

Abstract
NPM1 (nucleophosmin 1) is a nucleolar phosphoprotein that regulates many cellular processes, including ribosome biogenesis, proliferation, and genomic integrity. Although its role in proliferating cell types and tissues has been extensively investigated, little is known about its function in neurons and in the brain where it is highly expressed. We report that NPM1 protein expression is increased selectively in the striatum in both the R6/2 transgenic and 3-nitropropionic acid-injected mouse models of Huntington's disease. Examination of the effect of ectopic expression on cultured neurons revealed that increasing NPM1 is toxic to otherwise healthy cerebellar granule and cortical neurons. Toxicity is dependent on its cytoplasmic localization and oligomerization status. Forced retention of NPM1 in the nucleus, as well as inhibiting its ability to oligomerize, not only neutralizes NPM1 toxicity but also renders it protective against apoptosis. Although not blocked by pharmacological inhibition of the pro-apoptotic molecules, JNK, glycogen synthase kinase 3 beta, or caspases, toxicity is blocked by compounds targeting cyclin-dependent kinases (CDKs), as well as by dominant-negative forms of CDK1 and CDK2 and the pan-CDK inhibitor, p21Cip1/Waf1. Although induced in in vivo Huntington's disease models, NPM1 protein levels are unchanged in cultured cerebellar granule and cortical neurons induced to die by low potassium or homocysteic acid treatment, respectively. Moreover, and counterintuitively, knockdown of its expression or inhibition of endogenous NPM1 oligomerization in these cultured neurons is toxic. Taken together, our study suggests that although neurons need NPM1 for survival, an increase in its expression beyond physiological levels and its translocation to the cytoplasm leads to death through abortive cell cycle induction.
Keywords: cell cycle, cell death, Huntington's disease, neurodegeneration, neuron, neuroprotection, neuronal death, neuronal survival, nucleophosmin
Introduction
NPM1, also known as B23, is an abundant, ubiquitously expressed, and evolutionarily conserved non-ribosomal nucleolar phosphoprotein (1, 2). It is one of three members of the nucleophosmin/nucleoplasmin family of proteins, the other two being NPM2 and NPM3. NPM1 is an important regulator of a variety of cellular processes including centrosome duplication, genomic stability, cell proliferation, and the response to genotoxic stress. Inactivation of the NPM1 gene in mice causes death at midgestation, indicating its requirement for normal development. Fibroblasts cultured from NPM1 knock-out mice display genomic instability and reduced growth and cell proliferation (3). Other studies have shown that NPM1 can shuttle between the nucleolus and the nucleoplasm and from the nucleus to the cytoplasm. Nuclear export is believed to be required for cellular proliferation (4, 5). Mutations of NPM1 are associated with cancers and are responsible for approximately a third of cases of acute myeloid leukemia in humans (3, 6–8). These mutations, which result in an altered C terminus containing an addition of a nuclear export signal, have been reported to aberrantly localize NPM1 to the cytoplasm (8). Overexpression of NPM1, which results in elevated cytoplasmic localization, has been shown to promote cell transformation (9, 10).
Much of the research on NPM1 has been performed using proliferating cells and tissue types, particularly in the context of cancer (1–3). However, it is highly expressed in the brain and in neurons, where its function is poorly understood. Emerging evidence indicates that the nucleolus, of which NPM1 is a prominent resident, does play an important role in neuronal development and maintenance, and nucleolar dysfunction has been implicated in various neurodegenerative diseases (11, 12). This suggests that NPM1 may influence the regulation of neuronal survival. Indeed, two separate studies have described anti-apoptotic effects of NPM1 when overexpressed in cell lines of neuronal origin (13, 14). However, the contribution of NPM1 to the regulation of survival in primary neurons is less clear. We have examined this issue using two types of CNS neurons: cerebellar granule and embryonic cortical neurons. Our results suggest that NPM1 plays complex roles in the regulation of neuronal survival that are dependent on its level of expression, subcellular localization, and the extent to which it oligomerizes.
Results
NPM1 Is Up-regulated in the Striatum in Mouse Models of Huntington's Disease
Because nucleolar dysfunction is a feature of neurodegenerative diseases (12, 16, 17) and given that NPM1 is a key component of the nucleolus, we investigated the status of NPM1 expression in mouse models of HD.2 We first examined R6/2 transgenic mice, which are the most commonly used mouse model of HD (18, 19). These mice express an N-terminal fragment of the huntingtin gene (HTT) with ∼120 CAG repeats. Locomotor deficits can be observed as early as 7 weeks of age, with severe striatal atrophy and motor impairment by 12 weeks followed by death at ∼15 weeks (18, 19). Whereas NPM1 expression in R6/2 mice is elevated in the cortex, it is comparable in the rest of the brain to wild-type littermates at 6 weeks (Fig. 1A). However, at 10 weeks it is elevated in both the cortex and striatum, coinciding with the onset of neuropathology (Fig. 1A). Interestingly, in extra-striatal tissue (see other brain parts (OBP)), where there is limited or no neuropathology, expression is unchanged (Fig. 1A). We also used the 3-nitropropionic acid (3-NP) model, a chemical model of HD (20, 21). Administration of 3-NP to mice causes robust and selective striatal neurodegeneration and faithful recapitulation of key features of HD. In this model, degeneration is obvious in the striatum 3 days after 3-NP administration and is severe by 5 days. NPM1 expression is significantly elevated in the striatum within 1 day after 3-NP administration and is greatest at 3 days when degeneration is robust (Fig. 1B). Again, expression is unchanged in OBP after 3-NP administration (Fig. 1B).
FIGURE 1.

Induction of NPM1 expression in mouse models of Huntington's disease. A, NPM1 protein expression in the cortex (CTX), striatum (STR), cerebellum (CBM), and rest of the brain (OBP) from WT and transgenic (Tg) R6/2 littermates at 6, 10, and 12 weeks of age. α-Tubulin serves as a loading control. Graphs show densitometric analysis presented as means ± S.D. (n = 3). B, protein expression of NPM1 from the striatum (STR) and the rest of the brain (OBP) of wild-type mice injected with either saline control (C) or 3-NP for 1 day (D1), 3 days (D3), or 5 days (D5). α-Tubulin serves as a loading control. Graphs show densitometric analysis presented as means ± S.D. (n = 3).
Overexpression of NPM1 Kills Neurons
Because NPM1 expression is increased in mouse models of HD, we examined whether increasing its expression ectopically impacted the viability of normally healthy neurons. For this, we used CGNs, which are healthy in culture when maintained in depolarizing medium (high potassium or HK) but die when switched to non-depolarizing medium (low potassium or LK). Elevated NPM1 expression in CGNs causes death even in HK while not influencing cells already undergoing apoptosis (Fig. 2A). Induction of apoptosis by NPM1 was confirmed through cleaved caspase-3 co-staining (Fig. 2B). To confirm that this result was not specific to CGNs, we utilized a distinct neuronal type of embryonic cortical neurons, which can be induced to die through oxidative stress by treatment with homocysteic acid (HCA). Similarly, forced expression of NPM1 resulted in the death of otherwise healthy cortical neurons and did not exacerbate death caused by HCA (Fig. 2C). In both types of neurons, ectopic NPM1 localized predominantly in the nucleus and within nucleoli when the neurons were healthy (Fig. 2D). However, in neurons that were dying, NPM1 was localized to the cytoplasm (Fig. 2E), including within neurites (Fig. 2E). Quantitation of NPM1 localization in relation to neuronal viability confirmed this localization pattern in both CGNs (Fig. 2F) and cortical neurons (Fig. 2G).
FIGURE 2.

