

Abstract
Unlike Saccharomyces cerevisiae, the methylotrophic yeast Pichia pastoris can assimilate amino acids as the sole source of carbon and nitrogen. It can grow in media containing yeast extract and peptone (YP), yeast nitrogen base (YNB) + glutamate (YNB + Glu), or YNB + aspartate (YNB + Asp). Methanol expression regulator 1 (Mxr1p), a zinc finger transcription factor, is essential for growth in these media. Mxr1p regulates the expression of several genes involved in the utilization of amino acids as the sole source of carbon and nitrogen. These include the following: (i) GDH2 encoding NAD-dependent glutamate dehydrogenase; (ii) AAT1 and AAT2 encoding mitochondrial and cytosolic aspartate aminotransferases, respectively; (iii) MDH1 and MDH2 encoding mitochondrial and cytosolic malate dehydrogenases, respectively; and (iv) GLN1 encoding glutamine synthetase. Synthesis of all these enzymes is regulated by Mxr1p at the level of transcription except GDH2, whose synthesis is regulated at the level of translation. Mxr1p activates the transcription of AAT1, AAT2, and GLN1 in cells cultured in YP as well as in YNB + Glu media, whereas transcription of MDH1 and MDH2 is activated in cells cultured in YNB + Glu but not in YP. A truncated Mxr1p composed of 400 N-terminal amino acids activates transcription of target genes in cells cultured in YP but not in YNB + Glu. Mxr1p binds to Mxr1p response elements present in the promoters of AAT2, MDH2, and GLN1. We conclude that Mxr1p is essential for utilization of amino acids as the sole source of carbon and nitrogen, and it is a global regulator of multiple metabolic pathways in P. pastoris.
Keywords: amino acid, glutamate dehydrogenase, transcription factor, transcription regulation, yeast, methanol expression regulator 1, Pichia pastoris, amino acids, aspartate aminotransferase, malate dehydrogenase
Introduction
Saccharomyces cerevisiae can utilize NH4+ or amino acids such as l-glutamate as the source of nitrogen but not as the sole source of carbon and energy. When S. cerevisiae is cultured in media containing NH4+ as the source of nitrogen, glutamate dehydrogenase encoded by GDH1 or GDH3 functions as an anabolic enzyme catalyzing the reaction between NH4+ and α-ketoglutarate to generate glutamate with NADPH acting as a co-factor (1). When cultured in media containing glutamate as the sole source of nitrogen, glutamate dehydrogenase encoded by GDH2 acts as a catabolic enzyme by converting glutamate to α-ketoglutarate and NH4+ in the presence of NADH. The NH4+ thus generated is used for the biosynthesis of glutamine (2). Unlike S. cerevisiae, certain yeasts can utilize l-amino acids such as glutamate, aspartate, and proline as the sole source of carbon as well as nitrogen. These include the following: Scheffersomyces stipitis (also known as Pichia stipitis), Candida albicans, Candida maltosa, Candida shehatae, Candida glabrata, Candida reukaufii, Candida utilis, Debaryomyces hansenii, Kluyveromyces lactis, Kluyveromyces marxianus, Lodderomyces elongisporus, Meyerozyma guilliermondii, Pichia capsulata, Yarrowia lipolytica, Rhodotorula rubra, and Trichosporon beigelii (3). The assimilation of glutamate requires the activity of NAD-dependent glutamate dehydrogenase 2 (referred to as GDH2 in this study), and a Δgdh2 strain cannot utilize glutamate or aspartate as either a carbon or a nitrogen source as demonstrated in the case of S. stipitis (3).
In addition to GDH2, enzymes such as aspartate aminotransferase (AAT),2 malate dehydrogenase (MDH), and glutamine synthetase (GLN1) also play key roles in the metabolism of amino acids (4). In S. cerevisiae, AAT1 and AAT2 encode AAT localized in mitochondria (mAAT) and cytoplasm (cAAT). cAAT catalyzes the reversible conversion of glutamate and oxaloacetate into α-ketoglutarate and aspartate (4). The oxaloacetate thus generated is converted to malate by MDH present in the cytoplasm (cMDH) encoded by MDH2. Malate enters mitochondria via α-ketoglutarate-malate exchanger protein, which also transports α-ketoglutarate in the opposite direction. Malate is oxidized to oxaloacetate by the mitochondrial MDH (mMDH) encoded by MDH1, resulting in the formation of NADH, which enters the electron transport chain and generates energy (4). The mitochondrial oxaloacetate is converted to aspartate by mAAT, which is transported to cytoplasm via the aspartate-glutamate exchanger. Glutamate present in the cytoplasm is converted to glutamine by GLN1 (4).
P. pastoris, a methylotrophic yeast, is extensively used for the production of recombinant proteins. Being a respiratory yeast, P. pastoris completely oxidizes sugars, avoiding formation of ethanol, and this results in efficient utilization of carbon sources yielding high biomass. During high cell density fermentation of P. pastoris, ammonium sulfate or organic nitrogen sources such as amino acids are used as nitrogen sources. Several transcription factors that regulate carbon metabolism have been identified in P. pastoris. These include the following: Mxr1p, Rop1p, Trm1p, Mit1p, and Nrg1p (5–14). The role of these transcription factors in the regulation of amino acid metabolism has not been examined. Here, we demonstrate that P. pastoris can utilize amino acids as the sole source of carbon, and Mxr1p but not Trm1p or Rop1p is essential for this process. Mxr1p regulates the synthesis of several key enzymes involved in amino acid metabolism such as GDH2, AAT1, AAT2, MDH1, MDH2, and GLN1 at the transcriptional or post-transcriptional level.
Results
Enzymes Essential for the Utilization of Amino Acids as the Sole Source of Carbon by P. pastoris and Regulation of Their Biosynthesis by Mxr1p
The ability of P. pastoris to utilize amino acids as the sole source of carbon and nitrogen has not been investigated. We therefore examined the ability of P. pastoris GS115 strain to grow in media such as yeast nitrogen base (YNB) without amino acids and 0.5% ammonium sulfate supplemented with 2.0% glucose (YNBD), 1.0% glutamate (YNB + Glu), 1% aspartate (YNB + Asp) or YNB + Glu without ammonium sulfate. The results indicate that P. pastoris GS115 strain but not Δmxr1 strain (Table 1) can grow in these media (Fig. 1A) indicating that Mxr1p is essential for the utilization of amino acids as the sole source of carbon and nitrogen. Rop1 and Trm1p, which regulate the expression of genes of methanol utilization pathway (8, 9), have no role in the utilization of amino acids because growth of Δrop1 and Δtrm1 strains is not affected when cultured in YP medium (1.0% yeast extract and 2.0% peptone) (Fig. 1A). S. cerevisiae was unable to grow in YP medium as expected (Fig. 1B) (3). Subcellular localization studies employing P. pastoris strain expressing a FLAG-tagged Mxr1p (13) indicates that Mxr1p localizes to the nucleus of cells cultured in YP as well as YPM (YP + 2% methanol) but was cytosolic in cells cultured in YPD (YP + 2% glucose) (Fig. 1C). Cell lysates of GS115 and Δmxr1 strains cultured in YP were subjected to SDS-PAGE, and proteins were visualized by Coomassie Blue staining (Fig. 1D). Proteins a–f, which are differentially expressed in GS115 and Δmxr1, were selected; protein bands were excised and subjected to in-gel trypsin digestion, and the tryptic peptides were analyzed by MALDI-TOF mass spectrometry (supplemental data). Proteins a–c were identified as GDH2, alcohol oxidase, and formate dehydrogenase, respectively, and proteins d–f were identified as aconitase, malate synthase, and citrate synthase, respectively (Fig. 1D and supplemental data). To confirm their differential expression in GS115 and Δmxr1, qPCR analysis was carried out with RNA isolated from cells cultured in YP medium. The results indicate that AOXI and FDH transcripts are present in higher levels in GS115 than Δmxr1, whereas transcript levels of ACO, MS, and CS are higher in Δmxr1 than GS115 cultured in YP (Fig. 1E). However, GDH2 mRNA levels were comparable in GS115 and Δmxr1 cultured in YP (Fig. 1E). Because GDH2 has a key role in the utilization of amino acids in S. stipitis and GDH2 protein but not mRNA levels were consistently lower in Δmxr1 than GS115 in several independent experiments, a detailed study was undertaken. P. pastoris GS115 and Δmxr1 were transformed with pIB3-GDH2His, and expression of Gdh2pHis was examined in cells cultured in different carbon sources by Western blotting. GDH2His expression levels were higher in cells cultured in YP, YNB + Glu, and YNB + Asp than those cultured in YPD or YNBD (Fig. 1, F and G). Furthermore, GDH2His is expressed at higher levels in GS115 than Δmxr1 cultured in YP, YNB + Glu, and YNB + Asp media (Fig. 1, F and G). Immunofluorescence studies indicate that GDH2His is localized to the cytosol (Fig. 1H). Thus, Mxr1p is required for the synthesis of Gdh2p but not GDH2 mRNA suggesting that Mxr1p regulates GDH2 expression at the post-transcriptional level.
TABLE 1.
