

Abstract
Human NADPH-cytochrome P450 oxidoreductase (POR) gene mutations are associated with severe skeletal deformities and disordered steroidogenesis. The human POR mutation A287P presents with disordered sexual development and skeletal malformations. Difficult recombinant expression and purification of this POR mutant suggested that the protein was less stable than WT. The activities of cytochrome P450 17A1, 19A1, and 21A2, critical in steroidogenesis, were similar using our purified, full-length, unmodified A287P or WT POR, as were those of several xenobiotic-metabolizing cytochromes P450, indicating that the A287P protein is functionally competent in vitro, despite its functionally deficient phenotypic behavior in vivo. Differential scanning calorimetry and limited trypsinolysis studies revealed a relatively unstable A287P compared with WT protein, leading to the hypothesis that the syndrome observed in vivo results from altered POR protein stability. The crystal structures of the soluble domains of WT and A287P reveal only subtle differences between them, but these differences are consistent with the differential scanning calorimetry results as well as the differential susceptibility of A287P and WT observed with trypsinolysis. The relative in vivo stabilities of WT and A287P proteins were also examined in an osteoblast cell line by treatment with cycloheximide, a protein synthesis inhibitor, showing that the level of A287P protein post-inhibition is lower than WT and suggesting that A287P may be degraded at a higher rate. Current studies demonstrate that, unlike previously described mutations, A287P causes POR deficiency disorder due to conformational instability leading to proteolytic susceptibility in vivo, rather than through an inherent flavin-binding defect.
Keywords: cytochrome P450, flavoprotein, membrane protein, protein crystallization, protein stability, reductase, POR deficiency, cytochrome P450 oxidoreductase, diflavin oxidoreductase
Introduction
NADPH-cytochrome P450 oxidoreductase (POR)9 is an electron-transferring diflavin enzyme residing in the endoplasmic reticulum of cells in various tissues. POR catalyzes the one-at-a-time two-electron transfer from NADPH in the cytochrome P450 (CYP)-mediated metabolism of many endobiotics, such as retinoic acid, vitamin D3, lanosterol, estrogens and androgens, as well as many xenobiotics, including both drugs and environmental toxins (1, 2). It can be predicted that alteration in POR activity or stability can lead to a myriad of defects.
All of the ∼50 human endoplasmic reticulum resident (microsomal) CYPs are dependent upon POR for electron shuttling between NADPH and the CYP-heme, the reduction of which is necessary for O2 binding. The biosynthesis of endobiotics, such as steroids, bile acids, and arachidonic acid metabolites, and the metabolism of many xenobiotics require cytochromes P450. POR is also essential for electron delivery to heme oxygenase (3), squalene mono-oxygenase (4), and fatty acid elongase (5). Mice in which POR was incapacitated exhibited multiple developmental defects and embryonic lethality (6). Although early embryogenesis was unaffected, many defects appeared in the mouse embryos by embryonic day 10.5. A mouse model in which POR was conditionally deleted in hepatocytes showed a marked induction of liver CYPs (up to 5-fold depending upon age of the mice) (7) and heme oxygenase (up to 9-fold) (8), as a compensatory mechanism, indicating the reliance of these redox partners on POR for activity. The mice were fertile and normal in gross appearance and growth rate, but as they aged, the homozygotes exhibited increases in liver weight, hepatic lipidoses, decreased cholesterol metabolism, and profound decreases in in vivo drug metabolism (7). These observations suggested that POR deficiency could have significant deleterious effects in humans.
Indeed, a multiplicity of POR mutations were identified in human patients in which embryonic development was severely affected (9) or patients presented with congenital adrenal hyperplasia, previously attributed to combined CYP17A1 and CYP21A2 deficiencies (10). In addition to severe steroidogenic defects, POR deficiency (PORD) also caused both craniofacial and long bone developmental anomalies, a bone phenotype resembling Antley-Bixler syndrome (ABS) (9). ABS with normal steroidogenesis is the result of gain-of-function mutations in fibroblast growth factor receptor type 2 (FGFR2), whereas ABS with accompanying steroidogenic defects is due to defects in POR. The disordered steroidogenesis in PORD is attributable to the inability of POR to capably transfer electrons to steroidogenic CYPs, but the role of POR in bone development remains of interest. A conditional knock-out of the POR gene in bone mesenchyme recapitulated the ABS-like bone phenotype in mice and indicated that POR may affect the FGF receptor signaling pathway (11).
Our long-standing biochemical research into the mechanism of this critical flavoprotein (12) led us to explore the structure/function relationships responsible for these phenotypes by expressing and purifying POR variants of interest reported in the literature (13–20). The variant A287P was reported as the most common mutation found in the Caucasian population (10, 14), and these studies were verified and extended to establish genotype-phenotype correlations in a cohort of patients from 11 countries (21). We and others have pursued the characterization of A287P (10, 14, 22, 23).
Unlike the recent report of flavin deficiency in the A287P protein (24), we found a complete complement of both flavins, FAD and FMN, determined by HPLC, and little compromise in a variety of enzymatic activities, either intrinsic to POR or requiring interaction with CYPs of various specificities. This result begged the question of how a POR mutation that appeared essentially normal in vitro could contribute to the phenotype seen in human patients. Difficulty in reproducibly isolating the full-length A287P mutant protein heterologously expressed in Escherichia coli suggested its inherent instability and led to the modification of our standard expression and purification protocol to achieve purification of the A287P variant. Once obtained in pure form, however, it remained active upon low temperature storage and during various measurements. Herein, we have applied a variety of biophysical techniques to evaluate the properties of the A287P variant of POR and expressed it in both bacterial and mammalian cell systems to determine its behavior in a more physiologically pertinent environment. In addition, we have performed differential scanning calorimetry (DSC) and compared the crystal structure of this mutant protein with the vulnerability of POR to proteolytic digestion by trypsin to explain the difference between the A287P POR variant and WT enzyme, demonstrating that subtle conformational changes in A287P may lead to protein instability and increased temperature sensitivity in vivo.
Results
A287P Variant Preparation Necessitates Modified Expression and Purification Conditions
We have expressed full-length WT human P450 reductase and 11 POR mutants in E. coli and purified them to homogeneity (22, 25–27).10 Our initial purification of the full-length A287P variant followed our established protocol, but the yield was extremely low and irreproducible as compared with WT. This behavior suggested that the A287P variant was somewhat less stable than any of the other polymorphic variants we had previously expressed and purified. Therefore, the established protocol was modified and optimized to express and purify A287P to homogeneity (Table 1). The WT enzyme, although successfully expressed and purified by the methods utilized previously, was also taken through the identical modified protocol for direct comparison. Specifically, the growth temperature was lowered to 23 °C, and a lower IPTG concentration (62.5 μm) was used for the induction of the protein. In addition, a higher detergent (0.5% Triton X-100) concentration was required to extract the full-length A287P as compared with the WT POR and other POR variants, suggesting a tighter association of A287P with the E. coli membrane. Although slower growth conditions were necessary for A287P expression in E. coli, but not for WT protein, the modified protocol improved the yield of WT protein as well. The yields of WT and A287P proteins purified via this protocol were similar and averaged ∼2.5 mg of protein/liter of culture.
TABLE 1.
Modifications to standard purification protocol for A287P
| Standard protocol | A287P modification | |
|---|---|---|
| Growth temperature | 28 °C | 23 °C |
| IPTG concentration | 500 μm | 62.5 μm |
| Growth time | 24 h | 48 h |
| Detergent concentration | 0.1% Triton X-100 | 0.5% Triton X-100 |
Characterization of A287P Protein, Spectral Properties and Flavin Content
The full-length A287P protein was expressed without tagging modification, purified as a single band on SDS-PAGE, similar to WT full-length POR (Fig. 1A), and characterized by its spectral properties (Fig. 1B) and flavin content (Table 2). Spectral analysis of A287P in the visible region, where protein-bound flavins absorb, showed no observable differences compared with WT protein (Fig. 1B). Protein-bound flavin content, both FAD and FMN, was measured using HPLC (25) for both WT and A287P. The ratio of FAD and FMN to protein was calculated to be ∼1:1:1, suggesting the mutation has, at most, a minimal effect on flavin content (Table 2). Although a recent publication reported that the A287P mutation in POR leads to deficiency in both FAD and FMN bound (24), it is not difficult to imagine that a point mutation could affect the conformation of the folded protein without affecting the binding affinity of either or both the flavins (see “Discussion”). Furthermore, the A287P mutation is not localized near either of the flavin-binding sites.
FIGURE 1.

Characterization of purified POR A287P protein. A, purified proteins (2 μg) on SDS-polyacrylamide gel, Coomassie-stained. B, absolute spectra of WT (left) and A287P (right) from 300 to 700 nm; 10 μm protein was measured. No difference is observed between WT and A287P.
TABLE 2.
Flavin content of POR WT and A287P purified proteins
| Protein | FAD | FMN |
|---|---|---|
| WT | 1.1 ± 0.2 | 1 ± 0.1 |
| A287P | 0.9 ± 0.2 | 1 ± 0.3 |
NADPH Km and NADP+ Ki Determinations
Despite the lack of dramatic effect of the A287P point mutation on the content of FAD, the entry flavin for electrons from NADPH, the Km for NADPH, and the Ki for NADP+ were determined to eliminate the possibility that any kinetic effects could result from alterations in the binding of the electron donor. The A287P mutation had little or no effect on these kinetically determined parameters using cytochrome c as the electron acceptor to measure electron flow-through the reductase molecule (Km NADPH = ∼5.9 μm and ∼5.0 μm for WT and A287P, respectively) or alteration in the inhibition constant of NADP+ (Ki NADP+ = ∼16.1 μm and ∼18.4 μm for WT and A287P, respectively, determined at 25 °C). The data clearly show that any effect of the A287P mutation is minimal and that the behavior of this variant in vivo cannot be attributed to a defect in the hydride transfer step from NADPH to FAD.
Determination of Initial Rates of Flavin Reduction by Stopped-flow Spectrophotometry
A further comparison of kinetic parameters between WT and A287P POR was made using stopped-flow spectrophotometry to measure the rates of flavin reduction at 450 nm. Table 3 shows the fast and slow phase rate constants for reduction of these POR enzymes by NADPH at 25 °C. The initial fast phase of flavin reduction, which reflects hydride transfer to FAD and one-electron reduction of FMN (28), was 34% lower for A287P compared with WT protein. The second slower phase of reduction, representing the conversion of two electron-reduced to four electron-reduced hydroquinone proteins (28), was 36% lower for A287P compared with WT. These data indicate that, despite full complementation with both flavins, other alterations in the A287P mutation are affecting the rate of electron transfer from NADPH into the entry flavin, FAD, to a minor degree. The degree of rate decrease may or may not affect the physiological rates of interaction with CYPs or other redox partners that involve much lower rates of electron transfer in catalytic turnover.
TABLE 3.
Rate constants for POR flavin reduction by NADPH
| WT | A287P | |
|---|---|---|
| s−1 | s−1 | |
| Fast phase, k1 | 14.31 ± 2.16 | 9.35 ± 1.01 |
| Slow phase, k2 | 1.59 ± 0.41 | 1.01 ± 0.44 |
Cytochrome c Reductase Activity
The rate of reduction of the artificial electron acceptor, cytochrome c, by POR, using NADPH as the electron donor under steady state conditions, was decreased 30% in the A287P mutant compared with the WT protein (1850 ± 240 and 2760 ± 270 min−1 for A287P and WT, respectively). Cytochrome c reductase activity not only measures the rate at which POR can donate electrons, but also reflects the internal rate of electron transfer between the flavins because electron transfer to this acceptor occurs from FMN. The observed decrease in reduction of cytochrome c observed for A287P compared with WT is in agreement with the stopped-flow data in which both rate constants are decreased 34–36% in A287P.
