

Abstract
Pancreatic ductal adenocarcinoma (Pdac) is a malignancy with a poor prognosis due to difficulties in early detection. Although promising biomarkers are increasingly reported, such methods are not yet easy to apply clinically, mainly due to their low reproducibility or technical difficulties. In this study, we developed a convenient and sensitive method for quantifying aberrantly expressed satellite repeat RNAs in sera, which can be used to efficiently detect patients with Pdac. Here, we introduce a Tandem Repeat Amplification by nuclease Protection (TRAP) method combined with droplet digital PCR (ddPCR) to detect human satellite II (HSATII) RNAs, which are specifically expressed in human Pdacs at greater levels than normal tissues but are difficult to measure due to their repetitive sequences and irregularities. HSATII RNA core sequence levels in sera were significantly higher in Pdac patients compared with noncancer patients (median copy number: 14.75 and 3.17 per μl in the training set and 17.35 and 2.9 in the validation set, respectively). In addition, patients with intraductal papillary mucinous neoplasm (IPMN), a precancerous lesion of Pdac, could also be efficiently detected. This method can be routinely applied to screen patients with Pdac and high-risk patients, facilitating the development of preventive medicine for this disease.
A newly developed method to measure human satellite RNA levels in sera may be useful for screening patients with pancreatic cancer.
Introduction
Pancreatic ductal adenocarcinoma (Pdac) is the fourth leading cause of cancer-related deaths (1) and its estimated incidence was 45,220 cases in 2013 in the United States (2, 3). The mortality/incidence rate is nearly 1.0, with a 5-year survival rate below 10% (4–6). One of the reasons for this poor prognosis is that most patients are diagnosed at the far-advanced stage and curative resection is not possible. Because the currently used clinical biomarkers, such as carbohydrate antigen 19-9 (CA19-9), are not adequate to efficiently detect Pdac, especially at the early stages of its development, more sensitive and specific biomarkers are urgently required to improve the prognosis.
Recently, the aberrant expression and functional importance of noncoding RNAs (ncRNAs) in cancer, including microRNAs and other long ncRNAs, have been demonstrated emerging (7, 8). These RNAs are differentially expressed in cancer tissues and are released into the bloodstream in relatively stable forms. Therefore, they can be used as novel biomarkers for various types of cancers (9). However, most results obtained so far cannot be simply applied clinically due to intrinsic problems with the methods, such as technical difficulties, low sensitivity, variation in the measurements because of the low expression levels, and undefined controls for normalization.
RNAs from repetitive genomic DNA sequences, which comprise a large portion of the human genome, have historically been excluded from transcriptome analyses mainly because of the technical difficulties in amplification and identification caused by their repetitiveness (10). Recently, a study using an RNA deep-sequencing method demonstrated that aberrant ncRNA expression from highly repetitive satellite DNA was detected in pancreatic tumors and that a subset of satellite RNAs, particularly human satellite II (HSATII) RNA, is specifically expressed at high levels in human Pdacs compared with expression in normal tissues (11). Because this satellite RNA expression occurs exclusively in cancer tissues, it may be useful clinically as a biomarker for cancer detection if its levels could be measured easily and reproducibly from the sera of patients.
In this study, we developed a convenient procedure to detect repetitive RNAs in the sera of patients by specifically amplifying tandem repeat transcripts, which were formerly difficult to quantify using conventional PCR procedures. The developed methods, combined with droplet digital PCR (ddPCR) analysis, provided for the highly sensitive detection and accurate quantification of HSATII RNA levels in the sera of the patients with Pdac and even in those with precancerous lesions. These methods can be routinely applied in clinical practice to facilitate the detection of cancers and may also allow for the identification of high-risk patients.
Results
HSATII RNA is specifically expressed in Pdac tissues.
Pericentromeric satellite HSATII RNA had the greatest differential expression between Pdac and normal pancreatic tissues among the overexpressed satellite ncRNAs in cancer tissues (11). Therefore, we examined HSATII RNA expression levels in the total RNA extracted from human Pdac tissues, adjacent noncancerous tissue, and normal pancreatic tissues from patients without cancer by Northern blotting. Consistent with previous reports (11), HSATII RNA was detected in cancerous tissues and, to a lesser extent, in adjacent noncancerous lesions, whereas almost no expression was detected in normal tissues (Supplemental Figure 1A; supplemental material available online with this article; doi:10.1172/jci.insight.86646DS1). As reported, HSATII RNA was detected as a broad smear band ranging from approximately 150–8,000 nucleotides by Northern blotting, indicating that HSATII RNA is a series of heterogeneous transcripts (12).
Development of a sensitive method for quantifying HSATII RNA levels.
Because HSATII RNA is exclusively expressed in pancreatic cancerous tissues and not in normal tissues, its expression may be useful as a biomarker to discriminate between patients with and without cancers if it can be conveniently and reproducibly quantified in the serum of patients. This led us to try to develop methods to detect HSATII RNA in the sera of patients with Pdac. Although reverse transcription PCR (RT-PCR) methods have been applied in several previous studies to measure satellite RNAs (13–16), the melting curves in our RT-PCR formed multiple peaks when measuring HSATII RNA, even though the reported primers were used (Supplemental Figure 1B). The products of the semi-quantitative RT-PCR showed smear or ladder bands after gel electrophoresis (Supplemental Figure 1C), which suggests that PCR primers possibly hybridize with multiple parts in the transcripts with various sizes, resulting in the production of various PCR amplicons with heterogeneous lengths due to their repeated sequences.
To overcome the unreliabilities of the simple PCR method when measuring repeated RNAs, we tried to develop a novel method that can precisely quantitate tandem repeat RNAs by taking advantage of their repetitive nature, which we named the Tandem Repeat Amplification by nuclease Protection (TRAP) method (Figure 1A). In this method, RNAs were first hybridized with 21-mer biotinylated RNA probes, which are exactly complementary to the HSATII core sequences. Next, unhybridized HSATII regions, other RNAs, and residual probes were completely digested by RNase A/T1, which selectively digests unhybridized single-stranded RNA. Through these procedures, only the core sequences from the HSATII RNAs with various lengths were gathered together with hybridized probes, which facilitated their subsequent detection. Finally, biotinylated probes were visualized at the same position relative to the probe length after gel separation, indicating the amount of the perfectly matched HSATII RNA core sequences present. Alternatively, instead of using gel separation, TaqMan real-time PCR was performed after the reverse transcription step using primers with loop sequences (Figure 1A). Using primers specific for the loop primers and the core sequences with the TaqMan probes, PCR was performed for HSATII RNA quantitation. Because it is unclear which gene should be used as an inner control for the quantification of cell-free RNA in sera (9) and because the unhybridized transcripts are digested in these protection steps, we added a set of 40-mer forward RNAs (as spike-in RNAs) and their biotinylated-reverse RNA probes derived from the bacterial dapB gene as an external control for quantification.