P. pastoris strains used in this study
| Strain | Genotype | Ref. |
|---|---|---|
| GS115 | his4 | 5 |
| Δmxr1 | GS115, Ppmxr1Δ::Zeocinr | 13 |
| Δprm1 | GS115, Ppprm1Δ::Zeocinr | 10 |
| Δrop1 | GS115, Pprop1Δ::Zeocinr | 9 |
| Δgdh2 | GS115, Ppgdh2Δ::Zeocinr | This study |
| Δaat1 | GS115, Ppaat1Δ::Zeocinr | This study |
| Δaat2 | GS115, Ppaat2Δ::Zeocinr | This study |
| Δmdh1 | GS115, Ppmdh1Δ::Zeocinr | This study |
| Δmdh2 | GS115, Ppmdh2Δ::Zeocinr | This study |
| Δgdh2-GDH2His | Δgdh2, his4:: (PGDH2PpGDH2-His) | This study |
| GS115-GDH2His | GS115, his4:: (PGDH2PpGDH2-His) | This study |
| Δmxr1-GDH2His | Δmxr1, his4:: (PGDH2PpGDH2-His) | This study |
| MXR1Myc-OE | Δmxr1, Blasticidinr:: (PGAPDHPpMXR1-Myc) | 13 |
| MXR1Myc-OE-GDH2His | MXR1Myc-OE, his4:: (PGDH2PpGDH2-His) | This study |
| MXR1N400-OE | Δmxr1, Blasticidinr:: (PGAPDHPpMXR1N400-Myc) | 13 |
| MXR1N400-OE-GDH2His | MXR1N400-OE, his4:: (PGDH2PpGDH2-His) | This study |
| Δmxr1-GDH2-OE1 | Δmxr1, Zeocinr:: (PGAPDHPpGDH2-Myc) | This study |
| Δmxr1-GDH2-OE2 | Δmxr1, Zeocinr:: (PCUPPpGDH2-Myc) | This study |
| AAT1Myc | GS115, Zeocinr:: (PAAT1PpAAT1-Myc) | This study |
| AAT2Myc | GS115, Zeocinr:: (PAAT2PpAAT2-Myc) | This study |
| MDH1His | GS115, his4:: (PMDH1PpGDH2-His) | This study |
| MDH2His | GS115, his4:: (PMDH2PpMDH2-His) | This study |
| GLN1Myc | GS115, Zeocinr:: (PGLN1PpGLN1-Myc) | This study |
FIGURE 1.

Growth of P. pastoris GS115 and Δmxr1 in media containing amino acids as the sole source of carbon and regulation of GDH2 expression by Mxr1p. A, growth of P. pastoris GS115 and Δmxr1 strains in media containing amino acids as the sole source of carbon and nitrogen. In YNB + Glu-(NH4)2SO4 medium, glutamate serves as the sole source of carbon as well as nitrogen. B, comparison of growth of P. pastoris and S. cerevisiae in YP medium. C, subcellular localization of FLAG-tagged Mxr1p in cells cultured in different media as analyzed by immunofluorescence using anti-FLAG antibodies. DAPI was used to stain the nucleus. D, protein profile of lysates of cells cultured in YP medium. Proteins were resolved on SDS-polyacrylamide gel and stained with Coomassie Brilliant Blue R. Proteins a–f, which are differentially expressed in GS115 and Δmxr1, are identified by mass spectrometry (see supplemental data). E, qPCR validation of genes differentially expressed in GS115 and Δmxr1. F, Western blotting analysis of lysates of GS115-GDH2His and Δmxr1-GDH2His strains expressing Gdh2pHis using anti-His tag antibodies. Cells were cultured in different media as indicated. ZTA1 (23) served as loading control. G, quantitation of data presented in F. H, cytosolic localization of Gdh2pHis in cells cultured in YP as examined by immunofluorescence using anti-His tag antibodies. *, p < 0.05; **, p < 0.005; ***, p < 0.0005; ns, not significant.
To confirm whether GDH2 is required for the utilization of amino acids as the sole source of carbon, P. pastoris Δgdh2 strain was generated (Fig. 2, A and B), and its ability to grow in YPD, YP, YNB + Glu, and YNB + Asp media was examined. Growth of Δgdh2 strain was normal in YPD medium but severely impaired when cultured in YP, YNB + Glu, and YNB + Asp media (Fig. 2C). pGAPBA-MXR1 was constructed and transformed into Δmxr1 to generate MXR1Myc-OE strain. Similarly, pIB3-GDH2His was constructed and transformed into Δgdh2 to generate Δgdh2-GDH2His strain. Expression of Mxr1pMyc and Gdh2pHis was confirmed by Western blotting using anti-Myc and anti-His tag antibodies, respectively (Fig. 2, D and E). Growth of MXR1Myc-OE and Δgdh2-GDH2His strains in YP, YNB + Glu, and YNB + Asp media was comparable with that of GS115 (Fig. 2F) confirming that Mxr1p and GDH2 are essential for the utilization of amino acids as the sole source of carbon.
FIGURE 2.

Analysis of the role of GDH2 in the utilization of amino acids as the sole source of carbon. A, strategy for the generation of Δgdh2 strain. The restriction enzymes used for the digestion of genomic DNA for use in Southern analysis are indicated. P1, P2 primer pairs used in PCRs are indicated. B, PCR and Southern blotting analysis of genomic DNA isolated from GS115 (1) and Δgdh2 (2) strains. The Zeocin expression cassette is amplified by PCR only in Δgdh2 strain. M, DNA molecular weight markers (kb). The 4.4- and 2.5-kb bands hybridize to a radiolabeled probe (−571 to −24 bp of GDH2) in the Southern blot as expected. C, analysis of growth of GS115, Δmxr1, and Δgdh2 strains cultured in media containing different carbon sources. D, Western blotting analysis of Δmxr1 overexpressing Mxr1p from GAPDH promoter as Myc-tagged protein and Δgdh2 expressing Gdh2p from its own promoter as His-tagged protein. Anti-Myc and anti-His tag antibodies were used to detect Mxr1p and Gdh2p, respectively. E, quantitation of data presented in D. F, analysis of growth of different P. pastoris strains in various media as indicated.
Differential Regulation of GDH2 Expression by MXR1 and MXR1N400
The first 400 amino acids of Mxr1p (Mxr1pN400) contain 14-3-3 protein interaction region between amino acids 212 and 225 and a trans-activation domain between amino acids 246 and 280 (Fig. 3A) (13, 14). This trans-activation domain mediates carbon source-dependent repression and activation of Mxr1p-regulated genes (13, 14). To examine the role of this N-terminal trans-activation domain in the regulation of GDH2 expression, P. pastoris Δmxr1 strain overexpressing Mxr1pN400 was generated and named MXR1N400-OE (Fig. 3A). Expression of MXR1 and MXR1N400 mRNA in cells cultured in YP and YNB + Glu media was confirmed by qPCR (Fig. 3B). Overexpression of Mxr1pN400 resulted in partial restoration of growth of cells cultured in YP but not YNB + Glu (Fig. 3C). Overexpression of Mxr1p but not Mxr1pN400 results in the restoration of Gdh2pHis levels in Δmxr1 (Fig. 3, D and E). However, deletion or overexpression of Mxr1p or Mxr1pN400 had no significant effect on GDH2 mRNA levels (Fig. 3F). These results not only confirm post-transcriptional regulation of GDH2 expression by Mxr1p but also demonstrate that the N-terminal 400 amino acids alone are not sufficient for this process.
FIGURE 3.
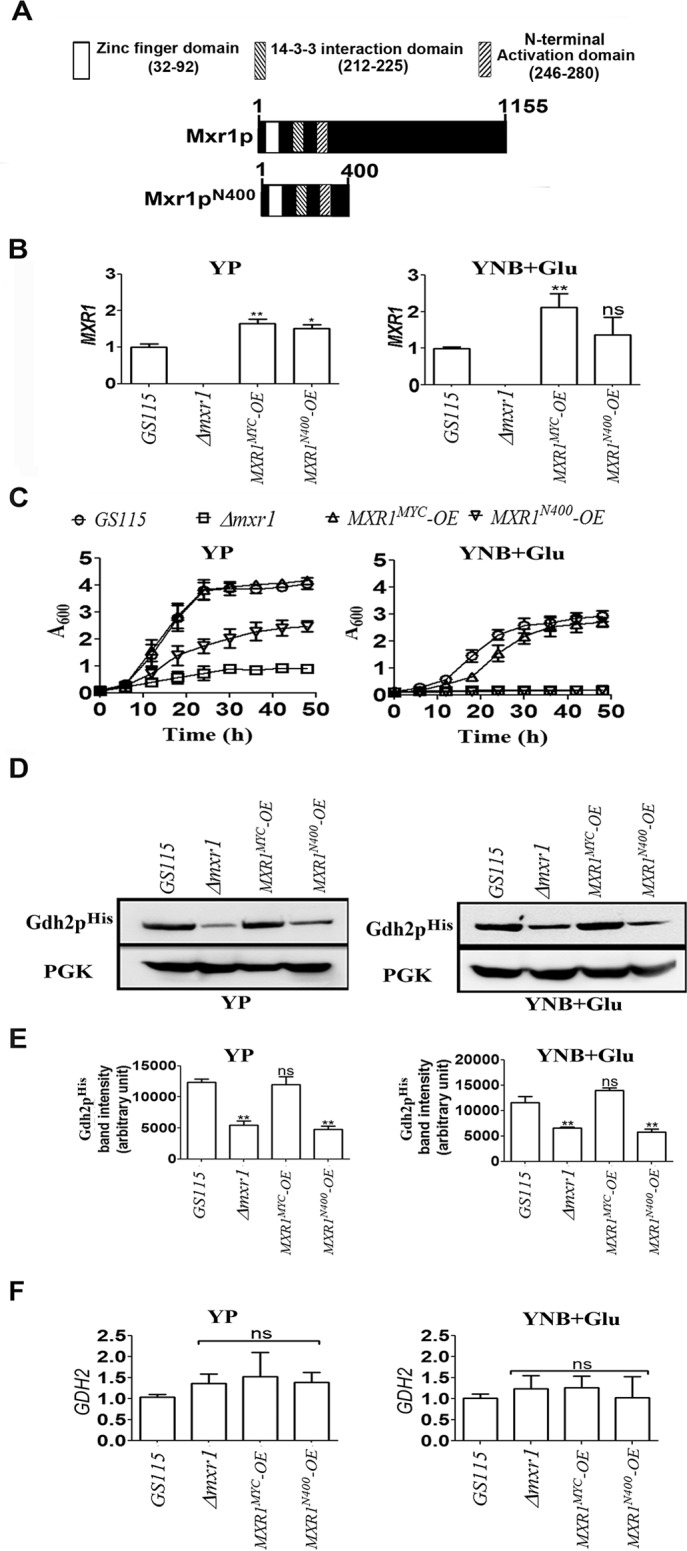
Regulation of GDH2 expression by Mxr1p and Mxr1pN400. A, schematic representation of Mxr1p and Mxr1pN400. The major domains within the 400 N-terminal amino acids are indicated. B, MXR1 and MXRN400 mRNA in different P. pastoris strains cultured in YP and YNB + Glu media as analyzed by qPCR. C, effect of overexpression of Mxr1p and Mxr1pN400 on the growth of Δmxr1 cultured in YP and YNB + Glu media. D, Western blotting analysis of Gdh2pHis levels in Δmxr1 overexpressing Mxr1p and Mxr1pN400. The gene encoding Gdh2pHis was expressed from its own promoter in different strains as indicated. E, quantitation of data presented in D. F, analysis of GDH2 mRNA levels by qPCR in different P. pastoris strains cultured in YP and YNB + Glu media. Error bars indicate mean ± S.D. *, p < 0.05; **, p < 0.005; ***, p < 0.0005; ns, not significant. One-way analysis of variance followed by Tukey's multiple comparison test was carried out (n = 3).