Interaction with Physiological Redox Partners
The ability of A287P to support CYP activity was tested in CYPs that metabolize two different classes of compounds, i.e. steroids and xenobiotics, to determine whether A287P might interact differently with various CYPs. Using reconstitution systems unique to each steroidogenic cytochrome P450, WT and A287P proteins were interacted with purified CYP17A1 and CYP21A2. The choice of these two CYPs was made to examine the possibility that the phenotype observed with A287P, i.e. sexual ambiguities in these patients (9, 10), was due to the inherent properties of this variant affecting its interactions with these particular steroidogenic CYPs. The data show that there are no significant differences in activity between WT and A287P in supporting CYP 17α- or 21-hydroxylase activities with progesterone (Fig. 2A). Furthermore, these constructs were tested with purified human aromatase (CYP19A1), the CYP responsible for estrogen biosynthesis from testosterone (Fig. 2B). As with CYP17A1 and CYP21A2, little or no differences were detected among activities supported by WT or A287P proteins. Thus, when assaying the ability of POR A287P to support these CYP activities in an in vitro assay system, POR A287P appeared to be essentially as competent as WT.
FIGURE 2.
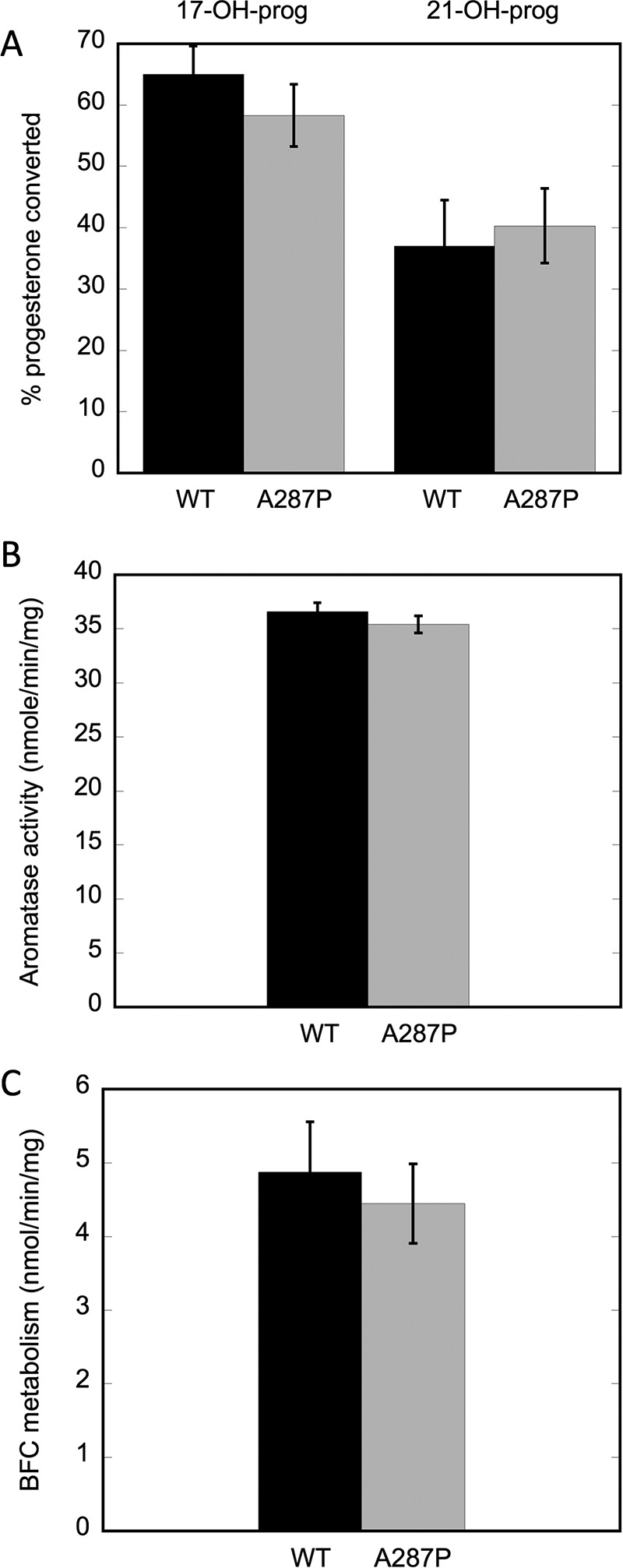
CYP17A1, CYP21A2, CYP19A1 (aromatase), and CYP3a11 activities supported by WT and A287P POR proteins. A, percentage of progesterone converted to 17-OH-progesterone by CYP17A1 and 21-OH progesterone by CYP21A2. B, aromatase activity as measured by conversion of androstenedione to estrone. C, conversion of BFC to HFC by xenobiotic-metabolizing CYPs. Black bars are WT POR and gray bars are A287P. Assays were repeated at least three times and were performed as described under “Experimental Procedures.”
Drug and xenobiotic metabolism by the WT and A287P proteins was also assessed. BFC is metabolized primarily by CYP3a11 in mouse liver, although other CYPs will also contribute to BFC conversion to HFC. Metabolism of BFC was measured in a reconstituted system using liver microsomes isolated from mice with liver-specific deletion of POR (8). This unique system allows us to measure CYP activity in a native milieu, eliminating any contribution of activity by endogenous POR. In addition, many CYPs are up-regulated in this mouse model, allowing robust CYP activity that is easily detectable. As shown in Fig. 2C, there is little to no difference between WT and A287P metabolism of BFC.
The cell model BTC1A2_POR was used for the co-expression of the POR variant A287P with human CYP1A2. The biplasmid system in BTC strains allows the expression of full-length proteins at a stoichiometry that reflects that found in vivo (23). Table 4 summarizes the characterization of the different BTC1A2_POR membranes, i.e. co-expressed with CYP1A2. Both the protein expression level and cytochrome c reduction activity of POR A287P were similar to those of WT. As CYP1A2 expression levels did not vary among the strains, a CYP/POR stoichiometry of 5:1 was utilized for both BTC1A2_PORWT and BTC1A2_PORA287P. Table 5 shows that the capacity to sustain CYP1A2 in methoxyresorufin O-demethylase activity was not significantly different between POR A287P and WT expressed with CYP1A2 in these cells, as demonstrated by their Michaelis-Menten kinetic parameters.
TABLE 4.
POR and CYP1A2 contents and cytochrome c reduction activity of BTC1A2_POR membranes
| BTC1A2_POR | Cytochrome c reduction | PORa | CYP1A2 | CYP/POR |
|---|---|---|---|---|
| nmol/[min·mg protein] | pmol/mg protein | pmol/mg protein | ||
| WT | 36.0 ± 2.5 | 11.2 ± 0.8 | 58 ± 4 | 5:1 |
| A287P | 37.7 ± 1.4 | 11.8 ± 0.4 | 66 ± 7 | 5:1 |
| Null | 0.6 ± 0.2 | 0.2 ± 0.1 | 56 ± 3 |
a Data were calculated with kcat of 3200 min1 and corrected for background activities observed with corresponding null membranes; null contains CYP1A2 only, no POR.
TABLE 5.
Methoxyresorufin O-dealkylation activities
| BTC1A2_POR | kcat | Km |
|---|---|---|
| min−1 | μm | |
| WT | 1.23 ± 0.03 | 0.63 ± 0.04 |
| A287P | 1.16 ± 0.02 | 0.67 ± 0.03 |
| Null |
In addition, the ability of the A287P variant to bioactivate three CYP1A2-dependent pre-mutagens, 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK), 2-amino-3-methylimidazo(4,5-f)quinoline (IQ), and 2-amino-anthracene (2AA) was tested. The POR variant A287P was as effective in sustaining CYP1A2-mediated mutagen formation from these three compounds as was WT (Fig. 3), although a slight trend toward lower efficiency was obtained with CYP1A2 in combination with variant A287P, in particular for IQ. Thus, both WT and A287P POR proteins demonstrate similar enzymatic and mutagenic activities in the bacterial expression system BTC1A2 POR.
FIGURE 3.
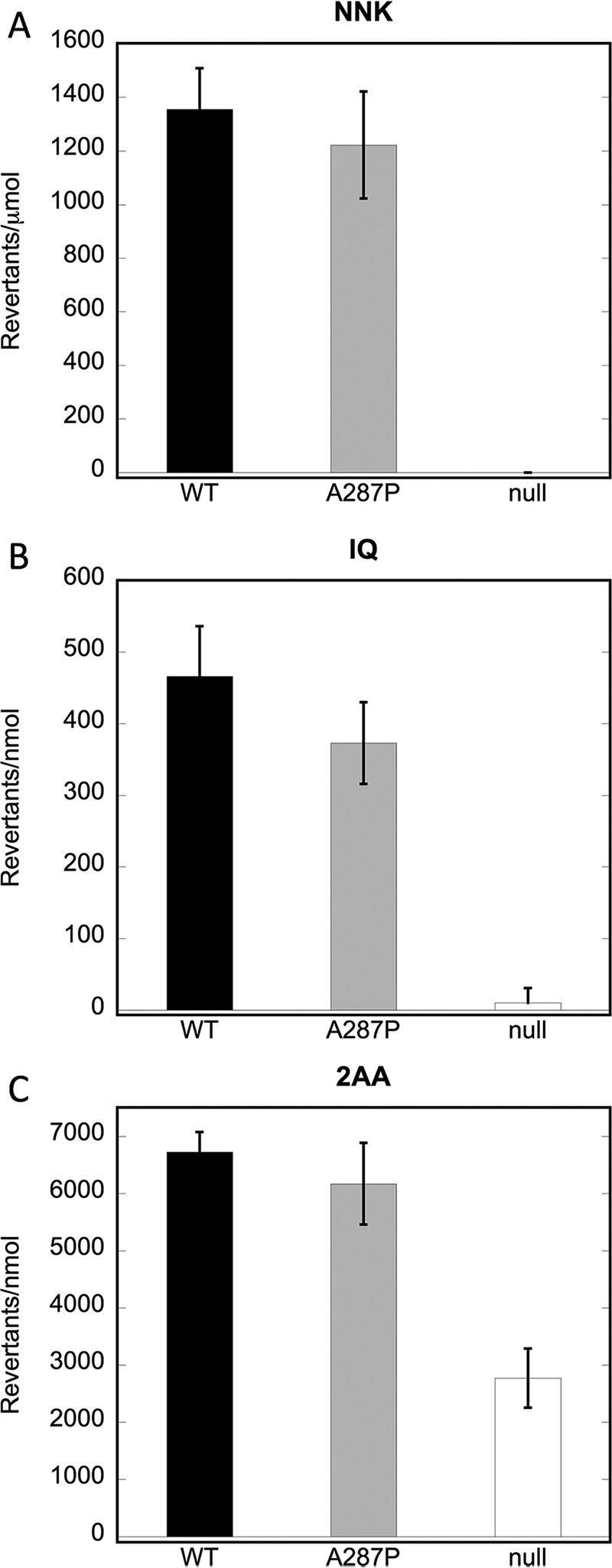
Mutagenic activities of BTC1A2_POR strains. A, revertants/μmol NNK. B, revertants/nmol IQ; C, revertants/nmol 2AA. WT POR, A287P POR, and POR null strains were tested as described under “Experimental Procedures”; n = 3.
Relative Stability of WT and A287P Proteins as a Function of Temperature
Because the optimal growth temperature of cells expressing A287P was lower than that of WT or other POR variants, it seemed that A287P protein might be less stable at higher temperatures. To investigate the effect of increased culturing temperature of the BTC constructs on the protein stability of the POR protein, cells expressed with either WT or A287P were cultured at 28 or 32 °C. (As stated previously, CYP expression at higher temperatures is negatively affected in the BTC bacterial expression system, so temperatures higher than 32 °C were not employed.) When the WT and POR A287P variant were expressed in the BTC bacterial system without human CYP (strains BTC_PORWT and BTC_PORAP; see Table 6), at 28 and 32 °C, immunoblot detection of POR in the BTC_PORWT membranes displayed, at most, a slightly lower expression level of WT POR at 32 °C when compared with 28 °C (Fig. 4). The A287P variant, however, demonstrated a substantially lower expression level when expressed at 32 °C versus 28 °C, consistent with lower protein stability than WT.