Increased expression of NPM1 is toxic to otherwise healthy neurons. A, CGNs transfected with either EGFP or NPM1 and treated with HK or LK media as described under “Experimental Procedures.” Viability of transfected cells was quantified by immunocytochemistry with GFP or FLAG antibodies. ***, p < 0.001 as compared with EGFP HK (n = 4). B, CGNs transfected as performed in A. Viability was quantified by immunocytochemistry with GFP or FLAG antibodies co-stained for cleaved caspase-3. ***, p < 0.001 as compared with EGFP HK (n = 3). C, cortical neurons transfected with either EGFP or NPM1 for 8 h and then either left untreated (Un) or treated with HCA for 15–16 h. Viability was quantified as done in A. ***, p < 0.001 as compared with untreated EGFP (n = 3). D, representative image showing localization of ectopic NPM1 expression in living cells. NPM1 was transfected into CGNs (left panels) or HT22 cells (right panels) for 24 h. CGNs were then treated with HK medium for an additional 24 h. The cells were then fixed and DAPI-stained, and NPM1 was imaged by EGFP autofluorescence. E, representative image showing localization of ectopic NPM1 in apoptotic neurons from B. NPM1 displayed a nucleolar and nucleoplasmic localization in healthy living cells and a cytoplasmic localization in dead cells. F and G, quantitation of NPM1 localization in relation to cell viability from CGNs (F) transfected in B and cortical neurons (G) transfected in C. Each graph represents ≥800 cells counted. ***, p < 0.001 (n = 3).
NPM1 Is Neurotoxic When Localized to the Cytoplasm
NPM1 is known to shuttle between the nucleolus, nucleoplasm, and cytoplasm, at least in proliferating cell types (4, 5). As described above, in dying neurons NPM1 localized preferentially to the cytoplasm. To investigate the contribution of intracellular localization to neurotoxicity, we utilized four mutant NPM1 constructs, denoted as NLSD, NESD, NESM, and NoLSM, whose localization was restricted to either the cytoplasm or nucleus (22). NLSD contains a 17-amino acid deletion (residues 141–157) of the bipartite nuclear localization signal of NPM1, whereas NESD has a 9-amino acid deletion (residues 94–102) and NESM has L100A and L102A mutations of the nuclear export signal (Fig. 3A). NoLSM contains W288G and W290G mutations in the nucleolar localization signal of NPM1 (Fig. 3A). As described previously by Wang et al. (22), the localization of NLSD is cytoplasmic and nucleolar but absent from the nucleoplasm, whereas NESD, NESM, and NoLSM are nuclear (Fig. 3B). When expressed in CGNs, NLSD displayed toxicity similar to wild-type NPM1 under both HK and LK conditions (Fig. 3C). In contrast, restricting NPM1 to the nucleus using NESD or NESM reduced the toxicity of NPM1 in HK substantially (Fig. 3C). Indeed, both NESD and NESM showed a significant level of protection in LK-treated neurons. Mutation of its nucleolar localization signal rendered NPM1 less toxic in HK, however, and offered modest protection under LK conditions (Fig. 3C). Similarly, these effects of the mutants on neuronal viability were also seen in cortical neurons treated with HCA (Fig. 3D). Along with the cytoplasmic localization of NPM1 in dying neurons, these results suggest that neurotoxicity by NPM1 requires its cytoplasmic location.
FIGURE 3.
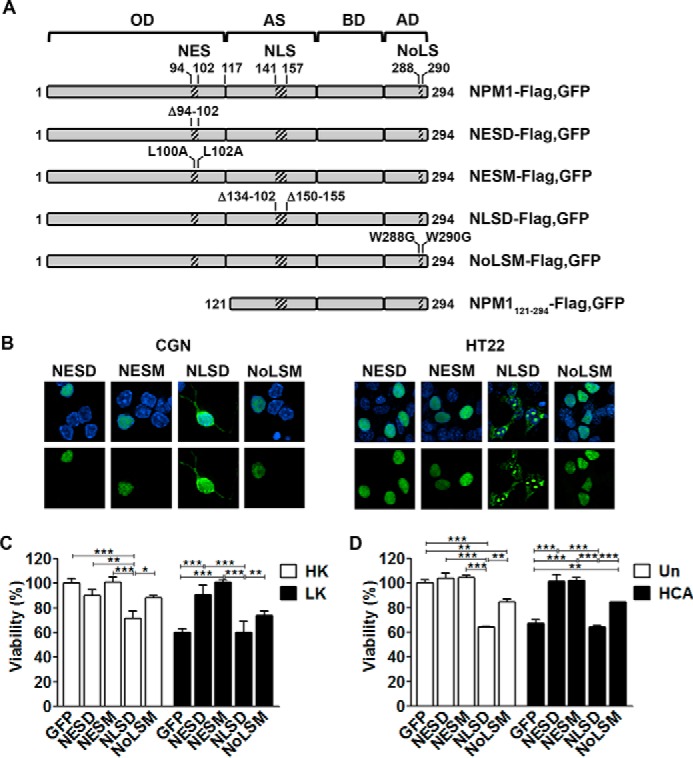
NPM1 toxicity is dependent on subcellular localization. A, schematic detailing the constructs used in this study (adapted from Wang et al. (22)). OD, N-terminal oligomerization domain; AS, acidic stretches; BD, basic domain; AD, aromatic domain. B, representative images showing the localization of NPM1 mutants in CGNs (left panel) and HT22s (right panel). NESD, NESM, NLSD, and NoLSM mutants were transfected into CGNs for 24 h, followed by 24-h HK treatment, or HT22s for 24 h. The cells were then fixed and DAPI-stained, and NPM1 localization was imaged by EGFP autofluorescence. C, CGNs transfected with EGFP, NESD, NESM, NLSD, or NoLSM and treated with HK/LK medium. Viability was quantified by immunocytochemistry with GFP or FLAG antibodies and DAPI staining. *, p < 0.05; **, p < 0.01; ***, p < 0.001 (n = 3). D, cortical neurons transfected with EGFP and the four NPM1 mutants for 8 h followed by HCA treatment for 15–16 h. Viability was quantified as just described. **, p < 0.01; ***, p < 0.001 (n = 3).
Oligomerization of NPM1 Promotes Neuronal Death
NPM1 forms pentameric oligomers through an oligomerization domain mapped to residues 15–118 within the protein (23, 24). Aptamers have been identified that inhibit NPM1 oligomerization both in vitro and in cells (25). Expression of these aptamers in cancer cells not only increases their sensitivity to DNA-damaging agents but is sufficient to promote apoptosis of these cells (25). To investigate the significance of NPM1 oligomerization to its regulation of neuronal viability, we utilized two aptamers previously shown to block NPM1 oligomerization by Jian et al. (25). We independently confirmed that both of these aptamers, 1A1 and 1A11–40 (designated here as 1A1Trunc) inhibited NPM1 oligomerization (Fig. 4A). Although neither aptamer had an influence on the viability of CGNs undergoing apoptosis, both had a slight toxic effect on the viability of healthy neurons, which was only significant with 1A1Trunc (Fig. 4B). However, when co-expressed with NPM1, not only was NPM1 no longer toxic in HK, it fully protected CGNs from LK-induced cell death (Fig. 4C), suggesting that whereas elevated expression by itself was not toxic, the oligomerization of NPM1 transformed it from a protective molecule to a toxic one. To further investigate this issue, we expressed an NPM1 incapable of oligomerizing because of a deletion of its N-terminal oligomerization domain (NPM1121–294) (Fig. 3A). Consistent with oligomerization being needed for toxicity rather than elevated levels, overexpression of monomeric NPM1 no longer induced death in healthy CGNs (Fig. 4D) and cortical neurons (Fig. 4E). As was seen by co-expression with the aptamers, NPM1121–294 was fully protective under apoptotic conditions (Fig. 4, D and E). We tested the ability of NESD, NESM, NLSD, NoLSM, and NPM1121–294 to oligomerize. As expected, NPM1121–294 failed to oligomerize (Fig. 4F). In comparison with wild-type NPM1, both NoLSM and NLSD displayed a similar ability to oligomerize (Fig. 4F). In contrast, NESD and NESM showed no oligomerization (Fig. 4F). Taken together, these results suggest that oligomerization of NPM1 occurs in the cytoplasm and that it is necessary for the neurotoxicity of NPM1.
