

Abstract
Amino-terminal acetylation is a critical co-translational modification of the newly synthesized proteins in a eukaryotic cell carried out by six amino-terminal acetyltransferases (NATs). All NATs contain at least one catalytic subunit, and some contain one or two additional auxiliary subunits. For example, NatE is a complex of Naa10, Naa50, and Naa15 (auxiliary). In the present study, the crystal structure of human Naa50 suggested the presence of CoA and acetylated tetrapeptide (AcMMXX) that have co-purified with the protein. Biochemical and thermal stability studies on the tetrapeptide library with variations in the first and second positions confirm our results from the crystal structure that a peptide with Met-Met in the first two positions is the best substrate for this enzyme. In addition, Naa50 acetylated all MXAA peptides except for MPAA. Transcriptome analysis of 10 genes that make up six NATs in humans from eight different cell lines suggests that components of NatE are transcribed in all cell lines, whereas others are variable. Because Naa10 is reported to acetylate all amino termini that are devoid of methionine and Naa50 acetylates all other peptides that are followed by methionine, we believe that NatE complex can be a major contributor for amino-terminal acetylation at the ribosome exit tunnel.
Keywords: acetyltransferase, enzyme kinetics, peptides, protein stability, protein structure
Introduction
Co-translational protein modifications are important biochemical events. Prominent among them are amino-terminal modifications such as methionine cleavage, acetylation, and myristoylation (1–3). Initiator methionine in eukaryotic proteins is removed co-translationally by the enzyme methionine aminopeptidase (MetAP)3 when the second amino acid is small and uncharged (1). About 70% of the matured proteins do not retain their amino-terminal methionine (4). Similarly, about 90% of all newly synthesized cytosolic proteins undergo amino-terminal acetylation (5). Protein amino-terminal acetylation is carried out by six amino-terminal acetyltransferases (NATs) named sequentially NatA to NatF, classified based on substrate preference (6–8). NatA acetylates the peptides with serine, alanine, threonine, cysteine, and valine on the amino termini that are formed after MetAP action at the ribosome exit tunnel (5, 9), whereas NatB acts on methionine followed by acidic residues in the second position (10, 11). NatC, NatE, and NatF act on methionine that is followed by hydrophobic amino acids like leucine, phenylalanine, isoleucine, and tryptophan (8, 10, 12, 13). Additionally, NatF also acetylates methionine that is followed specifically by a lysine (14). NatD acetylates the amino termini of Ser-Gly of histones H2A and H4 (15).
Each of the NATs (NatA–NatF) is a complex of one or two of the enzymes labeled serially as Naa10, Naa20, Naa30, Naa40, Naa50, and Naa60 attached to one or two of the auxiliary proteins named Naa15, Naa25, Naa35, and Naa38 (7, 9). Some NATs are known to bind to the ribosome at the exit tunnel through their auxiliary subunits (11, 16). Protein amino-terminal acetylation has potential effects on cell metabolism. A single nucleotide polymorphism observed in Naa10 results in Ogden syndrome, a lethal X-linked disorder of infancy whose symptoms include an aged appearance, craniofacial anomalies, hypotonia, global developmental delays, cryptorchidism, cardiac arrhythmias, and large eyes leading to death within a year after birth (17). N-Acetylation was shown as a degradation signal for individual proteins and inhibits the targeting of proteins to the endoplasmic reticulum (18, 19). Knockdown of Naa10 results in decreased cell proliferation and induction of apoptosis (20). Naa50 knockdown impaired sister chromatid cohesion and chromosome condensation (21, 22), emphasizing the important role of NATs in human health (23).
Liszczak et al. (9) reported the first structure of a heterodimeric NatA complex that consists of Naa10 and Naa15 with an inhibitor that is the conjugate of coenzyme A (CoA) and the substrate-like peptide. Naa50 associates with NatA, and the complex is named as NatE because of differences in the activity compared with NatA (24). Several studies aiming to understand substrate specificities of NATs have been reported (5, 10, 13–15, 25, 26). Understanding substrate specificity of multiple NAT complexes present in a cell is important in identifying their specific physiological and pathological function.
Because all the known structures of Naa50 found in the Protein Data Bank are complex structures (codes 2OB0, 2PSW, and 3TFY) with CoA and/or peptide, we have attempted to determine the apo structure (27, 28). However, CoA and an acetylated tetrapeptide were identified in the active site that might have co-purified along with the enzyme. The sequence of the tetrapeptide was unique and was not in the preferred list of substrates for Naa50 reported earlier. This observation prompted us to revisit the substrate specificity of this enzyme. Here, we report the biochemical and thermal shift assays and mass spectroscopic studies of human Naa50 using 40 designed tetrapeptides with variations in the first and second positions. Results from our studies have provided new insights into the substrate specificity of Naa50 in addition to confirming most of the previous observations. Transcriptome analysis of eight different human cell lines for all 10 genes that make up different NAT complexes suggests that a few are expressed in all cell types, whereas others are specific. Note that Naa50 is also called San, NAT5, Naa50p, and NAT13 (5, 23, 29).
Results and Discussion
Ternary Complex Shows Inherent Density for MMXX Peptide and CoA
Human Naa50 was crystallized in space group P212121 with a single molecule in the asymmetric unit (Table 1 and Fig. 1a). Three residues in the amino terminus and 16 residues in the carboxyl end could not be traced due to poor density. The overall structure revealed a mixed α/β-fold with a conserved acetyl-CoA core region composed of one α-helix (α3) and three β-strands (β2–β4). Crystal structures reported earlier were in the P21 space group with three molecules in the asymmetric unit (27, 28). However, the structure of individual monomers is similar between the current and reported structures. For example, the root mean square deviations are 0.47 and 0.42 Å, respectively, with 2PSW (CoA-bound) and 3TFY (CoA- and substrate peptide-bound) (27). Note that neither the cofactor nor the substrate peptides were added either during protein purification or at the crystallization stage. However, to our surprise Fo − Fc map displayed clear density for the CoA and acetylated tetrapeptide in the active site (Fig. 1, a and b). The complete CoA molecule except for the thioethanol part could be modeled. Similarly, density was clear for four amino acids in the substrate-binding pocket. Short contiguous density near the amino terminus of the substrate peptide was indicative of acetylation, and this was modeled and refined. To the best of our knowledge, this is the first structure of human Naa50 with the coenzyme A and product bound at the same time. The identity of the methionine residues at the first two positions was established based on the strong density in the Fo − Fc map near the sulfur atoms (Fig. 1b).
TABLE 1.
Crystallographic statistics
| Naa50 complex | |
|---|---|
| Cell parameters | |
| Space group | P212121 |
| a (Å) | 44. 61 |
| b (Å) | 53.19 |
| c (Å) | 67.58 |
| Data collection | |
| X-ray source | Rigaku Micromax 007 rotating anode x-ray generator |
| Wavelength (Å) | 1.54 |
| Resolution range (Å) | 28.52–2.49 (2.58–2.49)a |
| Collected reflections, total/unique | 26,542/5,836 (508) |
| Completeness (%) | 97.90 (88.04) |
| I/σ(I) | 16.38 (1.97) |
| Rsym (%) | 6.3 (36.2) |
| Refinement statistics | |
| Rwork (%) | 20.0 (29.2) |
| Rfree (%) | 26.5 (35.5) |
| Wilson B-factor | 59.9 |
| Mean B-factor (Å2) | 61.5 |
| Macromolecule | 61.4 |
| Solvent | 60.6 |
| Clash score | 17.5 |
| Ramachandran plot | |
| Most favored (%) | 96.1 |
| Additional allowed (%) | 3.2 |
| Disallowed (%) | 0.6 |
| r.m.s.b bonds (Å) | 0.01 |
| r.m.s. angles (°) | 1.48 |
| Protein Data Bank code | 4X5K |
a Highest resolution shell in parentheses.
b r.m.s., root mean square.