Mxr1p Is a Key Regulator of Aspartate Aminotransferase
Because the growth of Δgdh2 can be restored by the expression of GDH2, it was of interest to examine whether overexpression of GDH2 in Δmxr1 can restore the growth in YP and YNB + Glu media. GDH2 was expressed as a Myc-tagged protein (Gdh2pMyc) at high levels from the GAPDH promoter or moderate levels from S. cerevisiae CUP1 promoter (15) in Δmxr1 to generate Δmxr1-GDH2-OE1 and Δmxr1-GDH2-OE2 strains respectively. Gdh2pMyc expression in cells cultured in YP medium was confirmed by Western blotting with anti-Myc tag antibodies (Fig. 4, A and B). Overexpression of Gdh2pMyc from either of these promoters was unable to restore growth of Δmxr1 in YP and YNB + Glu media (Fig. 4C). The fact that expression of GDH2 results in the restoration of growth of Δgdh2 (Fig. 2F) but not Δmxr1 strain indicates that Mxr1p may regulate the expression of other enzymes that are required for utilization of amino acids as the sole source of carbon in P. pastoris.
FIGURE 4.

Analysis of the ability of Gdh2p to restore the growth of Δmxr1. A, Western blotting analysis of lysates of Δmxr1, Δmxr1 overexpressing GDH2 from GAPDH promoter, or CUP1 promoter. B, quantitation of data presented in A. C, analysis of ability of Gdh2p overexpressed from GAPDH or CUP1 promoters to restore the growth of Δmxr1 cultured in YP and YNB + Glu media.
To examine whether mAAT and cAAT are required for the utilization of amino acids as the sole source of carbon in P. pastoris, they were expressed as Myc-tagged proteins, and their localization in mitochondria and cytoplasm, respectively, was confirmed by immunofluorescence using anti-Myc tag antibodies (Fig. 5A). P. pastoris Δaat1 and Δaat2 strains were generated, and their growth was examined in media containing different carbon sources. The results indicate that Δaat2 strain is unable to grow in YP, YNB + Glu, and YNB + Asp media indicating that cAAT is essential for the growth of P. pastoris in these media (Fig. 5B). qPCR analysis indicates that AAT1 and AAT2 are expressed at low levels in Δmxr1 strain (Fig. 5C). Overexpression of either Mxr1p or Mxr1p-N400 in Δmxr1 strain results in up-regulation of AAT1 and AAT2 expression in cells cultured in YP medium. However, only Mxr1p but not Mxr1pN400 restores the expression of these genes in cells cultured in YNB + Glu (Fig. 5C).
FIGURE 5.

Function and regulation of mAAT and cAAT encoded by AAT1 and AAT2, respectively, in P. pastoris cells cultured in YP, YNB + Glu, and YP + Asp media. A, localization of Myc-tagged mAAT and cAAT in the mitochondria and cytoplasm, respectively, by immunofluorescence using anti-Myc tag antibodies. Hsp70 and DAPI were used as mitochondrial and nuclear markers, respectively. B, effect of deletion of genes encoding cAAT and mAAT on the growth of P. pastoris in YP, YNB + Glu, and YP + Asp media. C, quantification of AAT1 and AAT2 mRNAs by qPCR in various P. pastoris strains as indicated. Error bars indicate mean ± S.D. *, p < 0.05; **, p < 0.005; ***, p < 0.0005. One-way analysis of variance followed by Tukey's multiple comparison test was carried out (n = 3).
Mxr1p Regulates the Expression of Genes Encoding mMDH, cMDH, and GLN1
In view of the crucial role of mMDH and cMDH in the transport of malate between cytoplasm and mitochondria (4), a detailed study was undertaken to examine their role during the utilization of amino acids as the sole source of carbon in P. pastoris. Immunofluorescence studies indicate that mMDH and cMDH are indeed localized in mitochondria and cytoplasm, respectively, in P. pastoris cultured in YP medium (Fig. 6A). P. pastoris Δmdh1 and Δmdh2 strains were generated, and their growth was examined in different media. The results indicate that the growth of Δmdh1 is severely impaired in cells cultured in media containing glucose or amino acids as the sole source of carbon (Fig. 6B). However, the growth of Δmdh2 is affected only in cells cultured in YP or YNB + Glu media (Fig. 6B). Thus, the generation of malate in the cytoplasm and its conversion to oxaloacetate in the mitochondria are important for the normal growth of cells cultured in YP and YNB + Glu media. The expression of MDH1 as well as MDH2 is regulated by Mxr1p only in cells cultured in YNB + Glu but not YP medium as evident from qPCR studies (Fig. 6C). Mxr1p also regulates the expression of GLN1 in cells cultured in YNB + Glu as well as YP media (Fig. 6D). GLN1 was expressed from its own promoter as a Myc-tagged protein, and its localization in the cytoplasm was confirmed by immunofluorescence using anti-Myc tag antibodies (Fig. 6E). The function of GLN1 could not be examined further as a Δgln1 strain could not be generated, despite several attempts. Thus, overexpression of Mxr1p in Δmxr1 strain results in the restoration of synthesis of several key enzymes involved in amino acid metabolism in cells cultured in YP as well as YNB + Glu media. In contrast, Mxr1pN400 restores the expression of only AAT1 and AAT2 in cells cultured in YP medium. It does not activate the transcription of any other target gene of Mxr1p in cells cultured in YNB + Glu medium. These results are summarized in Table 3.
FIGURE 6.

Function and regulation of mMDH and cMDH in P. pastoris cells cultured in YP and YNB + Glu media. A, localization of his-tagged mMDH and cMDH in the mitochondria and cytoplasm, respectively, by immunofluorescence using anti-His tag antibodies. The genes were expressed from their own promoters. Hsp70 was used as mitochondrial marker. DAPI was used as nuclear as well as mitochondrial marker. B, effect of deletion of genes encoding mMDH and cMDH on the growth of P. pastoris in YPD, YNB + 2% glucose (YNBD), YP, and YNB + Glu media. C and D, quantification of MDH1, MDH2, and GLN1 mRNAs by qPCR in various P. pastoris strains as indicated. Error bars indicate mean ± S.D. *, p < 0.05; **, p < 0.005; ***, p < 0.0005; ns, not significant. One-way analysis of variance followed by Tukey's multiple comparison test was carried out (n = 3). E, localization of Myc-tagged glutamine synthetase (GLN) in the cytoplasm by immunofluorescence using anti-Myc tag antibodies. GLN1 was expressed from its own promoter. DAPI was used to stain nuclei.
TABLE 3.
Restoration of mRNA/protein levels of target genes in Δmxr1 strain by Mxr1p/Mxr1pN400 in cells cultured in YP or YNB + Glu media
| Transcription factor | Target genes |
|||||
|---|---|---|---|---|---|---|
| GDH2a | AAT1 | AAT2 | MDH1 | MDH2 | GLN1 | |
| YP medium | ||||||
| Mxr1p | + | + | + | − | − | + |
| Mxr1pN400 | − | + | + | − | − | − |
| YNB + Glu medium | ||||||
| Mxr1p | + | + | + | + | + | + |
| Mxr1pN400 | − | − | − | − | − | − |
a Mxr1p regulates the synthesis of GDH2 protein but not GDH2 mRNA.