Figure 1. Tandem repeat amplification with nuclease protection method.

(A) Conceptual scheme of the tandem repeat amplification with nuclease protection (TRAP) method. Human satellite II (HSATII) RNAs are heterogeneous transcripts containing random repeats of the core sequences (upper panel). After hybridizing complementary probes to the core sequences, single-stranded RNAs including unhybridized probes are digested by nuclease A and T1 (lower panel). Protected core sequences can be accumulated and visualized by gel electrophoresis or quantified by TaqMan reverse transcription PCR (RT-PCR, right panels). (B) Detection of synthesized HSATII transcripts by gel electrophoresis after the TRAP method. Ten-fold serial dilutions from 1 pg to 100 ng of HSATII templates were examined. Spike-in probes were used as normalized controls and were detected at the size of 40 nucleotides (nt) and HSATII probes at the level of 21 nt. The lower panel shows high-contrast images of the bands of HSATII RNA in the upper panel. n.s., nonspecific bands. (C) A standard curve reflecting the accurate quantitation of the synthesized HSATII transcripts. Serial dilutions from 1 pg to 1 μg of HSATII RNA templates were quantified by TaqMan real-time PCR after performing the TRAP method. The standard curve is shown as a black line. Data represent the mean ± SD of 3 independent experiments.
Using this procedure, serial 10-fold dilutions between 1 pg and 100 ng of HSATII RNA templates were tested to determine which were synthesized by in vitro transcription using the pCDH-HSATII-Fw vector containing 5 repeats of the HSATII core sequence as a template. HSATII RNA at and above 1 ng could be detected after gel separation, showing the presence of fully protected core sequences as the same length as the RNA probes (Figure 1B). In addition, using the TaqMan RT-PCR procedure, a proper standard curve could be produced with a range from 1 μg to 1 pg, implying that the TRAP method can be used to accurately measure repetitive core sequence levels from transcripts with very low levels of expression (Figure 1C).
HSATII RNA is released into the peripheral blood.
It is commonly believed that RNAs expressed in various types of human cells are released to the blood stream with an unexpected stability by liposomal cargo vesicles, such as exosome or microvesicles, or by protein complexes, such as the RNA-induced silencing complex (RISC). The expression profiles of such cell-free RNAs change dramatically depending on the pathological conditions of the patients (17). Therefore, we examined whether HSATII RNA is also released from cancer cells. For currently unknown reasons, satellite RNA expression does not occur in standard 2D cell cultures in vitro, even when culturing cells that express satellite RNAs in xenografts (11, 12, 18). Therefore, we ectopically overexpressed HSATII RNA by lentiviral transduction in pancreatic cancer cells (BxPC3) and in human TERT–immortalized (hTERT-immortalized) normal human pancreatic ductal cells (hTERT-HPNE) (19). The isolated exosome fraction from the collected cell cultured media was subjected to the TRAP method, followed by the quantification of HSATII RNA by TaqMan real-time PCR. HSATII RNA could be detected in the exosome fraction of the culture media (Supplemental Figure 2, A and B). CD9 and CD63 were measured in BxPC3 and hTERT-HPNE cells as exosome markers, which were expressed in each cell lysate, respectively. The detection of equal amounts of the markers confirmed that equal amounts of extracted exosome fraction had been collected from the control and HSATII RNA-expressing cells. Furthermore, 3D cultures of BxPC3 cells, forming tumor spheroids, restored their endogenous HSATII RNA expression, which had not been detected in standard 2D culture. HSATII RNA was also detected in exosome fractions from 3D culture media using the TRAP real-time PCR method (Supplemental Figure 2C).
Next, we compared HSATII RNA levels between the exosome fraction and the residual fraction of the serum, collected as the supernatant, from a patient with Pdac. Although HSATII RNA was detected mainly in the exosome fraction, it could be observed in the residual fraction, as well (Supplemental Figure 2D), suggesting that the total sera should be used for the measurement.
HSATII RNA is significantly detectable in the sera of Pdac patients.
Based on these results, we examined HSATII RNA expression in 400 μl of human sera using the TRAP method. To evaluate the efficacy of determining serum HSATII RNA levels as a possible marker for Pdac, 20 patients with Pdac and 20 controls with no evidence of cancer were first recruited as a training set (Table 1). In addition to HSATII RNA, other satellite repeat RNAs, satellite III (satIII) and α satellite (α-sat), in sera were also examined. Although all satellite RNAs showed a tendency to increase in the sera of the patients with Pdac, HSATII RNA showed higher specificity and sensitivity for the detection of Pdac, with the highest AUC among all the satellite repeat RNA tested (Supplemental Figure 3, A–F).
Table 1. Patient data from the training set.