FIGURE 1.
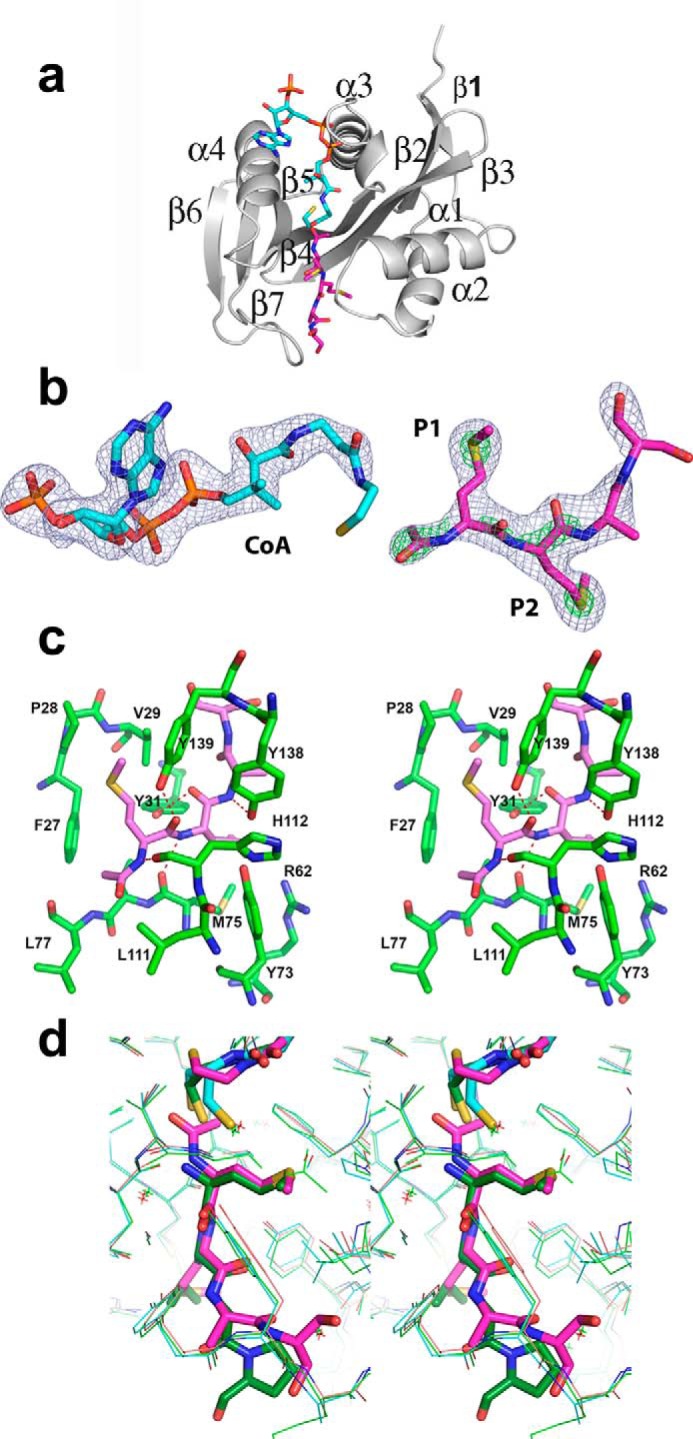
Crystal structure of Naa50. a, ribbon representation of the Naa50 with cofactor (cyan) and peptide (pink) bound in the active site. b, 2Fo − Fc map coverage at σ = 1.5 (blue) around CoA and the tetrapeptide. The density in green is the 2Fo − Fc map at 5.0 σ for sulfur atoms and 3.0 σ for acetylation. Clear density at higher contour levels around the sulfur atom confirms the methionine at P1 and P2 positions. c, stereodiagram near the active site shows residues surrounding the peptide (light pink). The peptide makes good hydrophobic and hydrophilic interactions with the enzyme. d, comparison of the hNaa50 bound to acetylated peptide and CoA (pink), the peptide and CoA bound in the active site of the hNaa50 (green), and only CoA bound to the hNaa50 (cyan) structure. Note that the acetylated peptide slides toward the cofactor along with residues that form hydrogen bonds.
From the first cycle of the refinement, the density for the first two residues in P1 and P2 subsites was suggestive of the amino acid methionine (Fig. 1b). The peptide backbone forms a series of hydrogen bonds with surrounding residues in the active site of the protein. The side chain hydroxyl groups of Tyr-31 and Tyr-139 and the backbone carbonyl atoms of Met-75 and His-112 interact with the peptide region of the first two residues (Fig. 1c). The P1 pocket where the amino-terminal methionine binds is hydrophobic in nature and is composed of Phe-27, Pro-28, and Val-29 from the α1-α2 loop and Tyr-139 and Ile-142 from the β6-β7 hairpin loop. The protein region where the P2 Met binds (P2 pocket) is amphiphilic and exposed to the solvent. Tyr-31 and Phe-35 from the α1-α2 loop region, Tyr-138 from the β6-β7 hairpin loop, Arg-62 in β3 strand, and Tyr-73 and Met-75 from the β4 strand form the P2 pocket (Fig. 1c). Further refinement revealed that residues in the third and fourth position (P3 and P4) could be alanine and serine (Fig. 1b). However, due to ambiguity in density, we refer to them as XX. Major parts of both these residues are solvent-exposed and make very little contact with the protein surface.
Comparison of the present structure with the two previous structures indicates that there is not much difference in the coordinates of either CoA or the protein surrounding it except near the thioethanol part. Some changes were noticed in the substrate (3TFY) and the product peptides (present structure) (Fig. 1d). The acetylated peptide moves by about 0.6 Å toward CoA. Similar concerted movement was observed in the side chains of residues that are involved in hydrogen bonding with the peptide. In contrast, no appreciable changes were observed either in the side chains or in the main chain atoms that form hydrogen bonds between holo (2PSW) and CoA and substrate-bound structure (3TFY) (Fig. 1d). This explains why there were no chemical shifts noted in the previous NMR study when substrate peptide was added to the enzyme in the absence of cofactor (29).
Peptide with Met in the P1 and P2 Positions Is the Best Substrate for Naa50
Numerous studies to understand the substrate specificities of different NAT complexes have been reported as reviewed by Van Damme et al. (30). However, most of these studies used limited number of synthetic peptides that mimic the natural substrates, whereas some others used the whole cell proteome to understand the substrate specificity (5, 13, 24). Based on these studies, it was established that different NAT complexes have preferences for different amino termini. For example, NatA, which is a complex of Naa10 and Naa15, has been implicated in acetylating peptides that have small and uncharged amino acids at the amino terminus that are formed after the removal of initiator methionine by MetAP (8). Similarly, Naa50 acetylates the amino-terminal methionine that is followed by leucine, isoleucine, phenylalanine, and tryptophan (8, 13). Because our x-ray crystal structure suggests Met-Met in the first two positions, we designed a series of tetrapeptides to explore the substrate specificity of Naa50.