Identification of Mxr1p Response Elements (MXREs) in the Promoters of AAT2, MDH2, and GLN1
Mxr1p regulates transcription of target genes by binding to MXREs present in their promoters. The MXREs identified so far contain a core motif whose consensus sequence is 5′-CYCCNY-3′ (Fig. 7A) (6, 7, 9, 13). A sequence similar to this core MXRE is present between −151 and −146 bp (site A) of the promoter of AAT2, −213 and −208 bp of the promoter of MDH2, and −528 and −523 bp of the promoter encoding GLN1 (Fig. 7, B, D, and E). In the region between −118 and −113 bp (site B) of AAT2 promoter, the sequence 5′-CCCCGA-3′ is present (Fig. 7B), which is similar to the consensus sequence 5′-CYCCNR-3′. Studies with AOX1 promoter have shown that Mxr1p binds only to 5′-CYCCNY-3′ but not 5′-CYCCNR-3′ (6). To examine the ability of Mxr1p to bind to site A or site B of the AAT2 promoter, oligonucleotides carrying mutations in either site B (pAAT2-MXRE-M1), site A (pAAT2-MXRE-M2), or both (pAAT2-MXRE-M3) were synthesized (Fig. 7C), and their ability to bind to recombinant His-tagged Mxr1pN150 (6, 9) was examined by EMSA. Similarly, oligonucleotides carrying point mutations within the putative core MXREs of promoters encoding MDH2 and GLN1 were synthesized (Fig. 7, D and E), and their ability to bind to recombinant His-tagged Mxr1pN150 was examined by EMSA. The results indicate that Mxr1p150 binds specifically to pAAT2-MXRE as well as pAAT2-MXRE-M1 but not pAAT2-MXRE-M2 and pAAT2-MXRE-M3 (Fig. 7F). Thus, Mxr1p binds specifically to site A but not site B of the AAT2 promoter. Addition of anti-His tag antibodies resulted in the supershift of the DNA-protein complexes, confirming the presence of recombinant Mxr1pN150 in these complexes (Fig. 7F). Mxr1pN150 also binds specifically to pMDH2-MXRE and pGLN1-MXRE but not to pMDH2-MXRE-M1 and pGLN1-MXRE-M1carrying point mutations within the core MXREs (Fig. 7F).
FIGURE 7.

Analysis of Mxr1p binding to MXREs present in the promoters of target genes by EMSA and ChIP. A, consensus sequence of core MXRE (6, 7, 13). B, nucleotide sequence of AAT2 promoter region between −105 and −162 bp. The nucleotide sequence of site A exactly matches with the pAOXI-MXRE consensus sequence (6) and that of site B differs by one nucleotide. C, nucleotide sequence of pAAT2-MXRE oligonucleotides used in EMSA. Mutated bases are indicated by arrows. D, oligonucleotides encompassing the MDH2 promoter region between −191 and −230 bp. The nucleotide sequence between −208 and −213 bp exactly matches with the MXRE consensus sequence. Mutated base is indicated by an arrow. E, oligonucleotides encompassing the GLN1 promoter region between −477 and −536 bp. The nucleotide sequence between −528 and −523 bp exactly matches with the MXRE consensus sequence. Mutated base is indicated by an arrow. F, EMSA with radiolabeled oligonucleotides and histidine-tagged recombinant Mxr1p containing 150 N-terminal amino acids encompassing the DNA binding domain (Mxr1p150) as indicated. Mxr1p150 has been described (6, 7). Supershift of DNA-Mxr1p150 complex by the addition of anti-His antibodies is shown. G, Western blotting analysis of lysate of MXR1N400-OE strain cultured in YP medium using anti-Myc antibodies. H, schematic representation of location of primers used in the PCRs for the amplification of promoter regions of AAT2 and GLN1 following ChIP. ChIP was carried out using anti-Myc antibody. Black boxes indicate core MXREs (5′-CYCCNY-3′). I, analysis of enrichment of ChIP-ed DNA over the reference sample by qPCR (21, 22). The data are expressed as Mxr1pN400 binding (ChIP/input) to AAT2 and GLN1 relative to binding (ChIP/input) at the TEL (telomere) region. TEL was used as a reference (14). Input refers to the amount of chromatin used in ChIP. The data represent the average of two independent experiments. J, table depicting correlation between promoter occupation by Mxr1pN400 in vivo and transcript levels of target genes (Table 3) in MXR1N400-OE strain cultured in YP medium.
Chromatin immunoprecipitation (ChIP) studies were carried out to study the ability of Mxr1pN400 to bind to MXREs present in the promoters of AAT2 and GLN1 in cells cultured in YP medium because Mxr1pN400 activates the transcription of AAT2 but not GLN1 under these culture conditions (Table 3). Extracts were prepared from MXR1N400-OE strain cultured in YP, and the presence of Myc-tagged Mxr1pN400 was confirmed by Western blotting (Fig. 7G). Mxr1pN400 was cross-linked to chromatin and immunoprecipitated with anti-Myc antibodies. DNA was extracted, and PCR was carried out with primers that amplify AAT2 and GLN1 promoter regions containing MXREs (Fig. 7H). Only AAT2 but not GLN1 promoter was amplified by PCR (Fig. 7I) indicating that Mxr1pN400 binds to the promoter of AAT2 but not GLN1 in cells cultured in YP medium. Thus, there is good correlation between promoter occupation in vivo and transcriptional activation by Mxr1pN400 (Fig. 7J).
Discussion
The integration of carbon and nitrogen metabolism with energy production is crucial for the survival of living organisms. Although yeasts such as S. cerevisiae utilize amino acids primarily for nitrogen metabolism, respiratory yeasts such as P. pastoris and P. stipitis have evolved pathways for efficient utilization of amino acids both as a source of carbon and nitrogen. The generation of TCA cycle intermediates such as α-ketoglutarate and oxaloacetate from glutamate and aspartate is the first step in the utilization of amino acids as the sole source of carbon. While examining the role of Mxr1p in the regulation of various metabolic pathways, it was observed that P. pastoris GS115 but not Δmxr1 can grow in media containing amino acids as the sole source of carbon. This led us to study the enzymes involved in the utilization of amino acids as well as to examine the role of Mxr1p in the regulation of their biosynthesis. We demonstrate that enzymes such as GDH2, cAAT, mAAT, cMDH, and mMDH, which play a crucial role in the inter-conversion of amino acids and keto acids in the cytosolic and mitochondrial compartments as well as glutamine synthetase involved in glutamine synthesis, are important for the utilization of amino acids as the sole source of carbon and nitrogen in P. pastoris. Disruption of genes encoding cAAT and cMDH impairs the ability of P. pastoris to grow in media containing amino acids as the sole source of carbon. Although mAAT is not required for growth in YP, its disruption results in growth retardation in YNB + Glu medium. Disruption of MDH1 severely impairs growth in media containing not only amino acids but also glucose as the sole source of carbon.
An important aspect of this study is the identification of Mxr1p as a regulator of synthesis of key enzymes involved in the utilization of amino acids. Biosynthesis of AAT1, AAT2, MDH1, MDH2, and GLN1 is regulated by Mxr1p at the level of transcription as evident from the significant reduction in mRNA levels of genes encoding these enzymes in Δmxr1 strain. Promoters of AAT2, MDH2, and GLN1 contain MXREs to which recombinant Mxr1p binds specifically in vitro. Using AAT2 and GLN1 as examples, we have demonstrated the importance of promoter occupancy by Mxr1p in vivo for transcriptional activation of these genes. Key enzymes identified in this study, whose synthesis is regulated by Mxr1p, are depicted schematically in Fig. 8A. We conclude that Mxr1p functions as a global regulator of multiple metabolic pathways in P. pastoris (Fig. 8B).
FIGURE 8.
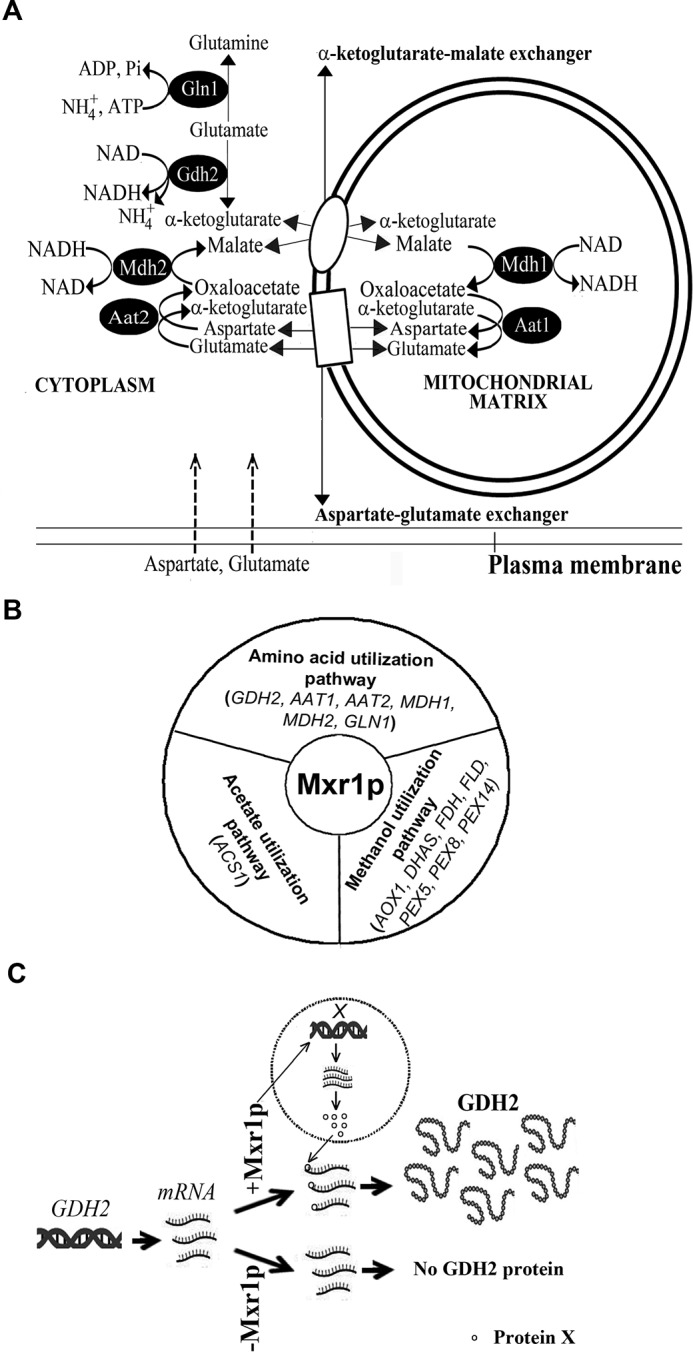
A, schematic representation of key enzymes involved in the utilization of amino acids as the sole source of carbon in P. pastoris whose expression is regulated by Mxr1p. B, schematic diagram depicting Mxr1p as a global regulator of multiple metabolic pathways. Genes regulated by Mxr1p in various metabolic pathways are indicated. Mxr1p target genes in methanol and acetate utilization pathways have been described (5, 13). C, model for the translational regulation of GDH2 by Mxr1p in cells cultured in YP or YNB + Glu media. We hypothesize that Mxr1p regulates the transcription of a gene encoding protein X, which is required for the translation of GDH2 mRNA. See under “Discussion” for details.
