

Abstract
Agp1 is a canonical biliverdin-binding bacteriophytochrome from the soil bacterium Agrobacterium fabrum that acts as a light-regulated histidine kinase. Crystal structures of the photosensory core modules (PCMs) of homologous phytochromes have provided a consistent picture of the structural changes that these proteins undergo during photoconversion between the parent red light-absorbing state (Pr) and the far-red light-absorbing state (Pfr). These changes include secondary structure rearrangements in the so-called tongue of the phytochrome-specific (PHY) domain and structural rearrangements within the long α-helix that connects the cGMP-specific phosphodiesterase, adenylyl cyclase, and FhlA (GAF) and the PHY domains. We present the crystal structures of the PCM of Agp1 at 2.70 Å resolution and of a surface-engineered mutant of this PCM at 1.85 Å resolution in the dark-adapted Pr states. Whereas in the mutant structure the dimer subunits are in anti-parallel orientation, the wild-type structure contains parallel subunits. The relative orientations between the PAS-GAF bidomain and the PHY domain are different in the two structures, due to movement involving two hinge regions in the GAF-PHY connecting α-helix and the tongue, indicating pronounced structural flexibility that may give rise to a dynamic Pr state. The resolution of the mutant structure enabled us to detect a sterically strained conformation of the chromophore at ring A that we attribute to the tight interaction with Pro-461 of the conserved PRXSF motif in the tongue. Based on this observation and on data from mutants where residues in the tongue region were replaced by alanine, we discuss the crucial roles of those residues in Pr-to-Pfr photoconversion.
Keywords: conformational change, crystal structure, histidine kinase, photoreceptor, signal transduction, tertiary structure, Agrobacterium, phytochrome, quaternary structure
Introduction
Phytochromes are a family of red light-absorbing photosensory proteins that covalently bind chromophores of the bilin type, which undergo light-induced conversion between the parent red light-absorbing (Pr)4 and the far-red light-absorbing (Pfr) forms. In plants, phytochromes have evolved to the dominating photoreceptors, controlling processes such as seed germination, seedling development, shade avoidance, or flower induction. DNA sequencing led to the discovery of phytochromes also in cryptophytes, heterokonts, green algae, fungi, and many bacteria (1–4), where they modulate diverse physiological processes in response to light conditions, hence enabling organisms to adapt to their environment. In Agrobacterium fabrum C58 (formerly Agrobacterium tumefaciens C58), the species of this study, phytochromes control bacterial conjugation (5). Typically, phytochromes have an N-terminal Per Arnt Sim (PAS) domain, followed by a GAF (cGMP-specific phosphodiesterases, adenylyl cyclases, and FhlA) (6) and a phytochrome-specific (PHY) domain, which together comprise the N-terminal sensory region that is referred to as the PCM. At the C terminus, most bacterial phytochromes have a histidine kinase (HK) module that consists of a dimerization and histidine phosphotransfer domain and a catalytic and ATP binding domain (7–9). A large variety of phytochrome-like proteins that contain a chromophore-binding GAF domain but do not follow the PAS-GAF-PHY arrangement has been found in cyanobacteria, such as RcaE (10, 11) or PixJ (12). These proteins are often termed cyanobacteriochromes (10), although the terms phytochrome or “group II phytochrome” have been used for SyA-Cph1 or SyB-Cph1, proteins with GAF and PHY domains (13) and Cph2 with a tandem GAF domain (14).
In phytochromes and cyanobacteriochromes, the natural bilin chromophore is covalently attached to the protein via a characteristic Cys residue. Fungal and most bacterial phytochromes incorporate a biliverdin (BV) chromophore that is bound to a Cys residue at the N terminus of the PAS domain (15–17). Most cyanobacterial phytochromes and cyanobacteriochromes incorporate a phycocyanobilin (PCB) chromophore, which is bound to a Cys in the GAF domain. The homologous Cys is also used in plant phytochromes, which incorporate a phytochromobilin (PΦB) chromophore (18). In prototypical phytochromes, the dark state is Pr, but some bacterial phytochromes termed bathy phytochromes have a thermodynamically stable Pfr state (19–21).
A wealth of structural knowledge of phytochromes has accumulated because of the original structural determination of the chromophore binding domain (CBD) of Deinococcus radiodurans phytochrome DrBphP, which contains the PAS and the GAF domains (22). Two structures of complete PCMs (containing PAS, GAF, and PHY domains) were simultaneously published in 2008, namely Cph1 from Synechocystis PCC 6803 (23) and the bathy phytochrome PaBphP from Pseudomonas aeruginosa (21). These two phytochromes were crystallized in their thermodynamically stable states Pr and Pfr, respectively. The arrangement of two protein subunits in the crystals is parallel in the case of PaBphP and anti-parallel in the case of Cph1. A parallel arrangement was also found for the CBD (24) and the PCM (25) of DrBphP, the PCMs of plant phytochrome (26) and of RpBphP2 from Rhodopseudomonas palustris (27), and other CBD or PCM structures (28). Anti-parallel orientations were observed in crystals of Cph2 (14) and of RpBphP3 from R. palustris (27).
Similarities between two different types of phytochromes as represented by Cph1 and PaBphP already indicated conserved structural features of phytochromes in general that are also valid for phytochrome structures described later, such as the conserved topologies in the PAS, GAF, and PHY domains and effective solvent shields that surround the bilin moieties (21, 23). The bilin chromophore is embedded in the GAF domain, which is connected with the PAS domain in a knotted structure. The PHY domain connects with the GAF domain via a long helix and a motif of the PHY domain termed the “arm” or the “tongue” that folds back to the chromophore pocket (21, 23). Comparison of the Cph1 Pr and PaBphP Pfr structures revealed also numerous differences that were shown later to be generally valid for the respective photochromic states of phytochromes. The conserved Asp of the PXXDIP motif in the GAF domain interacts with the chromophore via its backbone carbonyl group in Pr structures (22, 23) but interacts with ring D of the chromophore via its carboxylate side chain in PaBphP (21). The tongue of the PHY domain includes at least one anti-parallel β-sheet in all phytochrome crystal structures in the Pr state, with Cph1 being the first example (23). On the contrary, it is coils and an α-helix that comprise the tongue in all Pfr and Pfr-like structures, which are of PaBphP (21), RpBphP1 (28), and DrBphP (29, 30). These differences might reflect general differences between Pr and Pfr and thereby provide information about protein conformational changes during photoconversion. Yang et al. (31) found for a mutant of PaBphP that crystallized in a mixed Pr/Pfr state with the chromophore in ZZZssa and ZZEssa configuration/conformation that both photochromic forms have very similar global conformations. This result speaks against a rearrangement of secondary structure in the tongue in PaBphP. A model for the conformational changes that are associated with phytochrome signaling termed “tryptophan switch” was proposed based on a study on the group II phytochrome Cph2 and was suggested to be relevant for typical phytochromes as well (14). This model includes a large scale conformational change during photoconversion from Pr to Pfr at the tongue of the GAF2 domain in group II phytochromes, which structurally plays a similar role as the PHY domain in canonical phytochromes. Further detailed insights into structural changes during photoconversion were provided by x-ray crystallographic and small angle x-ray scattering studies on the PCM of DrBphP by Takala et al. (29) and Burgie et al. (25, 30). Although the former group could crystallize this PCM in the Pr state and as an illuminated form that was enriched in Pfr, the latter group managed to obtain crystal structures of the wild-type PCM in the Pr form and of the Pfr form after introducing a single site substitution in the middle of the tongue of the PHY domain. These studies show two anti-parallel β-strands in the tongue of the Pr and a short α-helix in the tongue of the Pfr structure. Although the Pfr subunit arrangements are significantly different in both studies and demand further investigation, the rearrangement of secondary structure in the tongue of the PHY domain during photoconversion seems to be a universal feature of bacterial phytochromes. The PHY domain connects directly to the HK of bacterial phytochromes and is the domain most relevant for intramolecular signal transduction from the PCM to the C terminus. How these and other protein conformational changes are induced by light triggered isomerization of the chromophore from ZZZssa to ZZEssa stereochemistry and how the signal is transmitted to the HK are questions that are still not answered on the structural level.
The phytochromes Agp1 and Agp2 from Agrobacterium have been extensively used as model systems for biochemical and spectroscopic studies related to chromophore protein interaction, light-induced conformational changes, or HK regulation, and it is desired to combine results from these studies with structural data (7, 32–37). Crystallization of the PAS-GAF-PHY fragment of Agp1 (Agp1-PCM) with the wild-type sequence has been reported previously (38), but crystallographic structure refinement could not be carried out satisfactorily (7). Here, we describe how we employed the surface entropy reduction (SER) strategy that was devised by Derewenda and co-workers (39–41) and how we obtained crystals of high diffraction quality. We could in this way determine the crystal structure of an “SER mutant” of the PCM of Agp1 at 1.85 Å resolution. With the high resolution structure of the Agp1-PCM SER mutant available, it was then possible to solve and refine the structure of Agp1-PCM to a resolution of 2.7 Å. The SER mutant crystals contain dimer subunits in anti-parallel arrangement, whereas dimer subunits are oriented parallel in the wild-type Agp1-PCM crystals. In addition to the crystal structures, we present biochemical analyses on Agp1 single-site mutants where highly conserved residues in the tongue of the PHY domain were substituted by alanine. Collective evidence from previously published work and our data suggests that these residues play similar roles in stabilizing the photochromic forms in both prototypical and bathy phytochromes.
Results and Discussion
Crystal Structure Analysis of the Agp1 Photosensory Core Module
We used crystals of the selenomethionine derivative (space group I422, unit cell parameters a = b = 171 Å, c = 81 Å, α = β = γ = 90°) of the adduct formed between the sterically locked BV derivative 15Za and Agp1-PCM (38, 42) to obtain a preliminary structural model of Agp1-PCM in a Pr-like state. The crystallographic phase problem was solved by a combination of molecular replacement using as search model the PAS-GAF fragment of the prototypical bacteriophytochrome DrBphP (22) and single wavelength anomalous dispersion (43). Crystallographic refinement at 3.2 Å resolution, however, was unsatisfactory, resulting in poorly defined electron densities for a significant number of amino acid side chains and the chromophore (7). We were then able to grow crystals of the Agp1-PCM adduct with the natural chromophore BV diffracting maximally to 2.7 Å resolution. These crystals belong to the same space group and have similar unit cell parameters as those of the locked chromophore adduct, indicating the same type of crystal packing (Table 1). Crystallographic refinement ended up with the same problems as observed with the locked chromophore adduct, even if solving the phase problem was solely based on molecular replacement, using in addition the PAS-GAF bidomain of DrBphP (22) and the PHY domain of the cyanobacterial phytochrome Cph1 (23) as search models.
TABLE 1.