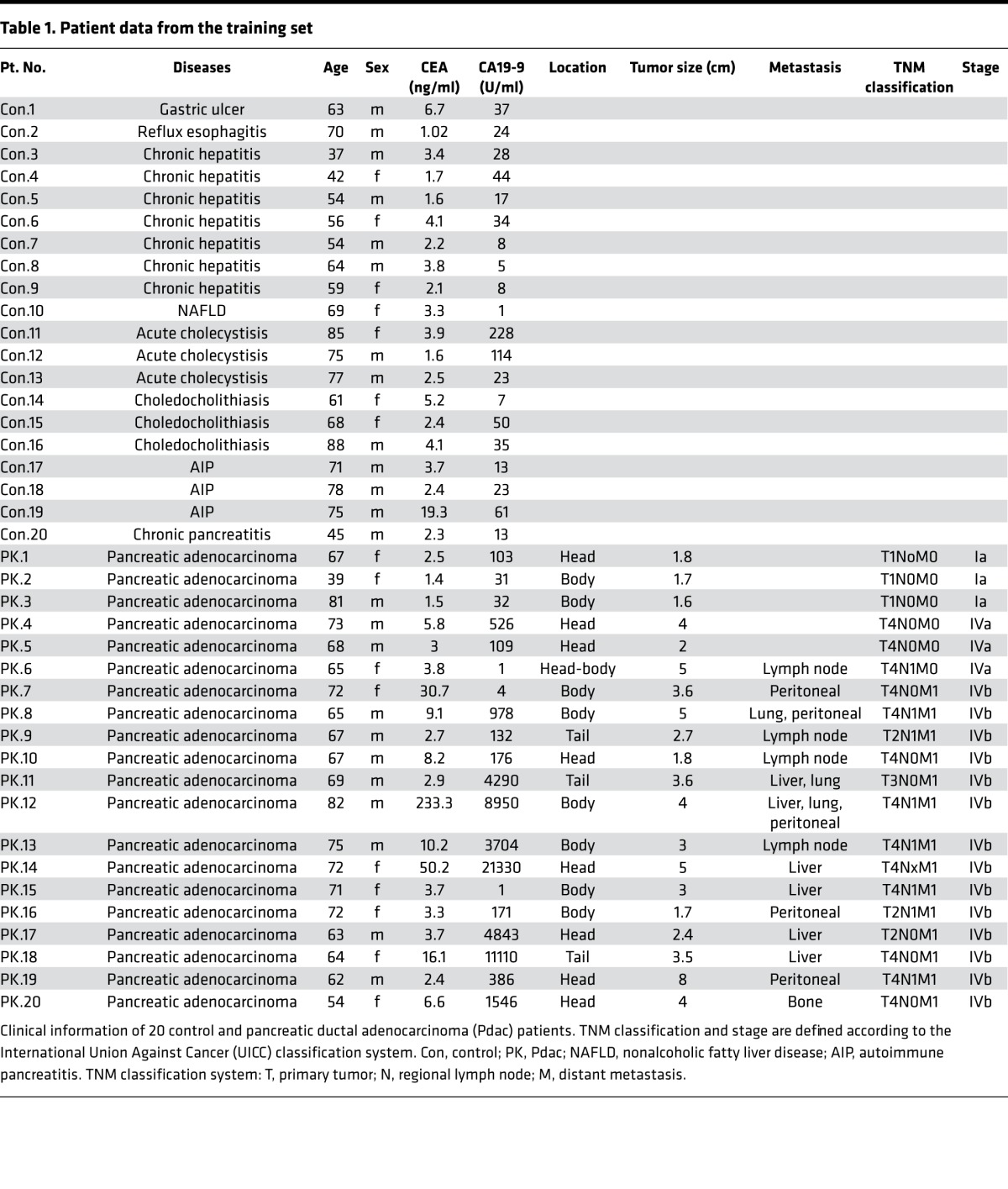
While HSATII RNA showed the highest specificity and sensitivity in detecting Pdac, its absolute levels in sera were relatively low, even after preparation with the TRAP method, compared with the levels of α-sat RNA (Supplemental Figure 3, A–C). Because ddPCR is superior to real-time PCR with respect to the quantitative accuracy for nucleic acids from minute specimens (20), we compared the standard deviations obtained with real-time PCR and with ddPCR in measuring serum HSATII levels of 40 samples from the training set. Consistent with the previous report, the standard deviations associated with ddPCR were much smaller than those from real-time PCR, especially in the samples with low expression levels (Supplemental Figure 4, A and B). While the lowest limits of detection were comparable between ddPCR and real-time PCR in this assay, likely due to the limitations of the TRAP procedure, the deviations were smaller in ddPCR (Supplemental Figure 4C). Because of these results, we decided to measure serum HSATII RNA levels in the total sera of the patients in the training set using the TRAP method followed by ddPCR. The absolute number of copies of HSATII RNA core sequences was significantly higher in the sera of the patients with Pdac than it was in the sera of noncancer patients (median 14.75 versus 3.17). In addition, HSATII had a higher AUC value (0.91 versus 0.80) than CA19-9 (Figure 2, A and B), which is currently commonly used as a serum tumor marker for Pdac, when using 10 copies/μl as a threshold. These results suggested that the measurement of HSATII RNA levels in sera using the TRAP with ddPCR method may be useful for the detection of Pdac.
Figure 2. Human satellite II (HSATII) RNA levels in the sera of the training set determined by tandem repeat amplification with nuclease protection–droplet digital PCR (TRAP-ddPCR).

(A) HSATII RNA levels in the sera of controls (n = 20) and patients with pancreatic ductal adenocarcinoma (Pdac) (n = 20) in the training set. Bee swarm dot plots and box plots are shown. The horizontal line in the middle of each box indicates the median, whereas the top and bottom borders of the box mark the 75th and 25th percentiles, respectively. The whiskers above and below each box extend to the most extreme point that is no more than 1.5 times the interquartile range of the box. *P < 0.001, by 2-tailed Welch’s t test. (B) Receiver operating characteristic (ROC) curve analysis for diagnosing Pdac. blue, CA19-9; red, HSATII RNA.
Serum HSATII levels can be used for the detection of Pdac and IPMN.
To confirm the usefulness of the measurement of HSATII RNA in sera for the detection of Pdac, we next examined HSATII RNA levels in the sera of another 10 noncancer control patients and 10 patients with Pdac to serve as a validation set (Table 2). In this set, 10 patients with intraductal papillary mucinous neoplasm (IPMN) were also included to provide samples associated with precancerous lesions (Figure 3, A–D). Consistent with the results of the training set, absolute HSATII values in the sera of Pdac patients were significantly higher than those of the control patients (Figure 3A, median 17.35 versus 2.9) with almost the same AUC values (0.90 versus 0.93) as those of the training set (Figure 3B), although the AUC value of CA19-9 was unexpectedly higher than previous reports (21). In addition, while only 2 cases could be examined from the cohorts in this study, elevated HSATII RNA levels in the sera of Pdac patients who underwent surgery significantly decreased after surgery in both cases (Supplemental Figure 5). These results confirmed the usefulness of the measurement of HSATII RNA levels in sera for detecting patients with Pdac. Interestingly, HSATII RNA levels in the sera of IPMN patients (median 17.15) were also significantly higher than those in control patients. The AUC values of control versus IPMN (0.75 versus 0.55) and of control versus IPMN + Pdac (0.82 versus 0.69) when using HSATII RNA levels as a marker were substantially higher than the AUC values when using CA19-9 (Figure 3, C and D). In particular, one IPMN patient with nodules, who underwent surgery and was pathologically diagnosed with mixed-type IPMN with intermediate-grade dysplasia, had low CA19-9 levels but the highest HSATII RNA level among IPMN patients (Table 2 and Figure 3A). These results suggest that serum HSATII RNA levels may be used not only as a detecting marker for Pdac, but also as a highly sensitive screening marker for the patients with precancerous lesions.
Table 2. Patient data from the validation set.