We have tested the activity of Naa50 with 20 designed tetrapeptides (XAAA) summarized in the Fig. 2 and supplemental Fig. S1. As expected, from the XAAA series, MAAA was the best substrate for Naa50 (Fig. 2c). Although the affinity (Km) for MAAA and LAAA peptides (from the first series) was almost the same (132.9 and 126.1 μm, respectively), the turnover rate of MAAA is 5 times higher compared with LAAA peptide, making it the best substrate (Fig. 2c).
FIGURE 2.
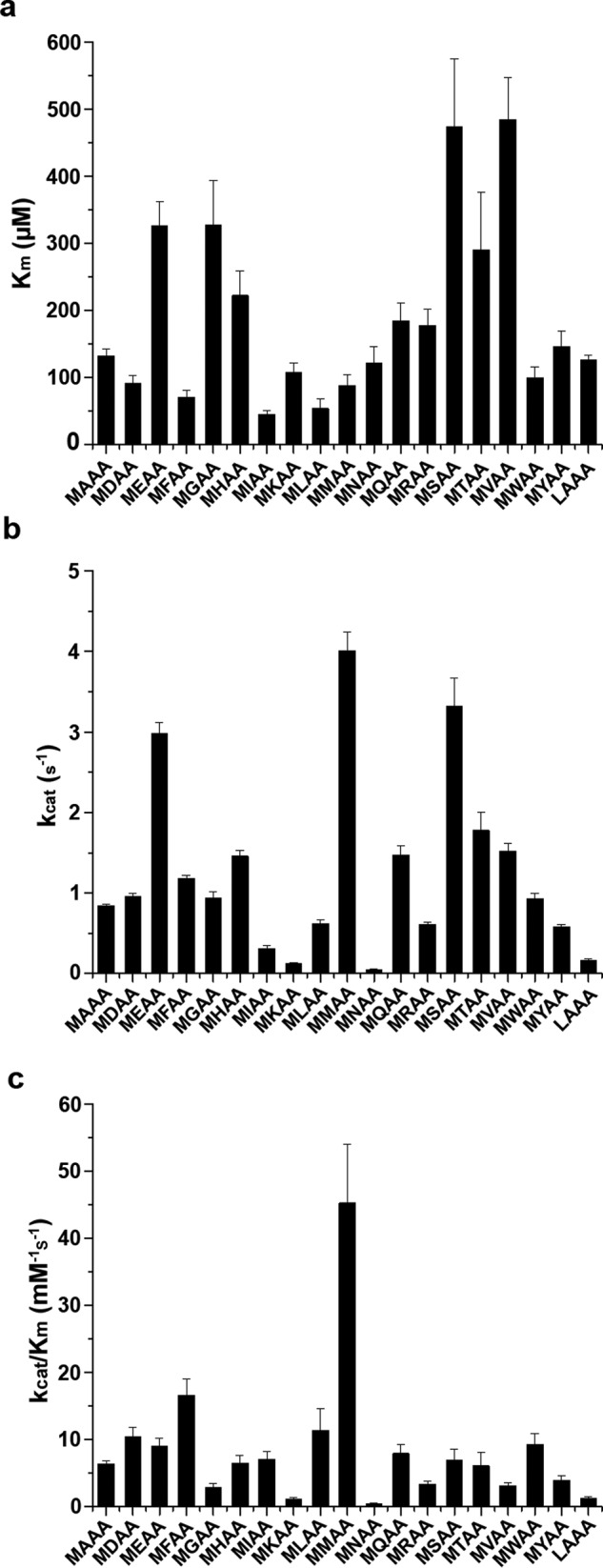
Kinetic characterization of N-acetyltransferase activity of Naa50 against 40 synthetic tetrapeptides. A comparison of Km (a), kcat (b), and Km/kcat (c) values of different synthetic peptides of MXAA and XAAA series against Naa50 is shown. Data of XAAA peptides are limited to only those that are active. Error bars represent S.D.
Because methionine is the most preferred P1 residue, in the next round of design it was fixed, and the second amino acid was varied (MXAA peptides) (Fig. 2). Among the MXAA tetrapeptides, most surprisingly and in line with our structural data, MMAA peptide was acetylated with highest efficiency. Peptides with leucine, tryptophan, phenylalanine, isoleucine, alanine, aspartic acid, glutamic acid, glutamine, serine, threonine, valine, and tyrosine in the second position are also acetylated at least 50% as efficiently as the MMAA peptide (Fig. 2c).
Based on the nature of the second amino acid, MXAA peptides were subclassified as follows: hydrophobic (when X is Met, Leu, Ile, Phe, Tyr, and Trp), positively charged (His, Lys, and Arg), negatively charged (Glu and Asp), and small and/or uncharged amino acids (Gly, Ala, Val, Ser, Thr, Cys, Pro, Gln, and Asn). Kinetic parameters summarized in Fig. 2 suggest that Naa50 acetylates peptides with hydrophobic and acidic amino acids in the second position with higher efficiency. In contrast, peptides with smaller amino acids have low affinity, whereas positively charged amino acids have medium affinity. As expected, MMAA peptide displayed the highest turnover number. Similarly, most of the peptides with hydrophobic and acidic residues are processed 3–4-fold more slowly than the MMAA peptide. Naa50 did not acetylate MPAA peptide. Although activity was observed on the MCAA peptide, kinetic parameters could not be obtained because of heavy background resulting from the reaction between the cysteine side chain in the substrate and 5,5′-dithiobis(2-nitrobenzoic acid) (DTNB).
As proline is a cyclic amino acid, the conformation of the backbone of the peptide is different from other MXAA peptides, and thereby it may not bind properly in the active site, and hence no activity was observed. However, it is not clear why asparagine (MNAA) is not on the preferred list, whereas glutamine (MQAA) is at the top. It is well known that the side chain of asparagine bends onto the backbone, forming a hydrogen bond with the preceding carbonyl, thereby adapting a cyclic conformation (31, 32). Because of this cyclic conformation of asparagine, MNAA peptide may adopt a shape similar to that of MPAA, resulting in reduced activity. Acetylation status of several peptides was monitored by positive ion electrospray ionization mass spectrometry analysis (Table 2).
TABLE 2.