FIGURE 4.
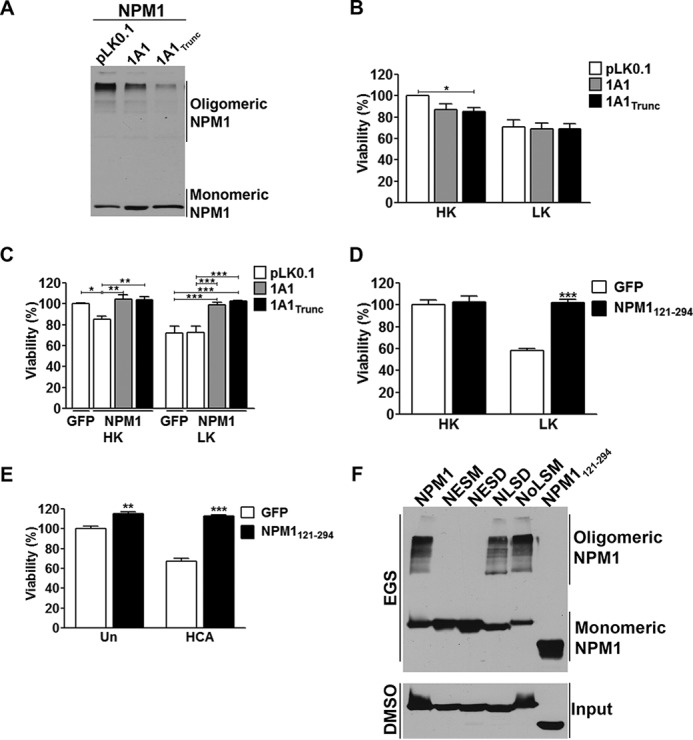
Effect of NPM1 on viability is dependent on its oligomerization status. A, NPM1 RNA aptamers inhibit NPM1 oligomerization. NPM1 was co-transfected into HEK293T cells with either a control plasmid (pLK0.1), 1A1, or 1A1Trunc in a 1:2 ratio for 24 h. The cells were then lysed and subjected to cross-linking as described under “Experimental Procedures.” B, CGNs were transfected with either pLK0.1 co-expressing with EGFP in a 6.5:1 ratio, 1A1, or 1A1Trunc for 48 h followed by HK/LK treatment. Viability was quantified based on either EGFP (pLK0.1) or DsRed (aptamers) autofluorescence. *, p < 0.05 (n = 3). C, CGNs transfected with EGFP and pLK0.1 or NPM1 and pLK0.1, 1A1, or 1A1Trunc in a 1:2 ratio for 48 h followed by HK/LK. Viability was quantified by immunocytochemistry based on EGFP or NPM1 transfection with GFP or FLAG antibodies. *, p < 0.05; **, p < 0.01; ***, p < 0.001 (n = 3). D and E, EGFP or NPM1121–294 were transfected into CGNs and treated with HK/LK (D) or cortical neurons and either left untreated (Un) or treated with HCA (E) as previously described. Viability quantification was performed as done in C. **, p < 0.01 untreated NPM1 transfected neurons compared with untreated EGFP transfected neurons; ***, p < 0.001 NPM1121–294 transfected neurons compared with EGFP transfected neurons in LK and HCA (n = 3). F, cross-linking of NPM1 mutants transfected into HEK293T cells for 24 h as described under “Experimental Procedures.” EGS, ethylene glycol bis-succinimidylsuccinate.
Overexpressed NPM1 Kills by Promoting Abortive Cell Cycle Re-entry
Several studies performed in cell lines have found that NPM1 promotes cell cycle progression, particularly when it is in the cytoplasm. In fact, nuclear export is necessary for cell cycle progression by NPM1 (4, 5, 10). Abortive cell cycle re-entry is a well described mechanism of neuronal death (26–28). We therefore investigated whether NPM1-induced toxicity involved activation of the cell cycle machinery. Treatment with both roscovitine, a widely used and broad spectrum CDK inhibitor (29), as well as another CDK inhibitor, HSB13 (30), protected against NPM1 neurotoxicity (Fig. 5A). Similarly, co-expression of dominant-negative forms of either CDK1 or CDK2 inhibited NPM1 neurotoxicity (Fig. 5B). Furthermore, the toxicity from NLSD expression is blocked by both treatment with roscovitine and expression of dominant-negative CDK1 and CDK2 (Fig. 5, D and E). Activation of caspases, JNK, and glycogen synthase kinase 3 beta have also been shown to be important for neuronal death (31–33). In contrast to the CDK inhibitors, however, inhibitors targeting these pro-apoptotic molecules had no protective effect (Fig. 5A). These results suggest that the toxic effect of elevated NPM1 in neurons is mediated by cell cycle activation.
FIGURE 5.
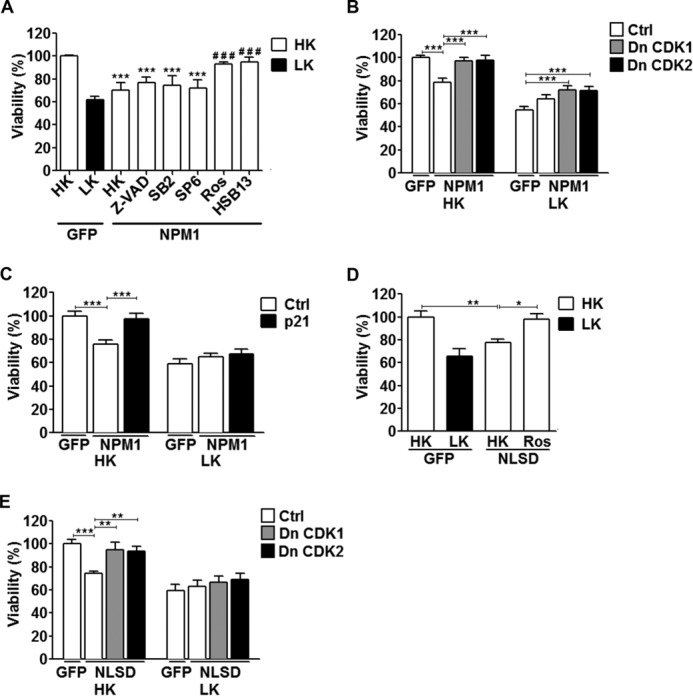
Toxicity by increased NPM1 expression is cell cycle-dependent. A, CGNs transfected with either EGFP or NPM1 were then treated with either HK medium alone or HK medium supplemented with the following inhibitors: Z-VAD (50 μm), SB216763 (5 μm), SP600125 (10 μm), roscovitine (50 μm), or HSB13 (25 μm). Viability was quantified by immunocytochemistry with a GFP or FLAG antibody. ***, p < 0.001 as compared with GFP in HK; ###, p < 0.001 as compared with NPM1 treated with HK (n = 3). B and C, CGNs were transfected with either EGFP and a control vector (Ctrl, pK3HA) or NPM1 and pK3HA (Ctrl), DnCDK1, DnCDK2 (B), or p21 (C) in a 1:2 ratio followed by HK/LK treatment. Viability was quantified based on NPM1 fluorescence by immunocytochemistry with a GFP antibody. ***, p < 0.001 (n = 3). D, CGNs transfected with EGFP or NLSD. The cells were then treated with either HK medium alone or HK medium supplemented with roscovitine (50 μm). Viability was quantified as described for A. *, p < 0.05; **, p < 0.01 (n = 3). E, CGNs transfected with EGFP and pK3HA (Ctrl) or NLSD and pK3HA (Ctrl), DnCDK1, or DnCDK2 in a 1:2 ratio followed by HK/LK treatment. Viability was quantified as described for B and C. **, p < 0.01; ***, p < 0.001 (n = 3). Ctrl, control.