TABLE 6.
Strains and plasmids
| Description | Ref. | |
|---|---|---|
| Strains | ||
| PD301 | E. coli mutagenicity tester strain, without heterologous expression | 57 |
| BTC1A2_PORWT | PD301/pCWh1A2/pLCM_PORWT | 57 |
| BTC1A2_PORAP | PD301/pCWh1A2/pLCM_PORAP | This study |
| BTC1A2_PORnull | PD301/pCWh1A2/pLCM | This study |
| BTC_PORWT | PD301/pCWΔ/pLCM_PORWT | This study |
| BTC_PORAP | PD301/pCWΔ/pLCM_PORAP | This study |
| BTC° | PD301/pCWΔ/pLCM | This study |
| Plasmids | ||
| pCWh1A2 | Medium high copy number expression plasmid for human CYP1A2, with tandem pTAC promotor; lacIq repressor; AmpR | 59 |
| pCWΔ | pCWh1A2, but without human CYP1A2 cDNA | 59 |
| pLCM | Low copy number mutator plasmid; mucAB operon; KanR | 60 |
| pLCM_PORWT | pLCM but containing POR cDNA for POR WT, expressed via a single pTAC promotor | 58 |
| pLCM_PORAP | pLCM but containing POR cDNA for POR A287P, expressed via a single pTAC promotor | 23 |
FIGURE 4.

Immunodetection of POR in BTC membrane fractions at 28 and 32 °C. Left panel, 1st and 2nd lanes are WT POR in the BTC expression system, grown at 28 and 32 °C, respectively. 3rd and 4th lanes are A287P in the BTC expression system, grown at 28 and 32 °C, respectively. 5th and 6th lanes are purified WT POR, and 7th and 8th lanes are the BTC expression system without POR or A287P. Lanes contain 5 μg of membrane protein, except for C1 and C2, which contain 10 and 25 ng of purified WT POR enzyme, respectively. Right panel, quantitation of left panel using Image Studio Lite (LI-COR Biosciences). Data shown are density relative to that of WT at 28 °C and are the result of n = 2 experiments.
Differential Scanning Calorimetry to Determine Tm of WT and A287P Proteins
To assess differences in structural stability between the WT and mutant proteins, DSC was performed. Fig. 5 shows the thermograms derived for WT POR (solid line) and A287P (dotted line). It is clear that these two proteins behave very differently in response to the increasing temperature gradient. The data for the WT protein show a major peak at 52.4 ± 0.2 °C (average of two separate experimental trials). The thermogram for A287P protein, however, is more complex. The thermogram appears to have at least two peaks, a major one at 47.2 ± 0.3 °C and a smaller one at 52.6 ± 0.3 °C (average of two trials). Thus, the melting temperature (Tm) represented by the major peak observed for A287P protein is shifted ∼5 °C cooler than that of WT.
FIGURE 5.
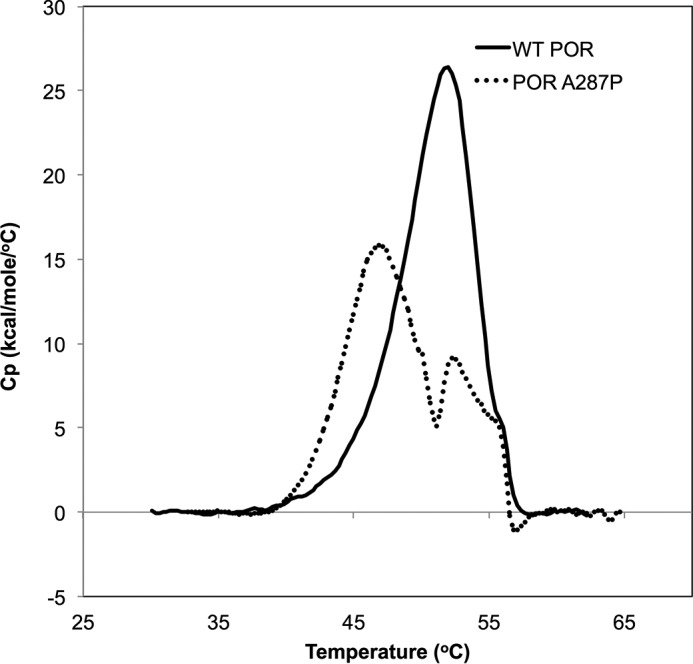
Determination of protein stability by DSC. Thermograms for WT (solid line) and A287P (dotted line). Two trials were performed with each protein and shown is a representative trace of one of the trials. WT protein was scanned from 13 to 90 °C, and protein concentration was 6.5 μm for both trials. A287P protein was scanned from 17 to 75 °C, and protein concentration was 6.9 and 6.7 μm for the first and second trials, respectively. Data were analyzed in the range 30–70 °C for all experiments.
Limited Trypsinolysis of WT and A287P Proteins
The DSC experiments and the behavior of the expressed and purified A287P and that expressed in the BTC system suggest that the A287P mutation has perturbed the conformation of the protein in some manner. The effect of this mutation was examined using limited proteolysis by trypsin performed on the full-length enzyme. Limited proteolysis experiments have been used to probe the three-dimensional architecture, as well as conformational flexibility, of proteins (29). Local fluctuations in protein structure can also be studied by limited proteolysis (30–32). Both wild type and A287P holoenzymes were digested using trypsin in a molar ratio of 800:1 (POR/trypsin) at two different temperatures, 30 and 37 °C. The digested products were analyzed between 0 and 180 min, using SDS-PAGE, and revealed very similar digestion patterns between WT and A287P at 30 °C; however, at 37 °C (human body temperature) the A287P protein is digested more completely over time (Fig. 6A), indicated by the persistence of the ∼55-kDa band at 30 °C for both WT and A287P and for WT only at 37 °C. The ∼55-kDa band virtually disappeared upon limited trypsinolysis of A287P at 37 °C. Consistent with the DSC experiments, the WT protein was more resistant to thermal perturbation as the banding pattern upon trypsin digestion at both 30 and 37 °C was similar (Fig. 6A). Taken as a whole, the results of trypsin digestion of WT and A287P holoenzymes under similar conditions suggest that the resulting A287P protein exhibited increased structural flexibility upon exposure to trypsin digestion at 37 °C; the mutation did not lead to misfolded protein, as both full-length proteins were expressed and purified.
FIGURE 6.

Determination of protein stability by limited trypsinolysis. A, thermal stability. 1st lane, 2 μg of purified POR protein; 2nd and 8th lanes, molecular mass markers; 3rd to 7th lanes, 0, 10, 60, 120, and 180 min of trypsinolysis, respectively. Left panels are WT protein, and right panels are A287P protein. Upper panels are reactions performed at 30 °C, and lower panels are reactions performed at 37 °C. The a, b, and c labels in the lower right panel correspond to the bands of 20, 17, and 14 kDa, respectively, as discussed in the text. B, stability with 2′-AMP. 1st lane, 2 μg of protein; 2nd and 8th lanes, molecular mass markers; 3rd to 7th lanes, 0, 10, 60, 120, and 180 min of trypsinolysis, respectively. Left panels are WT protein, and right panels are A287P protein. Upper panels are reactions without 2′-AMP, and lower panels are reactions with 5 mm 2′-AMP. Reactions were performed at 37 °C. C, stability with DLPC. 1st lane, 2 μg of protein; 2nd and 8th lanes; molecular mass markers, 3rd to 7th lanes, 0, 10, 60, 120, and 180 min of trypsinolysis, respectively. Left panels are WT protein, and right panels are A287P protein. Upper panels are reactions performed without DLPC, and lower panels are reactions performed in the presence of DLPC. Reactions were performed at 37 °C. Trypsinolysis was performed as described under “Experimental Procedures.” Trysinolysis experiments (A) were performed three times, with the same results; figure shown is representative.
Further probing of the differences between WT and A287P susceptibility to trypsinolysis was conducted in the presence of 2′-AMP (Fig. 6B), which contains the unique 2′-phosphate substituent on the adenine ribose moiety of NADP+ and therefore binds in the FAD/NADP+-binding domain of POR. The presence of 2′-AMP was shown to be protective during trypsinolysis for both WT and A287P but showed a more dramatic effect with the A287P variant.
Because our POR proteins were full-length with a hydrophobic N terminus, dilauroylphosphatidylcholine (DLPC), prepared as 100 nm extruded vesicles, was added as a possible mimic of its interaction with the endoplasmic reticulum membrane to both WT and A287P proteins to determine its effect on trypsinolysis. As can be seen in Fig. 6C, the bands at both ∼70 and 55 kDa, which disappear almost completely in the case of A287P at 180 min, are partially protected from trypsinolysis by DLPC addition. The difference in trypsin susceptibility between the WT and the A287P variant in the presence of DLPC could indicate subtle conformational differences. These differences and possible trypsin digestion sites will be discussed in view of the crystal structure of A287P (see below).
Structural Characterization of A287P POR in the Vicinity of Ala-287 (the Mutation Site)
The human POR A287P crystal structure of the soluble form, in which the N-terminal 66 residues that anchor it to the endoplasmic reticulum membrane were removed (as reported for crystallographic studies of the Δ66 constructs of other human POR proteins by Xia et al. (27)), was solved and compared with our published wild type structure (PDB code 3QE2) (Fig. 7) (27). As discussed under “Experimental Procedures,” both of these structures also contained two common polymorphic mutations, P228L and A503V, both of which were in the original wild type POR clone obtained from ATCC. To ensure that these extra mutations do not cause any additional structural perturbations, the structure of the “true wild type” POR (i.e. Pro-228 and Ala-503, Δ66-WT in Table 7) was also solved. When the true wild type structure (this work; 2.3 Å resolution) is compared with the previously published higher resolution structure (WT-P228L/A503V; 3QE2, 1.75 Å resolution), the two structures are essentially identical with a root mean square deviation of 0.3 Å for all 603 visible Cα carbons. More importantly, there are no significant differences in the vicinity of the Ala-287 residue in these two structures. Therefore, it is concluded that the two common polymorphic mutations, P228L and A503V, do not cause any significant structural deviations and that any structural changes observed in the structure of A287P/P228L/A503V are due to the mutation of A287P and not influenced by the P228L and A503V mutations. Thus, structural comparisons of A287P discussed below are made with the higher resolution WT-P228L/A503V structure 3QE2.
FIGURE 7.