High resolution MS data of acetylated peptides
| Compound | Observed ion | Molecular formula | Theoretical mass | Observed mass | Error |
|---|---|---|---|---|---|
| m/z | m/z | ppm | |||
| LAAA + Naa50 | [M(Ac) + Na]+ | C17H30O6N4Na | 409.2063 | 409.2055 | 8 |
| MEAA + Naa50 | [M(Ac) + K]+ | C18H30O8N4SK | 501.1421 | 501.1418 | 3 |
| MKAA + Naa50 | [M(Ac) + K]+ | C19H35O6N5SK | 500.1945 | 500.1940 | 5 |
| MSAA + Naa50 | [M(Ac) + K]+ | C16H28N4O7SK | 459.1316 | 459.1310 | 6 |
| MHAA + Naa50 | [M(Ac) + K]+ | C19H30N6O6SK | 509.1585 | 509.1579 | 6 |
Hydrophobic S1 and Amphiphilic S2 Pockets Support the Binding of MM Type Peptides
To establish the chemical basis of substrate selectivity, we analyzed the electronic nature of the S1 and S2 pockets where the first two amino acids of the substrate bind. Electrostatic maps of the surface of these two pockets demonstrate their distinct nature (Fig. 3). S1 pocket is wide near the Cγ atom but becomes narrow near the Sδ atom and hydrophobic in nature, supporting the binding of linear methionine and γ-branched leucine. Conversely, S2 region is amphiphilic in nature with 50% of the pocket exposed to the solvent. The protein surface in the S2 pocket is mostly hydrophobic with a partial positive charge contributed by Arg-62. Except for proline, all the other amino acids irrespective of the charge or size in the P2 position are substrates for Naa50 with different strengths. Methionine, by virtue of its linear shape and flexibility, will adjust in the hydrophobic region of the P2 pocket without being exposed to the solvent, thereby gaining the best substrate status. A similar trend is observed for other hydrophobic residues in the P2 position such as Leu, Ile, Phe, and Trp. The partial positive charge of this pocket tilts the balance toward the binding of negatively charged amino acids like Glu and Asp when compared with Arg, Lys, and His that are positively charged. In addition, smaller amino acids like Ala, Ser, Val, and Thr find a place in the list of preferred residues probably because of crowding of Tyr-138, Tyr-129, and Lys-140 in the P2 pocket. The protein region where the P3 and P4 residues bind is featureless both in terms of structure and electronic nature, suggesting very little role of this region in substrate selectivity.
FIGURE 3.
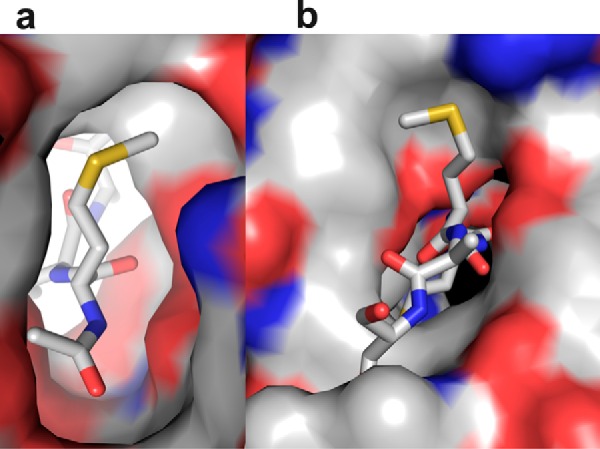
Electrostatic surface diagram of Naa50 in the P1 and P2 pockets using the crystal structure. a, the S1 pocket is hydrophobic in nature and can support the binding of methionine and leucine. b, the S2 site is amphiphilic, i.e. partly hydrophobic and partly charged. This subsite is exposed to the solvent. Because of this mixed environment, almost all amino acids can be accommodated in this pocket.
Crystals of cofactor and the peptide appeared in the first batch of protein purification using His affinity purification alone. Several attempts were made to reproduce this result using different batches of protein with no success. All biochemical characterizations were made using the protein produced in subsequent batches. Thermal shift and mass spectrometry data analyses on subsequent batches have suggested that the small molecules did not co-purify along with the enzyme. If Naa50 binds to CoA the denaturing temperature should be about 55 °C. However, each batch we purified showed that the denaturation temperature is about 52 °C, suggesting that the cofactor and the peptide did not bind to the enzyme. Although our first crystal structure looks like an accident, it has provided important information about the substrate preference of Naa50, forming the basis for studies described here.
MMAA Peptide Provides the Highest Stability to Naa50
It is well established that ligands alter the stability of proteins (33, 34). To estimate the binding ability of 40 peptides and their influence on the stability of Naa50, thermal shift (Tm) assays were performed (35) (Fig. 4). The stability of Naa50 improved by 6 °C in the presence of MAAA peptide alone (apo, 52.74 ± 0.59), 5 °C in the presence of CoA alone, and 10 °C in the presence of this peptide and CoA together. From these data, it is concluded that the contribution of the peptide toward the thermal stability of Naa50 is independent of CoA. This is in contrast to the observations made in an NMR study where they did not notice any chemical shift changes upon addition of substrate alone (29). Because there is a correlation between the biochemical data and thermal shift assays, we believe that substrate alone could bind to the enzyme without the assistance of AcCoA/CoA. Based on this observation, the rest of the experiments were carried out without CoA (Fig. 4). Except for the MAAA peptide, the increase in stability was marginal for other XAAA peptides (Fig. 4a). Surprisingly, peptides with negatively charged amino acids on the amino terminus (DAAA and EAAA) systematically destabilized the enzyme by about 3 °C. Similarly, asparagine- and arginine -based peptides destabilized the enzyme.
FIGURE 4.
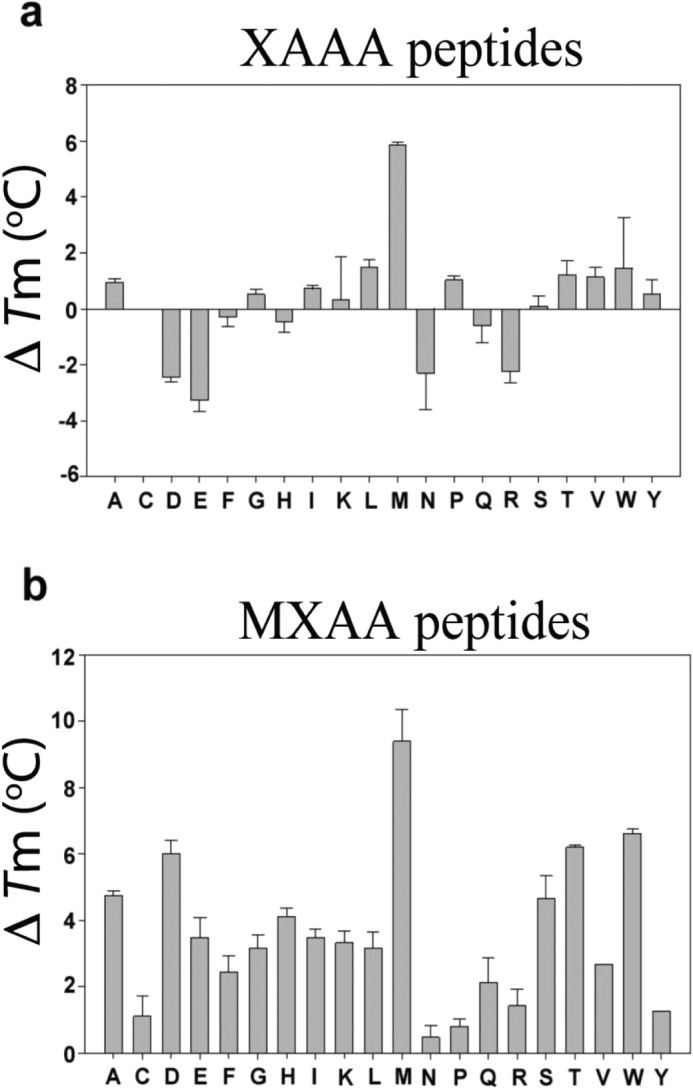
Thermal shift assay for XAAA and MXAA series of peptides. The ΔTm values were obtained by subtraction of the Tm value of apo-Naa50 with that of the peptide-bound form. a, in the XAAA series, in line with biochemical data, only methionine based peptide showed appreciable protein stability followed by leucine. Surprisingly several other peptides destabilized the protein. b, in the MXAA series, all peptides displayed good thermal stability of Naa50. Error bars represent S.D.
