydroxysulphosuccinimide (sulpho-NHS)-biotin (Thermo Fisher Scientific) for 1 hour at room temperature. Bacteria was immersed in 1 M glycine to quench the reaction and then rinsed with PBS. Streptavidin-coated grids were then incubated with biotinylated bacteria for 30 minutes and rinsed in PBS. Bacteria-coated grids were placed on the airway surface in pigs in vivo. After removal from the airway surface, they were immediately (2–3 seconds) rinsed with PBS and then immersed in PBS containing the fluorescent indicators SYTO 9 and propidium iodide (Live/Dead BacLight Bacterial Viability assay, Invitrogen). After 15 minutes, the grids were rinsed with PBS and placed on slides for imaging with a laser-scanning confocal microscope (Olympus FV1000). Images were automatically counted using ImageJ plugin ITCN (76).
Measurement of ASL pH
To assess pH in vivo, we used noninvasive dual lifetime referencing to interrogate a 3 × 3 mm planar optode (pH sensitive foil, PreSens GmbH) (77) applied directly to the tracheal surface as previously described (1). Briefly, a single channel pH meter (pH-1 mini; PreSens GmbH) was used to transmit and receive the excitation and emission light. The tip of the fiber optic pH meter was kept at the same distance from the tracheal surface in all samples and was confirmed by recording the amplitude registered by the device. Calibration before each set of measurements was done by placing the planar optode on the surface of a flat filter soaked in standard pH buffers. To minimize alterations in CO2 during placement of the probe, the experiments were done in an environment of 5% CO2.
Statistics
Unless otherwise noted, all data were evaluated using GraphPad Prism software. Error bars represent ±SD. Statistical significance was determined by Mann-Whitney nonparametric t test.
Study approval
All animal studies were reviewed and approved by the University of Iowa Animal Care and Use Committee and were carried out in accordance with institutional guidelines and regulations. All human lung tissues were obtained from the University of Iowa Cell Culture Core Facility, which acquired tissue from donors under an organ research donation protocol that was approved by the IRB of the University of Iowa.
Author contributions
BS, DDD, and KJDAE designed research studies, conducted experiments, acquired data, analyzed data, and wrote the manuscript. JB and JRW generated the AAV library and amplified the library after selection. XL, MHAA, VSS, and DCB conducted experiments, acquired data, and analyzed data. ZY and JFE developed the synthetic enhancer/promoter. JZ did study design, data interpretation, and manuscript writing. LSP and NDG acquired data, analyzed data, and managed animals. PLS handled study design, AAV viral production, and manuscript editing. LSO designed research studies, and was responsible for immunocytochemistry. DAS, MJW, and DVS did study design, data interpretation, and manuscript editing.
Acknowledgments
This work was supported by grants from the NIH (HL051670) and the University of Iowa Center for Gene Therapy (DK054759).
Footnotes
Conflict of interest: KJDAE is a member of the scientific advisory board, and DVS is the cofounder, acting CSO, and cochairman of 4D Molecular Therapeutics. MJW is a founder of Exemplar Genetics. DAS, MJW, and the University of Iowa Research Foundation have applied for a patent related to genetically modified pigs. MJW, JFE, and JZ are founders of and hold equity in Talee Bio.
Reference information:JCI Insight. 2016;1(14):e88728. doi:10.1172/jci.insight.88728.
Contributor Information
Benjamin Steines, Email: bensteines@gmail.com.
Jamie Bergen, Email: jamie.bergen@gmail.com.
Xiaopeng Li, Email: xiaopeng-li@uiowa.edu.
Ziying Yan, Email: ziying-yan@uiowa.edu.
John F. Engelhardt, Email: john-engelhardt@uiowa.edu.
Joseph Zabner, Email: joseph-zabner@uiowa.edu.