Comparison of the crystal structures of WT and A287P POR. A, overlay of the structures of A287P mutant and wild type POR. The superposition was done using the entire 603 visible residues. Wild type POR is colored gray, and the A287P structure is colored by domains as follows: the FMN-binding domain (blue), connecting domain (cyan), FAD-binding domain (gold and red), NADP(H)-binding domain (green), and the hinge between the two flavin-binding domains is shown as a dotted line with its last residue Arg-246 marked as a small red ball. The mutation site, A287P, is shown with a blue ball. The region in the FAD-binding domain showing minor but significant structural changes caused by the A287P mutation is shown in red. Following the nomenclature used for the structure of rat POR (12), secondary structural elements in this region are listed in red. β7 (Phe-285–Asn-296) is antiparallel to β16 (Leu-511–Lys-518), and part of these two β-strands are packed onto the αN-helix (Gly-491–Ala-499). The red balls are possible trypsin digestion sites consistent with the results of both limited trypsinolysis experiments and the mass spectrometry (MS) analyses of trypsinized fragments, except that the meshed red ball (Lys-558) site has not been confirmed by the MS analysis. See under “Results” for the relationship between possible trypsin cleavage sites and the cleavage pattern. B, two enlarged views of the vicinity of the mutation site in the structure of A287P (the red region shown in A). Left panel, a similar view to A. In the mutant structure, the A287P mutation causes a slight bending of the N-terminal side of β7, which pushes the neighboring β16 and part of the connected loop-500 outward. Although these changes are small (the distance between Cα carbons of Pro-287 of the mutant and Ala-287 of WT is 1.6 Å, and the distance between two Arg-509 Cα carbons is ∼2 Å), the loop-500 in A287P was then stabilized by a neighboring molecule (gray mass shown in the left panel, molecule B) in the crystal packing, making loop-500 of molecule A in A287P visible in the crystal structure. It is possible that, without the crystal packing restriction in solution, Loop-500 and part of β16 can be pushed out further, so that residues in β16 in the A287P mutant make closer contact with Trp-496, Leu-497, and Lys-500 in helix αN (Gly-491–Ala-499). Thus, the mutation at Ala-287 to proline can cause a disturbance of the αN-helix, which propagates to the entire FAD domain of the mutant molecule, resulting in the unstable A287P protein. Right panel, view with ∼130° rotation from the left panel view. For clarity, the part of the neighboring molecule (molecule B) covering Loop-500 (the gray cloud represented in the left panel) is not shown in this view. In the mutant structure, the A287P mutation causes a slight bending of the N-terminal side of β7, which pushes the neighboring β16 and part of the connected Loop-500 outward. Although these changes are small, the root mean square distance between Cα carbons of A287P and WT for this part of β16 (residues Arg-509–Pro-513) is 1.9 Å (corresponding value for 603 residues of the entire molecule is 0.3 Å). Loop-500 in A287P is then stabilized by a neighboring molecule (molecule B) in the crystal packing, resulting in Loop-500 of molecule A in the A287P structure visible in the mutant structure. It is certainly possible that, in solution without the crystal packing restriction, Loop-500 and part of β16 can be pushed further to make close contact with residues Trp-496, Leu-497, and Lys-500 in helix-αN (Gly-491–Ala-499), thus separating the two subdomains, FAD-binding and NADPH-binding domains, and making the mutant protein unstable and more susceptible to proteolysis (see Figs. 6A and Fig. 7A). C, electron density maps for Loop-500. Comparison of 2Fo − Fc electron density maps contoured at 1σ level (gray mesh) corresponding to Loop-500 in the mutant (left panel) and wild type structures (right panel).
TABLE 7.
Data collection and refinement statistics
r.m.s.d. means root mean square deviation; ASU means asymmetric unit.
| Protein | Human POR A287P/P228L/A503V (PDB code 5EMN) | Wild Type Human POR (PDB code 5FA6) |
|---|---|---|
| Data collectiona | ||
| Resolution, Å | 50–2.20 (2.28–2.20) | 50–2.30 (2.38–2.30) |
| No. of measured refs. | 344,576 | 522,824 |
| No. of unique refs. | 65,852 | 57,865 |
| Completeness (%) | 97.4 (87.2) | 97.5 (89.0) |
| Redundancy | 5.3 (4.3) | 9.1 (8.7) |
| I/σ(I) | 24.8 (1.9) | 31.9 (2.7) |
| Unit cell, a, b, c, Å | 69.7, 118.1, 157.2 | 70.0, 119.3, 156.9 |
| α, β, γ, ○ | 90, 90, 90 | 90, 90, 90 |
| Space group | P212121 | P212121 |
| R_symm | 0.060 (0.610) | 0.062 (0.572) |
| No. of molecules in ASU | 2 | 2 |
| Vm | 2.28 Å3/Da | 2.31 Å3/Da |
| Refinement | ||
| Resolution range | 50–2.2 | 50–2.30 |
| Rcrystal | 0.222 | 0.225 |
| Rfree | 0.278 | 0.279 |
| r.m.s.d. bond (Å) | 0.007 | 0.008 |
| r.m.s.d. angle (o) | 1.2 | 1.3 |
| No. of protein atoms | 9618 | 9638 |
| No. of water molecules | 232 | 199 |
| B factors, Å2 | ||
| Wilson B | 41 | 46 |
| Average B, protein/solvent | 51/45 | 55/45 |
| Average B, cofactorsb | 58 | 56 |
| Ramachandran analysis, % | ||
| Most favored | 88.9 | 86.8 |
| Allowed | 10.9 | 13.0 |
| Generously allowed | 0.2 | 0.2 |
| Disallowed | 0 | 0 |
a Data were collected at SBC 19ID, Advanced Photon Source, Argonne National Laboratory. Values in the parentheses are those for the highest resolution shell.
b Cofactors include FMN, FAD, and the 2′,5′-ADP moiety of NADP+.
For the purpose of the following discussion, the conventional FAD-binding domain is subdivided into two subdomains, the NADPH-binding domain (green) and the FAD-binding domain (gold and red) (Fig. 7A). Both WT-P228L/A503V and A287P/P228L/A503V POR crystals are isomorphic, i.e. they have the same space group with the same unit cell dimensions (Table 7 and Ref. 27). In the crystal structure of WT-P228L/A503V human POR, there are two molecules in the asymmetric unit, A and B molecules. Molecule A is always a better defined structure with lower B factor values than molecule B in various human POR structures (27). In the WT-P228L/A503V structure, Loop-500 (residues Lys-500–Ala-510) is not well defined, Loop-500 of molecule A is mostly disordered, and in molecule B, only the main chain atoms can be barely traced (Fig. 7A). In fact, this is the case in other known structures of human POR, including V492E and R457H. However, in the A287P/P228L/A503V structure, Loop-500 of molecule A is clearly defined (Fig. 7, B and C, red), whereas that in molecule B is disordered.
These facts indicate the following. 1) Loop-500 is flexible, and its conformation, as observed in various POR crystal structures, is dependent on the crystal packing, and thus the loop would be even more flexible in the solution state. 2) The A287P mutation influences/alters the structure of vicinal strands, i.e. the mutation changes conformations of β7 (Phe-285–Asn-296), β16 (Leu-511–Lys-518), and αN (Gly-491–Ala-499). These subtle structural changes can propagate to other parts of the molecule, as observed in the structure of the V492E POR mutant. In the case of V492E (27), the mutation causes the long β-finger (containing Val-479–Gly-491) to become more mobile, so that intramolecular contacts in molecule B produce conformational changes leading to proteolysis and subsequent release of FAD. It is important to note that these subtle dissimilarities are seen between the WT-P228L/A503V and A287P/P228L/A503V structures, despite the fact that both contain bound NADP+, as this was the form that crystallized in both cases. It was reported above that 2′-AMP, the adenine-containing moiety of NADP+, with the requisite 2′-phosphate group, actually stabilizes the A287P/P228L/A503V structure and renders it less susceptible to trypsinolysis. In fact, attempts to crystallize the mutant protein in the absence of either NADP+ or 2′-AMP have not been successful. Even in the presence of the nucleotide, the mutant protein is unstable such that more often crystals of only the FMN domain have been obtained under the same conditions that generate crystals of the WT protein.
In the mutant structure (Fig. 7), the β-strand, in which Ala-287 is located, has a slight bend due to the mutation from alanine to proline, which pushes the neighboring strand (Leu-511–Leu-523), thus destabilizing the preceding loop (Lys-500–Ala-510, Loop-500) and rendering the Arg-509 residue more trypsin-sensitive. Furthermore, due to the tight interaction between residues in the β16-strand (Leu-511–Lys-518) and the αN-helix (Gly-491–Ala-499), the A287P mutation may further disturb the αN-helix, which would subsequently affect FAD/NADP+ binding and render Arg-487 and/or Arg-509 trypsin-sensitive. This scenario is very similar to the V492E perturbation (27), except that the degree of disorder/instability is much more subtle. In the V492E mutation, which lacks FAD, the αN-helix (Gly-491–Glu-501) is disordered, and Arg-487 in the preceding loop becomes sensitive to trypsin.
The relationship between the limited trypsin digestion patterns (Fig. 6A) and the structure of POR (Fig. 7) is important to explore. WT POR (with and without 2′-AMP) and A287P (only with 2′-AMP) exhibit similar trypsin digestion patterns. The ∼70-kDa band in the SDS-polyacrylamide gels from trypsin-digested WT enzyme must have resulted from a cleavage at residue Lys-59, confirmed by mass spectrometry analysis, which would eliminate the membrane-binding N-terminal 59 residues and yield a 70.3-kDa fragment, producing the cytosolic domain of the POR protein. Further cleavage produces two additional bands at ∼14.3 and 55.6/53.3 kDa (Fig. 6A). The lower band is below the 17-kDa marker (∼14 kDa), suggesting that Lys-558 could be the trypsin site in this case (Fig. 6A, band c). In the case of the A287P protein, in the absence of 2′-AMP, the protein is degraded more rapidly. However, the initial cleavage patterns are similar to that in the wild type protein. In addition to the accumulation of the ∼14-kDa band, however, the ∼55-kDa band disappears rapidly with the concomitant accumulation of the ∼17- and ∼20-kDa bands (Fig. 6A), consistent with the fact that Arg-487 and/or Arg-509 serve as the cleavage sites in the mutant protein, resulting in a rapid degradation of the main part of the FAD-binding domain. Mass spectrometry analysis of the Coomassie-stained SDS-polyacrylamide gel bands in the 60–75- and 14–20-kDa regions positively identified Lys-59, Arg-487, and Arg-509 as sites cleaved in our limited trypsinolysis experiments. This cleavage process, i.e. the earlier fragmentation of the FAD domain in the mutant protein as compared with the WT, is consistent with the fact that crystals of the FMN-binding domain were more frequently obtained during our crystallization trials with A287P/P228L/A503V but not with WT-P228L/A503V.
It is not surprising that both wild type and A287P proteins are more resistant to trypsin digestion in the presence of 2′-AMP. Note that NADPH binds at the interface between the two subdomains, i.e. between the green and gold/red subdomains (Fig. 7A). In the absence of 2′-AMP (or NADP(H)), this region of the polypeptide may be less ordered in A287P/P228L/A503V, thus rendering the two subdomains more easily separable and allowing the polypeptide to be more easily degraded by trypsin. Furthermore, it has been shown that the POR molecule adopts two conformations, open and closed forms (33, 34), and it has been suggested that binding NADP(H)/2′-AMP favors the closed form (33–35). In the absence or diminished occupancy of the nucleotide, the POR molecule would more likely be in an open state, which would be more susceptible to trypsinolysis. Therefore, the in vivo consequences would depend on the exposure of either a WT or A287P protein to a potentially proteolytic environment, the latter variant being the more susceptible to degradation.
Differential Stability of Stably Expressed WT and A287P in Mammalian Osteoblast MC3T3 Cells
Although the frequency of the A287P human mutation in the human population has been reported to be highest among Caucasians and is associated with sexual dimorphisms as well as bone defects (14), A287P can be purified to homogeneity with fully complemented flavins, both FAD and FMN. Furthermore, the measured CYP activities mediated by A287P are not significantly different from WT as to explain the ambiguous external genitalia or the defects in bone development. Our data indicate that A287P protein may be less stable than WT, particularly when elevated to human body temperature. Thus, the observed sexual dimorphisms and bone phenotype in humans harboring the A287P mutation can be explained if the A287P protein is less stable within the cellular milieu than WT, thus creating POR deficiency. To examine whether differential stabilities exist for WT versus A287P under more physiological conditions, MC3T3 E1 C4 osteoblast cells were stably transfected with plasmids (pIRES eGFP2) carrying either WT or A287P cDNAs. The persistence of A287P protein was compared with that of WT protein after cells were treated with cycloheximide (3 μg/ml), a protein synthesis inhibitor, for 12 h. Cell lysates from cycloheximide- and vehicle-treated cells overexpressing WT and A287P proteins were examined by immunoblot analysis (Fig. 8). Densitometric analysis of these immunoblots revealed that cycloheximide-treated cells overexpressing A287P had 38 ± 2% (n = 2) less POR protein compared with vehicle-treated cells, whereas no significant difference was noticed between cycloheximide- and vehicle-treated WT-overexpressing cells. This suggests that the stability of A287P is likely to be compromised in the mammalian cellular milieu compared with WT POR.