Data collection and refinement statistics
| Agp1-PCM-SER13a | Wild-type Agp1-PCMa | |
|---|---|---|
| Data collection | ESRF, ID 14.4 | ESRF, ID 14.4 |
| Wavelength (Å) | 1.0718 | 0.9395 |
| Space group | P 43212 | I 4 2 2 |
| Cell dimensions | ||
| a, b, c (Å) | 69.35, 69.35, 236.84 | 173.02, 173.02, 78.82 |
| α, b, g (°) | 90.0, 90.0, 90.0 | 90.0, 90.0, 90.0 |
| Resolution (Å) | 34.67–1.85 (1.95–1.85)b | 46.55–2.70 (2.85–2.70) |
| Rmerge | 0.048 (0.560) | 0.061 (0.640) |
| I/σI | 18.2 (2.8) | 20.0 (3.3) |
| Completeness (%) | 98.6 (99.8) | 96.7 (87.7) |
| Redundancy | 4.8 (4.7) | 7.3 (6.1) |
| Refinement | ||
| Resolution (Å) | 34.31–1.85 | 46.55–2.70 |
| No. of reflections | 46,971 | 15,275 |
| Rwork/Rfree (%) | 18.3/22.4 | 21.6/27.9 |
| No. of atoms/residues (1 monomer per asymmetric unit) | ||
| Protein | 3830/488 | 3558/480 |
| Others (excluding water) | 55/4 | 61/4 |
| Ligands | 1 biliverdin | 1 biliverdin |
| 1 calcium (Ca2+) | 1 calcium (Ca2+) | |
| 1 glycerol | 1 glycerol | |
| 1 imidazole | 1 Tris | |
| Water | 349/349 | 23/23 |
| Mean B-factor (all atoms; Å2) | 37.8 | 77.1 |
| r.m.s.d.s | ||
| Bond lengths (Å) | 0.020 | 0.009 |
| Bond angles (°) | 2.048 | 1.447 |
| Ramachandran plotc | ||
| % most favored | 98.4 | 97.0 |
| % allowed | 1.4 | 2.7 |
| % disallowed | 0.2 | 0.2 |
| PDB code | 5HSQ | 5I5L |
a One crystal was used.
b Highest resolution shell is shown in parentheses.
c Data are as defined in the program RAMPAGE (68).
Attempts to optimize the crystal quality by streak-seeding, re-crystallization, or introducing point mutations did not lead to any significant improvements. Because the quality of the refined structural model was much poorer than expected for the given resolution, we considered that an unresolved disorder or symmetry associated with the I422 crystal form could be the cause of the unsatisfactory structure refinement. To grow crystals of improved diffraction quality, we carried out protein surface engineering according to the SER strategy that was designed by Derewenda and co-workers (39–41), intending to produce mutant proteins that crystallize with a packing that is different from the previously observed form of the wild-type protein. This strategy consists in replacing at the surface of the protein structure amino acid residues with long and flexible side chains within clusters of at least two by smaller residues. Three clusters were identified by the SERp server (44) as potential targets. Surface exposure of these clusters was confirmed by using the then available preliminary Agp1-PCM model and a homology model. The clusters that were selected are cluster 1 with Glu-86 and Glu-87, cluster 2 with Glu-96, Lys-97 and Lys-98, and cluster 3 with Glu-336 and Lys-337. We replaced the large amino acids of single clusters or combinations of two or three clusters by Ala; the resulting proteins are designated Agp1-PCM-SER followed by the numbers of the clusters that were replaced. Agp1-PCM-SER1, Agp1-PCM-SER2, Agp1-PCM-SER3, Agp1-PCM-SER13, and Agp1-PCM-SER23 could be expressed, purified, and assembled with BV, whereas Agp1-PCM-SER12 and Agp1-PCM-SER123 exhibited greatly diminished solubility. BV adducts of the Agp1-PCM-SER mutants were spectrally indistinguishable from the wild-type Agp1-PCM holoprotein (Fig. 1, A and B). Further control experiments with the full-length versions of Agp1-SER13 and wild-type Agp1 revealed highly similar dark reversion kinetics (Fig. 1D), similar retention times in size exclusion chromatography (data not shown), and only slightly reduced autophosphorylation activities in both Pr and Pfr forms of Agp1-SER13 compared with the wild-type protein (Fig. 1C).
FIGURE 1.

Effect of SER mutations on spectral characteristics, histidine kinase function, and dark reversion kinetics in Agp1-PCM-SER13. A and B, UV-visible absorption spectra of full-length Agp1 wild-type (WT; A) and SER13 mutant (B) in their dark states (Dark; solid line) and after illumination with red light (After R; dashed line). The difference spectrum (Diff; dotted line) obtained by subtracting the spectrum after red light illumination from the dark spectrum shows that the wild-type and the mutant protein photoconvert into a mixture of similar proportions of Pr and Pfr. C, autoradiograph (upper panel) after SDS-PAGE of wild-type Agp1 and its SER13 mutant after incubation with [γ-32P]ATP in the dark (Pr) and illumination with red light (R). Similar to the wild-type, the mutant protein shows pronounced autophosphorylation activity in the dark that decreases after red light illumination. The lower panel shows the same section of the gel after staining with Coomassie Blue. D, dark reversion of wild-type Agp1 (■, solid line) and the SER13 mutant (○, dashed line) as measured by the time dependence of the relative absorbance at 750 nm (A750).
Screening revealed the mutants Agp1-PCM-SER1, Agp1-PCM-SER2, and Agp1-PCM-SER3 to crystallize under similar conditions as the wild-type Agp1-PCM with PEG3350 as the precipitant (38), resulting in crystals of similar morphology. X-ray diffraction showed that Agp1-PCM-SER1 and Agp1-PCM wild-type crystals had the same space group and unit cell parameters, but the diffraction quality was not improved in Agp1-PCM-SER1. Novel crystal forms were identified for Agp1-PCM-SER2 and Agp1-PCM-SER23 under (NH4)2SO4-based conditions and for Agp1-PCM-SER13 under PEG8000-based conditions. Crystals of Agp1-PCM-SER13, belonging to space group P43212 (unit cell parameters a = b = 69.3 Å, c = 236.8 Å, α = β = γ = 90°), exhibited markedly improved diffraction data, from which the structure of Agp1-PCM could be determined and refined to 1.85 Å resolution (Table 1), the highest resolution for a PCM structure so far. With the high resolution structure of Agp1-PCM-SER13 as search model for molecular replacement, the structure of the wild-type Agp1-PCM could be improved significantly and refined satisfactorily to a resolution of 2.7 Å (Table 1).
Overall Structures
An overview of both structures is given in Fig. 2. Different from wild-type Agp1-PCM, which crystallizes with parallel dimer subunits where the two monomers are related by a crystallographic 2-fold symmetry axis that is oriented almost parallel to the long axis of the PCM, the Agp1-PCM-SER13 crystal structure consists of anti-parallel dimers (Fig. 2, C and D). Thus, Agp1 is the only phytochrome that has been crystallized in both dimerization modes with anti-parallel and parallel subunits. Glu-87 in cluster 1 stabilizes the parallel dimer of the wild-type protein by forming a hydrogen bond with Ser-132 of the second monomer and is involved in an ionic interaction with Lys-135 of the second dimer subunit. Although mutation of Glu-87 into Ala is expected to result in destabilization of the parallel dimer, the same crystal form was found for wild-type Agp1-PCM and Agp1-PCM-SER1, indicating that the SER1 mutant crystals still contain parallel dimers. Different from cluster 1, cluster 3 does not form part of the dimer interface in WT Agp1-PCM. Replacement of Lys-337 in cluster 3 by Ala, however, enables formation of a small new interface between monomers of neighboring anti-parallel dimers in crystals of Agp1-PCM-SER13. This interface cannot form with the wild-type protein because this would lead to electrostatic repulsion with Lys-220 of the neighboring monomer. Obviously, only a combination of mutations in clusters 1 and 3 makes the new crystal form with anti-parallel dimers thermodynamically favorable by destabilizing the interface in the parallel dimer and at the same time establishing a new crystallographic interface between neighboring dimers that would not form with the wild-type protein. Notably, the interface in the anti-parallel dimer buries a larger area (1891 Å2) than the parallel dimer interface (1193 Å2) (45).
FIGURE 2.

Structure of the PCM of Agp1 in the Pr state. A, ribbon representation of the Agp1-PCM-SER13 monomer. The chromophore BV and its attachment site Cys-20 are shown as balls and sticks, and carbon atoms are colored in yellow. PAS, GAF, and PHY domains are depicted in green, purple, and blue, respectively. The two SER clusters of mutated amino acid residues (SER1, E86A/E87A; SER3, E336A/K337A) are indicated by dotted circles and depicted as balls and sticks. B, rotated close-up view (same color code as in A) of a section of the chromophore binding pocket showing the potential hydrogen bond network that links biliverdin to the protein environment, including mediating water molecules. The four pyrrole rings of biliverdin are labeled A to D, and the propionate side chains of rings B and C are labeled propB and propC, respectively. Some of the potential hydrogen bonds have been omitted for clarity. The complete set of binding interactions in the chromophore binding pocket is visualized schematically in Fig. 5C. The highly conserved PRXSF motif of the tongue region from the PHY domain interacts with the chromophore via van der Waals interactions between Pro-461 and ring A of BV and stabilizes the chromophore binding pocket by a salt bridge contact between Arg-462 and Asp-197. C and D, structures of crystallographic dimers of wild-type Agp1-PCM (C) and Agp1-PCM-SER13 (D). In each dimer one monomer is colored, with PAS, GAF, and PHY domains depicted in green, purple, and orange, respectively, in C, and in D the color code is the same as in A. The second monomer (labeled SYM) related by a crystallographic symmetry axis to the first is shown in gray in C and in D. In the parallel dimer formed by the wild-type protein, the PAS, GAF, and PHY domains are juxtaposed along the symmetry axis, which is roughly parallel to the long axis of each monomer and perpendicular to the view direction in C. In the anti-parallel dimers in which the SER13 mutant protein forms in the crystals, the quaternary structure is stabilized by large interfaces comprising the GAF and PHY domains of different monomers, and the symmetry axis is perpendicular to the long axes of the monomers and parallel to the view direction in D.
In DrBphP (46), the full-length protein forms parallel dimers with strongly interacting parallel HK domains as found in many HK structures (47, 48). That the full-length version of Agp1-SER13 shows autophosphorylation activity in the dark-adapted Pr state (Fig. 1C) can only be reconciled with the protein forming parallel dimers in solution, and significant decrease of this activity upon red light illumination indicates that during the Pr-to-Pfr photoconversion conformational changes in the mutant protein are transmitted from the PCM to HK module by the same mechanism as in the WT protein. The slightly lower activity of Agp1-SER13 in the Pr and Pfr states in comparison with WT Agp1 may at least for the Pr state be due to the reduced stability of the dimer interface in the mutant protein as a consequence of the loss of the stabilizing effect of Glu-87. Assuming a similar quaternary structure in Agp1 as in DrBphP, the different dimer subunit arrangements found in the two Agp1-PCM crystal forms indicate only weak interactions between two subunits such that the mode of subunit interaction in the crystal depends to a significant extent on the crystallization conditions. Cross-linking and size exclusion experiments have indeed shown that full-length Agp1 forms stable dimers, irrespective of the photochromic state (Pr or Pfr), whereas the PCM in the Pr and Pfr forms is predominantly monomeric and dimeric, respectively (34). For Cph1-PCM, also weak and strong subunit interactions in the Pr and Pfr forms, respectively, have been observed by ultracentrifugation (49). Interestingly, the crystals of Agp1-PCM-SER13 and of Cph1-PCM (23) share the same space group (P43212) with one molecule per asymmetric unit and comparable unit cell parameters, indicative of similar crystal packing. Buffer conditions and pH during crystal growth were also similar. Another anti-parallel arrangement, but within a different crystal packing, has recently been described for the PCM of RpBphP3 from R. palustris in the Pr form (27). Of the 13 available PCM crystal structures of different phytochromes in different photochromic states, 10 are in the Pr state, and three of these assume an anti-parallel arrangement, whereas of three Pfr state or Pfr-like structures, two exhibit a parallel subunit arrangement, the only exception being R. palustris phytochrome RpBphP1 (28), which is not an HK. RpBphP1 has an unusual C terminus with PAS/PAC and HOS domains; the PAS/PAC domain is present in the crystal structure. Taken together, these observations seem to underline the weak and strong subunit interactions reported for Pr and Pfr PCMs in solution, respectively. The substantial differences found between the structures of the parallel PCM dimers from DrBphP in its Pfr-enriched and from PaBphP in its Pfr state may be interpreted such that even in the Pfr states the dimer interfaces of PCMs are altered during crystallization (30). Similarly, a comparison of the wild-type Agp1-PCM dimer structure with other parallel PCM dimers in their Pr states shows significant differences between these quaternary structures (Fig. 3). This is most obviously due to the different bending of the long GAF-PHY helices (see below), which results in the dimer structures of DrBphP and RpBphP2 being significantly different from that of Agp1 in the region defined by the pair of PHY domains. These structural differences are also reflected by the variation of the surface areas buried in the dimers and by the different orientations between the PAS-GAF bidomains of the dimer subunits as defined by the angles formed between the C-terminal helical segments of the GAF domains (Table 2). Because there is no universal pattern of subunit arrangement in PCM crystals, the quaternary structures do not necessarily represent the situation in the full-length proteins in solution, even if the subunits are in a parallel orientation in the crystal. Other experimental data need to be considered when deciding whether the assembly found in a crystal structure is biologically relevant.