References
- 1.Pezzulo AA, et al. Reduced airway surface pH impairs bacterial killing in the porcine cystic fibrosis lung. Nature. 2012;487(7405):109–113. doi: 10.1038/nature11130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tang XX, et al. Acidic pH increases airway surface liquid viscosity in cystic fibrosis. J Clin Invest. 2016;126(3):879–891. doi: 10.1172/JCI83922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shah VS, et al. Airway acidification initiates host defense abnormalities in cystic fibrosis mice. Science. 2016;351(6272):503–507. doi: 10.1126/science.aad5589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rowe SM, Miller S, Sorscher EJ. Cystic fibrosis. N Engl J Med. 2005;352(19):1992–2001. doi: 10.1056/NEJMra043184. [DOI] [PubMed] [Google Scholar]
- 5.Stoltz DA, Meyerholz DK, Welsh MJ. Origins of cystic fibrosis lung disease. N Engl J Med. 2015;372(4):351–362. doi: 10.1056/NEJMra1300109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wagner JA, et al. A phase II, double-blind, randomized, placebo-controlled clinical trial of tgAAVCF using maxillary sinus delivery in patients with cystic fibrosis with antrostomies. Hum Gene Ther. 2002;13(11):1349–1359. doi: 10.1089/104303402760128577. [DOI] [PubMed] [Google Scholar]
- 7.Moss RB, et al. Repeated aerosolized AAV-CFTR for treatment of cystic fibrosis: a randomized placebo-controlled phase 2B trial. Hum Gene Ther. 2007;18(8):726–732. doi: 10.1089/hum.2007.022. [DOI] [PubMed] [Google Scholar]
- 8.Mingozzi F, High KA. Therapeutic in vivo gene transfer for genetic disease using AAV: progress and challenges. Nat Rev Genet. 2011;12(5):341–355. doi: 10.1038/nrg2988. [DOI] [PubMed] [Google Scholar]
- 9.Alton EW, et al. Repeated nebulisation of non-viral CFTR gene therapy in patients with cystic fibrosis: a randomised, double-blind, placebo-controlled, phase 2b trial. Lancet Respir Med. 2015;3(9):684–691. doi: 10.1016/S2213-2600(15)00245-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Keswani SG, et al. Pseudotyped AAV vector-mediated gene transfer in a human fetal trachea xenograft model: implications for in utero gene therapy for cystic fibrosis. PLoS One. 2012;7(8):e43633. doi: 10.1371/journal.pone.0043633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Limberis MP, Vandenberghe LH, Zhang L, Pickles RJ, Wilson JM. Transduction efficiencies of novel AAV vectors in mouse airway epithelium in vivo and human ciliated airway epithelium in vitro. Mol Ther. 2009;17(2):294–301. doi: 10.1038/mt.2008.261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bals R, Xiao W, Sang N, Weiner DJ, Meegalla RL, Wilson JM. Transduction of well-differentiated airway epithelium by recombinant adeno-associated virus is limited by vector entry. J Virol. 1999;73(7):6085–6088. doi: 10.1128/jvi.73.7.6085-6088.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sirninger J, et al. Functional characterization of a recombinant adeno-associated virus 5-pseudotyped cystic fibrosis transmembrane conductance regulator vector. Hum Gene Ther. 2004;15(9):832–841. doi: 10.1089/hum.2004.15.832. [DOI] [PubMed] [Google Scholar]
- 14.Zabner J, et al. Adeno-associated virus type 5 (AAV5) but not AAV2 binds to the apical surfaces of airway epithelia and facilitates gene transfer. J Virol. 2000;74(8):3852–3858. doi: 10.1128/JVI.74.8.3852-3858.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fischer AC, et al. Expression of a truncated cystic fibrosis transmembrane conductance regulator with an AAV5-pseudotyped vector in primates. Mol Ther. 2007;15(4):756–763. doi: 10.1038/sj.mt.6300059. [DOI] [PubMed] [Google Scholar]