FIGURE 8.

Effect of cycloheximide on expression of WT POR or A287P in MC3T3 cells. Stably transfected MC3T3 cells overexpressing either WT or A287P POR were treated with 3 μg per ml of cycloheximide, a protein synthesis inhibitor, for 12 h. Left, level of WT or A287P POR upon treatment with cycloheximide (+) was compared with cells treated with vehicle (−) by immunoblotting. Equal amounts of whole cell lysate (15 μg) were loaded in each lane, and β-actin was used as the loading control. Experiment was repeated twice and showed the same result; figure shown is representative. Right, densitometric analysis of immunoblot: % expression of WT POR and A287P POR in 2T3 stable cells treated with cycloheximide (solid bars). Expression level is normalized against untreated cells (empty bars). Values are mean ± S.D. (n = 2).
Discussion
The frequency of the A287P POR mutation in human subjects with PORD has been reported to be highest among Caucasians (28%) and is associated with both disordered sex development as well as bone defects (14), both of which are devastating conditions. If the phenotype could be prevented at an early stage of development by the addition of riboflavin in utero, a treatment that has been successful in treating short-chain acyl-CoA dehydrogenase deficiency (36, 37), it would be a boon to the treatment of POR deficiency. At this juncture, the only human mutations that have been shown to be reversible at the molecular level, when examined by addressing the purified, homogeneous enzymes with biophysical techniques, have been those exhibiting flavin deficiencies (22, 25, 26, 38). However, it has become apparent from this work that certain missense mutations could lead to conformational alterations resulting in unstable or more readily degradable proteins, thereby leading to diminished activity, but not necessarily directly affecting flavin or NADPH binding. This applies in the present case of A287P, which in this report is identified as a temperature-sensitive mutant when in a cellular milieu, even at levels elevated only to body temperature at 37 °C. After purification and removal from the cellular environment, this enzyme was stable, as all activity measurements performed in vitro at 37 °C were very similar to those of the WT protein. In this case, the absence of proteases and the presence of substrate and redox partners probably afford protection of POR.
With our full-length preparations, any effect of the A287P point mutation on the protein structure did not result in decreased flavin content or weakened NADPH binding in the purified full-length enzyme preparations. The FAD content of A287P, in particular, does not correspond to that previously reported (Table 2) (24). Similarly, the measured Km values for NADPH for WT and A287P were very similar, as was the Ki value for NADP+ (Table 3). Also, measurements of enzyme activities with the three steroidogenic cytochrome P450 partners CYP17A1, CYP19A1, and CYP21A2 did not show differences between A287P and WT POR preparations (Fig. 2, A and B). Similarly, measurements of enzyme activities with the xenobiotic CYPs revealed no differences between POR WT and A287P (Figs. 2C and 3 and Table 5). Because we saw little to no differences between POR WT and A287P in any measurement of catalytic activity, whether with electron acceptors to measure the catalytic efficiency of the flavoprotein or with CYPs as redox partners, we concluded that the full-length purified POR A287P protein was a fully competent enzyme, able to interact with and transfer electrons to the measured CYPs.
Although the A287P enzyme is functional, difficulties with its initial purification led us to question its stability, particularly when exposed to more physiologically relevant parameters. To directly examine the stability of the WT and A287P proteins, DSC experiments were performed to determine melting temperatures (Fig. 5). As predicted, the main peak of the thermogram for the A287P protein is about 5 °C cooler than WT, indicating its lower thermal stability. In addition, the A287P thermogram is very complex compared with WT, exhibiting multiple peaks. Although these thermograms cannot by themselves elucidate the mechanism of unfolding of these proteins, it is interesting to speculate about several different models that might potentially explain the presence of multiple peaks. There may be two or more unfolding domains in the proteins, e.g. the FMN-binding and FAD/NADPH-binding domains, with different temperature stabilities, particularly in A287P protein. Because the crystallization experiments indicate that the FMN domain of the mutant protein appears to be more stable than that of the FAD/NADPH domain, the first peak (Tm = 47 °C) of the A287P thermogram may correspond to the unfolding of the FAD/NADPH domain, although the second peak with a similar melting point to that of the WT (Tm = 52 °C) may reflect the unfolding of the FMN domain.
An alternative speculation is that the POR proteins exist in solution as populations of conformers in equilibrium, with some being more stable than others. The majority of WT POR then spends the most time in a conformer with a Tm of ∼52 °C, although A287P preferentially adopts at least two main conformers, the majority of molecules being in a less stable conformation than that of WT. That A287P might adopt more relaxed conformations in solution compared with WT would explain its greater sensitivity to trypsinolysis and to in vivo proteolysis and is supported by crystal structure data that show regions in A287P that are inherently less ordered than in WT.
To probe potential temperature-dependent conformational differences between WT and A287P, we utilized limited trypsinolysis. By comparing trypsinolysis at 30 °C versus 37 °C, we determined that sensitivity of POR to trypsin was modulated by temperature, more so for A287P than WT. Time-dependent studies revealed that the rate of proteolysis was greater with A287P than WT at either temperature (Fig. 6A). The results show dramatically that the WT preparation is much more resistant to degradation at the higher temperature than the A287P variant. Similarly, other attempts to stabilize A287P by the addition of 2′-AMP and incorporation into DLPC vesicles led to decreased susceptibility to trypsin, similar to that of WT (Fig. 6, B and C), demonstrating differences in flexibility of the preparations that could be altered by the addition of factors that may stabilize the enzyme structure.
Differences in stability between WT and A287P were also probed in more physiologically relevant systems, i.e. intracellularly. In the BTC bacterial system, the A287P variant demonstrated a substantially lower expression level when expressed at 32 °C versus 28 °C (Fig. 4), suggesting a decreased protein stability compared with WT. Furthermore, overexpression of both WT and A287P constructs in mammalian osteoblast MC3T3 cells, treated with cycloheximide, showed that less A287P protein was detected than WT protein (Fig. 8), so that in the mammalian cellular environment, the polymorphic variant is less stable.
This indicated to us that the human variant A287P was unstable in the cellular environment, even at the slightly elevated temperature of 32 °C in the bacterial expression system. Under these circumstances, conformational differences would make this mutation more susceptible to protein degradation in the cell, ultimately leading to release of flavins from the enzyme. This occurrence could explain the results obtained by Jin et al. (24) in which the conclusion is drawn that the A287P mutation affects flavin binding per se. Other potential explanations of the differences between the Jin et al. report (24) and the data herein could relate to the enzyme preparations used; those used in this study were full-length, unmodified, and purified to a high degree of homogeneity (Fig. 1).
Based on our data, the human phenotype of A287P can be explained by POR having a structural defect resulting in a semi-stable conformational state that increases proteolytic susceptibility, leading to loss of function of this protein. This loss could potentially result in disruption of cellular POR-protein homeostasis, causing the failure of a multitude of critical cellular processes that are dependent upon CYP metabolites.
Because the crystal structures have been obtained with preparations containing both FAD and FMN flavins (without any addition of exogenous flavins) and NADP+, these structures are minimally disrupted with respect to their binding sites, but subtle differences are nevertheless apparent compared with the wild type structure. It is clear from these subtle deviations that the potential trypsin-sensitive sites become more exposed (Fig. 7), and the SDS gels in Fig. 6A reveal the estimated cleaved fragment sizes expected from trypsinolysis at those residues. Therefore, it is reasonable to conclude from these structural comparisons that A287P can behave normally when not exposed to a hostile environment. Also, although loss of flavin can occur under these less than optimal conditions, the A287P variant does not have an inherently defective binding capacity for its prosthetic flavins, but instead, it is unstable at even normal human body temperature and is susceptible to degradation under these circumstances when in a cellular environment.
These studies provide ample evidence for the continued careful examination of human mutations in POR to determine possible therapeutic approaches that could revert the deficiencies displayed in the human population. In the case of A287P, mere supplementation of the polymorphic variant with riboflavin in vivo would not be sufficient, because its native instability will not have been addressed. This new paradigm in examining the many and varied mutations in human POR should be kept in mind when therapeutically addressing these devastating genetic deficiencies.
Experimental Procedures
Plasmids
The expression plasmids for CYPs 17A1 and 21A2 were generous gifts obtained from the following investigators: modified human (Gly3His6)-CYP17A1 in pCWori+ from Professor Walter L. Miller (University of California, San Francisco); human CYP21A2 in pET-17b was from Professor Michael R. Waterman (Vanderbilt University, Nashville, TN), which was modified as described (39), i.e. the N terminus was truncated and residues altered to reflect the CYP 2C3 N-terminal region, and the C terminus was His6-tagged.
POR Protein Expression and Purification
Full-length human POR was expressed as a recombinant OmpA3 fusion protein in the pCWori plasmid in Overexpress C41(DE3) (Lucigen) E. coli. A colony was selected from freshly transformed C41 cells and grown in stages, starting with 10 ml of LB + ampicillin (100 μg/ml) at 37 °C for 8 h, which was added to 250 ml of LB + ampicillin (100 μg/ml) at 37 °C overnight. An aliquot (25 ml) of this culture was added to a final volume of 1 liter of TB + ampicillin (100 μg/ml) and then grown at a reduced temperature (24 °C) until the A600 = 1.6. Protein expression was induced with 65 μm IPTG, and cultures were supplemented with ∼2 mg of riboflavin and then grown at 23 °C for 48 h and harvested.
Cell harvest and lysis were as described previously (25) except that the detergent concentration was increased from 0.1 to 0.5% Triton X-100, and the extraction volume ratio was 0.2 mg of original pellet weight/1 ml of 0.5% Triton X-100 extraction buffer to facilitate the extraction of the A287P protein. The extracted protein was affinity-purified on a 2′,5′-ADP-Sepharose 4B (GE Healthcare) column as described previously (40), with the following modifications. Bound protein was washed in buffer with 0.1% Triton X-100 followed by an additional wash with buffer containing 5 mm adenosine. POR was eluted with a gradient of 2′-AMP (0–5 mm) in buffer with 0.1% Triton X-100.
Fractions were analyzed by SDS-PAGE, and those with a single band at 77 kDa were pooled and concentrated. The semiquinone was oxidized by titrating 2 mm potassium ferricyanide into the concentrated sample until the absorbance of the semiquinone peak at 600 nm reached baseline absorbance. The oxidized protein was dialyzed in buffer (50 mm Tris-HCl, pH 7.7, 0.1 mm EDTA, 0.05 mm DTT, 10% glycerol + 0.1% Triton X-100) to remove excess 2′-AMP and potassium ferricyanide. Final protein concentration was determined by both the microbicinchoninic acid (BCA) protein assay (Pierce), according to standard protocol, and spectral analysis of oxidized flavin absorbance at 454 nm, ϵ = 21.4 mm−1, accounting for 2 flavins per mol of protein.
The wild type and A287P mutant proteins used for the crystal structure analyses were the soluble proteins, i.e. the N-terminal 66-residue membrane-binding domain was deleted (Δ66-WT, Δ66-WT-P228L/A503V, and Δ66-A287P/P228L/A503V). All three proteins were expressed and purified as other soluble human POR proteins described previously (27). Our initial “WT” POR clone purchased from ATCC contained two common polymorphic variations, P228L and A503V. Therefore, the previously published WT structure (PDB code 3QE2) and Δ66 mutants contained these polymorphisms. We altered our expression plasmids for the full-length WT and POR variants to the residues considered to be WT, i.e. Pro-228 and Ala-503. All experiments other than crystallography were performed with full-length WT and A287P POR containing Pro at 228 and Ala at 503.