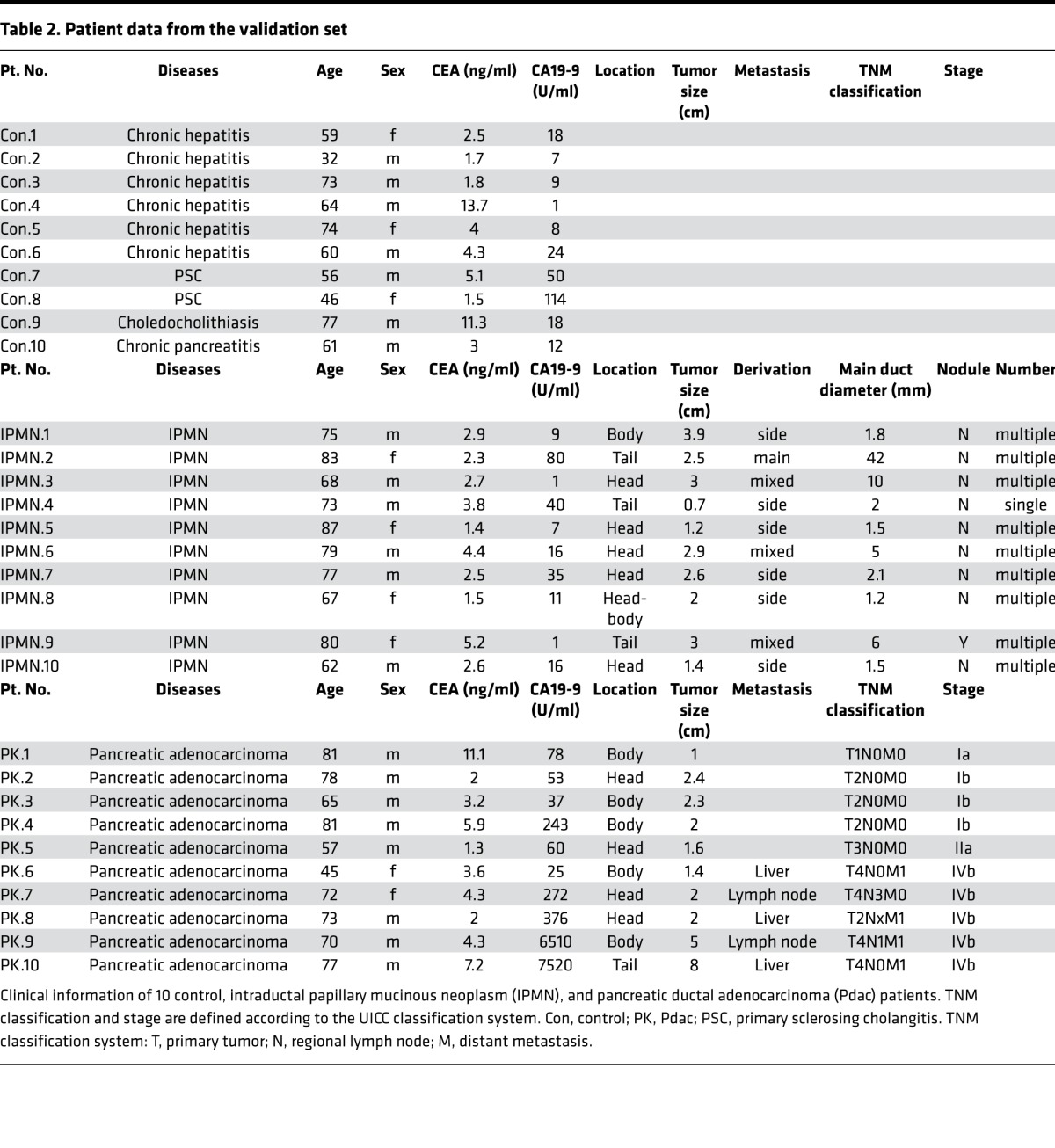
Figure 3. Human satellite II (HSATII) RNA levels in the sera of the validation set determined by tandem repeat amplification with nuclease protection–droplet digital PCR (TRAP-ddPCR).

(A) HSATII RNA levels in the sera of the controls and patients with intraductal papillary mucinous neoplasm (IPMN) or pancreatic ductal adenocarcinoma (Pdac) in the validation cohort. Bee swarm dot plots and box plots are shown as described in the legend of Figure 2. *P < 0.05, by 2-tailed Welch’s t test. (B–D) Receiver operating characteristic (ROC) curve analyses for distinguishing Pdac from the control group (B), IPMN from the control group (C), and IPMN and Pdac from the control group (D). blue, CA19-9; red, HSATII RNA.
Discussion
We demonstrate here that HSATII RNA, which has been recently reported to be aberrantly expressed in human pancreatic cancer (11), was accurately and conveniently detectable in the sera of patients with pancreatic cancer using our TRAP procedure with TaqMan RT-PCR and ddPCR.
HSATII RNA is a series of heterogeneous, noncoding transcripts with various lengths that carry multiple arrays of tandem-repeat core sequences. Therefore, they are difficult to correctly quantitate because of the difficulties in establishing appropriate primers that amplify a single product using simple PCR procedures (16). Northern blotting may be useful for the quantitation because probes for the core sequences can possibly hybridize to multiple parts in the transcript. However, this is not suitable for clinical use because of the requirement of large amounts of RNAs as the starting material and also because it is a time-consuming and difficult process. Comprehensive RNA sequencing using a high-throughput technique, which was initially applied to quantify the aberrantly expressed RNAs in pancreatic cancerous tissues (11), may be an ideal alternative. However, it may not be suitable for the measurement of large numbers of clinical samples because of its high cost, the time required, and the necessity for the complicated analysis of the data obtained. Considering these issues, our TRAP procedure can produce a single product by collecting the protected core sequences of the same length from heterogeneous RNAs with various lengths of the repeated sequences. In addition, because a single transcript contains multiple core sequences, the TRAP procedure itself raises the sensitivity of the subsequent measurement of RNA levels. It means the repetitive nature of these sequences, which is a drawback for other methods, is an advantage for the TRAP method.
In this study, we added external control RNAs, derived from the bacterial dapB gene, for technical normalization during the protection procedure. At present, there is no consensus about which gene is appropriate as an internal control for the measurement of circulating cell-free RNA. Although some microRNAs or other small ncRNAs — such as U6 small-nuclear RNA — are frequently used as controls, those RNAs are also significantly altered in certain pathological states (9, 22). Because ddPCR can calculate absolute copy numbers even without reference genes and because HSATII RNA levels in the sera of control patients are extremely low (median 3.0 copies/μl), the quantitation of HSATII RNA levels by ddPCR shown here may be more advantageous for the accurate quantitation of relevant biomarkers compared with the results obtained by the quantitation of circulating microRNAs or other ncRNAs in sera, which are determined by the Ct values after normalization in comparison with the levels of the selected internal controls.