In contrast, all MXAA peptides stabilized the enzyme, and this effect was almost proportional to the rate of enzyme catalysis (Fig. 4b). MMAA peptide imparted the highest thermal stability to the enzyme (by about 10 °C). MNAA, MPAA, and MRAA peptides had the least stabilizing effect. MYAA, however, is a good substrate but did not improve the thermal stability proportionately.
Broad Substrate Specificity of NatE May Account for Majority of the Co-translational Amino-terminal Acetylation
MetAP cleaves the initiator methionine that is followed by small and uncharged amino acids including proline, which accounts for 70% of the proteome. The rest of the proteins retain the initiator methionine. NatE complex that is composed of Naa10, Naa15, and Naa50 is reported to be associated with the ribosome (16). From earlier reports, it is clear that Naa10 acetylates the amino termini of all peptides with small and uncharged amino acids (5, 8). In the present study, we have shown that human Naa50 acetylates the methionine followed by any residue except for proline. Therefore, we conclude that NatE complex may account for the major co-translational amino-terminal acetylation demands of a human cell.
To investigate this hypothesis, we carried out gene transcription profiling of all NATs. Gene-specific primers for 10 genes in humans that account for six NAT complexes (A–F) were designed to estimate the transcription levels from eight different human cell lines using quantitative RT-PCR (qRT-PCR) (Fig. 5). These include a pair of lung normal (MRC-5) and lung cancer (A-549) cell lines and human embryonic kidney cell line (HEK 293). The other cancer cell lines used were breast (MDA-MB-231), liver (HepG-2), cervical (HeLa), neuronal (SK-N-SH), and prostate (DU-145). A variable transcription profile was noticed for several genes between different cell lines except for the components of NatE complex (Naa10, Naa15, and Naa50), which are transcribed in all cell lines. The universal presence of NatE components adds strength to our prediction that NatE complex would take care of most of the N-acetylation of peptides at the ribosome exit tunnel. Earlier studies demonstrated that NatF is an organellar acetyltransferase specific to Golgi, whereas NatD is a histone acetyltransferase and NatC has a role in acetylating viral proteins, which is important for viral particle assembly (13, 36, 37). However, there is an elevated expression level of most of the NAT genes in cancer cell lines, suggesting a critical role of these enzymes in pathological conditions. We are in the process of expanding the scope of these transcriptome data to understand the patho/physiological specific expression of individual NATs in different cancers.
FIGURE 5.
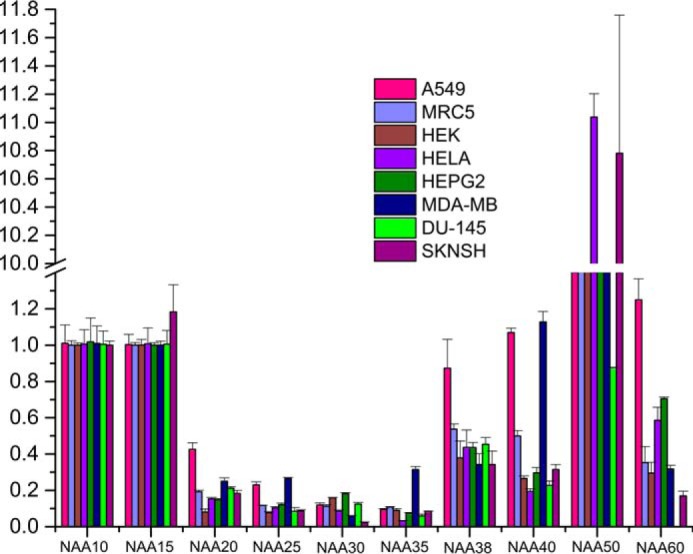
Quantitative RT-PCR analysis of all Naa constituents from eight different human cell lines: relative expression of Naa genes (normalized to GAPDH gene). Note that NatE components (Naa10, Naa15, and Naa50) are transcribed uniformly in all cell lines, whereas others have differential transcriptional behavior. Values depict mean ± S.E. of three independent experiments. Vertical lines on each bar represent S.D.
Role of Naa50 in Humans and Yeast Could be Different
Recent studies suggest that knockdown of Naa50 function in yeast does not affect the acetylation pattern of peptides such as ML/MI/MD/ME/MQ and does not show any phenotypic behavior unlike deleterious affects noted in humans and Drosophila (21, 22, 38). It is also important to note that the level of acetylation increases when human Naa50 is overexpressed in yeast, whereas a similar pattern was not noted with yeast Naa50 overexpression (39). Together, these data suggest that yeast may have an alternative path of acetylation compared with humans.
Conclusions
Based on the unexpected result from the crystal structure, we decided to explore the substrate specificity of Naa50 in detail. To achieve this, we designed 40 tetrapeptides with variations in the first and second positions of the amino termini. Biochemical and thermal shift assays suggested that the peptides with methionine in the first position and any other amino acid in the second position except for proline are the substrates for Naa50. Among all 40 peptides tested, MMAA was found to be the best substrate in terms of catalysis and thermal stabilization of the enzyme. Transcriptome analysis of eight different human cell lines for 10 genes that are components of various NAT complexes suggested that Naa10, Naa15, and Naa50 (components of NatE complex) are uniformly transcribed in all cell lines, whereas others are cell type-specific. Based on the literature and from the current study, we propose that NatE complex could take care of the majority of the co-translational acetylation demands in a human cell.
Experimental Procedures
Materials
All peptides were purchased from Genpro Biotech (New Delhi, India). Oligonucleotides for molecular biology were from Eurofins Biotech (India). The cDNA synthesis kit and restriction enzymes were from Fermentas (Canada). The plasmid isolation kit was from Macherey & Nagel (Germany). Acetyl-CoA and other chemicals were procured from Sigma-Aldrich. All peptides were HPLC-purified (better than 95%) and characterized by mass spectrometry.
Cell Culture
Human lung normal (MRC-5), human lung carcinoma (A-549), human breast adenocarcinoma (MDA-MB-231), human cervix adenocarcinoma (HeLa), human embryonic kidney (HEK 293), human prostate carcinoma (DU-145), human hepatocellular carcinoma (HepG-2), and human neuroblastoma (SK-N-SH) cells were grown in high glucose Dulbecco modified Eagle's medium (DMEM; Sigma-Aldrich) containing l-glutamine and supplemented with penicillin-streptomycin and 5 or 10% fetal bovine serum (FBS; Invitrogen), respectively. All cell lines were obtained from the American Type Culture Collection (ATCC). Cells were maintained under a 5% CO2 atmosphere at 37 °C.