FIGURE 3.

Comparison of parallel dimer structures in prototypical phytochromes in the Pr state. A, crystallographic dimer of wild-type Agp1-PCM is shown in ribbon representation (PDB code 5I5L; same color code as in Fig. 2C). B, crystallographic dimer of DrBphP-PCM (PDB code 4Q0J) is shown, with one monomer in dark blue and the symmetry-related monomer in gray. C, non-crystallographic dimer of RpBphP2-PCM (PDB code 4R6L) is shown, with one monomer in firebrick-red and the second in orange. In all three panels the chromophore biliverdin is depicted as balls and sticks, and the structures are shown in identical views after superpositioning of the PAS-GAF bidomains. The dimer structures are similar in the regions defined by the PAS and GAF domains only. Because of the different helical spines and in particular the different bending of the long GAF-PHY helices, however, the size of the gaps between the PHY domains along the symmetry axes and the relative orientations of the C-terminal helices leading to the His kinase modules are significantly different between the three structures.
TABLE 2.
Comparison of parallel PCM dimers in their Pr states
| Agp1 (5I5L)a | DrBphP (4Q0J) | RpBphP2 (4R6L) | |
|---|---|---|---|
| Superpositioning of PAS-GAF monomers (upper right) and dimers (lower left) | |||
| Agp1 (5I5L) | 1.27b (270)c | 1.38 (277)/1.29 (261)d | |
| DrBphP (4Q0J) | 2.87 (536) | 1.47 (276)/1.16 (258) | |
| RpBphP2 (4R6L) | 2.28 (536) | 2.72 (521) | |
| Angles formed between the C-terminal helical segments of the GAF domains within a dimer | |||
| Agp1 (5I5L) | 42° (289–304)e | ||
| DrBphP (4Q0J) | 50° (299–314) | ||
| RpBphP2 (4R6L) | 47° (294–309) | ||
| Surface area (Å2) buried at the dimer interface | |||
| Agp1 (5I5L) | 1193 | ||
| DrBphP (4Q0J) | 1350 | ||
| RpBphP2 (4R6L) | 1313 | ||
a PDB code is shown in parentheses.
b r.m.s.d. values are in Å.
c Number of Cα atoms used in structural alignment is shown in parentheses.
d Data shown are with first (chain A) and second copy (chain B) of the polypeptide chain.
e Range of residues used to define the helix axis is shown in parentheses.
Although the available data speak for parallel subunit arrangements in the full-length proteins under natural conditions, it should be noted that such an arrangement is only strictly required for those phytochromes in which the function of the C terminus is dependent on such a dimerization, like HKs in which phosphorylation requires dimerization of the DHp (dimerization and histidine phosphotransfer) domain (50). In this context it is also relevant that R. palustris phytochrome RpBphP1 (28) with an unusual C terminus forms anti-parallel dimers in the crystals.
Consistent with the PAS-GAF-PHY architecture, the overall structures of the Agp1-PCM-SER13 and Agp1-PCM subunits (Figs. 2, A, C and D, and 4) resemble the known PCM structures of canonical phytochromes, i.e. the prototypical phytochromes Cph1 (23); PhyB from Arabidopsis (25); RpBphP2 and RpBphP3 (27); DrBphP (25, 29); and the bathy phytochromes PaBphP (21) and R. palustris RpBphP1 (28). This includes the figure of eight knot formed by PAS and GAF domains (22), a long helix connecting the GAF and PHY domains, a long helix at the C terminus that connects with the HK, and the “tongue of the PHY domain,” a structure that folds back on and closes the chromophore pocket that is largely formed by the GAF domain (Fig. 2A).
FIGURE 4.

Structural comparison of the monomers in the crystal structures of wild-type Agp1-PCM and Agp1-PCM-SER13. A, superposition of the PAS-GAF bidomains of wild-type Agp1-PCM and Agp1-PCM-SER13. The complete PCMs are shown, with PAS and GAF domains of both structures colored green and purple, respectively, and the PHY domain is colored orange for the wild-type and blue for the SER13 mutant protein. B and C, the same superposition as in A shown in different orientations. Two potential hinge regions, hinge region 1, around Met-308 in the long GAF-PHY helix, and hinge region 2, around Trp-445 and Trp-468 in the middle of the tongue of the PHY domain, are shown in the close-up views in B and C, respectively. Met-308, Trp-445, and Trp-468 are shown in stick and sphere representations in all three panels. The different positions and orientations that the PHY domains adopt with respect to the PAS-GAF bidomains in the two structures can be described by a rotation of ∼23° about the hinges mentioned above and correlate with the different helical spines of the long GAF-PHY helix, which is almost straight in the wild-type protein but strongly bent in the SER13 mutant (B). A definition of the angles that were used to describe the relative orientations of the GAF and PHY domain is given in Fig. 8.
Chromophore-binding Site
In both Agp1-PCM structures, the chromophore-binding sites are nearly identical. The chromophore adopts a 5Zs10Zs15Za (often abbreviated as ZZZssa) stereochemistry found in all other Pr state crystal structures of phytochromes. The direct hydrogen-bonding and ionic interactions between the BV chromophore and the protein are highly similar to DrBphP as found in the structures of its chromophore binding domain (CBD) (22) and its PCM (25). The overall conformation of the BV chromophore (Figs. 2B and 5) is similar to that reported for DrBphP, if the Agp1-PCM-SER13 and DrBphP-CBD structures are compared. Rings A, B, and C are roughly co-planar, the tilt angle between rings A and B being smaller (8° in Agp1; 14.0° in DrBphP) and the tilt between the central rings B and C being significantly larger (11° in Agp1; 2° in DrBphP) in Agp1 than in DrBphP. As in all phytochrome structures, the tilt between rings C and D is the largest (51° in Agp1 and DrBphP), which certainly impairs conjugation between the π-electron systems of these rings. Similarly, large angles between rings C and D as seen in BV-binding phytochromes were also found for the plant phytochrome PhyB (25), which binds PΦB (60° and 56°), and for the PCB binding phytochrome Cph2 (55° and 57°) (14), whereas a significantly smaller angle (26°) is found in Cph1, which binds PCB (23). The origin of the strong C-D ring tilt is probably 2-fold, steric repulsion between the hydrogen atoms of the neighboring methyl group at ring C and the NH group of ring D in the 5Zs10Zs15Za stereochemistry, as well as interactions between the chromophore and the surrounding protein matrix of the GAF domain. There is no particular role of the PHY domain in controlling the geometry of the ring D of Agp1-PCM, because there is no direct contact with this part of the chromophore in the Pr structures (Fig. 5C).
FIGURE 5.

Electron density of the BV chromophore in the Agp1-PCM-SER13 crystal structure at 1. 85 Å resolution. A, BV and Asp-197 are shown as sticks with carbon atoms in yellow and purple, respectively. Water molecules (W777, W883, and W918) are depicted as blue spheres, and all other surrounding amino acid residues as thin lines. The blue mesh represents a weighted 2Fo − Fc map (contoured at 1.5 σ) where the weights were calculated using REFMAC5 (64). B, 2Fo − Fc simulated annealing σA-weighted omit map (green), calculated using the program PHENIX (65) for the omitted chromophore BV in the Agp1-PCM-SER13 crystal structure. C, analysis of the potential hydrogen bonds and van der Waals contacts between BV and its protein and water molecule environment. Potential hydrogen bonds were analyzed using HBPLUS (72) as implemented in the program LigPlot+ (73), which was used to draw this schematic view. Residues with closest distances less than 4 Å are considered to be in van der Waals contact.
The PHY domain, however, interacts with ring A of the chromophore, as in other PCM Pr structures (23). Because the resolution is high enough to resolve the electron densities of all chromophore side chains, details of this interaction can be seen in the Agp1-PCM-SER13 crystal structure that have not been reported before for any other PCM structure (Figs. 5, A and B, and 6, A and B). The tongue of the PHY domain is in van der Waals contact via Pro-461 of the conserved PRXSF motif (23) with the C21 methyl group on C2 and the carbonyl group of ring A of the chromophore (Figs. 5C and 6, A–C, and see Fig. 7 for atom numbering). The electron density at ring A is fully consistent with an endocyclic C2=C3 double bond and an exocyclic C3–C31 single bond as described for bathy phytochromes like PaBphP (21). This “BV-like” structure of the chromophore in Agp1, however, is in contrast with the “PΦB-like” structure with a chiral C2 carbon and an exocyclic C3=C31 double bond that BV was found to adopt in DrBphP-CBD (Fig. 7B) (24). To test whether our data are sufficient to discriminate between the two alternative chromophore structures, we applied tight geometric restraints during crystallographic refinement to ring A forcing it to adopt a PΦB-like structure with a tetrahedral C2 carbon. This results in negative difference density appearing in the Fo − Fc maps at the C21 methyl group (Fig. 6, A and B), proving that the position of this group is wrong. In the PΦB-like structure the methyl group is ∼0.5 Å closer to Pro-461 in comparison with the BV-like structure where the methyl group is already in van der Waals contact with Pro-461. This suggests that formation of a tetrahedral C2 carbon after assembly of BV with the apoprotein following 1,4-addition of the Cys-20 sulfhydryl group to the conjugated double bond system C2=C3–C31=C32 would result in a highly sterically strained geometry at ring A, which is alleviated if covalent attachment of Cys-20 to BV occurs via 1,2-addition that leaves the C2=C3 double bond unchanged. It is noticeable, however, that the electron density can unambiguously be fitted only with the exocyclic C3–C31 bond being in an almost perfectly ecliptic, sterically strained conformation (Fig. 5C), the torsion angle C2-C3-C31-C32 being 6° and the bond angle at C31 (∠(C3-C31-C32) = 122°) deviating significantly from ideal tetrahedral geometry (109.5°). Taken together, the particular geometry at ring A and the thioether linkage in Agp1-PCM-SER13, which has not been explicitly recognized in other phytochromes, appear to be under steric strain even if the chromophore is modeled as a BV-like structure. We attribute the strained geometry to a combination of the tight interaction between the C21 methyl group at C2 and Pro-461 from the PRXSF motif and the steric restraints imposed by the covalent thioether linkage between Cys-20 and the chromophore. Pro-461 is located at the C terminus of the β-hairpin structure at the tip of the tongue and confers rigidity to the polypeptide backbone at this position (Fig. 6C). It may be possible that Z-to-E photoisomerization at the C15=C16 double bond is immediately followed by smaller movements and relaxation even of ring A within the chromophore binding pocket or that rapid rotation of ring A in an excited electronic state of Pr caused by a twist of the C4=C5 double bond even precedes Z-to-E isomerization at C15=C16 as has been proposed based on ultrafast spectroscopic measurements on Agp2 (51). Such movements would in turn further increase the steric strain in this particular region, thereby lowering the activation barrier for the transition toward one of the subsequent intermediate states that is accompanied by structural reorganization of the tongue region. Increase of the steric strain is only possible with a Pro in the position described here, because it cannot evade the increase in conformational energy by adjusting the polypeptide torsion angles in the same way as any other amino acid residue could do, which may finally drive large scale conformational changes as observed for the tongue region during photoconversion.