Amino acids enhance growth rate as well as recombinant protein production in several yeast species when added to media containing conventional carbon sources, primarily by serving as precursors for the synthesis of proteins as well as participating in anaplerotic reactions (16–19). In the case of P. pastoris, mixed feeds of methanol and a multicarbon source instead of methanol as the sole carbon source have been shown to improve recombinant protein production (20). However, metabolic flux analysis during methanol metabolism in the presence of amino acids as a second source of carbon has not been examined thus far. The various P. pastoris strains described in this study can be exploited for these studies. Such studies may provide new insights into the potential interactions between methanol metabolism and amino acid metabolism on heterologous protein production, leading to novel biotechnological applications.
A surprising result of this study is the regulation of GDH2 expression by Mxr1p at the translational level rather than the transcriptional level as evident from the reduction in GDH2 protein but not mRNA levels in the Δmxr1 strain. Furthermore, expression of Mxr1p but not Mxr1pN400 in Δmxr1 restores GDH2 protein levels indicating that the N-terminal region of Mxr1p alone is not sufficient for the post-transcriptional regulation of GDH2. GDH2 is the first example of a gene whose expression is regulated by Mxr1p at the post-transcriptional level (Fig. 8C). Mxr1p may not be directly involved in the regulation of translation of GDH2 mRNA because it localizes to the nucleus of cells cultured in YP medium. We hypothesize that Mxr1p activates the transcription of a gene whose product (protein X) is required for the efficient translation of GDH2 mRNA (Fig. 8C).
Experimental Procedures
Media and Culture Conditions
P. pastoris (GS115, his−) was cultured in either minimal medium containing 0.17% yeast nitrogen base (YNB) without amino acids and 0.5% ammonium sulfate supplemented with 2.0% glucose (YNBD), 1.0% glutamate (YNB + Glu), or 1% aspartate (YNB + Asp) or nutrient-rich YP medium (1.0% yeast extract and 2.0% peptone) alone or YP medium containing 2.0% glucose (YPD) or 2% methanol (YPM). S. cerevisiae BY4741 strain (Euroscarf) was cultured in YPD/YP medium. Yeast strains were grown at 30 °C in an orbital shaker at 180 rpm. For growth assays, colonies were first cultured overnight in YPD medium, then washed with sterile water, and shifted to different media with initial A600 of ∼0.1. To assay growth in liquid medium, P. pastoris cells grown overnight in YPD were diluted in duplicate with fresh YPD to A600 of 0.1, and aliquots of cells were removed at regular intervals, and A600 was measured. Escherichia coli DH5α and BL21 (DE3) strains were used for the isolation of recombinant plasmids and expression of recombinant proteins, respectively. Bacterial and yeast transformations were done by CaCl2 and electroporation method (Gene Pulser, Bio-Rad) respectively.
Antibodies and Other Reagents
Oligonucleotides and anti-FLAG antibodies were purchased from Sigma (Bangalore, India). Anti-His tag and anti-c-Myc tag antibodies were purchased from Thermo Fisher Scientific (Bangalore, India) and Merck Millipore (Bangalore, India) respectively.
Generation of P. pastoris Strain Expressing GDH2His from Its Own Promoter
The gene encoding GDH2 along with 545 bp of its promoter was cloned into pIB3 vector (Addgene) as a histidine-tagged protein (GDH2His). GDH2 along with −545 bp of promoter was amplified from genomic DNA of P. pastoris by the primer pair 5′-CCGGAATTCCTCTCATGTTCGATATTCCAGCGGCTTTC-3′ and 5′-CCGCTCGAGCTAATGATGATGATGATGATGCAATCCCCGAGACTTGTAC-3′. EcoRI and XhoI sites are underlined. The histidine tag-encoding region is shown in italics. The PCR product was digested with EcoRI and XhoI and cloned into the pIB3 vector at the EcoRI and XhoI sites to obtain pIB3-GDH2His. After linearization with BspHI, the plasmid was transformed to P. pastoris GS115 and Δmxr1 strains by electroporation to obtain GS115-GDH2His and Δmxr1-GDH2His strains, respectively.
Generation of P. pastoris Δmxr1 Strain Overexpressing Mxr1p, Mxr1pN400, or GDH2myc
P. pastoris Δmxr1 strain in which MXR1 coding region was replaced by a Zeocin expression cassette has been described (13). P. pastoris Δmxr1 strain overexpressing full-length Mxr1p (MXR1-OE) or a truncated Mxr1p containing 400 N-terminal amino acids (MXR1N400-OE) has been described (13). To generate Δmxr1 strain expressing Myc-tagged GDH2 (GDH2Myc) from the GAPDH promoter, the gene encoding GDH2 was amplified by PCR from P. pastoris genomic DNA using the primer pair 5′-CCGGAATTCATTATGGTCGACAGATCCAAG-3′ and 5′-CGGGGTACCCAATCCCCGAGACTTGTAC-3′. EcoRI and KpnI sites in the primers are underlined. The gene was cloned at the EcoRI and KpnI sites of the pGAPZA vector, and the recombinant plasmid thus obtained (pGAPDH-GDH2myc) was transformed into Δmxr1 strain to obtain Δmxr1-GDH2-OE1. The pCUP1-GDH2 expression cassette containing S. cerevisiae copper-inducible promoter (pCUP1) (15) was generated by a series of overlapping PCRs. The S. cerevisiae CUP1 promoter region between −460 and −1 bp was amplified by PCR from S. cerevisiae genomic DNA using the primer pair 5′-CCGGAATTCGCCGATCCCATTACCGAC-3′ (−460 to −443 bp of CUP promoter, EcoRI site is underlined) and 5′-GACACTTGGAGTCTGTCGACCATTTTATGTGATGATTGATTGATTGATTG-3′ (region from +1 to +23 bp of GDH2 and −32 to −1 bp of CUP1 promoter (italics)). In another PCR, GDH2 was amplified from P. pastoris genomic DNA using the primer pair 5′-CAATCAATCAATCAATCATCACATAAATGGTCGACAGACTCCAAGTGTC-3′ (−32 to −1 bp of CUP promoter, +1 to+23 bp of GDH2 (italics)) and 5′-CGGGGTACCCAATCCCCGAGACTTGTAC-3′ (BamHI site is underlined). The products of the two PCRs were used as templates in the final PCR and amplified using the primer pair 5′-CCGGAATTCGCCGATCCCATTACCAC-3′ and 5′-CGGGGTACCCAATCCCCGAGACTTGTAC-3′ (EcoRI and BamHI sites are underlined). The pCUP1-GDH2 expression cassette thus obtained was digested with EcoRI and KpnI and cloned into pGAPZA vector to obtain pCUP-GDH2myc plasmid. The recombinant plasmid was linearized with AvrII and transformed to P. pastoris Δmxr1 strain by electroporation to obtain Δmxr1-GDH2-OE2.
Construction of P. pastoris Δgdh2, Δaat1, Δaat2, Δmdh1, and Δmdh2 Strains
The coding regions of genes encoding GDH2, AAT1, AAT2, MDH1, and MDH2 were deleted by homologous recombination method, using resistance to Zeocin (ZeocinR) as selection marker. The gene deletion constructs for each of these genes consisted of ∼1 kb of promoter, ∼1.2 kb of Zeocin expression cassette, and ∼1 kb of 3′-flanking sequences. The promoters and 3′-flanking regions of respective genes were amplified by PCR from P. pastoris GS115 genomic DNA, and the Zeocin expression cassette was amplified from pGAPZA (Invitrogen).