Measurement of HSATII RNA levels in sera of the patients using the TRAP procedure followed by ddPCR could efficiently discriminate between control and Pdac patients. In particular, 5 of 7 Pdac patients of both groups whose CA19-9 levels were within normal range (37 U/ml below) had higher HSATII RNA levels and were detected correctly, suggesting that measurement of HSATII RNA levels is more sensitive than CA19-9 levels to detect Pdac patients. Because the control groups in this study predominantly included chronic liver disease patients who came to the outpatient clinic serially, one cannot exclude the possibility that the disease deviation in the control samples might work as a confounder. Also, 400 μl of serum was used as the starting material in this study. If larger serum volumes were available and subjected to TRAP-ddPCR assays, then the dynamic range of the method could be expanded and more efficient discrimination between control and Pdac patients may be obtained. Therefore, prospective studies with larger cohorts with unbiased disease inclusion and the use of an appropriate starting volume of serum are required to precisely determine the usefulness of HSATII RNA as a biomarker.
Although the number of patients tested here was relatively small, it is noteworthy that HSATII RNA was also detected in the sera of patients with IPMN and that it had a higher AUC value than CA19-9 (0.75 versus 0.55), which was not elevated in almost all IPMN cases in this study. Six of 9 IPMN patients with normal CA19-9 levels were correctly detected by measuring HSATII RNA. IPMN is a pathological state regarded as a high risk for the occurrence of Pdac, and periodical imaging inspection is clinically needed (23). The incidence of Pdac development in IPMN patients has been reported to be 0.68%–1.1% per year in several retrospective studies (24). Therefore, it is important to use noninvasive screening examinations to identify patients who may be at risk of Pdac and who may require diagnostic imaging, such as magnetic resonance cholangiopancreatography (MRCP) or CT. HSATII RNA detection may be useful in such cases, as well.
Although our TRAP-ddPCR method showed a high detectability and accurate quantitation with small standard deviations, some caution should be taken in the interpretation of the levels of HSATII RNA determined. That is, quantitation by this method represents the exact core sequences, and it is not always proportional to the exact copy number of the transcripts with various tandem repeats. Because the longer HSATII RNA may contain a greater number of core sequences, if there are tissues that are predominantly producing longer RNA, the amounts of the core sequences determined by this method will result in apparently higher levels. In addition, because the nucleotide length of RNAs in the exosome fraction is shorter than those that exist in the cells (25), the ratio of the HSATII RNA released in the exosome may affect the levels determined. Some cases that seemed to have exceptionally high HSATII RNA levels in this study may reflect these conditions. Examining the similarities and differences in the spectrum of the lengths of HSATII transcripts and the released forms of HSATII RNA outside the cells in various pathological conditions will be required.
At present, it is unclear how satellite RNAs are aberrantly transcribed during the stages of cancer development, as can be observed in various epithelial cancers (11, 12, 26). In addition, the biological significance of these aberrantly expressed satellite RNAs have not been fully determined, although several procarcinogenic factors induced by these satellite RNAs have been reported, such as the increase in mitotic errors (14), production of proinflammatory cytokines (26), and progressive elongation of pericentromeric regions by intrinsic reverse transcriptases (12). In any case, the development of a convenient quantitation of HSATII RNAs, which are expressed exclusively in cancerous and precancerous tissues, in patient sera may provide novel biology-based clues for the detection of the cancerous and precancerous lesions.
In summary, we developed a convenient method for the measurement of HSATII RNA levels in sera, which may be useful for detecting patients with Pdac and IPMN at a low cost. Although prospective studies with larger cohorts are still required to precisely determine the usefulness of HSATII RNA as a biomarker and to clarify relationships between its levels and tumor burden or the malignant potential of the lesions, the TRAP procedure with ddPCR may prove clinically valuable and may contribute to the development of preventive strategies against pancreatic cancer.
Methods
Clinical tissues and serum samples.
Total RNAs from patient-matched Pdacs and their adjacent noncancerous tissues were purchased from Biochain Institute Inc. (catalog R8235188-P). Normal human pancreatic total RNA was purchased from Ambion (catalog AM7954). Serum samples for the training set were obtained from 20 patients with Pdac and 20 control patients with no evidence of malignant diseases who were hospitalized or came to the outpatient clinic at the University of Tokyo Hospital between March 2014 and April 2015. For the validation set, serum samples were obtained from 10 patients with Pdac, 10 control patients with no malignant diseases, and 10 patients with IPMN who were hospitalized or came to the outpatient clinic at the University of Tokyo Hospital between May 2015 and November 2015. The diagnosis of Pdac and IPMN were confirmed pathologically using tissues obtained by fine-needle aspiration or by typical radiologic findings from CT and MRCP.
Cells.
Human Pdac cells, BxPC3 cells obtained from the American Type Culture Collection (ATCC, catalog CRL-1687), were maintained in RPMI supplemented with 10% FBS. hTERT-HPNE cells, which are immortalized normal pancreatic duct cells that express hTERT (19), were purchased from ATCC (catalog CRL-4023). 293TN cells were purchased from System Biosciences (SBI, catalog LV900A-1). 293TN and hTERT-HPNE cells were maintained in DMEM with 10% FBS. All cells were incubated under 37°C with 20% O2 and 5% CO2. To develop tumor spheroids as a 3D culture, 1 × 106 BxPC3 cells were seeded on ultra-low adherent Nunclon Sphera 90-mm plates (Thermo Fisher Scientific) and cultured with the same medium used for 2D cultures.
Constructs.
For HSATII RNA overexpression, 170 bp segments of the HSATII consensus sequences — which contain 5 tandem repeats of the 21-mer core sequence (5′-CATTCGATTCCATTCGATGAT-3′), as reported in the Repbase database (http://www.girinst.org/repbase/index.html, GenBank: X03460) — were synthesized as artificial oligonucleotides (Eurofins Genomics) and were subcloned into the pCDH-CMV-MCS-EF1-Neo lentiviral vector (pCDH-vector) (SBI) using the EcoRI and BamHI sites (pCDH-HSATII-Fw). To generate lentiviruses for constructing polyclonal cells that stably expressed HSATII RNA, the Lentivirus Packaging System (SBI) was used according to the manufacturer’s protocol. Briefly, 1.0 μg of pCDH-vectors or pCDH-HSATII-Fw plasmids and 5.0 μg of pPACKH1 Packaging Plasmid mix (SBI) were transfected into 293TN cells using the Effectene Transfection Reagent (Qiagen). After 48 hours, cultured media was collected and incubated with PEG-it Reagent (SBI) for 24 hours at 4°C and then centrifuged at 1,500 g for 30 minutes. The pellet was resuspended in 1 ml of 1× PBS, and aliquots were kept at –80°C. Viruses were transduced into 1 × 104 HPNE cells (HPNE-HSATII) and BxPC3 cells (BxPC-HSATII) with 1 μg/ml Polybrene Reagent (Santa Cruz Biotechnology Inc.), followed by selection under 400 μg/ml of G418 (Wako Pure Chemical Industries Ltd.).