- 16.Song Y, et al. Functional cystic fibrosis transmembrane conductance regulator expression in cystic fibrosis airway epithelial cells by AAV6.2-mediated segmental trans-splicing. Hum Gene Ther. 2009;20(3):267–281. doi: 10.1089/hum.2008.173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Excoffon KJ, et al. Directed evolution of adeno-associated virus to an infectious respiratory virus. Proc Natl Acad Sci U S A. 2009;106(10):3865–3870. doi: 10.1073/pnas.0813365106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li W, et al. Generation of novel AAV variants by directed evolution for improved CFTR delivery to human ciliated airway epithelium. Mol Ther. 2009;17(12):2067–2077. doi: 10.1038/mt.2009.155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dickey DD, et al. Enhanced sialic acid-dependent endocytosis explains the increased efficiency of infection of airway epithelia by a novel adeno-associated virus. J Virol. 2011;85(17):9023–9030. doi: 10.1128/JVI.05154-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jang JH, et al. An evolved adeno-associated viral variant enhances gene delivery and gene targeting in neural stem cells. Mol Ther. 2011;19(4):667–675. doi: 10.1038/mt.2010.287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Maheshri N, Koerber JT, Kaspar BK, Schaffer DV. Directed evolution of adeno-associated virus yields enhanced gene delivery vectors. Nat Biotechnol. 2006;24(2):198–204. doi: 10.1038/nbt1182. [DOI] [PubMed] [Google Scholar]
- 22.Koerber JT, Klimczak R, Jang JH, Dalkara D, Flannery JG, Schaffer DV. Molecular evolution of adeno-associated virus for enhanced glial gene delivery. Mol Ther. 2009;17(12):2088–2095. doi: 10.1038/mt.2009.184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li X, et al. Electrolyte transport properties in distal small airways from cystic fibrosis pigs with implications for host defense. Am J Physiol Lung Cell Mol Physiol. 2016;310(7):L670–L679. doi: 10.1152/ajplung.00422.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liu X, Luo M, Guo C, Yan Z, Wang Y, Engelhardt JF. Comparative biology of rAAV transduction in ferret, pig and human airway epithelia. Gene Ther. 2007;14(21):1543–1548. doi: 10.1038/sj.gt.3303014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ostedgaard LS, et al. A shortened adeno-associated virus expression cassette for CFTR gene transfer to cystic fibrosis airway epithelia. Proc Natl Acad Sci U S A. 2005;102(8):2952–2957. doi: 10.1073/pnas.0409845102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yan Z, et al. Optimization of Recombinant Adeno-Associated Virus-Mediated Expression for Large Transgenes, Using a Synthetic Promoter and Tandem Array Enhancers. Hum Gene Ther. 2015;26(6):334–346. doi: 10.1089/hum.2015.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kern A, et al. Identification of a heparin-binding motif on adeno-associated virus type 2 capsids. J Virol. 2003;77(20):11072–11081. doi: 10.1128/JVI.77.20.11072-11081.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Walters RW, et al. Binding of adeno-associated virus type 5 to 2,3-linked sialic acid is required for gene transfer. J Biol Chem. 2001;276(23):20610–20616. doi: 10.1074/jbc.M101559200. [DOI] [PubMed] [Google Scholar]
- 29.Li X, et al. CFTR is required for maximal transepithelial liquid transport in pig alveolar epithelia. Am J Physiol Lung Cell Mol Physiol. 2012;303(2):L152–L160. doi: 10.1152/ajplung.00116.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ji ZS, Brecht WJ, Miranda RD, Hussain MM, Innerarity TL, Mahley RW. Role of heparan sulfate proteoglycans in the binding and uptake of apolipoprotein E-enriched remnant lipoproteins by cultured cells. J Biol Chem. 1993;268(14):10160–10167. [PubMed] [Google Scholar]