For generating P. pastoris Δgdh2 strain, −930 bp of GDH2 promoter, 1.2 kb of coding region, and 1 kb of 3′-flanking region were amplified by PCR using the primer pair 5′-GCTGACCTATAGTTTGCTAGAACCGTTTTCTC-3′ (−930 to −898 bp of GDH2 promoter) and GCTATGGTGTGTGGGGGATCCGCAGGTAGTTTTAGTTTCTTTCTACTAATTG 3′ (+962 to +985 bp of pGAPZA and −52 to −23 bp of GDH2 promoter (italics)). In another PCR, the primer pair 5′-CAATTAGTAGAAAGAAACTAAAACTACCTGCGGATCCCCCACACACCATAGC-3′ (−52 to −24 bp of GDH2 promoter (italics),+962 to +985 bp of pGAPZA) and 5′-GATATCCCCATACTTTTCTTCTCTTTCCTTGCTCACATGTTGGTCTCCAGCTTGC-3′ (+3124 to +3152 bp of 3′-flanking region of GDH2 in reverse complement (italics) +2136 to +2160 bp in reverse complement of pGAPZA) was used. In the third PCR, the primer pair 5′-GCAAGCTGGAGACCAACATGTGAGCAAGGAAAGAGAAGAAAAGTATGGGGATATC-3′ (+2136 to +2160 bp of pGAPZA (italics) and 30 nucleotides of 3′ UTR from +3124 to +3152 bp of 3′-flanking region of GDH2) and 5′-CTTACTTCATTAAAAGAGGTCATTCTTCTGCTACTGC-3′ (+4071 to +4100 bp of 3′-flanking region of GDH2) were used, respectively. The three PCR products were purified and used as templates in the final PCR along with the primers 5′-GCTGACCTATAGTTTGCTAGAACCGTTTTCTC-3′ and 5′-CTTACTTCATTAAAAGAGGTCATTCTTCTGCTACTGC-3′ to generate the 3.2-kb GDH2 deletion construct that was transformed into P. pastoris GS115 by electroporation. Zeocin-resistant colonies were selected, and GDH2 deletion was confirmed by PCR and Southern blotting.
For generating P. pastoris Δaat1 strain, 1.025 kb of AAT1 promoter region was amplified by PCR using the primer pair 5′-CAATTGCAATGTGATACGGTGGTAC-3′ (−1025 to −1000 bp of AAT1) and 5′-GCTATGGTGTGTGGGGGATCCGCAGATGACTTGATATGGTCTGATTTGG-3′ (+962 to +985 bp of pGAPZA in reverse complement (italics), and 25 nucleotides of 5′ UTR from −25 to −1 bp of AAT1 in reverse complement). The 1.2-kb Zeocin expression cassette was amplified by PCR using the primer pair 5′-CCAAATCAGACCATATCAAGTCATCTGCGGATCCCCCACACACCATAGC-3′ (−25 to −1 bp of AAT1promoter (italics), +962 to +985 bp of pGAPZA) and 5′-CATTGGCAAGGTAGTCAACGTTTTGGCTCACATGTTGGTCTCCAGCTTG-3′ (+1230 to +1255 bp of 3′-flanking region of AAT1 in reverse complement (italics), +3124 to +3152 bp in reverse complement of pGAPZA). The 1.011 kb of 3′-flanking region of AAT1 was amplified by PCR using the primer pair 5′-CAAGCTGGAGACCAACATGTGAGCCAAAACGTTGACTACCTTGCCAATG-3′ (3124 to 3152 bp of pGAPZA (italics), +1230 to +1255 bp of 3′-flanking region of AAT1) and 5′-AGATCTCTCAAATACGATGGGGTC-3′(+2252 to +2275 bp of 3′-flanking region of AAT1). All three PCR products thus obtained were purified and used as templates in the final PCR along with the primers 5′-CAATTGCAATGTGATACGGTGGTAC-3′ and 5′-AGATCTCTCAAATACGATGGGGTC-3′ to generate the AAT1 deletion construct that was transformed to P. pastoris GS115. Zeocin-resistant colonies were selected, and deletion of AAT1 was confirmed by PCR using gene-specific primers.
For generating P. pastoris Δaat2 strain, 1.009 kb of AAT2 promoter region was amplified by PCR using the primer pair 5′-TATCACCATCAACTCTCGTTGATCTTTG-3′ (−1009 to −982 bp of AAT2) and 5′-GCTATGGTGTGTGGGGGATCCGCAGCTTAACGGTATGTTGACGAGTTC-3′ (+962 to +985 bp of pGAPZA in reverse complement (italics), −24 to −1 bp in reverse complement of AAT2). In another PCR, the 1.2-kb Zeocin expression cassette was amplified by PCR using the primer pair 5′-GAACTCGTCAACATACCGTTAAGCTGCGGATCCCCCACACACCATAGC-3′ (−24 to −1 bp of AAT2 (italics), +962 to +985 bp of pGAPZA) and 5′-CTTATCGTTTATAAATCACTCGTTGCTCACATGTTGGTCTCCAGCTTG-3′ (+1247 to +1270 bp in reverse complement of 3′-flanking region of AAT2 (italics), +3124 to +3152 bp in reverse complement of pGAPZA). In the third PCR, 1.045 kb of 3′-flanking region of AAT2 was amplified using the primer pair 5′-CAAGCTGGAGACCAACATGTGAGCAACGAGTGATTTATAAACGATAAG-3′ (+3124 to +3152 bp of pGAPZA (italics), +1247 to +1270 bp of 3′-flanking region of AAT2) and 5′-CTTCGTTGGTGAAAACCAAGATGCTTG-3′ (3′ UTR from +2257 to +2231 bp of 3′-flanking region of AAT2). All three PCR products were purified and used as templates in a final PCR along with the primers 5′-TATCACCATCAACTCTCGTTGATCTTTG-3′ and 5′-CTTCGTTGGTGAAAACCAAGATGCTTG-3′. The 3.2-kb PCR product thus obtained consisting of a Zeocin expression cassette flanked by ∼1 kb of AAT2 promoter and 1 kb of 3′-flanking region of AAT2 was transformed into P. pastoris GS115. Zeocin-resistant colonies were selected, and deletion of AAT2 was confirmed by PCR using gene-specific primers.
For generating P. pastoris Δmdh1 strain, ∼1 kb of MDH1 promoter was amplified by PCR using the primer pair 5′-TGTGTAGCTCTGAACTCGTTG-3′ (−921 to −900 bp of MDH1) and 5′-CTATGGTGTGTGGGGGATCCGCATGAAGGGATTATAATGGTTG-3′ (962 to 984 bp of pGAPZA in reverse complement (italics), −20 to −1 bp of MDH1 in reverse complement). In another PCR, the Zeocin expression cassette was amplified by PCR using the primer pair 5′-CAACCATTATAATCCCTTCATGCGGATCCCCCACACACCATAG-3′ (−20 to −1 bp of MDH1 (italics), +962 to +984 bp of pGAPZA) and 5′-CGTTCAATATCGTCACCCAGGAGCTCACATGTTGGTCTCCAGC-3′ (+1060 to +1081 bp in reverse complement of 3′-flanking region of MDH1 (italics), +3124 to +3147 bp in reverse complement of pGAPZA). In the third PCR, the 3′-flanking region of MDH1 was amplified using the primer pair 5′-GCTGGAGACCAACATGTGAGCTCCTGGGTGACGATATTGAACG-3′ (+3130 to +3151 bp of pGAPZA, +1060 to +1081 bp of 3′-flanking region of MDH1) and 5′-CGTGGGGATGGACATGATCGTGG-3′ (+1997 to +2019 bp in the reverse complement of 3′-flanking region of MDH1). All three PCR products were purified and used as templates in the final PCR along with the primer pair 5′-CGTGGGGATGGACATGATCGTGG-3′ and 5′-TGTGTAGCTCTGAACTCGTTG-3′ to obtain a 3.2-kb product consisting of Zeocin expression cassette flanked by ∼1 kb of MDH1 promoter and 1 kb of 3′-flanking region of MDH1 that was transformed into P. pastoris GS115. Zeocin-resistant colonies were selected, and deletion of MDH1 was confirmed by PCR using gene-specific primers.
For generating P. pastoris Δmdh2 strain, ∼1-kb MDH2 promoter was amplified by PCR using the primer pair 5′-GAGGACGTGTGGGATTAGAAAG-3′ (−1047 to −1026 bp of MDH2) and 5′-CTATGGTGTGTGGGGGATCCGCATTTGTTATTGTTCGTTGGTTGTAAC-3′ (+962 to +985 bp of pGAPZA in reverse complement (italics), −24 to −1 bp in reverse complement of MDH2). The Zeocin expression cassette was amplified by PCR using the primer pair 5′-GTTACAACCAACGAACAATAACAAATGCGGATCCCCCACACACCATAG-3′ (−24 to −1 bp of MDH2 (italics), +962 to +984 bp of pGAPZA) and 5′-GAGACCAGCCCAGAGTGCACAATGCTCACATGTTGGTCTCCAGC-3′ (+1232 to +1054 bp in reverse complement of 3′-flanking region of MDH2 (italics), +3124 to +3152 bp in reverse complement of pGAPZA). In the third PCR, the 3′-flanking region of MDH2 was amplified using the primer pair 5′-GCTGGAGACCAACATGTGAGCATTGTGCACTCTGGGCTGGTCTC-3′ (+3130 to +3151 bp of pGAPZA (italics), +2017 to +2040 bp of 3′-flanking region of MDH2) and 5′-AATATGCTAGTCATGTGACCATAG-3′ (+2017 to +2040 bp in reverse complement of 3′-flanking region of MDH2). All three PCR products were purified and used as templates in a final PCR along with the primer pair 5′-GAGGACGTGTGGGATTAGAAAG-3′ and 5′-AATATGCTAGTCATGTGACCATAG-3′. The 3.2-kb PCR product thus obtained consisting of Zeocin expression cassette flanked by ∼1 kb of MDH2 promoter and 1 kb of 3′-flanking region of MDH2 was transformed into P. pastoris GS115. Zeocin-resistant colonies were selected, and deletion of MDH2 was confirmed by PCR using gene-specific primers.