CYP19A1 (Aromatase) Purification
Aromatase was purified from human placenta to homogeneity by immunoaffinity chromatography using a well established procedure (41, 42) under the Institutional Review Board protocol 2011.1704, Crouse Hospital, Syracuse, NY.
CYP17A1 and CYP21A2 Protein Expression and Purification
Modified human (Gly3His6)-CYP17A1 was expressed in E. coli JM109 cells and purified as described (43, 44). Modified human CYP21A2 was expressed in E. coli BL21(DE3) cells and purified as described (45). GroEL/ES chaperones (pGro7 plasmid) were co-expressed with the P450s to increase expression of active enzyme.
Flavin Content Analysis
The analysis of POR flavin content was as described (26), with minor differences. A 50 × 4.6-mm Kinetex XB-C18 column from Phenomenex with an HPLC Krudkatcher ultra column in-line filter was used. The flow rate was 0.6 ml/min, and flavin peaks were monitored at 446 nm for FMN and 450 nm for FAD. Flavin concentrations were calculated by comparing respective peak areas to standard curves generated by known concentrations of FAD (ϵ450 nm = 11.3 mm−1) and FMN (ϵ446 nm = 12.2 mm−1), confirmed spectrophotometrically by their given extinction coefficients. Integration and analysis were performed using Agilent Technologies ChemStation software (revision B.03.02).
Limited Trypsinolysis
For each condition, 30 μg of purified full-length POR was incubated at 37 °C with trypsin (Sigma T1426) at a protein/trypsin molar ratio of 800:1, in 50 mm Tris-HCl, pH 7.7, 0.1 mm EDTA, 10% glycerol, 0.05 mm DTT, 0.1% Triton X-100, 20 mm CaCl2. Variations in conditions included the addition of 5 mm 2′-AMP or 60 μg of DLPC (0.4 μg/μl, in the form of 100 nm extruded vesicles (46)) in separate experiments. At indicated times, aliquots (150 μl) were removed, and the reaction was terminated by addition of soybean trypsin inhibitor followed by Laemmli sample buffer and boiling for 5 min. Samples (2 μg) were loaded for SDS-PAGE analysis on Bio-Rad TGX 4–15% polyacrylamide gels.
Mass Spectrometry Analysis
The mixture generated by limited trypsinolysis was separated by one-dimensional SDS-PAGE; the visible bands were excised, and proteins were digested in situ with chymotrypsin. The digests were analyzed by capillary HPLC-electrospray ionization-tandem mass spectrometry on an Orbitrap Velos Pro (ThermoFisher). Mascot (Matrix Science) was used for database searching of the human subset of the SwissProt database. Scaffold (Proteome Software) was then employed to conduct subset searches of the Mascot results with X! Tandem and to determine probabilities of peptide assignments and protein identifications. Experiments were performed by the Institutional Mass Spectrometry Core Laboratory, University of Texas Health Science Center, San Antonio, directed by Dr. Susan Weintraub.
DSC
WT or A287P proteins (0.5 mg/ml in 100 mm potassium phosphate, pH 7.7, 0.1% Triton X-100, 0.05 mm DTT, and 10% glycerol) were analyzed using a VP-DSC Microcalorimeter (Microcal, Northampton, MA) at the Center for Macromolecular Interactions, University of Texas Health Science Center, San Antonio. Samples were degassed prior to DSC measurement, and buffer was used as the reference sample to obtain the baseline. Measurements were performed at a scan rate of 1 °C/min from 13 to 90 °C for WT protein and 17–75 °C for A287P protein. The final thermogram was processed by subtracting the baseline thermogram and evaluated using the Microcal LLC DSC plugin for the Origin 7.0 software package provided with the equipment. Denaturation of POR proteins is not a reversible process, as expected for a large multidomain protein such as POR, so refolding thermograms were not generated.
Crystallization, Data Collection, and Structure Determination
Crystals of both WT and A287P Δ66 proteins were grown using the hanging drop method (47) and micro-seeding techniques (48). The initial seed crystals were obtained with the Δ66-WT protein, using the original WT clone obtained from ATCC, that had two common polymorphisms, i.e. P228L and A503V as described previously (27); this will be referred to as POR Δ66-WT-P228L/A503V. Using these seed crystals, crystals for both the Δ66-WT (without the P228L and A503V mutations) and of Δ66-A287P/P228L/A503V were obtained following an iterative seeding process (27). Briefly, purified Δ66-WT or Δ66-A287P/P228L/A503V protein was dialyzed in 50 mm MES buffer at pH 6.5 overnight. The dialyzed protein solution was concentrated to ∼12 mg/ml, and 1 mm NADP+ was added prior to the crystallization setup. For micro-seeding, 5 μl of the POR protein was mixed with 2 μl of reservoir solution (100 mm MES, pH 6.5, 14% PEG 4K, 50 mm CaAc2, and 100 mm NaCl) and equilibrated with the reservoir solution overnight at 19 °C. Then the crystallization drops were seeded with microcrystals of the human POR Δ66-WT-P228L/A503V crystals. This process was repeated, except that, in the second rounds of seeding, the seed crystals were those obtained from the first seeding, i.e. micro crystals of the mutant (Δ66-A287P/P228L/A503V) or the Δ66-WT. Diffraction-quality crystals were obtained several days after the second seeding. However, unlike other POR proteins, Δ66-A287P/P228L/A503V often resulted in the crystals of only the FMN domain of the POR protein in some of the crystallization dips, suggesting that the mutant protein was less stable even under the same crystallization conditions.
All diffraction data sets were collected at the SBC 19ID beam line, Advanced Photon Source, Argonne National Laboratory. The data were processed using HKL 2000 (49). Both the true wild type and the mutant crystals belong to the P212121 space group with two molecules in an asymmetric unit. The structure was solved by molecular replacement methods using MOLREP (50) in the CCP4 package (51) using the human POR Δ66-WT-P228L/A503V structure (PDB code 3QE2 (27)) as the search model. For the Δ66-WT structure, the difference Fourier method was used, because 3QE2 and the new WT crystals were isomorphic. A round of rigid body minimization, position minimization, simulated annealing at 3000 K, and individual isotropic B factor refinement was performed using CNS (52) with all cofactors omitted. Using the resulting electron density maps, cofactors (FAD, FMN, and NADP+) were added, and further refinements were performed with iterative rounds of model rebuilding with COOT (53) and refinement with CNS (52). The final Rwork and Rfree values were 22.2 and 27.8%, respectively, for the mutant structure. The corresponding R values for the WT structure were 22.5 and 27.9%. The data collection and refinement statistics are summarized in Table 1. All biochemical studies other than the crystallography were performed using the full-length WT or A287P proteins without the two additional mutations, P228L and A503V.
Rapid Kinetic Measurements
Stopped-flow experiments were performed aerobically at 25 °C in 50 mm Tris-HCl, pH 7.7, 0.1 mm EDTA, 0.05 mm DTT, 10% glycerol, and 0.1% Triton X-100, using an Applied Photophysics SX.18MV diode array stopped-flow spectrophotometer (Leatherhead, UK), with 1 μm (by BCA) oxidized, dialyzed, and full-length human POR and 500 μm NADPH (ϵ340 nm = 6.22 mm−1 cm−1). Reduction of POR was measured as a change in absorbance at 450 nm for 2 s. Each measurement was repeated ≥3 times using two independent protein preparations. The measured kinetics traces were fitted to a double exponential equation using the supplied SX.18MV instrument software.
Cytochrome c Reductase Assay
Cytochrome c reduction was assayed as described previously (25) using a Shimadzu UV-visible 2401 PC spectrophotometer.
Metabolism of BFC
Metabolism of BFC (Sigma 368512) to HFC (54, 55) was monitored in mouse microsomes isolated from mice with liver-specific deletion of POR (8). In this system, the endogenous POR background is eliminated, and a full complement of CYPs is present, making it ideal for monitoring POR activity in a physiological system. The reaction mixture, consisting of buffer (100 mm KPi, pH 7.4, 2.5 μm MgCl2), 50 μm BFC, 200 nm total P450 in POR-deleted mouse liver microsomes, and 30 nm POR, was preincubated 1 h at 37 °C, with mixing. Aliquots (190 μl) of reaction mixture were added to a pre-warmed black 96-well microtiter plate. Reaction was initiated with 10 μl of NADPH and an NADPH-regenerating system (final concentrations are 1 mm NADPH, 10 mm isocitrate, 183 milliunits of isocitrate dehydrogenase), giving a final volume of 200 μl/reaction. Samples were assayed in a TECAN Infinite M200 microplate reader. This real time kinetic assay gives linear rates with time and appropriate protein concentrations.
CYP19A1 (Aromatase) Activity Measurement
The specific activity of purified human placental aromatase in the presence of human full-length WT and A287P POR was determined by the release of tritiated water following the [1β-3H,4-14C]androstenedione-to-estrone aromatization reaction (56). Aromatase (400 nm) and POR (8 μm) were reconstituted in phospholipids and assayed in quadruplicate. The enzyme activity was adjusted by subtraction of background activity in the absence of NADPH. Any inconsistent data were re-measured, and outliers were identified as data two standard deviations from the mean.
CYP17A1 and CYP21A2 Activity Measurement
Either purified CYP17A1 or CYP21A2 (10 pmol) was reconstituted with 1 molar eq of full-length POR using a 73.5:24.5:2 mixture of phosphatidylcholine, phosphatidylethanolamine, and phosphatidylinositol in 2–3 μl. The contents were gently swirled and set at room temperature for 5 min. The mixture was diluted to 0.2 ml with 50 mm KPi buffer, pH 7.4, containing 20 μm progesterone. The resulting mixture was mixed gently and set at 37 °C for 1 min. NADPH (1 mm) was added, and the incubation was continued at 37 °C for 15 min. The products were extracted with 1 ml of dichloromethane, and the organic phase was collected and dried under a nitrogen flow. Steroids were analyzed using the Agilent 1260 Infinity HPLC system with UV detector set to 254 nm using Laura4 software (LabLogic, Tampa, FL). Steroids were dissolved in 20 μl of methanol, and 7-μl injections were resolved with a 50 × 2.1 mm, 2.6-μm particle size C8 Kinetex column (Phenomenex, Torrence, CA) equipped with a guard column at a flow of 0.4 ml/min. Aqueous methanol linear gradients were employed, 27% methanol from 0 to 0.5 min, jumped to 39% methanol, and gradient from 39 to 75% methanol over 30 min. Products were identified by retention times of external standards chromatographed at the beginning of the experiment. Data are analyzed as described previously (44). Percent product is calculated by product/(substrate + product), which is multiplied by the starting concentration to determine amount.
Reconstitution of CYP1A2 Activity in Bacterial Cells with WT and A287P Constructs
The E. coli cell model BTC was used for the co-expression of a human CYP variant with POR, using a biplasmid co-expression system, as described previously (57). The biplasmid system in BTC strains allows the expression of full-length proteins, which are correctly inserted in the membrane with a stoichiometry that reflects that found in vivo (57). POR A287P was co-expressed with human CYP1A2 to determine its capacity in sustaining CYP1A2-mediated activity. The effect of culturing temperature was examined with both WT and A287P protein to determine relative stability in these cells. The POR WT and A287P variant were also expressed in the BTC bacteria without human CYPs (strains BTC_PORWT and BTC_PORAP; see Table 2) and grown at either 28 or 32 °C. (The CYPs in general are not expressed well at higher temperatures.) Culture conditions, membrane preparation, protein content determination, cytochrome c reduction, POR immunodetection, methoxyresorufin-O-dealkylation assay, and the mutagenicity assay were performed as described previously (58).