An endogenous and physiological inhibitor of CDKs is p21Cip1/Waf1. Indeed, p21Cip1/Waf1 has been shown by a number of laboratories to have strong neuroprotective effects both in vitro and in vivo (34–36). Consistent with its well established protective activity, co-expression of p21Cip1/Waf1 blocked NPM1-induced neuronal death (Fig. 5C).
Expression and Significance of Endogenous NPM1 to Neuronal Viability
As described above, NPM1 expression is elevated in mouse models of HD. Ectopically increasing its levels in cultured neurons has a neurotoxic effect. To examine whether the expression of endogenous NPM1 is increased in cultured neurons induced to die, we looked at its protein levels in LK-treated neurons. As shown in Fig. 6A, there is no significant change in expression of NPM1 protein. Similarly, in cortical neurons induced to die by HCA treatment, NPM1 expression remains unaltered (Fig. 6B). Immunocytochemical analysis of expression showed no discernible changes in the number on NPM1-positive nucleoli or the intensity of staining of the nucleoli in neurons that appeared to be relatively healthy (data not shown). Interestingly and consistent with the idea that NPM1 localization to the cytoplasm induces neurotoxicity, NPM1 was cytoplasmic in dying neurons (Fig. 6C and supplemental Fig. S1).
FIGURE 6.
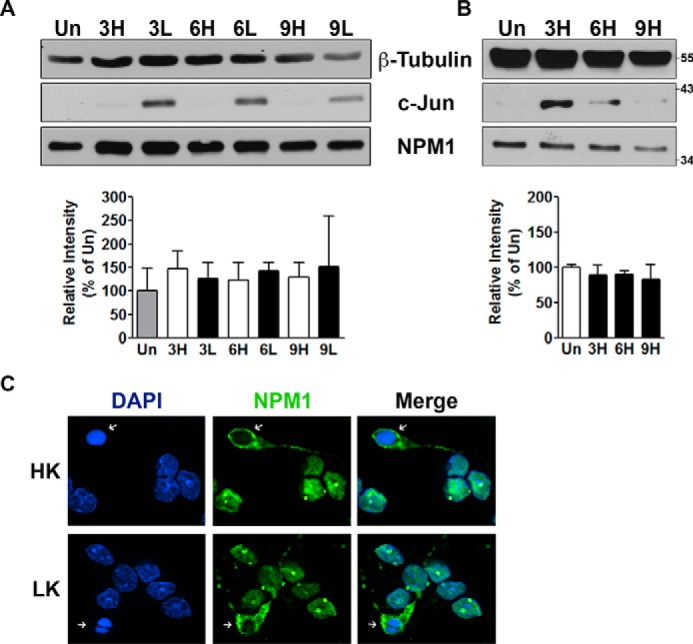
Expression pattern of endogenous NPM1. A and B, protein expression (top panels) and densitometric analysis (bottom panels) presented as means ± S.D. of endogenous NPM1 in CGN (A, n = 5) and cortical (B, n = 3) cultures. The cells were either left untreated (Un) or treated with HK/LK medium (CGNs) or HCA (corticals) for 3, 6, and 9 h. Endogenous NPM1 expression was analyzed as described under “Experimental Procedures.” β-Tubulin serves as a loading control and c-Jun serves as a marker for induction of apoptosis. C, representative image showing localization of endogenous NPM1 in 6-h HK/LK-treated CGNs. Arrows indicate NPM1 localized to the cytoplasm of apoptotic cells.
We proceeded to determine whether knocking down NPM1 influenced death of cultured neurons. We first tested the effectiveness of three different shRNAs to suppress NPM1 expression in HT22 cells, a mouse neuroblastoma cell line that is easily transfected. Two of the shRNA constructs tested, sh1 and sh2, were effective in knocking down NPM1 (Fig. 7A). When transfected into CGNs, both sh1 and sh2 induced death in otherwise healthy neurons (Fig. 7B). Knockdown of NPM1 with sh1 and sh2 also reduced survival of cortical neurons (data not shown). This suggested that although high levels of NPM1 are detrimental to neuronal survival, NPM1 at moderate levels is necessary for the viability of neurons.
FIGURE 7.

Knockdown of endogenous NPM1 is toxic to otherwise healthy neurons. A, HT22 cells were transfected with either a control (pLK0.1) or three commercially available shRNAs targeting NPM1 (sh1, sh2, and sh3) for 72 h. The cells were then lysed and subjected to Western blotting analysis with an antibody against NPM1. β-Tubulin serves as a loading control. B, viability of CGN cultures transfected with a control (pLK0.1), sh1, or sh2 along with EGFP in a 6.5:1 ratio and treated with HK/LK medium. Viability was quantified by EGFP autofluorescence. ***, p < 0.001 as compared with pLK0.1 in HK (n = 3).
Nuclear NPM1 Is Protective against mHTT-induced Cell Death
As described above, NPM1 expression is increased in the cortex and striatum of symptomatic R6/2 transgenic mice (Fig. 1A). We therefore examined whether mutant huntingtin (mHTT) itself had an effect on endogenous NPM1 oligomerization and localization. The mouse neuroblastoma Neuro2a (N2a) cell line is sensitive to mHTT (37). Expression of two mHTT constructs (Q138-GFP and Q138-RFP) into these cells showed no change in endogenous NPM1 oligomerization over a 72-h period (Fig. 8A).
FIGURE 8.
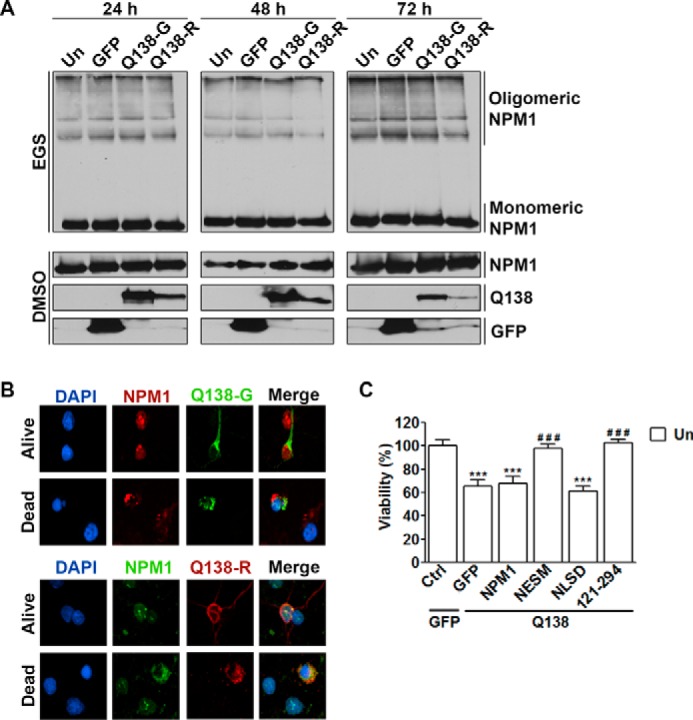
Nuclear and monomeric NPM1 protects against mHTT toxicity. A, N2a cells were either left untransfected (Un) or transfected with EGFP, Q138-GFP, or Q138-RFP. The cells were lysed 24, 48, or 72 h later and subjected to cross-linking as described under “Experimental Procedures.” The blots were probed for endogenous NPM1 and then reprobed with GFP and RFP antibodies. B, cortical neurons were transfected with either Q138-GFP (top panel) or Q138-RFP (bottom panel), and the cells were fixed 24 h later. Immunocytochemistry was performed by co-staining with NPM1 and either GFP or RFP antibodies. C, cortical neurons transfected in a 1:2 ratio with either EGFP and a control vector (pK3HA) or Q138-RFP and EGFP, NPM1, NESM, NLSD, or NPM1121–294. The cells were fixed 24 h later, and immunocytochemistry was performed with RFP and GFP antibodies. Viability was quantified by RFP or GFP fluorescence and DAPI staining. ***, p < 0.001 as compared with EGFP/control transfected cells; ###, p < 0.001 as compared with Q138-RFP/EGFP transfected cells (n = 3). Ctrl, control.