Generation of P. pastoris Strains AAT1Myc, AAT2Myc, MDH1His, MDH2His, and GLN1Myc
Expression cassettes consisting of genes encoding AAT1/AAT2 or MDH1/MDH2 along with ∼1 kb of their own promoters were cloned into pGAPZA or pIB3 vector (Addgene) and expressed in P. pastoris GS115 as Myc- or His-tagged proteins. AAT1 and AAT2 encoding mitochondrial (mAAT) and cytosolic AAT (cAAT), respectively, were cloned into pGAPZA vector, whereas MDH1 and MDH2 encoding mitochondrial MDH (mMDH) and cytosolic MDH (cMDH), respectively, were cloned into pIB3 vector. AAT1 was amplified from P. pastoris GS115 genomic DNA by the primer pair 5′-CGGGGTACCCAATTGCAATGTGATACGGTGGTAC-3′ and 5′-ATAAGAATGCGGCCGCTGTTGGTCGTAACCTCGTGG-3′. The KpnI and NotI sites in the primers are underlined. Because AAT2 had an intron from +716 to +800 bp, AAT2 cDNA was obtained by RT-PCR from RNA. The AAT2 expression cassette was generated as follows: AAT2 promoter (−1000 bp) was amplified by PCR using the primer pair 5′-CGGGGTACCTATCACCATCAACTCTCGTTGATCTTTG-3′ (KpnI site is underlined) and 5′-GATATTCTGCGTTGAAAACGACATGCTTAACGGTATGTTGACGAGTTC-3′ (+1 to +24 bp of AAT2, −24 to −1 bp of AAT2 in italics) from genomic DNA. AAT2 coding region was obtained by RT-PCR from P. pastoris RNA and the primer pair 5′-GAACTCGTCAACATACCGTTAAGCATGTCGTTTTCAACGCAGAATATC-3′ (−24 to −1 bp of AAT2 in italics, +1 to +24 bp of AAT2) and 5′-ATAAGAATGCGGCCGCTTACACGCACAACCTGATCAATG-3′ (NotI site is underlined). Products of the two PCRs were purified and used as templates in the final PCR along with the primer pair, 5′-CGGGGTACCTATCACCATCAACTCTCGTTGATCTTTG-3′ and ATAAGAATGCGGCCGCTTACACGCACAACCTGATCAATG-3′. AAT1and AAT2 expression cassettes were cloned into the KpnI and NotI sites of pGAPZA vector to obtain pGAPZA-AAT1Myc and pGAPZA-AAT2Myc vectors, respectively. These plasmids were transformed into P. pastoris GS115 to generate AAT1Myc and AAT2Myc, respectively.
MDH1 was amplified by PCR with the primer pair 5′-CGCGGATCCTGTGTAGCTCTGAACTCGTTG-3′ (EcoRI site is underlined) and 5′-CCGCTCGAGCTAATGATGATGATGATGATGTGGGTTTTGCTTAACAAACTC-3′ (XhoI site underlined, DNA encoding His tag is shown in italics). MDH2 was amplified by PCR using the primer pair 5′-CGCGGATCCGAGGACGTGTGGGATTAGAAAG-3′ (EcoRI site is underlined) and 5′-CCGCTCGAGCTAATGATGATGATGATGATGGTTGCCAGCAATGAAGGCAGTTC-3′ (XhoI site is underlined; DNA encoding His tag is shown in italics). P. pastoris genomic DNA was used as the template. The MDH1 and MDH2 expression cassettes were digested with EcoRI and XhoI and cloned into pIB3 vector to obtain pIB3-MDH1His and pIB3-MDH2His, respectively. These plasmids were transformed into P. pastoris GS115 to obtain MDH1His and MDH2His strains, respectively.
GLN1 along with −1000 bp of its own promoter was amplified by PCR with primer pairs 5′-CCGCTCGAGTCAGTATTAATCTTGTCACATGACCTAC-3′ and 5′-TAAGAATGCGGCCGCTATCAGATTCTCTCTTGTACTCCTTG-3′ fromP. pastoris genomic DNA. KpnI and NotI restriction sites are underlined. PCR product was digested with KpnI and NotI and cloned into pGAPZA vector. The recombinant plasmid (pGAPZA-GLN1Myc) was linearized and transformed to GS115 to obtain GLN1Myc strain. P. pastoris strains and plasmids used in this study are listed in Tables 1 and 2, respectively.
TABLE 2.
Plasmids used in this study
| Plasmid | Description | Source/Ref. |
|---|---|---|
| pIB3 | P. pastoris expression vector with HIS4 selection marker | Addgene |
| pIB3-GDH2His | pIB3 expressing GDH2His from − 545 bp of P. pastoris GDH2 promoter | This study |
| pGAPZA | P. pastoris expression vector with GAPDH promoter and ZeocinR selection marker | Invitrogen |
| pGAPBA | Modified pGAPZA vector with GAPDH promoter and BlasticidinR selection marker | 13 |
| pGAPDH-GDH2Myc | pGAPZA vector expressing Myc-tagged GDH2 from GAPDH promoter | This study |
| pCUP-GDH2Myc | pGAPZA vector expressing Myc-tagged GDH2 from CUP1 promoter | This study |
| pGAPBA-MXR1Myc | pGAPBA vector expressing Mxr1p from GAPDH promoter | 13 |
| pGAPBA-MXR1N400 | pGAPBA vector expressing Mxr1pN400 from GAPDH promoter | 13 |
| pGAPZA-AAT1Myc | pGAPZA vector expressing Myc-tagged AAT1 from − 1000 bp of AAT1 promoter | This study |
| pGAPZA-AAT2Myc | pGAPZA vector expressing Myc-tagged AAT2 from − 1000 bp of AAT2 promoter | This study |
| pIB3-MDH1His | pIB3 vector expressing His-tagged MDH1 from − 1000 bp of MDH1 promoter | This study |
| pIB3-MDH2His | pIB3 vector expressing His-tagged MDH2 from − 1000 bp of MDH2 promoter | This study |
| pGAPZA-GLN1Myc | pGAPZA vector expressing Myc-tagged GLN1 from − 1000 bp of GLN1 promoter | This study |
Western Blotting
Total protein extracts were prepared from P. pastoris by vortexing with glass beads, and cell lysates containing 50 μg of proteins were resolved by PAGE in the presence of SDS. The resolved proteins were transferred to a PVDF membrane using an electroblotting apparatus in transfer buffer (39 mm glycine, 48 mm Tris-HCl (pH 8.0), 20% methanol). The membrane was blocked overnight in 5% nonfat milk (HiMedia) prepared in 1× TBS (25 mm Tris-HCl, 125 mm NaCl (pH 8.0)). The protein of interest was detected by using antibodies raised against the specific protein or anti-epitope tag antibodies. Primary antibodies were detected by peroxidase-conjugated anti-rabbit/anti-mouse IgG secondary antibody (1:10,000 dilutions; Bangalore Genei, India). The blots were developed with chemiluminescence plus reagents (PerkinElmer Life Sciences) according to the manufacturer's instructions.
Subcellular Localization
Subcellular localization of epitope-tagged proteins was studied by immunofluorescence using a fluorescent microscope (Leica) and Zeiss confocal microscope (Zeiss 510 Meta). Immunofluorescence was carried out essentially as described (8).
Quantitative Real Time-PCR (qPCR)
RNA isolation from P. pastoris cells was carried out as described previously (8). qPCR was performed using iQ SYBR Green Super Mix and a iQ5 multi-Color real time PCR thermal cycler (iCycler, Bio-Rad). Levels of mRNA in mutant P. pastoris strains relative to GS115 were normalized to 18S mRNA. Data were analyzed by the comparative Ct method for relative quantification (ΔΔCt method), which describes the change in expression of the target genes in a test sample relative to a calibrator sample. The following primer pairs were used in qPCRs: MXR1, 5′-TGCTGAAACTTGGATGAAC-3′ and 5′-TCGGATATAATATAGGCTCTGAAT-3′; GDH2, 5′-CCGCAGTTGGTATGTTC-3′ and 5′-CCTCTTGGTGTGATTGGA-3′; AAT1, 5′-CTCTACAACCAGGACACTAA-3′ and 5′-CATGGCTTACCATTCTCATC-3′; AAT2, 5′-TTGGCTGGATTCTTGGAT-3′ and 5′-ATAGAGGAAGATGCTGGTAC-3′; MDH1, 5′-TCTCAATCTAAGCACAAGGA-3′ and 5′-GGCTCAACGACATCATTC-3′; MDH2, 5′-AGGAGTCGGTAAGGATCTA and 5′-TTAGCCAAGTCTCTGATGAT-3′; GLN1, 5′-TTCCATCCTAAGCCACTG and 5′-TCGTTGTCAGAACCATATAGA-3′; 18S rRNA, 5′-AATGAGGATTGACAGGATGA-3′ and 5′-AGGTCTCGTTATCG-3′.