- 31.Xie Q, et al. The atomic structure of adeno-associated virus (AAV-2), a vector for human gene therapy. Proc Natl Acad Sci U S A. 2002;99(16):10405–10410. doi: 10.1073/pnas.162250899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Opie SR, Warrington KH, Agbandje-McKenna M, Zolotukhin S, Muzyczka N. Identification of amino acid residues in the capsid proteins of adeno-associated virus type 2 that contribute to heparan sulfate proteoglycan binding. J Virol. 2003;77(12):6995–7006. doi: 10.1128/JVI.77.12.6995-7006.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Summerford C, Samulski RJ. Membrane-associated heparan sulfate proteoglycan is a receptor for adeno-associated virus type 2 virions. J Virol. 1998;72(2):1438–1445. doi: 10.1128/jvi.72.2.1438-1445.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shen X, Storm T, Kay MA. Characterization of the relationship of AAV capsid domain swapping to liver transduction efficiency. Mol Ther. 2007;15(11):1955–1962. doi: 10.1038/sj.mt.6300293. [DOI] [PubMed] [Google Scholar]
- 35.McCraw DM, O’Donnell JK, Taylor KA, Stagg SM, Chapman MS. Structure of adeno-associated virus-2 in complex with neutralizing monoclonal antibody A20. Virology. 2012;431(1-2):40–49. doi: 10.1016/j.virol.2012.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lochrie MA, et al. Mutations on the external surfaces of adeno-associated virus type 2 capsids that affect transduction and neutralization. J Virol. 2006;80(2):821–834. doi: 10.1128/JVI.80.2.821-834.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Girod A, et al. The VP1 capsid protein of adeno-associated virus type 2 is carrying a phospholipase A2 domain required for virus infectivity. J Gen Virol. 2002;83(Pt 5):973–978. doi: 10.1099/0022-1317-83-5-973. [DOI] [PubMed] [Google Scholar]
- 38.Chen JH, et al. Loss of anion transport without increased sodium absorption characterizes newborn porcine cystic fibrosis airway epithelia. Cell. 2010;143(6):911–923. doi: 10.1016/j.cell.2010.11.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Walters RW, Pilewski JM, Chiorini JA, Zabner J. Secreted and transmembrane mucins inhibit gene transfer with AAV4 more efficiently than AAV5. J Biol Chem. 2002;277(26):23709–23713. doi: 10.1074/jbc.M200292200. [DOI] [PubMed] [Google Scholar]
- 40.Akache B, Grimm D, Pandey K, Yant SR, Xu H, Kay MA. The 37/67-kilodalton laminin receptor is a receptor for adeno-associated virus serotypes 8, 2, 3, and 9. J Virol. 2006;80(19):9831–9836. doi: 10.1128/JVI.00878-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pillay S, et al. An essential receptor for adeno-associated virus infection. Nature. 2016;530(7588):108–112. doi: 10.1038/nature16465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Moss R, et al. A multicenter double-blind, placebo controlled, phase II study of aerosolized TGAAVCF in cystic fibrosis patients with mild lung disease. Peds Pulmonol. 2002;S24(2):250–251. [Google Scholar]
- 43.Zabner J, Couture LA, Gregory RJ, Graham SM, Smith AE, Welsh MJ. Adenovirus-mediated gene transfer transiently corrects the chloride transport defect in nasal epithelia of patients with cystic fibrosis. Cell. 1993;75(2):207–216. doi: 10.1016/0092-8674(93)80063-K. [DOI] [PubMed] [Google Scholar]
- 44.Ma W, Kahn RE, Richt JA. The pig as a mixing vessel for influenza viruses: Human and veterinary implications. J Mol Genet Med. 2008;3(1):158–166. [PMC free article] [PubMed] [Google Scholar]
- 45.Matsui H, et al. Evidence for periciliary liquid layer depletion, not abnormal ion composition, in the pathogenesis of cystic fibrosis airways disease. Cell. 1998;95(7):1005–1015. doi: 10.1016/S0092-8674(00)81724-9. [DOI] [PubMed] [Google Scholar]
- 46.Wine JJ, Joo NS. Submucosal glands and airway defense. Proc Am Thorac Soc. 2004;1(1):47–53. doi: 10.1513/pats.2306015. [DOI] [PubMed] [Google Scholar]