We proceeded to investigate whether the localization of NPM1 is important for mHTT-induced toxicity of cortical neurons. Although we find endogenous NPM1 localized to the cytoplasm of dead cells (Fig. 6C and supplemental Fig. S1), interestingly in neurons expressing either Q138-GFP or Q138-RFP, NPM1 showed a nuclear pattern in both living and dead cells (Fig. 8B). Toxicity in cortical neurons from mHTT expression occurs within 24 h. To examine the effect of NPM1 localization on this toxicity, mHTT was expressed along with the different NPM1 constructs. Toxicity by mHTT was not influenced by wild-type NPM1 but was slightly increased when co-expressed with the cytoplasmic localized NPM1 (Fig. 8C). However, nuclear-restricted and monomeric NPM1 (NESM and NPM1121–294) was highly protective against mHTT toxicity (Fig. 8C). These results suggest that mHTT does not directly influence NPM1 but instead localization of NPM1 may play a role in mHTT-induced neuronal death.
Discussion
In recent years, the importance of the neuronal nucleolus, the proteins that reside there, and the impact of nucleolar stress on neuronal health and during neurodegeneration have been gaining increasing attention (12). Because NPM1 is a key nucleolar protein and because of its high abundance and ubiquitous expression in the brain, as well as the cellular processes it is known to regulate, it is potentially an attractive molecule in the regulation of neuronal viability. Indeed, recent evidence has suggested this (13, 38). However, there remains to be a clear consensus as to what its function in this process is. In this study, we describe a complex role for NPM1 whereby neurons require a healthy balance of its expression as too much and too little of the protein can have negative consequences. Although the mechanism of death by knockdown requires additional elucidation, toxicity caused by increased expression is dependent upon the ability of NPM1 to oligomerize and subsequently by alteration of its subcellular localization and cell cycle activation.
In proliferating cells, changes in NPM1 expression have revealed both pro-survival and pro-apoptotic qualities, depending on cellular conditions (1). Thus far, neuronal NPM1 has been investigated in two independent studies in the context of excitotoxicity, a process that leads to the necrotic death of neurons. These studies showed that NPM1 mRNA is decreased in glutamate-treated cortical cultures from PS1 M146V mutant transgenic mice (39) and that its protein is down-regulated in degenerating neurons of the rat hippocampal CA1 region, as well as primary neuronal cultures following kainic acid treatment (13). We report, however, that NPM1 expression is increased in both the R6/2 transgenic and 3-NP chemical mouse models of Huntington's disease. This increase is more specific to the striatum and observed when neurological deficits emerge. Furthermore, comparatively we find that protein levels are relatively unchanged in cultures of both HK/LK treated CGNs, as well as HCA-treated cortical neurons. Thus, similar to what is seen in actively dividing cells, changes in neuronal NPM1 expression, as well as the effect this has, might be cell- and context-dependent. The mode of cell death may also influence the pattern of NPM1 expression in dying neurons. Although it is possible that the up-regulation of NPM1 occurs in surviving neurons, rather than dying cells, and may thus represent a neuroprotective response, results from the overexpression of NPM1 in primary neurons argue against this. Indeed, both in CGNs and cortical neurons, elevated expression of NPM1 induces death. Although we do find that NPM1 can protect against mHTT toxicity in cortical neurons, this was only true when restricted to the nucleus. Wild-type NPM1 was unable to afford this protection, and a cytoplasmic restriction slightly exacerbated death by mHTT. Thus, more investigation is required into the in vivo nature of NPM1 during both early and late symptomatic stages of HD.
Treatment with two pharmacological inhibitors, co-expression with dominant-negative forms of CDK1/2, and the overexpression of p21Cip1/Waf1, inhibits NPM1 neurotoxicity. We therefore conclude that death from NPM1 expression is the result of cell cycle activation, a process known to induce apoptosis in neurons. Activation of cell cycle proteins and evidence of abortive cell cycle re-entry has also been reported in vivo in various models of neurodegenerative disease (40–42). Both cyclin E/CDK2 and cyclin B/CDK1 have been reported to phosphorylate NPM1 (43, 44). It is possible that an increase in NPM1 expression results in phosphorylation by CDK1 or CDK2, thereby leading to an abortive re-entry into the cell cycle and consequently to cell death. However, it is plausible that CDK1/2 activation is a consequence of cytoplasmic accumulation of NPM1, thereby leading to cell cycle re-entry.
Although we report that ectopic expression induces death, we also find, as others (13, 38) have, that knockdown also induces neuronal death. However, although the mechanism by which knockdown of NPM1 kills neurons remains to be elucidated, this finding indicates that a physiological level of NPM1 is necessary for the survival of neurons. Interestingly, although inhibition of cell cycle machinery prevented death from forced expression in HK, only a slight protection is seen under apoptotic conditions. Thus, although important for neurons under normal conditions, NPM1 might not be necessary for the regulation of death under LK. It is possible that NPM1 provides this effect under HK conditions through cooperation with a partner whose expression decreases as neurons commit to apoptosis. Alternatively, under LK conditions and in the presence of elevated NPM1 expression, cell death pathways other than those regulated by cell cycle machinery could be activated.
NPM1 is typically found as higher order oligomers, the formation of which is important for its function. We investigated this fact with the use of RNA aptamers designed to interfere with oligomer formation. In line with physiological levels of NPM1 being necessary for healthy neurons and as others have reported (25) in cell lines, inhibiting endogenous oligomerization induces death. Interestingly, when co-expressed together, NPM1 was not only no longer toxic but also fully protective against LK-induced apoptosis. The death-inducing activity of ectopically expressed NPM1 is further blocked if it is constrained to the nucleus, and we show that NPM1 was unable to oligomerize when strictly nuclear, suggesting that this process required nuclear export. Less is known about monomeric NPM1. Inhibition of oligomerization by mutation of Cys21 blocks NPM1 chaperone activity (45). Additionally, NPM1 is known to bind DNA, as well as both single- and double-stranded RNA (46). Interestingly, it is reported to be more capable of binding DNA when there is a greater presence of monomers, whereas this is lost with greater amounts of oligomers (46). However, the mechanism by which monomeric is protective against LK- and HCA-induced apoptosis is unclear and requires further investigation.
In summary, we show for the first time that the nucleolar phosphoprotein, NPM1, is selectively up-regulated in the striatum of two models of Huntington's disease, the R6/2 transgenic and the 3-NP-injected mice. Both knockdown and ectopic expression have detrimental effects on otherwise healthy neurons. The toxic effect from increased expression is dependent upon the ability of NPM1 to translocate to the cytoplasm and oligomerize, which ultimately results in cell cycle activation. However, we have found that monomeric forms of NPM1 are fully protective against the induction of apoptosis. Further, NPM1 can protect against mHTT toxicity but only in its nuclear and monomeric form. Although thus far described as a protective molecule in neurons, our study sheds light on a more complicated role, thereby revealing that a more in-depth investigation is needed to fully elucidate the role of neuronal NPM1.
Experimental Procedures
Materials
Unless stated otherwise, all tissue culture medium was purchased from Invitrogen, and chemicals and reagents were purchased from Sigma-Aldrich. Poly-l-lysine for primary neuronal cultures was purchased from Trevigen (Gaithersburg, MD). Antibodies used in this study were as follows: anti-B23 (catalog no. B0556; Sigma-Aldrich), α-tubulin (catalog no. T5168; Sigma-Aldrich), β-tubulin (catalog no. 5568P; Cell Signaling, Danvers, MA), cleaved caspase-3 (catalog no. 9661S; Cell Signaling), c-Jun (catalog no. 9165S; Cell Signaling), FLAG (catalog no. F1804; Sigma-Aldrich), GFP (catalog no. SC-9996; Santa Cruz Biotechnology, Dallas, TX), and RFP (catalog no. R10367; Invitrogen). Primary antibodies were used at a concentration of 1:1,000, or 1:40,000 for α-tubulin, in 5% bovine serum albumin. HRP-conjugated secondary antibodies (Pierce) were used at concentrations of 1:10,000 to 1:40,000.