RNA isolation.
Total RNA was extracted from the adherent cells and 3D-cultured spheroids using the spin column protocol of the NucleoSpin RNA II kit (Macherey-Nagel) according to the manufacturer’s instructions. Human serum samples were centrifuged at 2,000 g for 30 minutes, and the supernatants were filtered through 450-μm nylon mesh to exclude cell debris. Total RNA was extracted from 400 μl of the filtered serum using the miRCURY RNA Isolation Kit (Exiqon) according to the manufacturer’s protocol.
Northern blotting.
Northern blotting of the RNAs derived from pancreatic tissues was performed as described previously (27). Briefly, 8 μg of total RNA were separated in 1% formaldehyde–denatured agarose gel at 100 V for 45 minutes and then hydrostatically transferred to Hybond N+ membranes (GE Healthcare). Membranes were UV-crosslinked and prehybridized in an ULTRAhyb-Oligo Hybridization Buffer (Ambion). Hybridization was performed overnight at 55°C in hybridization buffer containing biotinylated RNA probes that had been denatured at 90°C for 10 minutes. Membranes were washed at 55°C in 2× standard saline citrate (SSC) containing 0.1% SDS and in 0.1× SSC containing 0.1% SDS. The series was performed twice, and the bound probe was visualized using the BrightStar BioDetect kit (Ambion). Biotinylated probes were synthesized by in vitro transcription using a MEGAscript T7 transcription kit (Ambion), followed by biotinylation with a Brightstar Psoralen-Biotin nonisotopic labeling kit (Ambion). For construction of the probes to detect HSATII RNA, 170 bp segments of the HSATII consensus sequences were subcloned into the pcDNA 3.1 (–) vector (Invitrogen), which can be used as a template for in vitro transcription under the regulation of the T7 promoter. For the actin control probe, the human ACTB open reading frame was cloned by TA cloning into the pCR2.1-TOPO vector using the cDNA of hTRERT-HPNE cells, which was used as a template for in vitro transcription. The primers used were as follows: Fw: 5′-GACGAGGCCCAGAGCAAGAG-3′ and Rv: 5′-TGCGCTCAGGAGGAGCAATG-3′.
Semi-quantitative RT-PCR.
Total RNA from patient serum was reverse transcribed to cDNA using SuperScript III reverse transcriptase (Invitrogen). PCR was performed using FastStart universal SYBR green master with rox (Roche Diagnostics) with the StepOnePlus real-time PCR instruments (Invitrogen). PCR products were separated by electrophoresis in 1.5% agarose gel and visualized by ethidium bromide solution. The primers used were as follows: satII-Fw: 5′-CATCGAATGGAAATGAAAGGAGTC-3′ and satII-Rv: 5′-ACCATTGGATGATTGCAGTCAA-3′ (15).
Probes for RNase protection.
Complementary RNA probes for RNase protection were artificially synthesized with biotin-labeling in their 3′ ends (Eurofins Genomics). As a spike-in outer control, biotinylated reverse-strand probes and unmodified forward-strand probes based on the bacterial dapB gene (GenBank: NC_000964.3, 169-199) were synthesized (Eurofins Genomics). The sequences used were as follows: HSATII-Rv-bio: 5′-AUCAUCGAAUGGAAUCGAAUG-3′ (11), satIII-Rv-bio: 5′-CAUUCCACUCGGGUUGAUUCC-3′, α-sat-Rv-bio: 5′-AAUUCUCAGUAACUUCCUUGU-3′ (28), dapB-Rv-bio: 5′-GAUCAGUCCCGGAAGACGGACGCUGUGCAAGCGAAUACCG-3′, and dapB-Fw: 5′-CGGUAUUCGCUUGCACAGCGUCCGUCUUCCGGGACUGAUC-3′.
TRAP method.
During the RNA isolation steps, both strands of the spike-in probes (dapB-Fw: 0.02 pmol and dapB-RV-bio: 0.2 pmol) were added to the serum samples. Isolated total RNA in 200 μl of nuclease-free water was mixed with HSATII-Rv-bio probes (0.2 pmol) and incubated with a 1/10 volume of 5 M NH4OAc and 2.5 volumes of ethanol at –20°C for 30 minutes. Samples with probes were coprecipitated by centrifugation at 15,000 g for 15 minutes and resuspended in 10 μl of hybridization buffer (Ambion). This was followed by denaturing at 90°C for 3 minutes and incubation at 42°C overnight to hybridize the probes to their complementary sequence arrays. Subsequently, 12.5 U of RNase A/T1 mix in 150 μl of digestion buffer (Ambion) was added and incubated at 37°C for 30 minutes to degrade unhybridized single-stranded RNAs, followed by incubation with 225 μl of an RNase inactivation/precipitation solution (Ambion) and 150 μl of ethanol for 2 hours at –20°C. Protected double-stranded RNAs, including tandem repeat RNAs with complementary probe RNAs and outer spike-in RNAs, were precipitated by centrifugation at 15,000 g for 15 minutes and resuspended in 9 μl of nuclease-free water. While the TRAP method was performed as a single assay for each sample in this study due to the limited availability of serum stocks, we initially examined the reproducibility of the TRAP method using selected identical samples. The results were confirmed with high reproducibility, and differences between identical samples were within 1 Ct in real-time PCR experiments.
Detection of protected repeat RNAs by gel separation.
Protected RNAs were separated in 15% denaturing polyacrylamide gel and transferred to Hybond N+ membranes (GE Healthcare). After UV crosslinking with a Stratalinker UV Crosslinker (Stratagene), the biotinylated probes were visualized using the Chemiluminescent Nucleic Acid Detection Module (Thermo Fisher Scientific) according to the manufacturer’s protocol. Images were captured using a bioimaging analyzer (LAS-3000, Fujifilm)
TaqMan real-time PCR and ddPCR.