Cloning and Purification of Naa50
The human Naa50 coding sequence was amplified from an A549 (lung adenocarcinoma) cDNA library using the following primer pair: 5′-GGAATTCCATATGATGAAAGGTAGCCGGATCGA-3′ and 5′-CCGCTCGAGTCAGTTGTCTG TCTTTTGCA-3′). Naa50 was cloned into pET15b vector using NdeI and XhoI restriction sites; the vector encodes an amino-terminal poly-His tag, which was confirmed by DNA sequencing. Naa50 plasmid was transformed into Escherichia coli BL21(DE3) cells for expression of the protein. Two liters of LB medium containing ampicillin with 20 ml of overnight culture was incubated at 37 °C until A600 reached 1.0. The temperature of the culture was reduced to 25 °C followed by induction of protein expression with 1 mm isopropyl β-d-thiogalactopyranoside (final concentration) and further incubated for 16 h. Cells were harvested by centrifugation at 8000 × g and resuspended in 50 ml of lysis buffer (50 mm Hepes, pH 8.0, 0.5 m NaCl, 10% glycerol, 0.1% Triton X-100, 15 mg of lysozyme, 3 mg of protease inhibitor mixture, 5 mg of DNase, 5 mm MgCl2, and 5 mm imidazole). After passing through a constant cell disruptor at 25,000 p.s.i., the lysate was centrifuged at 35,000 × g for 45 min. The supernatant (50 ml) was passed through a +TG buffer (50 mm Hepes, pH 8.0, 0.5 m NaCl, 10% glycerol, 0.1% Triton X-100, and 5 mm imidazole)-pre-equilibrated nickel affinity column. The protein-loaded column was washed with sufficient volumes of +TG and −TG (50 mm Hepes, pH 8.0, 0.5 m NaCl, and 5 mm imidazole) buffers until the A280 reached zero. Finally, protein was eluted using 150 mm imidazole in −TG buffer followed by dialysis against protein storage buffer (50 mm Hepes, pH 8.0, and 150 mm NaCl). No attempts were made to remove the amino-terminal poly-His tag. Protein concentration was determined by Bradford assay, and the final yield was 30 mg/liter of culture.
Determination of Naa50 Activity
100 μl of reaction solution containing 50 mm Hepes, pH 7.5, 150 mm KCl, 2 μm recombinant Naa50, 200 μm acetyl-CoA, and 200 μm DTNB in a 96-well plate was incubated at 37 °C in the microplate multimode reader (TECAN Infinite 200) for 30 min. Similar conditions were used except for the presence of the enzyme as a control. The reaction was initiated by the addition of 200 μm substrate peptide. After the acetyltransferase activity, the free sulfhydryl group on the CoA reacts with DTNB and releases5-thio-2-nitrobenzoic acid that is yellow in color and has an absorption maxima at 412 nm. Based on the coenzyme A standard graph, product formation was computed by using the equation y = 0.0064x − 0.0032.
Thermal Shift Assay
The thermal shift assay was performed according to Gumpena et al. (35). The 20-μl experimental solution contained 5 μg of Naa50, 4 μl of SYPRO® Orange dye, 5 μl of 4× buffer (100 mm Hepes and 600 mm KCl), and 2.5% dimethyl formamide (DMF). All experiments were carried out in the presence of 2.5% DMF (apo and coenzyme A thermal shift assays; in the case of peptides, all peptides were dissolved in DMF). Whenever required, 250 μm substrate peptide and/or 250 μm CoA was introduced into the above mixture. Control experiments were performed without peptide and CoA. Denaturation experiments were conducted between 4 and 90 °C at an incremental rate of 1 °C min−1. Protein unfolding was monitored by following the change in the fluorescence intensity of SYPRO Orange at 570 nm (40). The inflection point in the temperature versus fluorescence curve was identified as the melting temperature (Tm). All experiments were performed on an ABI 7500 real time PCR machine.
Crystallization, Data Collection, and Structure Determination of Naa50
Protein concentration was adjusted to 12.5 mg/ml before setting up crystallization screens at 25 °C. Initial crystals appeared after 2 months (Index screen, Hampton Research). After optimization, crystals in the orthorhombic space group (P212121) were obtained in the hanging drop with 5 μl of protein and 5 μl of reservoir solution (20% PEG 3350 and 0.2 m sodium formate). Crystals with an average diameter of 150 μm in a lens shape were equilibrated for 5 min in 40% PEG 1050 and 0.2 m sodium formate before vitrification in the liquid nitrogen stream. X-ray data were collected on the home source diffractometer (Rigaku Micromax 007 with R-AXIS IV2+ image plate). Diffraction images were processed using HKL-3000 (41) (Table 1). Unit cell parameters were different from all previously reported structures, and the Matthews coefficient suggested the presence of a single molecule in the asymmetric unit. The structure was solved by molecular replacement (coordinates from Protein Data Bank code 2PSW) using MOLREP and refined by REFMAC5 from the CCP4 suite (28, 42–44) (Table 1). The quality of the final model was judged using PROCHECK (45). Structure figures were generated using the program PyMOL (46).
Enzyme Kinetics
40 different peptides (XAAA and MXAA type (X is any of the 20 natural amino acids) were tested against Naa50 in the same way as described above. Steady state kinetics were performed, and the initial velocities were calculated from the linear portion of the progression curves and fitted against substrate concentration using the Michaelis-Menten equation. Substrate concentrations in the range of 1.56–800 μm were used in these reactions. Each experiment was performed in triplicates, and the standard deviation was calculated. Final data were analyzed using SigmaPlot (Fig. 2).
qRT-PCR Analysis
Total RNA was extracted from the harvested cells using TRI reagent (Sigma) according to the manufacturer's instructions. RNA concentration was determined using a NanoDrop ND-100 spectrophotometer (Thermo Fisher Scientific), and 3 μg of RNA was used to make first strand cDNA synthesis. The reverse transcription reaction was carried out using an Enhanced Avian First Strand Synthesis kit (Sigma-Aldrich) according to the manufacturer's recommended protocol. To measure the expression levels of 10 genes of different NAT constituents, qRT-PCR was performed using a DyNAmo Flash SYBR Green qPCR kit (Thermo Scientific, Waltham, MA; F-416L) in an ABI 7500 thermal cycler (Applied Biosystems). Expression of the gene of interest was normalized to transcript levels of GAPDH, and -fold change was calculated using the Ct method (47). GAPDH Ct values were stable in all conditions and experiments. The final data are shown as average ± S.E. of three independent experiments. qPCR runs were normalized according to the ΔΔCt method using GAPDH as a reference gene. Primer sets used for qPCR analysis are provided in supplemental Table S1.
Mass Spectrometric Analysis of Tetrapeptides after Reaction with Naa50
The reaction mixture containing 25 mm Hepes, 150 mm KCl, pH 7.5, 2 μm recombinant Naa50, 200 μm DTNB, and 200 μm synthetic tetrapeptide was incubated for 1 h at 37 °C. The resulting solution was used for mass spectrometry analysis. 10 μl of the solution was diluted with 1.5 ml of methanol, and 10 μl of this solution was injected into the source of the mass spectrometer using an autosampler. Mass spectrometry experiments were carried on an Exactive Orbitrap mass spectrometer (Thermo Scientific) equipped with a heated electrospray ionization source. (Table 2).
Author Contributions
R. R., V. S., D. K. C., P. Sankoju, and P. Sripadi performed the experiments. R. R. and A. A. wrote the main manuscript text. All authors reviewed the manuscript.