- 47.Hoegger MJ, et al. Impaired mucus detachment disrupts mucociliary transport in a piglet model of cystic fibrosis. Science. 2014;345(6198):818–822. doi: 10.1126/science.1255825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Stoltz DA, et al. Cystic fibrosis pigs develop lung disease and exhibit defective bacterial eradication at birth. Sci Transl Med. 2010;2(29):29ra31. doi: 10.1126/scitranslmed.3000928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hoegger MJ, et al. Impaired mucus detachment disrupts mucociliary transport in a piglet model of cystic fibrosis. Science. 2014;345(6198):818–822. doi: 10.1126/science.1255825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Herzog RW, et al. Long-term correction of canine hemophilia B by gene transfer of blood coagulation factor IX mediated by adeno-associated viral vector. Nat Med. 1999;5(1):56–63. doi: 10.1038/4743. [DOI] [PubMed] [Google Scholar]
- 51.Pien GC, et al. Capsid antigen presentation flags human hepatocytes for destruction after transduction by adeno-associated viral vectors. J Clin Invest. 2009;119(6):1688–1695. doi: 10.1172/JCI36891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Beyersdorf N, et al. Selective targeting of regulatory T cells with CD28 superagonists allows effective therapy of experimental autoimmune encephalomyelitis. J Exp Med. 2005;202(3):445–455. doi: 10.1084/jem.20051060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Suntharalingam G, et al. Cytokine storm in a phase 1 trial of the anti-CD28 monoclonal antibody TGN1412. N Engl J Med. 2006;355(10):1018–1028. doi: 10.1056/NEJMoa063842. [DOI] [PubMed] [Google Scholar]
- 54.Aitken ML, et al. A phase I study of aerosolized administration of tgAAVCF to cystic fibrosis subjects with mild lung disease. Hum Gene Ther. 2001;12(15):1907–1916. doi: 10.1089/104303401753153956. [DOI] [PubMed] [Google Scholar]
- 55.Moss RB, et al. Repeated adeno-associated virus serotype 2 aerosol-mediated cystic fibrosis transmembrane regulator gene transfer to the lungs of patients with cystic fibrosis: a multicenter, double-blind, placebo-controlled trial. Chest. 2004;125(2):509–521. doi: 10.1378/chest.125.2.509. [DOI] [PubMed] [Google Scholar]
- 56.Ding W, Yan Z, Zak R, Saavedra M, Rodman DM, Engelhardt JF. Second-strand genome conversion of adeno-associated virus type 2 (AAV-2) and AAV-5 is not rate limiting following apical infection of polarized human airway epithelia. J Virol. 2003;77(13):7361–7366. doi: 10.1128/JVI.77.13.7361-7366.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Li X, et al. Integrin α6β4 identifies human distal lung epithelial progenitor cells with potential as a cell-based therapy for cystic fibrosis lung disease. PLoS One. 2013;8(12):e83624. doi: 10.1371/journal.pone.0083624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Cooney A, et al. Lentiviral-mediated phenotypic correction of cystic fibrosis pigs. JCI Insight. 2016;1(14):e88730. doi: 10.1172/jci.insight.88730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Stoltz DA, et al. Intestinal CFTR expression alleviates meconium ileus in cystic fibrosis pigs. J Clin Invest. 2013;123(6):2685–2693. doi: 10.1172/JCI68867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Karp PH, et al. An in vitro model of differentiated human airway epithelia. Methods for establishing primary cultures. Methods Mol Biol. 2002;188:115–137. doi: 10.1385/1-59259-185-X:115. [DOI] [PubMed] [Google Scholar]
- 61.Zabner J, Wadsworth SC, Smith AE, Welsh MJ. Adenovirus-mediated generation of cAMP-stimulated Cl- transport in cystic fibrosis airway epithelia in vitro: effect of promoter and administration method. Gene Ther. 1996;3(5):458–465. [PubMed] [Google Scholar]