For TaqMan RT-PCR, 5× looped RT primer, 20× PCR primer, and TaqMan probes were customized using the TaqMan assay design web tool (Applied Biosystems, https://www5.appliedbiosystems.com/tools/cadt/). Reverse transcription was performed with the TaqMan MicroRNA RT Kit (Applied Biosystems) with minor modifications. Briefly, 3.0 μl of amplified RNA templates obtained by the TRAP method and 2.0 μl each of the 5× RT primer for the HSATII RNAs and the spike-in RNAs were incubated at 85°C for 5 minutes, followed by immediate cooling at 4°C for 5 minutes. The reverse transcriptase reagent mix was added and incubated at 16°C for 30 minutes, followed by incubation at 37°C for 30 minutes and at 85°C for 5 minutes. For each sample, 1.0 μl of template was subjected to quantitative PCR using SYBR universal mix 2 (Applied Biosystems) with the StepOnePlus real-time PCR system. The Ct, at which the amount of amplified target reached a fixed threshold, was measured. Real-time PCR experiments were performed in triplicate for each sample. In analyzing 10-fold serial dilutions of the HSATII template, a standard curve was constructed using the average triplicate Ct of each sample with StepOne software (ver. 2.3, Applied Biosystems). The relative expression levels of exosome experiments and comparisons of PCR accuracy were determined by the ΔΔ-Ct method. The Δ-Ct value was calculated by subtracting the Ct value of dapB from that of HSATII.
ddPCR was performed using the QX100 Droplet Digital PCR system (Bio-Rad). Briefly, 1.0 μl of template cDNA reverse transcribed from protected RNAs with 20× primer and a TaqMan probe set were partitioned into approximately 20,000 droplets by the QX100 Droplet Generator. Cycling conditions were 95°C for 10 minutes, followed by 45 cycles of 94°C for 30 seconds and 56°C for 1.5 minutes and a final 10-minute incubation at 98°C. The droplets were subsequently read automatically by the QX10 droplet reader. Assays were performed in duplicate for each sample. The data were analyzed with QuantaSoft analysis software 1.3.2.0 (Bio-Rad). Duplicate data were combined into a single metawell before Poisson analysis.
Exosome isolation.
Exosomes from culture media were isolated using Total Exosome Isolation for Cell Culture Media (Invitrogen). Briefly, 5.0 × 106 BxPC3 and hTERT-HPNE cells overexpressing HSATII RNA or control vector cultured for 24 hours, or 1.0 × 106 BxPC3 cells forming tumor spheroids on the Nunclon Sphera plate, were cultured for 3 days, followed by the change of the culture media to 10 ml of serum-free DMEM or RPMI. After 24 hours, the cultured media were collected and centrifuged at 2,000 g for 30 minutes and filtered through 450-μm nylon mesh and incubated with 1 ml of isolation reagent at 4°C overnight according to the manufacturer’s protocol (Invitrogen). After centrifugation at 10,000 g for 1 hour, the pellet was resuspended in 200 μl of 1× PBS.
For isolation of exosomes in sera, after removing cells and debris by centrifugation and filtering, 500 μl of serum samples and 100 μl of isolation reagent for serum (Invitrogen) were incubated at 4°C for 2 hours. Samples were then centrifuged at 10,000 g for 30 minutes. The pellets were collected as the exosome fraction and the supernatant as the residual fraction.
Western blotting.
Western blotting for exosome lysates was performed using the NativePAGE Bis-Tris Gel System (Invitrogen). Briefly, each sample was lysed with a NativePAGE sample buffer containing 1% n-dodecyl-β-D-maltoside and then separated on 4%–16% Bis-Tris protein gel using the Blue-Native (BN) PAGE technique (29) followed by the electrical transfer to PVDF membranes (GE Healthcare). After blocking with 5% dry milk, membranes were probed with the appropriate primary antibodies diluted in Immunoshot Reagent 1 (Cosmo Bio) overnight at 4°C. HRP-conjugated secondary antibodies (catalogs NA931V and NA934V, GE Healthcare) were subsequently used. Bound antibodies were detected using Immunostar LD reagents (Wako). The antibodies used were as follows: anti-CD63 (catalog HPA010088, Sigma-Aldrich), anti-CD9 (catalog 10626D, Invitrogen), and anti-albumin (catalog A80-129A, Bethyl Laboratory).
Statistics.
Significant differences between groups were determined by 2-tailed Student’s t test when variances were equal. When variances were unequal, 2-tailed Welch’s t test was used instead. P values less than 0.05 were considered statistically significant. Bee swarm dot plots and box plots were visualized with a web tool (BoxPlotR, http://boxplot.tyerslab.com/). For comparisons of standard deviations between real-time PCR and ddPCR, a coefficient of variation (CV) was calculated for each sample, and significant differences were determined using the 2-tailed Welch’s t test. Receiver operating characteristic (ROC) curves were used to determine the sensitivity and specificity of the method and to compare the AUCs of each serum marker using the Delong method (30). The analyses were performed with a statistical software package (JMP, ver. 11; SAS Institute Japan).
Study approval.
Written consent for the serum sampling was obtained from all patients with disclosure of the study protocol, which was approved by the local ethical committee of the University of Tokyo (approval number: 500-[4]).
Author contributions
TK and M. Otsuka conceived the study, planned the research, and wrote the paper. TK, M. Otsuka, TY, M. Ohno, KY, NY, and AK performed the experiments. KK supervised the entire project.
Supplementary Material
Acknowledgments
This work was supported by Grants-in-Aid from the Ministry of Education, Culture, Sports, Science and Technology, Japan (grants 26860492, 16H05149, 16K19329, 150009038, and 24390183) (T. Kishikawa, M. Otsuka, T. Yoshikawa, N. Yamamoto, and K. Koike), by the Translational Research Network Program from Japan Agency for Medical Research and Development, AMED (to M. Otsuka), and by grants from the Japan Prize Foundation and from the Tokyo Biomarker Innovation Research Association (to T. Kishikawa).
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Reference information:JCI Insight. 2016;1(8):e86646. doi:10.1172/jci.insight.86646.
Contributor Information
Takahiro Kishikawa, Email: KISHIKAWAT-INT@h.u-tokyo.ac.jp.
Motoyuki Otsuka, Email: otsukamo-tky@umin.ac.jp.
Takeshi Yoshikawa, Email: YOSHIKAWAT-INT@h.u-tokyo.ac.jp.
Motoko Ohno, Email: onom-int@h.u-tokyo.ac.jp.
Keisuke Yamamoto, Email: yamamotok-int@h.u-tokyo.ac.jp.
Natsuyo Yamamoto, Email: yamamoton-int@h.u-tokyo.ac.jp.
Ai Kotani, Email: aikotani@k-lab.jp.
Kazuhiko Koike, Email: kkoikegastro@gmail.com.