FIGURE 6.

Structure of the biliverdin chromophore at pyrrole ring A and the PRXSF motif as directly interacting signal transducer module in bacterial phytochromes. A, electron density of ring A of the chromophore BV after, left panel, refinement with geometric restraints for a PΦB-like structure with an exocyclic C3=C31 double bond or, right panel, with restraints for a BV-like structure with an endocyclic C2=C3 double bond in ring A. The blue mesh represents a weighted 2Fo − Fc map contoured at 1.0 σ. That the PΦB-like chromophore structure is wrong is indicated by the negative electron density at the C21 methyl group, as shown by the red mesh that represents the weighted Fo − Fc map at −2.0 σ. B, similar representation as in A in a different orientation. In both panels the shortest distances between Pro-461 of the PRXSF motif and the C21 atom in the PΦB-like structure with an exocyclic double bond, left panel, and in the BV-like structure with an endocyclic double bond, right panel, are indicated by dashed lines. C–F, chromophore binding pockets in the crystal structures of different bacteriophytochromes are shown with BV, the highly conserved Asp of the conserved PXXDIP motif in the GAF domain, and the amino acids of PRXSF motif as balls and sticks or thin lines (x and Phe of the PRXSF motif). The polypeptide chains are depicted in ribbon representation. C and E, in the crystal structures of Agp1-PCM-SER13 (PDB code 5I5L) and DrBphP-PCM (PDB code 4Q0J), respectively, in their Pr states, the tongue of the PHY domain is folded as a β-hairpin loop over the chromophore binding pocket. In the Pr structure of Agp1-PCM-SER13, Pro-461 of the PRXSF motif in the tongue region (as well as Pro-465 in the Pr state of DrBphP-PCM) undergoes van der Waals interactions with ring A of the chromophore BV, thereby being poised to act as sensor of structural changes that occur at ring A after photoactivation of the Pr state. Arg-462 (Arg-466 in DrBphP) stabilizes Asp-197 (Asp-207 in DrBphP) via a salt bridge. Ser-464 (Ser-468 in DrBphP) is solvent-exposed and without any interaction with the chromophore region in the Pr state crystal structures. D and F, in the crystal structures of PaBphP-PCM (PDB code 3NHQ) and DrBphP-PCM (PDB code 5C5K) in their Pfr (D) or Pfr-enriched (F) states, respectively, the tongue of the PHY domain is folded as α-helical and loop structure over the chromophore binding pocket. Pro-456 of the PRXSF motif in PaBphP-PCM (or Pro-465 in the Pfr-enriched state of DrBphP-PCM) is in van der Waals contact with ring D of the chromophore BV, in a position to sense structural changes that occur at ring D after photoactivation of the Pfr state. Ser-459 (Ser-468 in DrBphP) stabilizes Asp-194 (Asp-207 in DrBphP) of the conserved PXXDIP motif via a hydrogen bond. Arg-457 in PaBphP and the corresponding residue Arg-466 in DrBphP are solvent-exposed and without any interactions with the chromophore regions in the crystal structures of the Pfr and the Pfr-enriched state, respectively. Parts of the secondary structures in front of the chromophore binding pocket were removed for clarity in C–F.
FIGURE 7.
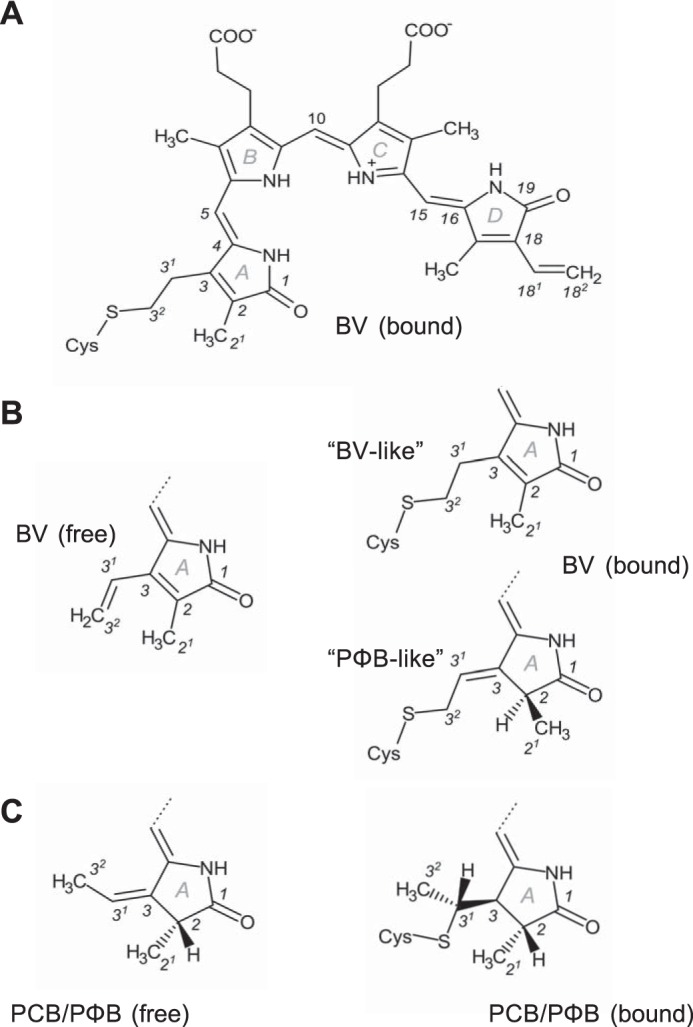
Structures of the bilin chromophores referred to in this work. A, complete structure of BV resulting from covalent attachment to the conserved Cys in bacteriophytochromes via 1,2-addition. The stereochemistry of this structure is 5Zs10Zs15Za (often abbreviated ZZZssa), indicating whether the central carbon atoms of the methine bridges that link adjacent pyrrole rings participate in single bonds that are either in syn (s) or anti (a) conformation and stating that all the corresponding double bonds are in Z configuration. Moreover, the structure contains an endocyclic C2=C3 double bond in ring A. Numbering is shown for all atoms that are mentioned in the text, tables, or figures. B and C, ring A structures of the free and bound chromophores BV, PCB, and PΦB as found in phytochrome crystal structures. Covalent attachment of BV (B) via 1,4-addition results in the PΦB-like structure with an exocyclic C3=C31 double bond in the C3 side chain that was found in the CBD structure of DrBphP (24) and is different from the BV-like structure also shown in A.
Our interpretation that the PHY domain directs chromophore assembly toward formation of a BV-like structure with an endocyclic C2=C3 double bond in ring A is consistent with the observation that the BV chromophore of the CBD of DrBphP (24), which lacks the PHY domain, forms the PΦB-like structure with an exocyclic C3=C31 double bond. The structures of DrBphP in the Pr state (25, 29) that were reported after the high resolution structure of DrBphP-CBD was published were modeled with chromophores in PΦB-like structures, but their resolutions have not been higher than 2.75 Å (25), which means that the true geometry at the C2 carbon could not be determined with certainty. In PCM Pr structures, the closest distance between the homologous Pro and ring A or its side chain is between 3.7 and 4.1 Å (Fig. 6, C and E; Table 3), consistent with the idea that this Pro is required for Pr-to-Pfr conversion in these phytochromes by a mechanism that is similar but not necessarily identical to the one suggested here for Agp1. In R. palustris RpBphP3, the homologous residue is a Thr that is 4.5 Å away from ring A of the BV chromophore (Table 3) (27). This phytochrome, however, converts into an atypical near-red light-absorbing state photoproduct with a shorter wavelength absorbance maximum, which according to our hypothesis is due to the lack of steric strain in the ring A region in the Pr state that would be required for photoconversion into a Pfr state, and therefore it results in the photo-activated state of RpBphP3 thermally relaxing via a different pathway into the near-red light-absorbing state. Notably, in all PCM Pfr structures the homologous Pro is in van der Waals contact with ring D or its vinyl side chain (Fig. 6, D and F; Table 3), due to movement of the PRXSF motif that is associated with the refolding of the tongue during photoconversion. We propose that at least in BV-binding bacteriophytochromes, the steric strain around ring A in the Pr form and its release during photoconversion to Pfr facilitates structural changes in this part of the chromophore binding pocket that together with the Z-to-E photoisomerization at the C15=C16 double bond in the C-D methine bridge would contribute to the propagation of conformational changes from the sensory module to the C-terminal signaling module. The fixed protein backbone at Pro-461 in Agp1 seems to be required to transmit the steric tension for complete Pfr formation (see also mutants below). Previous studies with Agp1 indicated that mobility of ring A is required for photoconversion as follows: Agp1 assembled with synthetic chromophores in which the methine bridge between rings A and B is arrested in Za or Zs stereochemistry photoconverted either to a blue-shifted adduct or to a species with unchanged absorbance maximum (52). Moreover, Agp1 with a fluorescein label at the position of the chromophore-binding site shows that the local environment of ring A changes significantly upon photoconversion (53).
TABLE 3.
Contacts between Pro (Thr) of the P(T)RXSF motif and rings A or D of the chromophore as well as their side chains

a a.u. is asymmetric unit.
bN is name of the corresponding atom in Pro(Thr) of the P(T)RXSF motif.
cCorresponding Pro missing in the structure in either chain A or B.
dThis contains TRXSF motif.
eThere is no distance < 4.5 Å for either chain A or B.
fStandard deviation is shown.
gDistances were measured only to BV in 15Ea stereochemistry.
Comparison of Both Protein Structures
Because the amino acids that were replaced in the SER13 mutant are not conserved and are most likely functionally irrelevant as shown for the PCM and full-length version of Agp1 and Agp1-PCM (Fig. 1), we assume that the solution structures of Agp1-PCM and Agp1-PCM-SER13 are not significantly different from each other. Differences between the crystal structures are therefore likely to be due to crystal packing effects. Alignments of both Agp1 structures show that the relative orientations between the PAS-GAF bidomain and the PHY domains are different. When the structures are aligned in the region of the GAF domains, PAS and GAF domains match well (root-mean-square deviation (r.m.s.d.) 0.63 Å for GAF domain, 187 Cα atoms; r.m.s.d. 0.62 Å for PAS and GAF domain, 290 Cα atoms), whereas the PHY domains diverge (Fig. 4). An alignment of PHY domains results in a poorer match with an r.m.s.d. of 1.29 Å if the tip of the tongue is excluded (169 Cα atoms), whereas PAS and GAF domains diverge. This observation is explained by a different bending of the long helix connecting the GAF and PHY domain and will be discussed in the context of other phytochrome structures below. The tip of the tongue aligns together with the GAF domains, indicative of a tight interaction of both regions (Fig. 4, B and C). The part of the tongue that moves together with the GAF domain can be clearly assigned to residues 445–468 (WGGDPHKTVQESGRIHPRKSFEIW; conserved residues in bold). This region contains two of the four β-strands of the tongue and bears highly conserved amino acids. The C-terminal helix that connects with the N-terminal helix of the HK also assumes a different orientation in both Agp1 structures (Fig. 4). Thus, our studies point to two hinge regions in the PCM of Agp1, hinge region 1 at Met-308 (Fig. 4B) and hinge region 2 at Trp-445 and Trp-468 (Fig. 4C).