- 62.Steimer A, Laue M, Franke H, Haltner-Ukomado E, Lehr CM. Porcine alveolar epithelial cells in primary culture: morphological, bioelectrical and immunocytochemical characterization. Pharm Res. 2006;23(9):2078–2093. doi: 10.1007/s11095-006-9057-7. [DOI] [PubMed] [Google Scholar]
- 63.Steimer A, Franke H, Haltner-Ukomado E, Laue M, Ehrhardt C, Lehr CM. Monolayers of porcine alveolar epithelial cells in primary culture as an in vitro model for drug absorption studies. Eur J Pharm Biopharm. 2007;66(3):372–382. doi: 10.1016/j.ejpb.2006.11.006. [DOI] [PubMed] [Google Scholar]
- 64.Koerber JT, Maheshri N, Kaspar BK, Schaffer DV. Construction of diverse adeno-associated viral libraries for directed evolution of enhanced gene delivery vehicles. Nat Protoc. 2006;1(2):701–706. doi: 10.1038/nprot.2006.93. [DOI] [PubMed] [Google Scholar]
- 65.Müller OJ, et al. Random peptide libraries displayed on adeno-associated virus to select for targeted gene therapy vectors. Nat Biotechnol. 2003;21(9):1040–1046. doi: 10.1038/nbt856. [DOI] [PubMed] [Google Scholar]
- 66.Maheshri N, Koerber JT, Kaspar BK, Schaffer DV. Directed evolution of adeno-associated virus yields enhanced gene delivery vectors. Nat Biotechnol. 2006;24(2):198–204. doi: 10.1038/nbt1182. [DOI] [PubMed] [Google Scholar]
- 67.Zhao H, Giver L, Shao Z, Affholter JA, Arnold FH. Molecular evolution by staggered extension process (StEP) in vitro recombination. Nat Biotechnol. 1998;16(3):258–261. doi: 10.1038/nbt0398-258. [DOI] [PubMed] [Google Scholar]
- 68.Stemmer WP. DNA shuffling by random fragmentation and reassembly: in vitro recombination for molecular evolution. Proc Natl Acad Sci USA. 1994;91(22):10747–10751. doi: 10.1073/pnas.91.22.10747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Koerber JT, Jang JH, Schaffer DV. DNA shuffling of adeno-associated virus yields functionally diverse viral progeny. Mol Ther. 2008;16(10):1703–1709. doi: 10.1038/mt.2008.167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Smith JJ, Karp PH, Welsh MJ. Defective fluid transport by cystic fibrosis airway epithelia. J Clin Invest. 1994;93(3):1307–1311. doi: 10.1172/JCI117087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Smith JJ, Travis SM, Greenberg EP, Welsh MJ. Cystic fibrosis airway epithelia fail to kill bacteria because of abnormal airway surface fluid. Cell. 1996;85(2):229–236. doi: 10.1016/S0092-8674(00)81099-5. [DOI] [PubMed] [Google Scholar]
- 72.Liu Y, et al. FoxM1 mediates the progenitor function of type II epithelial cells in repairing alveolar injury induced by Pseudomonas aeruginosa. J Exp Med. 2011;208(7):1473–1484. doi: 10.1084/jem.20102041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Yan Z, et al. Distinct classes of proteasome-modulating agents cooperatively augment recombinant adeno-associated virus type 2 and type 5-mediated transduction from the apical surfaces of human airway epithelia. J Virol. 2004;78(6):2863–2874. doi: 10.1128/JVI.78.6.2863-2874.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Chang EH, et al. Sinus hypoplasia precedes sinus infection in a porcine model of cystic fibrosis. Laryngoscope. 2012;122(9):1898–1905. doi: 10.1002/lary.23392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Chen JH, et al. Loss of anion transport without increased sodium absorption characterizes newborn porcine cystic fibrosis airway epithelia. Cell. 2010;143(6):911–923. doi: 10.1016/j.cell.2010.11.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nat Methods. 2012;9(7):671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Blossfeld S, Gansert D. A novel non-invasive optical method for quantitative visualization of pH dynamics in the rhizosphere of plants. Plant Cell Environ. 2007;30(2):176–186. doi: 10.1111/j.1365-3040.2006.01616.x. [DOI] [PubMed] [Google Scholar]