Supplementary Material
This work was supported by research fellowships from the Council of Scientific and Industrial Research and University Grants Commission, New Delhi, India (to R. R., V. S., and D. K. C.) and by Department of Science and Technology Grant SR/SO/BB-55/2008, Department of Biotechnology Grant BT-BRB-TF-2-2011, and the Council of Scientific and Industrial Research (SMiLE) New Delhi, India (to A. A.). The authors declare that they have no conflicts of interest with the contents of this article.
The atomic coordinates and structure factors (code 4X5K) have been deposited in the Protein Data Bank (http://wwpdb.org/).

This article contains supplemental Table S1 and Fig. S1.
- MetAP
- methionine aminopeptidase
- NAT
- amino-terminal acetyltransferase
- DTNB
- 5,5′-dithiobis(2-nitrobenzoic acid)
- DMF
- dimethyl formamide
- MRC-5
- human lung normal cell line
- A-549
- human lung carcinoma
- MDA-MB-231
- human breast adenocarcinoma
- HeLa
- human cervix adenocarcinoma
- DU-145
- human prostate carcinoma
- HepG-2
- human hepatocellular carcinoma
- SK-N-SH
- human neuroblastoma
- qRT-PCR
- quantitative RT-PCR
- qPCR
- quantitative PCR
- hNaa50
- human Naa50.
References
- 1. Frottin F., Martinez A., Peynot P., Mitra S., Holz R. C., Giglione C., and Meinnel T. (2006) The proteomics of N-terminal methionine cleavage. Mol. Cell. Proteomics 5, 2336–2349 [DOI] [PubMed] [Google Scholar]
- 2. Kramer G., Boehringer D., Ban N., and Bukau B. (2009) The ribosome as a platform for co-translational processing, folding and targeting of newly synthesized proteins. Nat. Struct. Mol. Biol. 16, 589–597 [DOI] [PubMed] [Google Scholar]
- 3. Wilcox C., Hu J. S., and Olson E. N. (1987) Acylation of proteins with myristic acid occurs cotranslationally. Science 238, 1275–1278 [DOI] [PubMed] [Google Scholar]
- 4. Bradshaw R. A., Brickey W. W., and Walker K. W. (1998) N-terminal processing: the methionine aminopeptidase and Nα-acetyl transferase families. Trends Biochem. Sci. 23, 263–267 [DOI] [PubMed] [Google Scholar]
- 5. Arnesen T., Van Damme P., Polevoda B., Helsens K., Evjenth R., Colaert N., Varhaug J. E., Vandekerckhove J., Lillehaug J. R., Sherman F., and Gevaert K. (2009) Proteomics analyses reveal the evolutionary conservation and divergence of N-terminal acetyltransferases from yeast and humans. Proc. Natl. Acad. Sci. U.S.A. 106, 8157–8162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Arnesen T. (2011) Towards a functional understanding of protein N-terminal acetylation. PLos Biol. 9, e1001074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Starheim K. K., Gevaert K., and Arnesen T. (2012) Protein N-terminal acetyltransferases: when the start matters. Trends Biochem. Sci. 37, 152–161 [DOI] [PubMed] [Google Scholar]
- 8. Giglione C., Fieulaine S., and Meinnel T. (2015) N-terminal protein modifications: Bringing back into play the ribosome. Biochimie 114, 134–146 [DOI] [PubMed] [Google Scholar]
- 9. Liszczak G., Goldberg J. M., Foyn H., Petersson E. J., Arnesen T., and Marmorstein R. (2013) Molecular basis for N-terminal acetylation by the heterodimeric NatA complex. Nat. Struct. Mol. Biol. 20, 1098–1105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Polevoda B., Norbeck J., Takakura H., Blomberg A., and Sherman F. (1999) Identification and specificities of N-terminal acetyltransferases from Saccharomyces cerevisiae. EMBO J. 18, 6155–6168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Van Damme P., Lasa M., Polevoda B., Gazquez C., Elosegui-Artola A., Kim D. S., De Juan-Pardo E., Demeyer K., Hole K., Larrea E., Timmerman E., Prieto J., Arnesen T., Sherman F., Gevaert K., et al. (2012) N-terminal acetylome analyses and functional insights of the N-terminal acetyltransferase NatB. Proc. Natl. Acad. Sci. U.S.A. 109, 12449–12454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Caesar R., Warringer J., and Blomberg A. (2006) Physiological importance and identification of novel targets for the N-terminal acetyltransferase NatB. Eukaryot. Cell 5, 368–378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hole K., Van Damme P., Dalva M., Aksnes H., Glomnes N., Varhaug J. E., Lillehaug J. R., Gevaert K., and Arnesen T. (2011) The human N-α-acetyltransferase 40 (hNaa40p/hNatD) is conserved from yeast and N-terminally acetylates histones H2A and H4. PLoS One 6, e24713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Van Damme P., Hole K., Pimenta-Marques A., Helsens K., Vandekerckhove J., Martinho R. G., Gevaert K., and Arnesen T. (2011) NatF contributes to an evolutionary shift in protein N-terminal acetylation and is important for normal chromosome segregation. PLoS Genet. 7, e1002169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Song O. K., Wang X., Waterborg J. H., and Sternglanz R. (2003) An Nα-acetyltransferase responsible for acetylation of the N-terminal residues of histones H4 and H2A. J. Biol. Chem. 278, 38109–38112 [DOI] [PubMed] [Google Scholar]
- 16. Gautschi M., Just S., Mun A., Ross S., Rücknagel P., Dubaquié Y., Ehrenhofer-Murray A., and Rospert S. (2003) The yeast Nα-acetyltransferase NatA is quantitatively anchored to the ribosome and interacts with nascent polypeptides. Mol. Cell. Biol. 23, 7403–7414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Rope A. F., Wang K., Evjenth R., Xing J., Johnston J. J., Swensen J. J., Johnson W. E., Moore B., Huff C. D., Bird L. M., Carey J. C., Opitz J. M., Stevens C. A., Jiang T., Schank C., et al. (2011) Using VAAST to identify an X-linked disorder resulting in lethality in male infants due to N-terminal acetyltransferase deficiency. Am. J. Hum. Genet. 89, 28–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Forte G. M., Pool M. R., and Stirling C. J. (2011) N-terminal acetylation inhibits protein targeting to the endoplasmic reticulum. PLoS Biol. 9, e1001073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hwang C. S., Shemorry A., and Varshavsky A. (2010) N-terminal acetylation of cellular proteins creates specific degradation signals. Science 327, 973–977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Gromyko D., Arnesen T., Ryningen A., Varhaug J. E., and Lillehaug J. R. (2010) Depletion of the human Nα-terminal acetyltransferase A induces p53-dependent apoptosis and p53-independent growth inhibition. Int. J. Cancer 127, 2777–2789 [DOI] [PubMed] [Google Scholar]
- 21. Williams B. C., Garrett-Engele C. M., Li Z., Williams E. V., Rosenman E. D., and Goldberg M. L. (2003) Two putative acetyltransferases, san and deco, are required for establishing sister chromatid cohesion in Drosophila. Curr. Biol. 13, 2025–2036 [DOI] [PubMed] [Google Scholar]