GAF-PHY Connecting Helix and C-terminal PHY Helix
The long helix that connects GAF and PHY domains is more bent in the Agp1-PCM-SER13 than in the wild-type Agp1-PCM structure. This is due to a kink around position 308, the transition point between both domains. A different bending has also been described for long GAF-PHY connecting helices in other phytochrome structures (27, 30). Because structural changes in the long helices connecting the GAF and the PHY domain as well as the PHY and the HK domain are thought to be crucial for the propagation of a signal from the sensory domain to the signal output domain (54), we compared the relative orientations of the long GAF-PHY connecting helix and the C-terminal helix (PHY helix) in all PCM structures to see whether a general pattern can be found. Structural alignment of only these segments (Fig. 8) reveals that the N termini of the long GAF-PHY helices point into different directions, indicative of hinge regions in the centers of the long GAF-PHY helices in all phytochrome structures. We therefore decided to describe in a simple way how the relative domain orientations differ in the known PCM structures by measuring for each structure the angle between the helix axes defined for the N-terminal part of the long GAF-PHY connecting helix and the PHY helix (Fig. 8). Strikingly, in all Pfr state structures the long GAF-PHY helices are more or less straight and oriented roughly parallel to the PHY helix, and consequently the angles between the helix segments as defined above fall into a narrow range between −11° and 2° (signs as defined in QHELIX (55)). In contrast, the bending in the long GAF-PHY connecting helix is more pronounced in the Pr structures, and the angles show larger variations; they range from −12° for plant phytochrome to −50° for the SER13 mutant of Agp1. Within this set of known Pr structures, the smallest angles indicating that the long helices are as straight as in Pfr structures are found for the PCMs of plant phytochrome and Cph2 (Fig. 8). This observation correlates with plant phytochrome lacking a functional HK module and a second GAF replacing a PHY domain in Cph2. Three conclusions arise from this comparison as follows. (i) In the Pr form, the angle at the hinge is variable, as seen by the pairwise comparison of Apg1-PCM and Agp1-PCM-SER13, Cph1-PCM and Cph1-PCM-Y263F, or by the comparison of all Pr structures. This flexibility would give rise to a dynamic Pr state that could have led to Agp1-PCM being trapped in two different conformations due to the different crystal packings and could be the origin of the heterogeneity that has been observed by NMR or other techniques in Cph1 before (56, 57). (ii) During photoconversion, the helix straightens at the same position and becomes more rigid, as deduced from the comparison of Pr and Pfr structures, especially the Pr- and Pfr-enriched structures of DrBphP-PCM (29). A limited proteolysis study with Cph1 is also consistent with this assumption (58). When comparing wild-type Agp1-PCM and Agp1-PCM-SER13 structures, the former comes closer to a Pfr structure. The hinge region in the GAF-PHY-connecting helix is characterized by weak homology. Within those phytochromes of which structures are available, Ile-306 and Leu-321 (in Agp1) are conserved, and all others are variable. Cph2 has an insertion of four amino acids, little more than another turn of the long helix.
FIGURE 8.

Variation in relative orientations between GAF and PHY domains in the known PCM structures of phytochromes. A, structural comparison of the long helices that connect the GAF and the PHY domains (GAF-PHY helices) and the C-terminal helices of the PHY domains (PHY helices) of PaBphP-PCM (black), wild-type Agp1-PCM (orange), and Agp1-PCM-SER13 (blue) shown in ribbon representation. The complete PCM structures are shown in surface representation in the background. The structural alignment was obtained by superpositioning the 10 C-terminal amino acid residues of the GAF-PHY helices and the PHY helices. B, for each structure the relative orientation between the GAF and the PHY domain is characterized as indicated here for chain A of PaBphP-PCM (PDB code 3C2W) by measuring the angle between the helix axes of the segment defined by the 20 N-terminal residues of the GAF-PHY helix on the one hand and the PHY helix on the other hand. The angles between helix axes were calculated by the Chou algorithm (77) using QHELIX (55). The signs of the angles are as defined in QHELIX. C–F, superpositions of helices of phytochrome PCM structures as in A, PDB codes indicated. For each structure shown, the text inset provides information on the type of phytochrome PCM together with the corresponding PDB code, the resolution and the photochromic state of the crystal structure, and the mean angle between helices (including its standard deviation if there is more than one monomer per asymmetric unit) as defined above. The color code is the same as in A–F.
Tongue of the PHY Domain
The most obvious structural differences between known Pr and Pfr structures are found in the tongue region of the PHY domain that protrudes from the core of the PHY domain and forms with its tip an interface with the GAF domain and the chromophore. The tongue adopts an extended and kinked β-hairpin structure only in the available Pr structures, whereas it consists of an extended loop and α-helical structure in all known Pfr structures. The rearrangement of the tongue's secondary structure during photoconversion has been proposed based on the DrBphP-PCM structures in the dark and illuminated forms (29) and is supported by all available Pfr and Pr PCM structures, including those of Agp1-PCM. Such a restructuring is also consistent with a limited proteolysis study on Cph1 where Arg-472 of the PRXSF motif was shown to become exposed upon Pr-to-Pfr photoconversion (58). The structure of the tandem-GAF bidomain of Cph2 showed that the C-terminal GAF domain assumes a similar fold as the PHY domain of bona fide phytochromes. It was shown by structural comparisons and site-directed mutagenesis in Cph2 targeting, in particular the conserved Trp residues that correspond to hinge region 2 (Fig. 4C) in the tongue domain of Agp1, that this structural element plays similar roles in photoconversion in Cph2 and canonical phytochromes (14).
The highly conserved Arg-462 of the PRXSF motif stabilizes the Pr state by forming a salt bridge with Asp-197 of the PXXDIP motif in the GAF domain (distance 2.9 Å in Agp1-PCM-SER13). This ionic bond must break during photoconversion when the tongue region refolds and moves relative to the chromophore. The role of Pro-461 can be described as a sensor of small structural changes that occur at pyrrole ring A after photoexcitation of the chromophore starting from the Pr state. According to our hypothesis, the increase in steric strain at Pro-461 and ring A that is then induced, due to local rigidity of the polypeptide backbone, causes relaxation of a state of high conformational energy by structural changes that can only occur in more remote regions of the tongue. The short distances found between the homologous Pro and ring D or its vinyl side chain in PCM Pfr structures (Table 3) may point to a similar role of this Pro as a sensor of the pronounced structural changes that occur in this region during photoisomerization starting from the Pfr state (Fig. 6, D and F).
Mutants in the Tongue of the PHY Domain
To study the role of the tongue of the PHY domain in Agp1-PCM or full-length protein more in detail, we modified four highly conserved amino acids and tested these mutants for Pr-to-Pfr photoconversion and dark reversion as follows; Trp-445, Pro-461, Ser-464, and Trp-468 were replaced by Ala, and in the resulting mutant proteins W445A, P461A, S464A, and W468A photoconversion and dark reversion were analyzed (Fig. 9). The S464A mutant of Agp1 has a normal Pr spectrum but photoconverts into a bleached species that does not evolve a new absorption maximum. Ser-464 is thus required for the stabilization of the Pfr form in Agp1. S464A is also characterized by mono-exponential dark reversion with a time constant of 18 min. In accordance with earlier studies (34), dark reversion of the wild-type protein was described by a bi-exponential function with time constants of 11 and 120 min. The Ser-464 homolog has also been mutagenized in the bathy phytochrome PaBphP; this mutant has a Pr dark-adapted state and a bleached Pfr-like photoproduct that undergoes rapid dark reversion (21). The Pr spectrum of P461A is again comparable with the wild type, whereas the photoproduct is again bleached. Dark reversion follows mono-exponential kinetics with a lifetime of 230 min. The bi-exponential and mono-exponential dark reversion of wild-type and mutant proteins could correspond to two or one steps of conformational changes, respectively. In earlier experiments it was found that the CBD fragment of Agp1, which lacks the PHY domain, also forms a bleached photoproduct and undergoes mono-exponential dark reversion (34).
FIGURE 9.

Photoconversion and dark reversion of Agp1 mutant proteins where selected residues of the tongue region of the PHY domain were replaced by Ala. A–E, UV-visible absorption spectra of full-length Agp1 wild-type (WT; A), as well as mutants W445A (B), P461A (C), S464A (D), and W468A (E) in their dark states (Dark; solid line) and after illumination with red light (After R; dashed line). Comparison of the difference spectra (Diff; dotted line) obtained for each sample by subtracting the spectrum after red light illumination from the dark spectrum shows that the W445A and the W468A mutant proteins photoconvert into a mixture of similar proportions of Pr- and Pfr-like states as the wild-type. Red light illumination of P461A and S464A that exhibit normal Pr-like dark spectra, however, generates bleached species that lack typical features of a Pfr state. F, dark reversion of wild-type Agp1 (■, solid line), the W445A (○, dashed line), and the W468A mutant (Δ, dotted line) as measured by the time dependence of the relative absorbance at 750 nm (A750). Dark reversion is incomplete for W445A, whereas Trp-468 exhibits complete reversion to the dark state. In contrast to dark reversion of wild-type Agp1, which can be described by bi-exponential kinetics with time constants of 13.0 ± 0.4 and 400 ± 20 min, the corresponding processes are characterized by mono-exponential kinetics with time constants of 190 ± 20 and 70 ± 4 min in W445A and W468A, respectively. G and H, dark reversion of wild-type Agp1 and the S464A and P461A mutants as measured by the time dependence of the increase in absorbance at 702 nm (A702). Data from the Agp1 wild-type (■, solid line) and the S464A mutant (▿, dashed line) are shown in G, and data from P461A (□, solid line) are shown in H. Although the data from the wild-type protein can be explained by bi-exponential kinetics with time constants of 11 ± 3 and 120 ± 10 min, the data of S464A and P461A were fitted by assuming only one kinetic phase for each mutant with time constants of 18.0 ± 0.5 and 230.0 ± 0.4 min, respectively.
Both mutants of Agp1 in which Trp residues in hinge region 2 of the tongue were replaced, W445A and W468A, have unchanged spectral characteristics of Pr-to-Pfr photoconversion. This result is in contrast to studies on the corresponding mutants of the tandem-GAF bidomain fragment of Cph2, which are characterized by bleached photoproducts (14). Similar to the other mutants presented here, however, the dark reversion of W468A is mono-exponential and has a lifetime of 72 min. The dark reversion of W445A is incomplete and can be described by mono-exponential kinetics with a lifetime of 210 min.
In earlier experiments, mutants where Arg-472 of the PRXSF motif in the tongue of Cph1 was replaced by two different amino acids (58) were found to exhibit wild-type-like Pr spectra and to form bleached photoproducts. Moreover, in these mutants regulation of HK activity was lost.