Expression Plasmids
Expression plasmids used in this study and purchased from Addgene (Cambridge, MA) are as follows: GFP- and FLAG-tagged NPM1 (catalog no. 17578), NESD (catalog no. 13283), NESM (catalog no. 13282), and NLSD (catalog no. 13287) plasmids were donated by Xin Wang (22), FLAG-p21 (catalog no. 16240) donated by Mien-Chie Hung (47), HA-tagged dominant-negative Cdc2/CDK1 (DnCDK1, catalog no. 1889), and CDK2 (DnCDK2, catalog no. 1885) plasmids were donated by van den Heuvel and Harlow (48). The mutant huntingtin (mHTT) constructs, Q138-GFP and Q138-RFP, were kind gifts from J. Troy Littleton at the Massachusetts Institute of Technology. NPM1 RNA aptamers, 1A1 and 1A11–40 (referred to in this study as 1A1Trunc), were a kind gift from C. Yang at the Institute of Genetics and Developmental Biology, Chinese Academy of Sciences. NPM1 mutant and deletion constructs, NoLSM and NPM1121–294, were created by using full-length NPM1 in pEGFP-C2 listed above as a template. The following primers were used to create these constructs: nucleolar localization signal mutant (W288G, W290G; NoLSM): NPM1fwd, 5′-TCGAATTCTGCAGTCGAC-3′, and NoLSMrev, 5′-TCCGGTGGATCCTTAAAGAGACTTCCTCCCCTGCCCGAG-3′ (NoLSM PCR product was digested with EcoRI and BamHI and then ligated into pEGFP-C2); and oligomerization domain mutant (NPM1121–294): NPM1121–294fwd, 5′-CAAGGATGACGACGACAAGCATATGGAAGATGCAGAG-3′, and NPM1-rev, 5′-TCCGGTGGATCCTTAAAGAGACTTCCTCCACTGCCAGAG-3′. This product was then used as a template for PCR with the primers: NPM1fwd: 5′-GGGCCCGGGATCCTGCCCGCACATGGACTACAAGGATGAC-3′, and NPM1-rev, 5′-TCCGGTGGATCCTTAAAGAGACTTCTCCACTGCCAGAG-3′. The resulting product was digested with BamHI and ligated into pEGFP-C2. All constructs were sequenced and then transfected in HEK293T cells to check for expression by EGFP autofluorescence. Protein lysates were subjected to Western blot analysis to check proper protein size.
Culture, Treatment, and Transfection of Neurons
Cerebellar granule neurons (CGNs) were cultured as previous described (49). Briefly, 7–8-day-old Wistar rats were euthanized, and cerebella were extracted and plated in 24-well plates (1 × 106 cells/well) or 60-mm dishes (12 × 106 cells) in culture medium (basal minimal Eagle's medium, supplemented with 10% FBS, 25 mm KCl, 2 mm glutamine, and 0.2% gentamycin). To prevent replication of non-neuronal cells, cytosine arabinoforanoside (10 μm) was added to the culture medium 18–22 h after plating. Transient transfections were performed on day 4–5 in vitro by the calcium phosphate method as previously described (50, 51) and allowed to express for 24 h (or 48 h in the case of shRNA). Cultures were then switched to serum-free culture medium (basal minimal Eagle's medium, 2 mm glutamine, and 0.2% gentamycin) supplemented with 25 mm KCl (HK) or without KCl (LK). For pharmacological inhibitor studies, at the time of medium switch cells were treated with either HK medium alone or HK medium supplemented with inhibitors of the following concentrations: Z-VAD at 50 μm, SB216763 at 5 μm, SP6001245 at 10 μm, roscovitine at 50 μm, or HSB13 at 25 μm. All pharmacological inhibitors were purchased from Calbiochem (Billerica, MA), and their ability to inhibit their targets at the doses listed above was confirmed in control experiments. After 24 h treatment, the cells were fixed, immunocytochemistry was performed, and cell viability was quantified based on cell morphology using DAPI staining as previously described (52, 53). The cells with condensed or fragmented nuclei were scored as dead. Co-staining for cleaved caspase-3 was used as a secondary method to confirm cell death.
Rat cortical cultures were prepared from the cerebral cortex of E16–17 Wistar rats as previously described (54–56). The cultures were maintained in neurobasal medium with 1% B27 supplement, 0.25% l-glutamine, 1% penicillin/streptomycin, 0.1% HEPES, and 1.1% sodium pyruvate without serum to minimize glial proliferation. The cultures were transfected on day 6 in vitro by the calcium phosphate method and allowed to express for 8 h followed by 15–16 h of treatment with 1 mm HCA. Viability was quantified as described above for CGNs. For cell lysates for Western blot analysis, 60-mm dishes were either left untreated or treated with 1mm HCA on day 7 in vitro. HCA induces death in cortical neurons through oxidative stress (57, 58).
Culture and Transfection of Cell Lines
The HEK293T (catalog no. CRL-11268) and Neuro2a (N2a, catalog no. CLL-131) cell lines were purchased from ATCC and maintained in DMEM supplemented with 10% FBS. The HT22 hippocampal neuroblastoma cell line was a kind gift from Dr. Rajiv Ratan (Burke Medical Research Institute) and was maintained in DMEM without sodium pyruvate supplemented with 10% FBS. All cell line transfections were performed using Lipofectamine 2000 (Life Technologies) diluted in Opti-Mem reduced serum medium by following the manufacturer's guidelines.
Western Blotting
The cells were lysed with 1× cell lysis buffer (20 mm Tris-HCl, pH 7.5, 150 mm NaCl, 1 mm Na2EDTA, 1 mm EGTA, 1% Triton, 2.5 mm sodium pyrophosphate, 1 mm β-glycerophosphate, 1 mm Na3VO4, 1 μg/ml leupeptin) containing a protease inhibitor mixture tablet (Roche), frozen at −80 °C for at least 1 h, thawed, and centrifuged at 14,000 × g for 10 min at 4 °C. The supernatant was collected as the whole cell soluble lysate, and protein concentration was determined by the Bradford assay (Bio-Rad). Unless stated otherwise, 40 μg of protein was mixed with 6× SDS sample buffer (375 mm Tris-HCl, pH 6.8, 12% SDS, 60% glycerol, 300 mm dithiothreitol, and 0.012% bromphenol blue), boiled at 95 °C for 5 min, and subjected to SDS-PAGE. The proteins were electrophoretically transferred from the gel to an enhanced PVDF membrane (Bio-Rad) at 4 °C overnight. Membranes were incubated in blocking buffer (1× TBS, 5% (w/v) nonfat dry milk and 0.05% Tween 20) at 25 °C for 1 h, and then subsequently incubated at 4 °C overnight with primary antibodies, which was followed by secondary antibody for 1 h at 25 °C. Immunoreactivity was developed by enhanced chemiluminescence and visualized by autoradiography using ECL or ECL prime reagents (GE Healthcare).
shRNA-mediated Knockdown
For knockdown experiments, the following shRNAs targeting NPM1 were purchased from Sigma-Aldrich: TRCN0000115427, TRCN0000115428, and TRCN0000115430 referred to here as sh1, sh2, and sh3, respectively. The pLK0.1-TRC (pLK0.1) control shRNA, which contains a non-hairpin 18-bp insert, was purchased from Addgene (catalog no. 10879) donated by David Root (59). To test knockdown efficiency, each shRNA was transfected into the HT22 neuroblastoma cell line using Lipofectamine 2000 according to the manufacturer's instructions and allowed to express for 72 h. The cells were then lysed in 1× cell lysis buffer and subjected to Western blotting with an antibody against endogenous NPM1. For viability studies, pLK0.1 or each shRNA was transfected into neuronal cultures along with EGFP in a 6.5:1 ratio as described above. For CGNs, the cells were transfected on day 4 in vitro. The medium was switched 48 h later to HK/LK medium for 24 h. Cortical neuronal cultures were transfected on day 6 in vitro and allowed to express for 72 h. The cells were then fixed and DAPI-stained, and viability was quantified based on EGFP fluorescence.