Statistical Analysis
Statistical tests were carried out by one-way analysis of variance followed by Tukey's multiple comparison. GraphPad Prism 5 software was used. Data are presented as mean ± S.D. p value summary is mentioned on the bar of each figure as follows: *, p < 0.05; **, p < 0.005; ***, p < 0.0005, ns, not significant.
DNA-Protein Interactions
Recombinant Mxr1pN150 consisting of 150 N-terminal amino acids of Mxr1p was expressed as His-tagged protein in E. coli and purified essentially as described (5). The ability of Mxr1pN150 to bind to 32P-labeled oligonucleotides containing MXREs of promoters encoding AAT2, MDH2, and GLN1 was examined by electrophoretic mobility shift assay (EMSA) essentially as described previously (5).
Chromatin Immunoprecipitation (ChIP)
ChIP was performed essentially as described (21). The following primer pairs were used for qPCR: AAT2, 5′-GTTTATCCGCTTGGACCGTG-3′ (−239 to −219 bp of AAT2 promoter) and 5′-GCTTAACGGTATGTTGACGAGTTC-3′ (−24 to −1 bp of AAT2 promoter); GLN1, 5′-GAACGACCATTTACTTTTTTAGATAGGC-3′ (−550 to −576 bp of GLN1 promoter) and 5′-GAACTTTCTGGAGAAGAAACGCTG-3′ (−390 to −367 bp of GLN1 promoter);TEL, 5′-CAGGGCCAGATGGAAAAATA-3′ and 5′-AACCGTTTACTACCGCATGG-3′ (14). ChIP qPCR results were analyzed based on the methodology described by Chakrabarti et al. (22). Binding of Mxr1p to the promoters of AAT2 and GLN1 was normalized with input. Telomeric (TEL) DNA was taken as control for the nonspecific DNA binding region (14).
Author Contributions
U. S. and P. N. R. designed the study, and U. S. performed all the experiments. P. N. R. wrote the paper. U. S. and P. N. R. analyzed the results and approved the final version of the manuscript.
Supplementary Material
This work was supported by Research Grant BT/PR3889/BRB/10/996/2011 from the Department of Biotechnology, New Delhi, and J. C. Bose Fellowship Grant SB/S2/JCB-025/2015 by the Science and Engineering Research Board, New Delhi, India (to P. N. R.). The authors declare that they have no conflicts of interest with the contents of this article.

This article contains supplemental data.
- AAT
- aspartate aminotransferase
- YNB
- yeast nitrogen base
- YP
- yeast extract and peptone
- m
- mitochondria
- c
- cytoplasm
- MDH
- malate dehydrogenase
- qPCR
- quantitative PCR.
References
- 1. Magasanik B. (2003) Ammonia assimilation by Saccharomyces cerevisiae. Eukaryot. Cell 2, 827–829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Miller S. M., and Magasanik B. (1990) Role of NAD-linked glutamate dehydrogenase in nitrogen metabolism in Saccharomyces cerevisiae. J. Bacteriol. 172, 4927–4935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Freese S., Vogts T., Speer F., Schäfer B., Passoth V., and Klinner U. (2011) C- and N-catabolic utilization of tricarboxylic acid cycle-related amino acids by Scheffersomyces stipitis and other yeasts. Yeast 28, 375–390 [DOI] [PubMed] [Google Scholar]
- 4. Easlon E., Tsang F., Skinner C., Wang C., and Lin S. (2008) The malate-aspartate NADH shuttle components are novel metabolic longevity regulators required for calorie restriction-mediated life span extension in yeast. Genes Dev. 22, 931–944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Lin-Cereghino G. P., Godfrey L., de la Cruz B. J., Johnson S., Khuongsathiene S., Tolstorukov I., Yan M., Lin-Cereghino J., Veenhuis M., Subramani S., and Cregg J. M. (2006) Mxr1p, a key regulator of the methanol utilization pathway and peroxisomal genes in Pichia pastoris. Mol. Cell. Biol. 26, 883–897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kranthi B. V., Kumar R., Kumar N. V., Rao D. N., and Rangarajan P. N. (2009) Identification of key DNA elements involved in promoter recognition by Mxr1p, a master regulator of methanol utilization pathway in Pichia pastoris. Biochim. Biophys. Acta 1789, 460–468 [DOI] [PubMed] [Google Scholar]
- 7. Kranthi B. V., Kumar H. R., and Rangarajan P. N. (2010) Identification of Mxr1p-binding sites in the promoters of genes encoding dihydroxyacetone synthase and peroxin 8 of the methylotrophic yeast Pichia pastoris. Yeast 27, 705–711 [DOI] [PubMed] [Google Scholar]
- 8. Kumar N. V., and Rangarajan P. N. (2011) Catabolite repression of phosphoenolpyruvate carboxykinase by a zinc finger protein under biotin and pyruvate carboxylase-deficient conditions in Pichia pastoris. Microbiology 157, 3361–3369 [DOI] [PubMed] [Google Scholar]
- 9. Kumar N. V., and Rangarajan P. N. (2012) The zinc finger proteins Mxr1p and repressor of phosphoenolpyruvate carboxykinase (ROP) have the same DNA binding specificity but regulate methanol metabolism antagonistically in Pichia pastoris. J. Biol. Chem. 287, 34465–34473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Sahu U., Krishna Rao K., and Rangarajan P. N. (2014) Trm1p, a Zn(II)2Cys6-type transcription factor, is essential for the transcriptional activation of genes of methanol utilization pathway, in Pichia pastoris. Biochem. Biophys. Res. Commun. 451, 156–164 [DOI] [PubMed] [Google Scholar]
- 11. Wang X., Cai M., Shi L., Wang Q., Zhu J., Wang J., Zhou M., Zhou X., and Zhang Y. (2016) PpNrg1 is a transcriptional repressor for glucose and glycerol repression of AOX1 promoter in methylotrophic yeast Pichia pastoris. Biotechnol. Lett. 38, 291–298 [DOI] [PubMed] [Google Scholar]
- 12. Wang X., Wang Q., Wang J., Bai P., Shi L., Shen W., Zhou M., Zhou X., Zhang Y., and Cai M. (2016) Mit1 transcription factor mediates methanol signalling and regulates alcohol oxidase 1 promoter in Pichia pastoris. J. Biol. Chem. 291, 6245–6261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Sahu U., and Rangarajan P. N. (2016) Regulation of acetate metabolism and acetyl Co-A synthetase 1 (ACS1) expression by methanol expression regulator 1 (Mxr1p) in the methylotrophic yeast, Pichia pastoris. J. Biol. Chem. 291, 3648–3657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Parua P. K., Ryan P. M., Trang K., and Young E. T. (2012) Pichia pastoris 14-3-3 regulates transcriptional activity of the methanol inducible transcription factor Mxr1 by direct interaction. Mol. Microbiol. 85, 282–298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Mascorro-Gallardo J. O., Covarrubias A. A., and Gaxiola R. (1996) Construction of a CUP1 promoter-based vector to modulate gene expression in Saccharomyces cerevisiae. Gene 172, 169–170 [DOI] [PubMed] [Google Scholar]
- 16. Bakker B. M., Overkamp K. M., van Maris A. J., Kötter P., Luttik M. A., van Dijken J. P., and Pronk J. T. (2001) Stoichiometry and compartmentation of NADH metabolism in Saccharomyces cerevisiae. FEMS Microbiol. Rev. 25, 15–37 [DOI] [PubMed] [Google Scholar]
- 17. Görgens J. F., van Zyl W. H., Knoetze J. H., and Hahn-Hägerdal B. (2005) Amino acid supplementation improves heterologous protein production by Saccharomyces cerevisiae in defined medium. Appl. Microbiol. Biotechnol. 67, 684–691 [DOI] [PubMed] [Google Scholar]
- 18. Görgens J. F., Passoth V., van Zyl W. H., Knoetze J. H., and Hahn-Hägerdal B. (2005) Amino acid supplementation, controlled oxygen limitation and sequential double induction improves heterologous xylanase production by Pichia stipitis. FEMS Yeast Res. 5, 677–683 [DOI] [PubMed] [Google Scholar]
- 19. Heyland J., Fu J., Blank L. M., and Schmid A. (2011) Carbon metabolism limits recombinant protein production in Pichia pastoris. Biotechnol. Bioeng. 108, 1942–1953 [DOI] [PubMed] [Google Scholar]
- 20. Jordà J., Jouhten P., Cámara E., Maaheimo H., Albiol J., and Ferrer P. (2012) Metabolic flux profiling of recombinant protein secreting Pichia pastoris growing on glucose:methanol mixtures. Microb. Cell Fact. 11, 57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Braunstein M., Rose A. B., Holmes S. G., Allis C. D., and Broach J. R. (1993) Transcriptional silencing in yeast is associated with reduced nucleosome acetylation. Genes Dev. 7, 592–604 [DOI] [PubMed] [Google Scholar]
- 22. Chakrabarti S. K., James J. C., and Mirmira R. G. (2002) Quantitative assessment of gene targeting in vitro and in vivo by the pancreatic transcription factor, Pdx1. Importance of chromatin structure in directing promoter binding. J. Biol. Chem. 277, 13286–13293 [DOI] [PubMed] [Google Scholar]
- 23. Kranthi B. V., Balasubramanian N., and Rangarajan P. N. (2006) Isolation of a single-stranded DNA-binding protein from the methylotrophic yeast, Pichia pastoris and its identification as ζ crystallin. Nucleic Acids Res. 34, 4060–4068 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.