- 22. Hou F., Chu C. W., Kong X., Yokomori K., and Zou H. (2007) The acetyltransferase activity of San stabilizes the mitotic cohesin at the centromeres in a shugoshin-independent manner. J. Cell Biol. 177, 587–597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Arnesen T., Anderson D., Torsvik J., Halseth H. B., Varhaug J. E., and Lillehaug J. R. (2006) Cloning and characterization of hNAT5/hSAN: an evolutionarily conserved component of the NatA protein N-α-acetyltransferase complex. Gene 371, 291–295 [DOI] [PubMed] [Google Scholar]
- 24. Evjenth R., Hole K., Karlsen O. A., Ziegler M., Arnesen T., and Lillehaug J. R. (2009) Human Naa50p (Nat5/San) displays both protein Nα- and Nϵ-acetyltransferase activity. J. Biol. Chem. 284, 31122–31129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Mullen J. R., Kayne P. S., Moerschell R. P., Tsunasawa S., Gribskov M., Colavito-Shepanski M., Grunstein M., Sherman F., and Sternglanz R. (1989) Identification and characterization of genes and mutants for an N-terminal acetyltransferase from yeast. EMBO J. 8, 2067–2075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Magin R. S., Liszczak G. P., and Marmorstein R. (2015) The molecular basis for histone H4- and H2A-specific amino-terminal acetylation by NatD. Structure 23, 332–341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Liszczak G., Arnesen T., and Marmorstein R. (2011) Structure of a ternary Naa50p (NAT5/SAN) N-terminal acetyltransferase complex reveals the molecular basis for substrate-specific acetylation. J. Biol. Chem. 286, 37002–37010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Berman H. M., Westbrook J., Feng Z., Gilliland G., Bhat T. N., Weissig H., Shindyalov I. N., and Bourne P. E. (2000) The Protein Data Bank. Nucleic Acids Res. 28, 235–242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Evjenth R. H., Brenner A. K., Thompson P. R., Arnesen T., Frøystein N. Å., and Lillehaug J. R. (2012) Human protein N-terminal acetyltransferase hNaa50p (hNAT5/hSAN) follows ordered sequential catalytic mechanism: combined kinetic and NMR study. J. Biol. Chem. 287, 10081–10088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Van Damme P., Arnesen T., and Gevaert K. (2011) Protein α-N-acetylation studied by N-terminomics. FEBS J. 278, 3822–3834 [DOI] [PubMed] [Google Scholar]
- 31. Addlagatta A., Krzywda S., Czapinska H., Otlewski J., and Jaskolski M. (2001) Ultrahigh-resolution structure of a BPTI mutant. Acta Crystallogr. D Biol. Crystallogr. 57, 649–663 [DOI] [PubMed] [Google Scholar]
- 32. Vasudev P. G., Banerjee M., Ramakrishnan C., and Balaram P. (2012) Asparagine and glutamine differ in their propensities to form specific side chain-backbone hydrogen bonded motifs in proteins. Proteins 80, 991–1002 [DOI] [PubMed] [Google Scholar]
- 33. Vedadi M., Niesen F. H., Allali-Hassani A., Fedorov O. Y., Finerty P. J. Jr., Wasney G. A., Yeung R., Arrowsmith C., Ball L. J., Berglund H., Hui R., Marsden B. D., Nordlund P., Sundstrom M., Weigelt J., et al. (2006) Chemical screening methods to identify ligands that promote protein stability, protein crystallization, and structure determination. Proc. Natl. Acad. Sci. U.S.A. 103, 15835–15840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Niesen F. H., Berglund H., and Vedadi M. (2007) The use of differential scanning fluorimetry to detect ligand interactions that promote protein stability. Nat. Protoc. 2, 2212–2221 [DOI] [PubMed] [Google Scholar]
- 35. Gumpena R., Kishor C., Ganji R. J., Jain N., and Addlagatta A. (2012) Glu121-Lys319 salt bridge between catalytic and N-terminal domains is pivotal for the activity and stability of Escherichia coli aminopeptidase N. Protein science 21, 727–736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Yang X., Yu W., Shi L., Sun L., Liang J., Yi X., Li Q., Zhang Y., Yang F., Han X., Zhang D., Yang J., Yao Z., and Shang Y. (2011) HAT4, a Golgi apparatus-anchored B-type histone acetyltransferase, acetylates free histone H4 and facilitates chromatin assembly. Mol. Cell 44, 39–50 [DOI] [PubMed] [Google Scholar]
- 37. Tercero J. C., and Wickner R. B. (1992) MAK3 encodes an N-acetyltransferase whose modification of the L-A gag NH2 terminus is necessary for virus particle assembly. J. Biol. Chem. 267, 20277–20281 [PubMed] [Google Scholar]
- 38. Zhang J., Shi X., Li Y., Kim B. J., Jia J., Huang Z., Yang T., Fu X., Jung S. Y., Wang Y., Zhang P., Kim S. T., Pan X., and Qin J. (2008) Acetylation of Smc3 by Eco1 is required for S phase sister chromatid cohesion in both human and yeast. Mol. Cell 31, 143–151 [DOI] [PubMed] [Google Scholar]
- 39. Van Damme P., Hole K., Gevaert K., and Arnesen T. (2015) N-terminal acetylome analysis reveals the specificity of Naa50 (Nat5) and suggests a kinetic competition between N-terminal acetyltransferases and methionine aminopeptidases. Proteomics 15, 2436–2446 [DOI] [PubMed] [Google Scholar]
- 40. Simpson R. J. (2010) SYPRO Orange fluorescent staining of protein gels. Cold Spring Harbor Protoc. 2010, pdb.prot5414 [DOI] [PubMed] [Google Scholar]
- 41. Minor W., Cymborowski M., Otwinowski Z., and Chruszcz M. (2006) HKL-3000: the integration of data reduction and structure solution—from diffraction images to an initial model in minutes. Acta Crystallogr. D Biol. Crystallogr. 62, 859–866 [DOI] [PubMed] [Google Scholar]
- 42. Winn M. D., Ballard C. C., Cowtan K. D., Dodson E. J., Emsley P., Evans P. R., Keegan R. M., Krissinel E. B., Leslie A. G., McCoy A., McNicholas S. J., Murshudov G. N., Pannu N. S., Potterton E. A., Powell H. R., et al. (2011) Overview of the CCP4 suite and current developments. Acta Crystallogr. D Biol. Crystallogr. 67, 235–242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Murshudov G. N., Skubák P., Lebedev A. A., Pannu N. S., Steiner R. A., Nicholls R. A., Winn M. D., Long F., and Vagin A. A. (2011) REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr. D Biol. Crystallogr. 67, 355–367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Vagin A., and Teplyakov A. (2010) Molecular replacement with MOLREP. Acta Crystallogr. D Biol. Crystallogr. 66, 22–25 [DOI] [PubMed] [Google Scholar]
- 45. Laskowski R. A., Rullmannn J. A., MacArthur M. W., Kaptein R., and Thornton J. M. (1996) AQUA and PROCHECK-NMR: programs for checking the quality of protein structures solved by NMR. J. Biomol NMR 8, 477–486 [DOI] [PubMed] [Google Scholar]
- 46. DeLano W. L. (2002) The PyMOL Molecular Graphics System, Schrödinger, LLC, New York [Google Scholar]
- 47. Zhang L., Yang J., Cai J., Song X., Deng J., Huang X., Chen D., Yang M., Wery J. P., Li S., Wu A., Li Z., Li Z., Liu Y., Chen Y., et al. (2013) A subset of gastric cancers with EGFR amplification and overexpression respond to cetuximab therapy. Sci. Rep. 3, 2992. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.