Cross-linking Analysis
Cross-linking was performed as previously described (60, 61). HEK293T or N2a cells were transfected using Lipofectamine 2000 and allowed to express for 24–72 h. The cells were then lysed in 150 μl of HEGNT buffer (20 mm HEPES, pH 7.5, 1 mm EDTA, 10% glycerol, 0.4 m NaCl, 1% Triton X-100) by freezing at −80 °C for at least 1 h, thawed, and centrifuged at 14,000 × g for 10 min. 50 μg of whole cell lysate was then incubated with either DMSO or 0.5 mm ethylene glycol bis-succinimidylsuccinate for 30 min at 25 °C. Cross-linking was quenched with the addition of 0.025 mm Tris-HCl, pH 7.5, for 15 min at 25 °C. The reactions were then boiled in 6× SDS at 95 °C for 5 min and then subjected to SDS-PAGE on an 8% gel.
The R6/2 Transgenic Mouse Model of HD
Female mice hemizygous for an ovarian transplant of exon 1 of the human huntingtin transgene containing 120 ± 5 CAG repeats were bred with WT B6CBAF1/J male mice (Jackson Laboratory). Genotyping was performed 5–7 days after birth following Institutional Animal Care and Use Committee-approved guidelines. For subsequent generations of breeding, transgenic (R6/2) male mice were bred to non-littermate WT females. At 6, 10, and 12 weeks of age, gender-matched WT and R6/2 littermates were euthanized, and brains were dissected into the following regions: striatum (STR), cortex (CTX), cerebellum (CBM), and the rest of the brain (OBP). Tissue was homogenized in 1× radioimmune precipitation assay buffer (20 mm Tris-HCl, pH 7.5, 150 mm NaCl, 1 mm Na2EDTA, 1 mm EGTA, 1% Nonidet P-40, 1% sodium deoxycholate, 2.5 mm sodium pyrophosphate, 1 mm β-glycerophosphate, 1 mm Na3VO4, 1 μg/ml leupeptin) containing a protease inhibitor mixture tablet and lysed by freezing at −80 °C for at least 1 h. Samples were then thawed, centrifuged at 14,000 × g for 10 min, and subjected to Western blotting.
The 3-NP Mouse Model of HD
10-week-old C57BL/6 male mice (Charles River Laboratories, Wilmington, MA) were administered 3-NP as previous described (15, 30, 62). Briefly, mice received 10 intraperitoneal injections of either saline control or 3-NP (50 mg/kg, pH 7.4) every 12 h for 5 days. Pairs of control and 3-NP mice were euthanized by CO2 inhalation after 1, 3, or 5 days of injections. Brains were dissected into either the STR or the rest of the brain (OBP). The tissue was then homogenized in 1× radioimmune precipitation assay buffer and lysed by freezing at −80 °C for at least 1 h. The samples were thawed, centrifuged at 14,000 × g for 10 min, and subjected to Western blotting.
Animal Usage
All of the procedures conducted using animals were reviewed and approved by the Southern Methodist University Institutional Animal Care and Use Committee.
Statistical Analysis
All graphs were created and statistical analysis was performed using the GraphPad Prism software, and all densitometric analysis was done using ImageJ. Student's t test was performed for statistical analysis. For comparing multiple data sets, one-way analysis of variance with Tukey's multiple comparisons posttest was used. The results are shown as the means ± standard deviation from at least three independent experiments. p values of p < 0.05 were deemed statistically significant. Asterisks are used to denote statistical significance: *, p < 0.05; **, p < 0.01; and ***, p < 0.001. Unless mentioned otherwise, all viability experiments were performed in duplicate and repeated three times. For each viability experiment, ≥200 transfected cells were counted.
Author Contributions
J. A. P. and S. R. D. designed the research and wrote the paper. J. A. P. performed the research and analyzed the data.
Supplementary Material
Acknowledgments
We thank Varun Rawat for performing 3-NP injections and Lulu Wang for technical assistance.
This work was supported by National Institutes of Health Grant R01 NS040408 (to S. R. D.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.

This article contains supplemental Fig. S1.
- HD
- Huntington's disease
- 3-NP
- 3-nitropropionic acid
- CDK
- cyclin-dependent kinase
- CGN
- cerebellar granule neuron
- HCA
- homocysteic acid
- OBP
- other brain parts
- HK
- high potassium
- LK
- low potassium
- NLS
- nuclear localization signal.
References
- 1. Pfister J. A., and D'Mello S. R. (2015) Insights into the regulation of neuronal viability by nucleophosmin/B23. Exp. Biol. Med. (Maywood) 240, 774–786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ye K. (2005) Nucleophosmin/B23, a multifunctional protein that can regulate apoptosis. Cancer Biol. Ther. 4, 918–923 [DOI] [PubMed] [Google Scholar]
- 3. Grisendi S., Mecucci C., Falini B., and Pandolfi P. P. (2006) Nucleophosmin and cancer. Nat. Rev. Cancer. 6, 493–505 [DOI] [PubMed] [Google Scholar]
- 4. Brady S. N., Yu Y., Maggi L. B. Jr., and Weber J. D. (2004) ARF impedes NPM/B23 shuttling in an Mdm2-sensitive tumor suppressor pathway. Mol. Cell. Biol. 24, 9327–9338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Yu Y., Maggi L. B. Jr., Brady S. N., Apicelli A. J., Dai M.-S., Lu H., and Weber J. D. (2006) Nucleophosmin is essential for ribosomal protein L5 nuclear export. Mol. Cell. Biol. 26, 3798–3809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Yohe S. (2015) Molecular genetic markers in acute myeloid leukemia. J. Clin. Med. 4, 460–478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Rau R., and Brown P. (2009) Nucleophosmin (NPM1) mutations in adult and childhood acute myeloid leukaemia: towards definition of a new leukaemia entity. Hematol. Oncol. 27, 171–181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Falini B., Sportoletti P., and Martelli M. P. (2009) Acute myeloid leukemia with mutated NPM1: diagnosis, prognosis and therapeutic perspectives. Curr. Opin. Oncol. 21, 573–581 [DOI] [PubMed] [Google Scholar]
- 9. Chan W. Y., Liu Q. R., Borjigin J., Busch H., Rennert O. M., and Tease L. A., and Chan P. K. (1989) Characterization of the cDNA encoding human nucleophosmin and studies of its role in normal and abnormal growth. Biochemistry 28, 1033–1039 [DOI] [PubMed] [Google Scholar]
- 10. Brady S. N., Maggi L. B. Jr., Winkeler C. L., Toso E. A., Gwinn A. S., Pelletier C. L., and Weber J. D. (2009) Nucleophosmin protein expression level, but not threonine 198 phosphorylation, is essential in growth and proliferation. Oncogene 28, 3209–3220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Erickson J. D., and Bazan N. G. (2013) The nucleolus fine-tunes the orchestration of an early neuroprotection response in neurodegeneration. Cell Death Differ. 20, 1435–1437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hetman M., and Pietrzak M. (2012) Emerging roles of the neuronal nucleolus. Trends Neurosci. 35, 305–314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Marquez-Lona E. M., Tan Z., and Schreiber S. S. (2012) Nucleolar stress characterized by downregulation of nucleophosmin: A novel cause of neuronal degeneration. Biochem. Biophys. Res. Commun. 417, 514–520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ahn J. Y., Liu X., Cheng D., Peng J., Chan P. K., Wade P. A., and Ye K. (2005) Nucleophosmin/B23, a nuclear PI(3,4,5)P3 receptor, mediates the antiapoptotic actions of NGF by inhibiting CAD. Mol. Cell. 18, 435–445 [DOI] [PubMed] [Google Scholar]