Generation of MC3T3 Cells That Stably Overexpress POR
MC3T3 cells were cultured in α-minimal essential media, 10% FBS, plus penicillin and streptomycin at 37 °C, and 5% CO2. POR WT and POR A287P cDNAs were amplified from the pCW bacterial expression system for POR WT and A287P, respectively, using the following primers: forward, TAT CGG CGA TCT CGA GAT GAT CAA CAT GGG AGA CTC CCA, and reverse, CCG ATC GCG ATG GAT CCC TAG CTC CAC ACG TCC AGG.
Cloning sites for XhoI and BamHI were included, which permitted cloning into the mammalian expression vector, pIRES2 eGFP (Clontech). 3T3 cells were plated overnight and then transfected with POR WT or POR A287P plasmid using Lipofectamine 2000 (Thermo Fisher Scientific, Waltham, MA) following the vendor-supplied protocol. Overexpression of POR was confirmed by immunoblot analysis. Stable cell lines expressing POR WT and POR A287P were created by using G418 (400 μg/ml) as a selection marker.
Cycloheximide Treatment of MC3T3 Cells
MC3T3 cells stably transfected with either WT and A287P POR, plated at a density of 20,000 cells/cm2, were treated with the protein synthesis inhibitor cycloheximide (3 μg/ml) for 12 h at 37 °C. Whole cell lysates were collected from untreated and treated WT- and A287P-overexpressing cells in 2× Laemmli sample buffer with benzonase and protease inhibitor mixture (Thermo Scientific, Waltham, MA). Samples were boiled for 5 min, and lysates (15 μg of protein, measured by BCA) were subjected to SDS-PAGE using 4–15% Tris-glycine polyacrylamide gels (Bio-Rad). Proteins were transferred to PVDF membranes and probed with the following: 1°Ab (Santa Cruz Biotechnology catalog no. 25270 mouse α-human POR (1:1000)); 2°Ab (Santa Cruz Biotechnology catalog no. 2031 goat α-mouse HRP (1:5000)); or 1°Ab (Sigma catalog no. A5441 mouse α-actin (1:5000)); 2°Ab (Santa Cruz Biotechnology catalog no. 2031 goat α-mouse HRP (1:10,000). Immobilon Western chemiluminescent substrate (Millipore, Billerica, MA) was used to detect antigen-antibody complexes on the membrane.
Author Contributions
K. M. M. and S. P. P. designed the protein purification, performed the biophysical characterizations, and contributed to the design and analysis of experiments. C. X. and J. J. P. K. crystallized the proteins, solved the structures, and analyzed the likely sites of trypsin digestion. M. K. designed the experimental approaches and together with D. M. performed experiments and analyzed the data using the BTC expression system. R. J. A. performed experiments and analyzed the data for CYP17A1 and CYP21A2 metabolism. E. M. L. supervised and aided in design and interpretation of the DSC experiments. D. G. supervised experiments and analyzed the data for CYP19A1 metabolism. P. M. aided in design and analysis of experiments. R. K. performed and analyzed the mammalian cell culture experiments. L. J. R., J. J. P. K., and B. S. M. aided in conception, design, and analysis of experiments and wrote the paper. All authors contributed to the preparation of figures, reviewed the results, and approved the final version of the manuscript.
Acknowledgments
We thank the staff at the Advanced Photon Source beamlines SBC 19ID for their assistance in data collection and Jessica Lo and Egbuta Chinaza for performing the aromatase assays. We gratefully acknowledge the support of the University of Texas Health Science Center at San Antonio Institutional Mass Spectrometry Core Laboratory and Center for Macromolecular Interactions, both of which are supported in part by the Cancer Therapy and Research Center, National Institutes of Health, NCI P30 Award CA054174, and by Texas State funds provided through the University of Texas Health Science Center at San Antonio Office of the Vice President for Research. The Mass Spectrometry Laboratory is also supported in part by National Institutes of Health Shared Instrumentation Grant S10RR025111 (to S.T. Weintraub), and the expert technical assistance of Sammy Pardo is gratefully acknowledged. We also thank Dr. Verna Frasca of Malvern Instruments for invaluable advice and guidance, and Liping Wang for technical expertise in performing the DSC experiments. Thanks are also due to Dr. Neal C. Robinson for the careful reading and sage advice regarding interpretation of the DSC experiments.
This work was supported in part by National Institutes of Health Grants GM081568 (to L. J. R. and M. K.), and GM097031 (to J. J. K.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
The atomic coordinates and structure factors (codes 5EMN and 5FA6) have been deposited in the Protein Data Bank (http://wwpdb.org/).
K. M. McCammon, C. C. Marohnic, S. P. Panda, B. S. Masters, and L. J. Roman, unpublished data.
- POR
- P450 oxidoreductase
- DSC
- differential scanning calorimetry
- CYP
- cytochrome P450
- PDB
- Protein Data Bank
- Ab
- antibody
- HFC
- 7-hydroxy-4-trifluoromethylcoumarin
- BFC
- 7-benzoyloxy-trifluoromethylcoumarin
- PORD
- POR deficiency
- ABS
- Antley-Bixler syndrome
- DLPC
- dilauroylphosphatidylcholine
- 2AA
- 2-amino-anthracene
- NNK
- 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone
- IQ
- 2-amino-3-methylimidazo(4,5-f)quinoline
- IPTG
- isopropyl 1-thio-β-d-galactopyranoside.
References
- 1. Pandey A. V., and Flück C. E. (2013) NADPH P450 oxidoreductase: structure, function, and pathology of diseases. Pharmacol. Ther. 138, 229–254 [DOI] [PubMed] [Google Scholar]
- 2. Riddick D. S., Ding X., Wolf C. R., Porter T. D., Pandey A. V., Zhang Q.-Y., Gu J., Finn R. D., Ronseaux S., McLaughlin L. A., Henderson C. J., Zou L., and Flück C. E. (2013) NADPH-cytochrome P450 oxidoreductase: roles in physiology, pharmacology, and toxicology. Drug Metab. Dispos. 41, 12–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Schacter B. A., Nelson E. B., Marver H. S., and Masters B. S. (1972) Immunochemical evidence for an association of heme oxygenase with the microsomal electron transport system. J. Biol. Chem. 247, 3601–3607 [PubMed] [Google Scholar]
- 4. Ono T., and Bloch K. (1975) Solubilization and partial characterization of rat liver squalene epoxidase. J. Biol. Chem. 250, 1571–1579 [PubMed] [Google Scholar]
- 5. Ilan Z., Ilan R., and Cinti D. L. (1981) Evidence for a new physiological role of hepatic NADPH:ferricytochrome (P-450) oxidoreductase. Direct electron input to the microsomal fatty acid chain elongation system. J. Biol. Chem. 256, 10066–10072 [PubMed] [Google Scholar]
- 6. Shen A. L., O'Leary K. A., and Kasper C. B. (2002) Association of multiple developmental defects and embryonic lethality with loss of microsomal NADPH-cytochrome P450 oxidoreductase. J. Biol. Chem. 277, 6536–6541 [DOI] [PubMed] [Google Scholar]
- 7. Henderson C. J., Otto D. M., Carrie D., Magnuson M. A., McLaren A. W., Rosewell I., and Wolf C. R. (2003) Inactivation of the hepatic cytochrome P450 system by conditional deletion of hepatic cytochrome P450 reductase. J. Biol. Chem. 278, 13480–13486 [DOI] [PubMed] [Google Scholar]
- 8. Gu J., Weng Y., Zhang Q.-Y., Cui H., Behr M., Wu L., Yang W., Zhang L., and Ding X. (2003) Liver-specific deletion of the NADPH-cytochrome P450 reductase gene: impact on plasma cholesterol homeostasis and the function and regulation of microsomal cytochrome P450 and heme oxygenase. J. Biol. Chem. 278, 25895–25901 [DOI] [PubMed] [Google Scholar]
- 9. Flück C. E., Tajima T., Pandey A. V., Arlt W., Okuhara K., Verge C. F., Jabs E. W., Mendonça B. B., Fujieda K., and Miller W. L. (2004) Mutant P450 oxidoreductase causes disordered steroidogenesis with and without Antley-Bixler syndrome. Nat. Genet. 36, 228–230 [DOI] [PubMed] [Google Scholar]
- 10. Arlt W., Walker E. A., Draper N., Ivison H. E., Ride J. P., Hammer F., Chalder S. M., Borucka-Mankiewicz M., Hauffa B. P., Malunowicz E. M., Stewart P. M., and Shackleton C. H. (2004) Congenital adrenal hyperplasia caused by mutant P450 oxidoreductase and human androgen synthesis: analytical study. Lancet 363, 2128–2135 [DOI] [PubMed] [Google Scholar]
- 11. Panda S. P., Guntur A. R., Polusani S. R., Fajardo R. J., Gakunga P. T., Roman L. J., and Masters B. S. (2013) Conditional deletion of cytochrome p450 reductase in osteoprogenitor cells affects long bone and skull development in mice recapitulating Antley-Bixler syndrome: role of a redox enzyme in development. PLoS ONE 8, e75638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Wang M., Roberts D. L., Paschke R., Shea T. M., Masters B. S., and Kim J. J. (1997) Three-dimensional structure of NADPH-cytochrome P450 reductase: prototype for FMN- and FAD-containing enzymes. Proc. Natl. Acad. Sci. U.S.A. 94, 8411–8416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Adachi M., Tachibana K., Asakura Y., Yamamoto T., Hanaki K., and Oka A. (2004) Compound heterozygous mutations of cytochrome P450 oxidoreductase gene (POR) in two patients with Antley-Bixler syndrome. Am. J. Med. Genet. A 128, 333–339 [DOI] [PubMed] [Google Scholar]
- 14. Huang N., Pandey A. V., Agrawal V., Reardon W., Lapunzina P. D., Mowat D., Jabs E. W., Van Vliet G., Sack J., Flück C. E., and Miller W. L. (2005) Diversity and function of mutations in p450 oxidoreductase in patients with Antley-Bixler syndrome and disordered steroidogenesis (see comment.). Am. J. Hum. Genet. 76, 729–749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Fukami M., Horikawa R., Nagai T., Tanaka T., Naiki Y., Sato N., Okuyama T., Nakai H., Soneda S., Tachibana K., Matsuo N., Sato S., Homma K., Nishimura G., Hasegawa T., and Ogata T. (2005) Cytochrome P450 oxidoreductase gene mutations and Antley-Bixler syndrome with abnormal genitalia and/or impaired steroidogenesis: molecular and clinical studies in 10 patients. J. Clin. Endocrinol. Metab. 90, 414–426 [DOI] [PubMed] [Google Scholar]
- 16. Adachi M., Asakura Y., Matsuo M., Yamamoto T., Hanaki K., and Arlt W. (2006) POR R457H is a global founder mutation causing Antley-Bixler syndrome with autosomal recessive trait. Am. J. Med. Genet. Part A 140, 633–635 [DOI] [PubMed] [Google Scholar]
- 17. Huang N., Agrawal V., Giacomini K. M., and Miller W. L. (2008) Genetics of P450 oxidoreductase: sequence variation in 842 individuals of four ethnicities and activities of 15 missense mutations. Proc. Natl. Acad. Sci. U.S.A. 105, 1733–1738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Flück C. E., Mullis P. E., and Pandey A. V. (2010) Reduction in hepatic drug metabolizing CYP3A4 activities caused by P450 oxidoreductase mutations identified in patients with disordered steroid metabolism. Biochem. Biophys. Res. Commun. 401, 149–153 [DOI] [PubMed] [Google Scholar]