References
- 1.Hidalgo M. Pancreatic cancer. N Engl J Med. 2010;362(17):1605–1617. doi: 10.1056/NEJMra0901557. [DOI] [PubMed] [Google Scholar]
- 2.Siegel R, Naishadham D, Jemal A. Cancer statistics, 2013. CA Cancer J Clin. 2013;63(1):11–30. doi: 10.3322/caac.21166. [DOI] [PubMed] [Google Scholar]
- 3.Yadav D, Lowenfels AB. The epidemiology of pancreatitis and pancreatic cancer. Gastroenterology. 2013;144(6):1252–1261. doi: 10.1053/j.gastro.2013.01.068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jemal A, et al. Cancer statistics, 2008. CA Cancer J Clin. 2008;58(2):71–96. doi: 10.3322/CA.2007.0010. [DOI] [PubMed] [Google Scholar]
- 5.Li D, Xie K, Wolff R, Abbruzzese JL. Pancreatic cancer. Lancet. 2004;363(9414):1049–1057. doi: 10.1016/S0140-6736(04)15841-8. [DOI] [PubMed] [Google Scholar]
- 6.Vincent A, Herman J, Schulick R, Hruban RH, Goggins M. Pancreatic cancer. Lancet. 2011;378(9791):607–620. doi: 10.1016/S0140-6736(10)62307-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gupta RA, et al. Long non-coding RNA HOTAIR reprograms chromatin state to promote cancer metastasis. Nature. 2010;464(7291):1071–1076. doi: 10.1038/nature08975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Volinia S, et al. A microRNA expression signature of human solid tumors defines cancer gene targets. Proc Natl Acad Sci U S A. 2006;103(7):2257–2261. doi: 10.1073/pnas.0510565103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kishikawa T, Otsuka M, Ohno M, Yoshikawa T, Takata A, Koike K. Circulating RNAs as new biomarkers for detecting pancreatic cancer. World J Gastroenterol. 2015;21(28):8527–8540. doi: 10.3748/wjg.v21.i28.8527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang Z, Gerstein M, Snyder M. RNA-Seq: a revolutionary tool for transcriptomics. Nat Rev Genet. 2009;10(1):57–63. doi: 10.1038/nrg2484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ting DT, et al. Aberrant overexpression of satellite repeats in pancreatic and other epithelial cancers. Science. 2011;331(6017):593–596. doi: 10.1126/science.1200801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bersani F, et al. Pericentromeric satellite repeat expansions through RNA-derived DNA intermediates in cancer. Proc Natl Acad Sci U S A. 2015;112(49):15148–15153. doi: 10.1073/pnas.1518008112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Maison C, et al. SUMOylation promotes de novo targeting of HP1alpha to pericentric heterochromatin. Nat Genet. 2011;43(3):220–227. doi: 10.1038/ng.765. [DOI] [PubMed] [Google Scholar]
- 14.Zhu Q, et al. BRCA1 tumour suppression occurs via heterochromatin-mediated silencing. Nature. 2011;477(7363):179–184. doi: 10.1038/nature10371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tilman G, et al. Cancer-linked satellite 2 DNA hypomethylation does not regulate Sat2 non-coding RNA expression and is initiated by heat shock pathway activation. Epigenetics. 2012;7(8):903–913. doi: 10.4161/epi.21107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kondratova VN, Botezatu, Shelepov VP, Likhtenshtein AV. Transcripts of satellite DNA in blood plasma: probable markers of tumor growth. Mol Biol (Mosk) 2014;48(6):999–1007. [PubMed] [Google Scholar]
- 17.Vickers KC, Remaley AT. Lipid-based carriers of microRNAs and intercellular communication. Curr Opin Lipidol. 2012;23(2):91–97. doi: 10.1097/MOL.0b013e328350a425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bouzinba-Segard H, Guais A, Francastel C. Accumulation of small murine minor satellite transcripts leads to impaired centromeric architecture and function. Proc Natl Acad Sci U S A. 2006;103(23):8709–8714. doi: 10.1073/pnas.0508006103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lee KM, Nguyen C, Ulrich AB, Pour PM, Ouellette MM. Immortalization with telomerase of the Nestin-positive cells of the human pancreas. Biochem Biophys Res Commun. 2003;301(4):1038–1044. doi: 10.1016/S0006-291X(03)00086-X. [DOI] [PubMed] [Google Scholar]
- 20.Hindson CM, et al. Absolute quantification by droplet digital PCR versus analog real-time PCR. Nat Methods. 2013;10(10):1003–1005. doi: 10.1038/nmeth.2633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ballehaninna UK, Chamberlain RS. The clinical utility of serum CA 19-9 in the diagnosis, prognosis and management of pancreatic adenocarcinoma: An evidence based appraisal. J Gastrointest Oncol. 2012;3(2):105–119. doi: 10.3978/j.issn.2078-6891.2011.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Williams GT, Farzaneh F. Are snoRNAs and snoRNA host genes new players in cancer? Nat Rev Cancer. 2012;12(2):84–88. doi: 10.1038/nrc3195. [DOI] [PubMed] [Google Scholar]
- 23.Matthaei H, Schulick RD, Hruban RH, Maitra A. Cystic precursors to invasive pancreatic cancer. Nat Rev Gastroenterol Hepatol. 2011;8(3):141–150. doi: 10.1038/nrgastro.2011.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tanaka M. International consensus on the management of intraductal papillary mucinous neoplasm of the pancreas. Ann Transl Med. 2015;3(19):e86646. doi: 10.3978/j.issn.2305-5839.2015.11.09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Valadi H, Ekstrom K, Bossios A, Sjostrand M, Lee JJ, Lotvall JO. Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat Cell Biol. 2007;9(6):654–659. doi: 10.1038/ncb1596. [DOI] [PubMed] [Google Scholar]
- 26.Tanne A, et al. Distinguishing the immunostimulatory properties of noncoding RNAs expressed in cancer cells. Proc Natl Acad Sci U S A. 2015;112(49):15154–15159. doi: 10.1073/pnas.1517584112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ohno M, et al. The flavonoid apigenin improves glucose tolerance through inhibition of microRNA maturation in miRNA103 transgenic mice. Sci Rep. 2013;3:e86646. doi: 10.1038/srep02553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tagarro I, Fernandez-Peralta AM, Gonzalez-Aguilera JJ. Chromosomal localization of human satellites 2 and 3 by a FISH method using oligonucleotides as probes. Hum Genet. 1994;93(4):383–388. doi: 10.1007/BF00201662. [DOI] [PubMed] [Google Scholar]
- 29.Schagger H, von Jagow G. Blue native electrophoresis for isolation of membrane protein complexes in enzymatically active form. Anal Biochem. 1991;199(2):223–231. doi: 10.1016/0003-2697(91)90094-A. [DOI] [PubMed] [Google Scholar]
- 30.DeLong ER, DeLong DM, Clarke-Pearson DL. Comparing the areas under two or more correlated receiver operating characteristic curves: a nonparametric approach. Biometrics. 1988;44(3):837–845. doi: 10.2307/2531595. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.