With regard to the spectral properties of phytochromes in the Pfr form, the tongue is clearly the most relevant part of the PHY domain, and the conserved amino acids Pro-461 and Ser-464 play dominant roles for light-triggered structural rearrangements of the tongue and other protein conformational changes during Pr-to-Pfr photoconversion. Incomplete photoconversion in a mutant results in an intermediate state or even a different state from which back-conversion to Pr is possible in one step, whereas dark reversion from Pfr to Pr requires at least two steps. The specific function of Ser-464 would be a stabilization of the Pfr conformation by participating in a hydrogen bonding network that includes Asp-197 of the PXXDIP motif as shown for the homologous residues in the dark state PCM structures of bathy phytochromes PaBphP and RpBphP1 as well as the Pfr-enriched PCM crystals of DrBphP and DrBphP-F469W (21, 28–30). The switch of the Ser side chain from a surface-exposed position in the Pr form (Fig. 6, C and E) to an interaction with the PXXDIP motif of the GAF domain (Fig. 6, D and F) seems central in the formation of Pfr. The D197A mutant of Agp1 is indeed also characterized by a bleached photoproduct (35). Arg-462 of the PRXSF motif in Agp1 like its homologs in all known PCM structures in Pr states stabilizes the Pr state by forming a salt bridge with Asp-197 of the PXXDIP motif, whereas the homologs of this residue assume surface-exposed positions in the structures of Pfr and Pfr-like states (Fig. 6, C–F). Collectively, structural comparison and mutagenesis data suggest that due to large scale movements of the PRXSF motif, the Ser and Arg residues of the PRXSF motif in canonical and bathy phytochromes swap their roles during photoconversion, having complementary functions in either stabilizing the Pr or the Pfr state. The specific function of Pro-461 lies possibly in locally restricting conformational changes during photoconversion, thereby contributing to the partial storage of the absorbed photon energy as conformational energy in an early intermediate state. The high energy content of this state may be crucial for Pr-to-Pfr photoconversion by being responsible for the low activation energy of the transition to the subsequent intermediate along the pathway that finally leads to reorganization of the tongue domain. Although in Agp1 Trp-445 and Trp-468 are not required for Pfr spectral integrity, they are relevant for dark reversion. If Pfr-to-Pr dark reversion is characterized by two steps, the apparent mono-exponential kinetics of these mutants is hard to understand. We assume that in each of these mutants one component is overlooked because it is either too rapid (in W468A) or too slow (in W445A).
Sequence of Structural Changes during Photoconversion
Pr-to-Pfr photoconversion is initiated by the Z-to-E isomerization of the chromophore around the C15=C16 double bond, followed by straightening of the long GAF-PHY connecting helix and refolding of the tongue region. In our Agp1-PCM crystals, the long helix is more stretched than in the Agp1-PCM-SER13 crystals indicating pronounced flexibility of this structural element, whereas the tip of the tongue remains bound to the same amino acids in the GAF domain and has the same secondary structure. We therefore propose that not straightening of the long helix but refolding of the tongue is the rate-limiting and crucial step for intramolecular signal transduction from the chromophore-binding site to the HK output module during Pr-to-Pfr conversion.
Conclusions
The two crystal structures of Agp1-PCM as wild-type and as mutant proteins show that due to the weak interactions between the subunits it is almost impossible to predict whether a PCM will crystallize as a parallel or anti-parallel dimer. Comparison of the protein subunits in the two Agp1-PCM and all other known PCM structures suggests pronounced structural flexibility of phytochromes in the Pr state, enabled by movement about hinges in the long GAF-PHY connecting helices and in the tongue region of the PHY domain. The detection of structural details in the chromophore binding pocket that was possible due to the high resolution of the Agp1-PCM-SER13 structure led to a hypothesis that provides a possible explanation for the crucial role of the Pro residue of the PRXSF motif during photoconversion. According to this hypothesis, an increase in steric strain at pyrrole ring A of the bilin chromophore and its protein environment at an early stage after photoexcitation is critical for the protein to overcome an activation barrier as required for the structural reorganization of the tongue region.
Experimental Procedures
Plasmid Constructs and Mutagenesis
An expression vector termed pNM19a, bearing the coding sequence corresponding to the PCM fragment (residues 10–504) of Agp1 cloned into pET21b (Novagen) via NdeI and XhoI sites, was utilized for the production of the PCM fragment of Agp1. Full-length Agp1 was produced using a pQE12 (Qiagen)-based vector termed pAg1 (16, 38). Mutations were introduced into the agp1 gene in a plasmid DNA according to the QuikChange mutagenesis protocol (Agilent Technologies).
The amino acid sequence of the Agp1 protein was analyzed by using the SERp server (44) to identify the clusters that contain sites suitable for mutation according to the SER strategy. The search resulted in identifying the following clusters: SER1, E86A/E87A; SER2, E96A/K97A/K98A; and SER3, E336A/K337A. A homology model based on the structures of the CBD of DrBphP (PDB code 2O9C) and the PHY domain of Cph1 (from PDB code 2VEA) that served to verify whether the corresponding amino acid residues are exposed at the surface of the protein was obtained using MODELLER (59). These clusters were mutated either singly or in combination with one or two other clusters such that all seven possible variations were obtained. Mutants of the Agp1-PCM fragment (amino acid residues 10–504) were subjected to crystallization trials along with the wild-type protein. The crystallization conditions and preliminary experiments for the latter have already been reported (38). The SER1 and SER3 clusters were also introduced simultaneously into the pAg1 plasmid that bears the full-length Agp1 coding sequence for producing the full-length Agp1 protein with the mutation clusters. The following mutations were also introduced into full-length Agp1 for biochemical analyses: W445A, P461A, S464A, and W468A.
Protein Sample Production
Escherichia coli cell lines XL1-blue and BL21(DE3) were used to produce the full-length and the PCM fragment of the Agp1 protein, respectively. The proteins were overexpressed and affinity-purified as apoprotein as described (38) and were subsequently assembled with BV with a molar ratio of 1:1.2. All subsequent sample manipulation was carried out in darkness or under the safe light condition using either filters (735 and/or 736, LEE filters) or green LEDs (NSPG510AS, Nichia, λ = 522 nm). A homogeneous population of holoprotein was obtained by separation on a 1 × 30-cm size exclusion column (Superdex 200 10/300 GL, GE Healthcare) using SEC buffer (20 mm Tris/HCl, 5 mm EDTA, pH 7.8). Holoprotein solutions were concentrated using Vivaspin 20 devices (Sartorius Stedim).
Spectroscopic Analyses
UV-visible absorption spectra were obtained using a U-3010 UV (Hitachi) or a Jasco V550 photometer. Dark reversion was monitored by measuring the absorption at specific wavelengths as a function of time. Dark reversions of P461A and S464A mutants were monitored by the recovery of Pr at 702 nm following red light illumination. Gradual decrease of Pfr was monitored at 750 nm for W445A and W468A mutants. The red light used for illuminating samples consisted of an array of LED lights (Nichia) with the peak emission at 660 nm.
Histidine Kinase Assay
The autophosphorylation assay was carried out essentially as reported previously (16). The phosphorylation assay was performed in darkness or green safelight. In a final reaction volume of 25 μl, 2.5 μm Agp1 (or mutant protein) was incubated with 0.5 mm ATP and [γ-32P]ATP adjusted to 2 μCi. At this stage, the sample was either irradiated with red light (20 μmol m−2 s−1 for 2 min) or kept in darkness. After incubation at 295 K for 17 min, the reaction was stopped by adding SDS-loading solution and subjected to SDS-PAGE followed by autoradiography.
Crystallization and Data Collection
Crystallization trials were carried out using a Mosquito robot (TTP Labtech) and MRC plates and screening kits (Molecular Dimensions). Crystallization screening by vapor diffusion in a sitting-drop setup was carried out by equilibrating 200 nl of protein (25 mg/ml) in SEC buffer and 200 nl of crystallization solution against 80 μl of the crystallization solution in the reservoir, followed by incubation in darkness at 292 K. Lead conditions identified from the screening were optimized by the hanging drop method using 24-well XRL crystallization plates (Molecular Dimensions), with 500 μl of crystallization solution in the reservoir and 2 μl each of protein solution and the crystallization solution in the hanging drop. Crystals typically grew to suitable sizes for diffraction analysis (approximate dimensions 0.15 × 0.15 × 0.8 mm3 for wild-type Agp1-PCM and 0.2 × 0.2 × 0.4 mm3 for Agp1-PCM-SER13) within 2 weeks of incubation following the setup. For data collection, crystals were either directly transferred from the mother liquor (Agp1-PCM-SER13) or vitrified after adding cryoprotectant solution (wild-type Agp1-PCM) consisting of 20% glycerol and 80% reservoir solution (v/v). Optimized crystallization conditions were follow: wild-type Agp1-PCM (0.2 m CaCl2, 16% PEG3350, 0.1 m Tris/HCl, pH 8.0); Agp1-PCM-SER13 (0.03 m diethyleneglycol, 0.03 m triethyleneglycol, 0.03 m tetraethyleneglycol, 0.03 m pentaethylene glycol, 20% glycerol, 10% PEG8000, 0.05 m MES, 0.05 m imidazole, pH 6.5); Agp1-PCM-SER2; and Agp1-PCM-SER23 (1.1 m (NH4)2SO4, 0.2 m Li2SO4, 0.1 m Tris/HCl, pH 8.5).
Structure Determination and Refinement
The best diffraction data from Agp1-PCM-SER13 and wild-type Agp1-PCM crystals were collected at beamline ID14-4 at the ESRF (Grenoble, France; Table 1). The data were indexed, integrated, and scaled using the XDS program package (60) and the CCP4 program SCALA (61, 62), respectively. For Agp1-PCM-SER13, the crystallographic phases were determined by molecular replacement by using PHASER (63) within CCP4 and a high resolution structure of DrBphP-CBD (PDB code 2O9C) (24) and the PHY domain of Cph1 (PDB code 2VEA, residues 331–514) (23) as search models. For the wild-type Agp1-PCM structure, solving the phase problem was only possible if the PAS-GAF fragment and the PHY domain from the refined Agp1-PCM-SER13 structure were used as separate search models in molecular replacement. Refinement was carried out by alternating cycles of automatic refinement with the CCP4 program REFMAC5 (64) and PHENIX (65) and manual rebuilding with COOT (66). TLS (translation, libration and screw-motion) parameters were included in the final stages of the refinement of both structures (67), and anisotropic temperature coefficients were used in the final refinement cycles of Agp1-PCM-SER13 only for a Ca2+ ion and the sulfur atoms of cysteine and methionine residues, unless their side chains were found to be in alternate conformations. In addition to COOT and PHENIX, programs RAMPAGE (68), SFCHECK (69), and PROCHECK (70) were used for structure validation. Relative orientations of helical segments and planes of pyrrole rings were determined with programs QHELIX (55) and Chimera (71). Potential hydrogen bonds and van der Waals contacts were analyzed using HBPLUS (72) and LigPlot+ (73). CCP4 programs LSQKAB (74) and SUPERPOSE (75) were used for structural alignment of polypeptide backbone Cα atoms. We used PISA (45) to calculate surface areas buried at protein-protein interfaces. All molecular graphics were generated using PyMOL (76).
Author Contributions
T. L. and N. K. conceived the project and coordinated it together with P. S.; S. N. designed the SER mutagenesis experiments and purified, characterized, and crystallized the resulting Agp1-PCM-SER variants; S. N. and T. L. performed biochemical assays on various Agp1-PCM mutants; S. N., K. Z., and N. M. generated the plasmids and produced recombinant proteins; P. S. and S. N. crystallized the Agp1-PCM wild-type protein; K. I. and N. M. contributed to establishing molecular biology and biochemical and crystallization protocols at the initial stage of the project; S. N., P. S., and N. K. performed crystallographic data collection and refinement of Agp1-PCM structures; S. N., P. S., T. L., and N. K. analyzed the data, wrote, and illustrated the paper with contributions from all authors.