- 19. Tomková M., Marohnic C. C., Gurwitz D., Seda O., Masters B. S., and Martásek P. (2012) Identification of six novel P450 oxidoreductase missense variants in Ashkenazi and Moroccan Jewish populations. Pharmacogenomics 13, 543–554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Tomková M., Panda S. P., Šeda O., Baxová A., Hůlková M., Siler Masters B. S., and Martásek P. (2015) Genetic variations in NADPH-CYP450 oxidoreductase in a Czech Slavic cohort. Pharmacogenomics 16, 205–215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Krone N., Reisch N., Idkowiak J., Dhir V., Ivison H. E., Hughes B. A., Rose I. T., O'Neil D. M., Vijzelaar R., Smith M. J., MacDonald F., Cole T. R., Adolphs N., Barton J. S., Blair E. M., et al. (2012) Genotype-phenotype analysis in congenital adrenal hyperplasia due to P450 oxidoreductase deficiency. J. Clin. Endocrinol. Metab. 97, E257–E267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Marohnic C. C., Huber Iii W. J., Patrick Connick J., Reed J. R., McCammon K., Panda S. P., Martásek P., Backes W. L., and Masters B. S. (2011) Mutations of human cytochrome P450 reductase differentially modulate heme oxygenase-1 activity and oligomerization. Arch. Biochem. Biophys. 513, 42–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Moutinho D., Marohnic C. C., Panda S. P., Rueff J., Masters B. S., and Kranendonk M. (2012) Altered human CYP3A4 activity caused by Antley-Bixler syndrome-related variants of NADPH-cytochrome P450 oxidoreductase measured in a robust in vitro system. Drug Metab. Dispos. 40, 754–760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Jin Y., Chen M., Penning T. M., and Miller W. L. (2015) Electron transfer by human wild-type and A287P mutant P450 oxidoreductase assessed by transient kinetics: functional basis of P450 oxidoreductase deficiency. Biochem. J. 468, 25–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Marohnic C. C., Panda S. P., Martásek P., and Masters B. S. (2006) Diminished FAD binding in the Y459H and V492E Antley-Bixler syndrome mutants of human cytochrome P450 reductase. J. Biol. Chem. 281, 35975–35982 [DOI] [PubMed] [Google Scholar]
- 26. Marohnic C. C., Panda S. P., McCammon K., Rueff J., Masters B. S., and Kranendonk M. (2010) Human cytochrome P450 oxidoreductase deficiency caused by the Y181D mutation: molecular consequences and rescue of defect. Drug Metab. Dispos. 38, 332–340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Xia C., Panda S. P., Marohnic C. C., Martásek P., Masters B. S., Kim J. J. (2011) Structural basis for human NADPH-cytochrome P450 oxidoreductase deficiency. Proc. Natl. Acad. Sci. U.S.A. 108, 13486–13491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Gutierrez A., Lian L. Y., Wolf C. R., Scrutton N. S., and Roberts G. C. (2001) Stopped-flow kinetic studies of flavin reduction in human cytochrome P450 reductase and its component domains. Biochemistry 40, 1964–1975 [DOI] [PubMed] [Google Scholar]
- 29. Fontana A., de Laureto P. P., Spolaore B., Frare E., Picotti P., and Zambonin M. (2004) Probing protein structure by limited proteolysis. Acta Biochim. Polonica Engl. Ed. 51, 299–322 [PubMed] [Google Scholar]
- 30. Heiring C., and Muller Y. A. (2001) Folding screening assayed by proteolysis: application to various cystine deletion mutants of vascular endothelial growth factor. Protein Eng. 14, 183–188 [DOI] [PubMed] [Google Scholar]
- 31. Arnold U., Köditz J., Markert Y., and Ulbrich-Hofmann R. (2005) Local fluctuations versus global unfolding of proteins investigated by limited proteolysis. Biocatalysis and Biotransformation 23, 159–167 [Google Scholar]
- 32. Royo M., Fitzpatrick P. F., and Daubner S. C. (2005) Mutation of regulatory serines of rat tyrosine hydroxylase to glutamate: effects on enzyme stability and activity. Arch Biochem. Biophys. 434, 266–274 [DOI] [PubMed] [Google Scholar]
- 33. Hamdane D., Xia C., Im S. C., Zhang H., Kim J. J., and Waskell L. (2009) Structure and function of an NADPH-cytochrome P450 oxidoreductase in an open conformation capable of reducing cytochrome P450. J. Biol. Chem. 284, 11374–11384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Aigrain L., Pompon D., Moréra S., and Truan G. (2009) Structure of the open conformation of a functional chimeric NADPH cytochrome P450 reductase. EMBO Rep. 10, 742–747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Pudney C. R., Khara B., Johannissen L. O., and Scrutton N. S. (2011) Coupled motions direct electrons along human microsomal P450 Chains. PLoS Biol. 9, e1001222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kmoch S., Zeman J., Hrebícek M., Ryba L., Kristensen M. J., and Gregersen N. (1995) Riboflavin-responsive epilepsy in a patient with SER209 variant form of short-chain acyl-CoA dehydrogenase. J. Inherit. Metab. Dis. 18, 227–229 [DOI] [PubMed] [Google Scholar]
- 37. Vergani L., Barile M., Angelini C., Burlina A. B., Nijtmans L., Freda M. P., Brizio C., Zerbetto E., and Dabbeni-Sala F. (1999) Riboflavin therapy. Biochemical heterogeneity in two adult lipid storage myopathies. Brain 122, 2401–2411 [DOI] [PubMed] [Google Scholar]
- 38. Nicolo C., Flück C. E., Mullis P. E., and Pandey A. V. (2010) Restoration of mutant cytochrome P450 reductase activity by external flavin. Mol. Cell. Endocrinol. 321, 245–252 [DOI] [PubMed] [Google Scholar]
- 39. Pallan P. S., Wang C., Lei L., Yoshimoto F. K., Auchus R. J., Waterman M. R., Guengerich F. P., and Egli M. (2015) Human cytochrome P450 21A2, the major steroid 21-hydroxylase: structure of the enzyme.progesterone substrate complex and rate-limiting C-H bond cleavage. J. Biol. Chem. 290, 13128–13143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Huber W. J. 3rd., Marohnic C. C., Peters M., Alam J., Reed J. R., Masters B. S., and Backes W. L. (2009) Measurement of membrane-bound human heme oxygenase-1 activity using a chemically defined assay system. Drug Metab. Dispos. 37, 857–864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Ghosh D., Griswold J., Erman M., and Pangborn W. (2009) Structural basis for androgen specificity and oestrogen synthesis in human aromatase. Nature 457, 219–223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Ghosh D., Griswold J., Erman M., and Pangborn W. (2010) X-ray structure of human aromatase reveals an androgen-specific active site. J. Steroid Biochem. Mol. Biol. 118, 197–202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Wang Y.-H., Tee M. K., and Miller W. L. (2010) Human cytochrome p450c17: single step purification and phosphorylation of serine 258 by protein kinase A. Endocrinology 151, 1677–1684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Yoshimoto F. K., Zhou Y., Peng H.-M., Stidd D., Yoshimoto J. A., Sharma K. K., Matthew S., and Auchus R. J. (2012) Minor activities and transition state properties of the human steroid hydroxylases cytochromes P450c17 and P450c21, from reactions observed with deuterium-labeled substrates. Biochemistry 51, 7064–7077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Yoshimoto F. K., Peng H.-M., Zhang H., Anderson S. M., and Auchus R. J. (2014) Epoxidation activities of human cytochromes P450c17 and P450c21. Biochemistry 53, 7531–7540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Mui B., and Hope M. J. (1993) Formation of Large Unilamellar Vesicles by Extrusion, in Liposome Technology (Gregoriadis G., ed) 3rd Ed., pp. 55–66, CRC Press, Boca Raton, FL [Google Scholar]
- 47. McPherson A. (1999) Crystallization of Biological Macromolecules, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- 48. Stura E. A., and Wilson I. A. (1990) Analytical and production seeding techniques. Methods 1, 38–49 [Google Scholar]
- 49. Otwinowski Z., and Minor W. (1997) Processing of x-ray diffraction data collected in oscillation mode. Methods Enzymol. 276, 307–326 [DOI] [PubMed] [Google Scholar]
- 50. Vagin A., and Teplyakov A. (2010) Molecular replacement with MOLREP. Acta Crystallogr. D Biol. Crystallogr. 66, 22–25 [DOI] [PubMed] [Google Scholar]
- 51. Winn M. D., Ballard C. C., Cowtan K. D., Dodson E. J., Emsley P., Evans P. R., Keegan R. M., Krissinel E. B., Leslie A. G., McCoy A., McNicholas S. J., Murshudov G. N., Pannu N. S., Potterton E. A., Powell H. R., et al. (2011) Overview of the CCP4 suite and current developments. Acta Crystallogr. D Biol. Crystallogr. 67, 235–242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Brünger A. T., Adams P. D., Clore G. M., DeLano W. L., Gros P., Grosse-Kunstleve R. W., Jiang J. S., Kuszewski J., Nilges M., Pannu N. S., Read R. J., Rice L. M., Simonson T., and Warren G. L. (1998) Crystallography & NMR system: a new software suite for macromolecular structure determination. Acta Crystallogr. D Biol. Crystallogr. 54, 905–921 [DOI] [PubMed] [Google Scholar]
- 53. Emsley P., and Cowtan K. (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 [DOI] [PubMed] [Google Scholar]
- 54. McLaughlin L. A., Dickmann L. J., Wolf C. R., and Henderson C. J. (2008) Functional expression and comparative characterization of nine murine cytochromes P450 by fluorescent inhibition screening. Drug Metab. Dispos. 36, 1322–1331 [DOI] [PubMed] [Google Scholar]
- 55. Makaji E., Trambitas C. S., Shen P., Holloway A. C., and Crankshaw D. J. (2010) Effects of cytochrome P450 inhibitors on the biotransformation of fluorogenic substrates by adult male rat liver microsomes and cDNA-expressed rat cytochrome P450 isoforms. Toxicol. Sci. 113, 293–304 [DOI] [PubMed] [Google Scholar]
- 56. Lala P., Higashiyama T., Erman M., Griswold J., Wagner T., Osawa Y., and Ghosh D. (2004) Suppression of human cytochrome P450 aromatase activity by monoclonal and recombinant antibody fragments and identification of a stable antigenic complex. J. Steroid Biochem. Mol. Biol. 88, 235–245 [DOI] [PubMed] [Google Scholar]
- 57. Duarte M. P., Palma B. B., Laires A., Oliveira J. S., Rueff J., and Kranendonk M. (2005) Escherichia coli BTC, a human cytochrome P450 competent tester strain with a high sensitivity towards alkylating agents: involvement of alkyltransferases in the repair of DNA damage induced by aromatic amines. Mutagenesis 20, 199–208 [DOI] [PubMed] [Google Scholar]
- 58. Kranendonk M., Marohnic C. C., Panda S. P., Duarte M. P., Oliveira J. S., Masters B. S., and Rueff J. (2008) Impairment of human CYP1A2-mediated xenobiotic metabolism by Antley-Bixler syndrome variants of cytochrome P450 oxidoreductase. Arch. Biochem. Biophys. 475, 93–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Fisher C. W., Caudle D. L., Martin-Wixtrom C., Quattrochi L. C., Tukey R. H., Waterman M. R., and Estabrook R. W. (1992) High-level expression of functional human cytochrome P450 1A2 in Escherichia coli. FASEB J. 6, 759–764 [DOI] [PubMed] [Google Scholar]
- 60. Kranendonk M., Mesquita P., Laires A., Vermeulen N. P., and Rueff J. (1998) Expression of human cytochrome P450 1A2 in Escherichia coli: a system for biotransformation and genotoxicity studies of chemical carcinogens. Mutagenesis 13, 263–269 [DOI] [PubMed] [Google Scholar]