Acknowledgments
We thank Iskander M. Ibrahim for carrying out the autophosphorylation assay and Sybille Wörner for helping with production and purification of recombinant protein. We are grateful to Uwe Müller, Manfred Weiss, and the scientific staff of the BESSY-MX/Helmholtz Zentrum Berlin für Materialien und Energie at beamlines BL14.1, BL14.2, and BL14.3 operated by the Joint Berlin MX-Laboratory at the BESSY II electron storage ring (Berlin-Adlershof, Germany) and the scientific staff of the European Synchrotron Radiation Facility (ESRF, Grenoble) at beamlines ID14-1 and ID14-4, where the final data were collected, for continuous support. We thank Antoine Royant and David von Stetten for extensive support with the usage of the microspectrophotometer on the crystals at the ID29S-Cryobench (ESRF, Grenoble, France).
This work was supported by Deutsche Forschungsgemeinschaft Research Grants SFB498-B2 (to T. L. and N. K.) and SFB1078-B6 (to P. S.), Cluster of Excellence “Unifying Concepts in Catalysis” Research Field D3/E3-1 (to P. S.), a Queen Mary University of London postgraduate research studentship (to S. N.), and by University of London Central Research Fund AR/CRF/B (to S. N.). The authors declare that they have no conflict of interest with the contents of this article.
The atomic coordinates and structure factors (codes 5HSQ and 5I5L) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- Pr
- red light-absorbing state
- Agp1 and Agp2
- bacteriophytochromes 1 and 2, respectively, from Agrobacterium fabrum
- AtPhyB
- phytochrome B from Arabidopsis thaliana
- BV
- biliverdin
- CA
- catalytic and ATP-binding
- CBD
- chromophore binding domain
- Cph1 and Cph2
- phytochrome 1 and 2, respectively, from Synechocystis
- DrBphP
- Deinococcus radiodurans bacteriophytochrome
- GAF
- cGMP-specific phosphodiesterase, adenylyl cyclases and FhlA
- HK
- histidine kinase
- PaBphP
- P. aeruginosa bacteriophytochrome
- PAS
- Per Arnt Sim
- PCB
- phycocyanobilin
- PCM
- photosensory core module
- PDB
- Protein Data Bank
- Pfr
- far red light-absorbing state
- PΦB
- phytochromobilin
- PHY
- phytochrome-specific
- r.m.s.d.
- root mean square deviation
- RpBphP1/2/3
- R. palustris bacteriophytochrome 1/2/3
- SER
- surface entropy reduction.
References
- 1. Auldridge M. E., and Forest K. T. (2011) Bacterial phytochromes: more than meets the light. Crit. Rev. Biochem. Mol. Biol. 46, 67–88 [DOI] [PubMed] [Google Scholar]
- 2. Ulijasz A. T., and Vierstra R. D. (2011) Phytochrome structure and photochemistry: recent advances toward a complete molecular picture. Curr. Opin. Plant Biol. 14, 498–506 [DOI] [PubMed] [Google Scholar]
- 3. Vierstra R. D., and Zhang J. (2011) Phytochrome signaling: solving the Gordian knot with microbial relatives. Trends Plant Sci. 16, 417–426 [DOI] [PubMed] [Google Scholar]
- 4. Duanmu D., Bachy C., Sudek S., Wong C. H., Jiménez V., Rockwell N. C., Martin S. S., Ngan C. Y., Reistetter E. N., van Baren M. J., Price D. C., Wei C. L., Reyes-Prieto A., Lagarias J. C., and Worden A. Z. (2014) Marine algae and land plants share conserved phytochrome signaling systems. Proc. Natl. Acad. Sci. U.S.A. 111, 15827–15832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bai Y., Rottwinkel G., Feng J., Liu Y., and Lamparter T. (2016) Bacteriophytochromes control conjugation in Agrobacterium fabrum. J. Photochem. Photobiol. B 161, 192–199 [DOI] [PubMed] [Google Scholar]
- 6. Aravind L., and Ponting C. P. (1997) The GAF domain: an evolutionary link between diverse phototransducing proteins. Trends. Biochem. Sci. 22, 458–459 [DOI] [PubMed] [Google Scholar]
- 7. Scheerer P., Michael N., Park J. H., Nagano S., Choe H. W., Inomata K., Borucki B., Krauß N., and Lamparter T. (2010) Light-induced conformational changes of the chromophore and the protein in phytochromes: bacterial phytochromes as model systems. ChemPhysChem 11, 1090–1105 [DOI] [PubMed] [Google Scholar]
- 8. Rockwell N. C., Su Y. S., and Lagarias J. C. (2006) Phytochrome structure and signaling mechanisms. Annu. Rev. Plant Biol. 57, 837–858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bhate M. P., Molnar K. S., Goulian M., and DeGrado W. F. (2015) Signal transduction in histidine kinases: insights from new structures. Structure 23, 981–994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hirose Y., Rockwell N. C., Nishiyama K., Narikawa R., Ukaji Y., Inomata K., Lagarias J. C., and Ikeuchi M. (2013) Green/red cyanobacteriochromes regulate complementary chromatic acclimation via a protochromic photocycle. Proc. Natl. Acad. Sci. U.S.A. 110, 4974–4979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Terauchi K., Montgomery B. L., Grossman A. R., Lagarias J. C., and Kehoe D. M. (2004) RcaE is a complementary chromatic adaptation photoreceptor required for green and red light responsiveness. Mol. Microbiol. 51, 567–577 [DOI] [PubMed] [Google Scholar]
- 12. Yoshihara S., Shimada T., Matsuoka D., Zikihara K., Kohchi T., and Tokutomi S. (2006) Reconstitution of blue-green reversible photoconversion of a cyanobacterial photoreceptor, PixJ1, in phycocyanobilin-producing Escherichia coli. Biochemistry 45, 3775–3784 [DOI] [PubMed] [Google Scholar]
- 13. Ulijasz A. T., Cornilescu G., von Stetten D., Kaminski S., Mroginski M. A., Zhang J., Bhaya D., Hildebrandt P., and Vierstra R. D. (2008) Characterization of two thermostable cyanobacterial phytochromes reveals global movements in the chromophore-binding domain during photoconversion. J. Biol. Chem. 283, 21251–21266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Anders K., Daminelli-Widany G., Mroginski M. A., von Stetten D., and Essen L. O. (2013) Structure of the cyanobacterial phytochrome 2 photosensor implies a tryptophan switch for phytochrome signaling. J. Biol. Chem. 288, 35714–35725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Bhoo S. H., Davis S. J., Walker J., Karniol B., and Vierstra R. D. (2001) Bacteriophytochromes are photochromic histidine kinases using a biliverdin chromophore. Nature 414, 776–779 [DOI] [PubMed] [Google Scholar]
- 16. Lamparter T., Michael N., Mittmann F., and Esteban B. (2002) Phytochrome from Agrobacterium tumefaciens has unusual spectral properties and reveals an N-terminal chromophore attachment site. Proc. Natl. Acad. Sci. U.S.A. 99, 11628–11633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Blumenstein A., Vienken K., Tasler R., Purschwitz J., Veith D., Frankenberg-Dinkel N., and Fischer R. (2005) The Aspergillus nidulans phytochrome FphA represses sexual development in red light. Curr. Biol. 15, 1833–1838 [DOI] [PubMed] [Google Scholar]
- 18. Rüdiger W. (1986) in Photomorphogenesis in Plants (Kendrick R. E., and Kronenberg G. H., eds) 1st Ed., pp. 17–33, Martinus Nijhoff Publishers, Dordrecht, The Netherlands [Google Scholar]
- 19. Giraud E., Fardoux J., Fourrier N., Hannibal L., Genty B., Bouyer P., Dreyfus B., and Verméglio A. (2002) Bacteriophytochrome controls photosystem synthesis in anoxygenic bacteria. Nature 417, 202–205 [DOI] [PubMed] [Google Scholar]
- 20. Karniol B., and Vierstra R. D. (2003) The pair of bacteriophytochromes from Agrobacterium tumefaciens are histidine kinases with opposing photobiological properties. Proc. Natl. Acad. Sci. U.S.A. 100, 2807–2812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Yang X., Kuk J., and Moffat K. (2008) Crystal structure of P. aeruginosa bacteriophytochrome: photoconversion and signal transduction. Proc. Natl. Acad. Sci. U.S.A. 105, 14715–14720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Wagner J. R., Brunzelle J. S., Forest K. T., and Vierstra R. D. (2005) A light-sensing knot revealed by the structure of the chromophore-binding domain of phytochrome. Nature 438, 325–331 [DOI] [PubMed] [Google Scholar]
- 23. Essen L. O., Mailliet J., and Hughes J. (2008) The structure of a complete phytochrome sensory module in the Pr ground state. Proc. Natl. Acad. Sci. U.S.A. 105, 14709–14714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wagner J. R., Zhang J., Brunzelle J. S., Vierstra R. D., and Forest K. T. (2007) High resolution structure of Deinococcus bacteriophytochrome yields new insights into phytochrome architecture and evolution. J. Biol. Chem. 282, 12298–12309 [DOI] [PubMed] [Google Scholar]
- 25. Burgie E. S., Wang T., Bussell A. N., Walker J. M., Li H., and Vierstra R. D. (2014) Crystallographic and electron microscopic analyses of a bacterial phytochrome reveal local and global rearrangements during photoconversion. J. Biol. Chem. 289, 24573–24587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Burgie E. S., Bussell A. N., Walker J. M., Dubiel K., and Vierstra R. D. (2014) Crystal structure of the photosensing module from a red/far-red light-absorbing plant phytochrome. Proc. Natl. Acad. Sci. U.S.A. 111, 10179–10184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Yang X., Stojković E. A., Ozarowski W. B., Kuk J., Davydova E., and Moffat K. (2015) Light signaling mechanism of two tandem bacteriophytochromes. Structure 23, 1179–1189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Bellini D., and Papiz M. Z. (2012) Structure of a bacteriophytochrome and light-stimulated protomer swapping with a gene repressor. Structure 20, 1436–1446 [DOI] [PubMed] [Google Scholar]
- 29. Takala H., Björling A., Berntsson O., Lehtivuori H., Niebling S., Hoernke M., Kosheleva I., Henning R., Menzel A., Ihalainen J. A., and Westenhoff S. (2014) Signal amplification and transduction in phytochrome photosensors. Nature 509, 245–248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Burgie E. S., Zhang J., and Vierstra R. D. (2016) Crystal structure of Deinococcus phytochrome in the photoactivated state reveals a cascade of structural rearrangements during photoconversion. Structure 24, 448–457 [DOI] [PubMed] [Google Scholar]
- 31. Yang X., Kuk J., and Moffat K. (2009) Conformational differences between the Pfr and Pr states in P. aeruginosa bacteriophytochrome. Proc. Natl. Acad. Sci. U.S.A. 106, 15639–15644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Velazquez Escobar F., Piwowarski P., Salewski J., Michael N., Fernandez Lopez M., Rupp A., Qureshi B. M., Scheerer P., Bartl F., Frankenberg-Dinkel N., Siebert F., Andrea Mroginski M., and Hildebrandt P. (2015) A protonation-coupled feedback mechanism controls the signalling process in bathy phytochromes. Nat. Chem. 7, 423–430 [DOI] [PubMed] [Google Scholar]
- 33. Salewski J., Escobar F. V., Kaminski S., von Stetten D., Keidel A., Rippers Y., Michael N., Scheerer P., Piwowarski P., Bartl F., Frankenberg-Dinkel N., Ringsdorf S., Gärtner W., Lamparter T., Mroginski M. A., and Hildebrandt P. (2013) Structure of the biliverdin cofactor in the Pfr State of bathy and prototypical phytochromes. J. Biol. Chem. 288, 16800–16814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Noack S., Michael N., Rosen R., and Lamparter T. (2007) Protein conformational changes of Agrobacterium phytochrome Agp1 during chromophore assembly and photoconversion. Biochemistry 46, 4164–4176 [DOI] [PubMed] [Google Scholar]
- 35. von Stetten D., Seibeck S., Michael N., Scheerer P., Mroginski M. A., Murgida D. H., Krauss N., Heyn M. P., Hildebrandt P., Borucki B., and Lamparter T. (2007) Highly conserved residues Asp-197 and His-250 in Agp1 phytochrome control the proton affinity of the chromophore and Pfr formation. J. Biol. Chem. 282, 2116–2123 [DOI] [PubMed] [Google Scholar]
- 36. von Stetten D., Günther M., Scheerer P., Murgida D. H., Mroginski M. A., Krauß N., Lamparter T., Zhang J., Anstrom D. M., Vierstra R. D., Forest K. T., and Hildebrandt P. (2008) Chromophore heterogeneity and photoconversion in phytochrome crystals and solution studied by resonance Raman spectroscopy. Angew. Chem. Int. Ed. Engl. 47, 4753–4755 [DOI] [PubMed] [Google Scholar]
- 37. Piwowarski P., Ritter E., Hofmann K. P., Hildebrandt P., von Stetten D., Scheerer P., Michael N., Lamparter T., and Bartl F. (2010) Light-induced activation of bacterial phytochrome Agp1 monitored by static and time-resolved FTIR spectroscopy. ChemPhysChem 11, 1207–1214 [DOI] [PubMed] [Google Scholar]
- 38. Scheerer P., Michael N., Park J. H., Noack S., Förster C., Hammam M. A., Inomata K., Choe H. W., Lamparter T., and Krauß N. (2006) Crystallization and preliminary x-ray crystallographic analysis of the N-terminal photosensory module of phytochrome Agp1, a biliverdin-binding photoreceptor from Agrobacterium tumefaciens. J. Struct. Biol. 153, 97–102 [DOI] [PubMed] [Google Scholar]
- 39. Cooper D. R., Boczek T., Grelewska K., Pinkowska M., Sikorska M., Zawadzki M., and Derewenda Z. (2007) Protein crystallization by surface entropy reduction: optimization of the SER strategy. Acta Crystallogr. D Biol. Crystallogr. 63, 636–645 [DOI] [PubMed] [Google Scholar]
- 40. Derewenda Z. S., and Vekilov P. G. (2006) Entropy and surface engineering in protein crystallization. Acta Crystallogr. D Biol. Crystallogr. 62, 116–124 [DOI] [PubMed] [Google Scholar]
- 41. Derewenda Z. S. (2004) Rational protein crystallization by mutational surface engineering. Structure 12, 529–535 [DOI] [PubMed] [Google Scholar]
- 42. Inomata K., Hammam M. A., Kinoshita H., Murata Y., Khawn H., Noack S., Michael N., and Lamparter T. (2005) Sterically locked synthetic bilin derivatives and phytochrome Agp1 from Agrobacterium tumefaciens form photoinsensitive Pr- and Pfr-like adducts. J. Biol. Chem. 280, 24491–24497 [DOI] [PubMed] [Google Scholar]
- 43. Wang B. C. (1985) Resolution of phase ambiguity in macromolecular crystallography. Methods Enzymol. 115, 90–112 [DOI] [PubMed] [Google Scholar]
- 44. Goldschmidt L., Cooper D. R., Derewenda Z. S., and Eisenberg D. (2007) Toward rational protein crystallization: a web server for the design of crystallizable protein variants. Protein Sci. 16, 1569–1576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Krissinel E., and Henrick K. (2007) Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 372, 774–797 [DOI] [PubMed] [Google Scholar]
- 46. Li H., Zhang J., Vierstra R. D., and Li H. (2010) Quaternary organization of a phytochrome dimer as revealed by cryoelectron microscopy. Proc. Natl. Acad. Sci. U.S.A. 107, 10872–10877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Casino P., Miguel-Romero L., and Marina A. (2014) Visualizing autophosphorylation in histidine kinases. Nat. Commun. 5, 3258. [DOI] [PubMed] [Google Scholar]
- 48. Gao R., and Stock A. M. (2009) Biological insights from structures of two-component proteins. Annu. Rev. Microbiol. 63, 133–154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Strauss H. M., Schmieder P., and Hughes J. (2005) Light-dependent dimerisation in the N-terminal sensory module of cyanobacterial phytochrome 1. FEBS Lett. 579, 3970–3974 [DOI] [PubMed] [Google Scholar]
- 50. Ashenberg O., Keating A. E., and Laub M. T. (2013) Helix bundle loops determine whether histidine kinases autophosphorylate in cis or in trans. J. Mol. Biol. 425, 1198–1209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Singer P., Wörner S., Lamparter T., and Diller R. (2016) Spectroscopic investigation on the primary photoreaction of bathy phytochrome Agp2-Pr of Agrobacterium fabrum isomerization in a pH-dependent H-bond network. ChemPhysChem 17, 1288–1297 [DOI] [PubMed] [Google Scholar]
- 52. Inomata K., Noack S., Hammam M. A., Khawn H., Kinoshita H., Murata Y., Michael N., Scheerer P., Krauß N., and Lamparter T. (2006) Assembly of synthetic locked chromophores with Agrobacterium phytochromes Agp1 and Agp2. J. Biol. Chem. 281, 28162–28173 [DOI] [PubMed] [Google Scholar]
- 53. Borucki B., and Lamparter T. (2009) A polarity probe for monitoring light induced structural changes at the entrance of the chromophore pocket in a bacterial phytochrome. J. Biol. Chem. 284, 26005–26016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Möglich A., Ayers R. A., and Moffat K. (2009) Design and signaling mechanism of light-regulated histidine kinases. J. Mol. Biol. 385, 1433–1444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Lee H. S., Choi J., and Yoon S. (2007) QHELIX: a computational tool for the improved measurement of inter-helical angles in proteins. Protein J. 26, 556–561 [DOI] [PubMed] [Google Scholar]
- 56. Song C., Psakis G., Lang C., Mailliet J., Gärtner W., Hughes J., and Matysik J. (2011) Two ground state isoforms and a chromophore D-ring photoflip triggering extensive intramolecular changes in a canonical phytochrome. Proc. Natl. Acad. Sci. U.S.A. 108, 3842–3847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Mailliet J., Psakis G., Feilke K., Sineshchekov V., Essen L. O., and Hughes J. (2011) Spectroscopy and a high-resolution crystal structure of Tyr263 mutants of cyanobacterial phytochrome Cph1. J. Mol. Biol. 413, 115–127 [DOI] [PubMed] [Google Scholar]
- 58. Esteban B., Carrascal M., Abian J., and Lamparter T. (2005) Light-induced conformational changes of cyanobacterial phytochrome Cph1 probed by limited proteolysis and autophosphorylation. Biochemistry 44, 450–461 [DOI] [PubMed] [Google Scholar]
- 59. Eswar N., Eramian D., Webb B., Shen M. Y., and Sali A. (2008) Protein structure modeling with MODELLER. Methods Mol. Biol. 426, 145–159 [DOI] [PubMed] [Google Scholar]
- 60. Kabsch W. (2010) XDS. Acta Crystallogr. D Biol. Crystallogr. 66, 125–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Collaborative Computational Project No. 4 (1994) The CCP4 Suite: Programs for protein crystallography. Acta Crystallogr. D Biol. Crystallogr. 50, 760–763 [DOI] [PubMed] [Google Scholar]
- 62. Evans P. (2006) Scaling and assessment of data quality. Acta Crystallogr. D Biol. Crystallogr. 62, 72–82 [DOI] [PubMed] [Google Scholar]
- 63. McCoy A. J., Grosse-Kunstleve R. W., Adams P. D., Winn M. D., Storoni L. C., and Read R. J. (2007) Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Vagin A. A., Steiner R. A., Lebedev A. A., Potterton L., McNicholas S., Long F., and Murshudov G. N. (2004) REFMAC5 dictionary: organization of prior chemical knowledge and guidelines for its use. Acta Crystallogr. D Biol. Crystallogr. 60, 2184–2195 [DOI] [PubMed] [Google Scholar]
- 65. Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L. W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., et al. (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Emsley P., and Cowtan K. (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr. D. Biol. Crystallogr. 60, 2126–2132 [DOI] [PubMed] [Google Scholar]
- 67. Winn M. D., Isupov M. N., and Murshudov G. N. (2001) Use of TLS parameters to model anisotropic displacements in macromolecular refinement. Acta Crystallogr. D Biol. Crystallogr. 57, 122–133 [DOI] [PubMed] [Google Scholar]
- 68. Lovell S. C., Davis I. W., Arendall W. B. 3rd, de Bakker P. I., Word J. M., Prisant M. G., Richardson J. S., and Richardson D. C. (2003) Structure validation by Cα geometry: φ,ψ and Cβ deviation. Proteins 50, 437–450 [DOI] [PubMed] [Google Scholar]
- 69. Vaguine A. A., Richelle J., and Wodak S. J. (1999) SFCHECK: a unified set of procedures for evaluating the quality of macromolecular structure-factor data and their agreement with the atomic model. Acta Crystallogr. D Biol. Crystallogr. 55, 191–205 [DOI] [PubMed] [Google Scholar]
- 70. Laskowski R. A., Moss D. S., and Thornton J. M. (1993) Main-chain bond lengths and bond angles in protein structures. J. Mol. Biol. 231, 1049–1067 [DOI] [PubMed] [Google Scholar]
- 71. Pettersen E. F., Goddard T. D., Huang C. C., Couch G. S., Greenblatt D. M., Meng E. C., and Ferrin T. E. (2004) UCSF Chimera—a visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612 [DOI] [PubMed] [Google Scholar]
- 72. McDonald I. K., and Thornton J. M. (1994) Satisfying hydrogen bonding potential in proteins. J. Mol. Biol. 238, 777–793 [DOI] [PubMed] [Google Scholar]
- 73. Laskowski R. A., and Swindells M. B. (2011) LigPlot+: multiple ligand-protein interaction diagrams for drug discovery. J. Chem. Inf. Model. 51, 2778–2786 [DOI] [PubMed] [Google Scholar]
- 74. Kabsch W. (1976) Solution for best rotation to relate 2 sets of vectors. Acta Crystallogr. Sect. A 32, 922–923 [Google Scholar]
- 75. Krissinel E., and Henrick K. (2004) Secondary-structure matching (SSM), a new tool for fast protein structure alignment in three dimensions. Acta Crystallogr. D Biol. Crystallogr. 60, 2256–2268 [DOI] [PubMed] [Google Scholar]
- 76. DeLano W. L. (2002) The PyMOL Molecular Graphics System, Version 0.99r, DeLano Scientific, San Carlos, CA [Google Scholar]
- 77. Chou K. C., Nemethy G., and Scheraga H. A. (1984) Energetic approach to the packing of α-helices. 2. General treatment of nonequivalent and nonregular helices. J. Am. Chem. Soc. 106, 3161–3170 [Google Scholar]