- 15. Chin P. C., Liu L., Morrison B. E., Siddiq A., Ratan R. R., Bottiglieri T., and D'Mello S. R. (2004) The c-Raf inhibitor GW5074 provides neuroprotection in vitro and in an animal model of neurodegeneration through a MEK-ERK and Akt-independent mechanism. J. Neurochem. 90, 595–608 [DOI] [PubMed] [Google Scholar]
- 16. Tsoi H., and Chan H. Y. (2013) Expression of expanded CAG transcripts triggers nucleolar stress in Huntington's disease. Cerebellum 12, 310–312 [DOI] [PubMed] [Google Scholar]
- 17. Lee J., Hwang Y. J., Ryu H., Kowall N. W., and Ryu H. (2014) Nucleolar dysfunction in Huntington's disease. Biochim. Biophys. Acta 1842, 785–790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Li J. Y., Popovic N., and Brundin P. (2005) The use of the R6 transgenic mouse models of Huntington's disease in attempts to develop novel therapeutic strategies. NeuroRx 2, 447–464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Bates G. P., Mangiarini L., Mahal A., and Davies S. W. (1997) Transgenic models of Huntington's disease. Hum. Mol. Genet. 6, 1633–1637 [DOI] [PubMed] [Google Scholar]
- 20. Borlongan C. V., Koutouzis T. K., and Sanberg P. R. (1997) 3-Nitropropionic acid animal model and Huntington' s disease. Neurosci. Biobehav. Rev. 21, 289–293 [DOI] [PubMed] [Google Scholar]
- 21. Brouillet E., Jacquard C., Bizat N., and Blum D. (2005) 3-Nitropropionic acid: a mitochondrial toxin to uncover physiopathological mechanisms underlying striatal degeneration in Huntington's disease. J. Neurochem. 95, 1521–1540 [DOI] [PubMed] [Google Scholar]
- 22. Wang W., Budhu A., Forgues M., and Wang X. W. (2005) Temporal and spatial control of nucleophosmin by the Ran-Crm1 complex in centrosome duplication. Nat. Cell Biol. 7, 823–830 [DOI] [PubMed] [Google Scholar]
- 23. Duan-Porter W. D., Woods V. L. Jr., Maurer K. D., Li S., and Rosen A. (2014) Dynamic conformations of nucleophosmin (NPM1) at a key monomer-monomer interface affect oligomer stability and interactions with granzyme B. PLoS One 9, e115062, [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Okuwaki M., Sumi A., Hisaoka M., Saotome-Nakamura A., Akashi S., Nishimura Y., and Nagata K. (2012) Function of homo-and hetero-oligomers of human nucleoplasmin/nucleophosmin family proteins NPM1, NPM2 and NPM3 during sperm chromatin remodeling. Nucleic Acids Res. 40, 4861–4878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Jian Y., Gao Z., Sun J., Shen Q., Feng F., Jing Y., and Yang C. (2009) RNA aptamers interfering with nucleophosmin oligomerization induce apoptosis of cancer cells. Oncogene 28, 4201–4211 [DOI] [PubMed] [Google Scholar]
- 26. Greene L. A., Liu D. X., Troy C. M., and Biswas S. C. (2007) Cell cycle molecules define a pathway required for neuron death in development and disease. Biochim. Biophys. Acta 1772, 392–401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Becker E. B., and Bonni A. (2004) Cell cycle regulation of neuronal apoptosis in development and disease. Prog. Neurobiol. 72, 1–25 [DOI] [PubMed] [Google Scholar]
- 28. Herrup K., and Yang Y. (2007) Cell cycle regulation in the postmitotic neuron: oxymoron or new biology? Nat. Rev. Neurosci. 8, 368–378 [DOI] [PubMed] [Google Scholar]
- 29. Verdaguer E., Jordá E. G., Canudas A. M., Jiménez A., Pubill D., Escubedo E., Camarasa J., Pallàs M., and Camins A. (2004) Antiapoptotic effects of roscovitine in cerebellar granule cells deprived of serum and potassium: a cell cycle-related mechanism. Neurochem. Int. 44, 251–261 [DOI] [PubMed] [Google Scholar]
- 30. Wang L., Ankati H., Akubathini S. K., Balderamos M., Storey C. A., Patel A. V., Price V., Kretzschmar D., Biehl E. R., and D'Mello S. R. (2010) Identification of novel 1,4-benzoxazine compounds that are protective in tissue culture and in vivo models of neurodegeneration. J. Neurosci. Res. 88, 1970–1984 [DOI] [PubMed] [Google Scholar]
- 31. Ham J., Eilers A., Whitfield J., Neame S. J., and Shah B. (2000) c-Jun and the transcriptional control of neuronal apoptosis. Biochem. Pharmacol. 60, 1015–1021 [DOI] [PubMed] [Google Scholar]
- 32. Borsello T., and Forloni G. (2007) JNK signalling: a possible target to prevent neurodegeneration. Curr. Pharm. Des. 13, 1875–1886 [DOI] [PubMed] [Google Scholar]
- 33. D'Mello S. R., and Chin P. C. (2005) Treating neurodegenerative conditions through the understanding of neuronal apoptosis. Curr. Drug Targets CNS Neurol. Disord. 4, 3–23 [DOI] [PubMed] [Google Scholar]
- 34. Harms C., Albrecht K., Harms U., Seidel K., Hauck L., Baldinger T., Hübner D., Kronenberg G., An J., Ruscher K., Meisel A., Dirnagl U., von Harsdorf R., Endres M., and Hörtnagl H. (2007) Phosphatidylinositol 3-Akt-kinase-dependent phosphorylation of p21(Waf1/Cip1) as a novel mechanism of neuroprotection by glucocorticoids. J. Neurosci. 27, 4562–4571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Jurk D., Wang C., Miwa S., Maddick M., Korolchuk V., Tsolou A., Gonos E. S., Thrasivoulou C., Saffrey M. J., Cameron K., and von Zglinicki T. (2012) Postmitotic neurons develop a p21-dependent senescence-like phenotype driven by a DNA damage response. Aging Cell 11, 996–1004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Mallick S., and D'Mello S. R. (2014) JAZ (Znf346), a SIRT1-interacting protein, protects neurons by stimulating p21 (WAF/CIP1) protein expression. J. Biol. Chem. 289, 35409–35420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Cisbani G., and Cicchetti F. (2012) An in vitro perspective on the molecular mechanisms underlying mutant huntingtin protein toxicity. Cell Death Dis. 3, e382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Lee S. B., Kim C. K., Lee K. H., and Ahn J. Y. (2012) S-Nitrosylation of B23/nucleophosmin by GAPDH protects cells from the SIAH1-GAPDH death cascade. J. Cell Biol. 199, 65–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Maezawa I., Wang B., Hu Q., Martin G. M., Jin L. W., and Oshima J. (2002) Alterations of chaperone protein expression in presenilin mutant neurons in response to glutamate excitotoxicity. Pathol. Int. 52, 551–554 [DOI] [PubMed] [Google Scholar]
- 40. Lim A. C., and Qi R. Z. (2003) Cyclin-dependent kinases in neural development and degeneration. J. Alzheimers Dis. 5, 329–335 [DOI] [PubMed] [Google Scholar]
- 41. Neve R. L., and McPhie D. L. (2006) The cell cycle as a therapeutic target for Alzheimer's disease. Pharmacol. Ther. 111, 99–113 [DOI] [PubMed] [Google Scholar]
- 42. Pelegrí C., Duran-Vilaregut J., del Valle J., Crespo-Biel N., Ferrer I., Pallàs M., Camins A., and Vilaplana J. (2008) Cell cycle activation in striatal neurons from Huntington's disease patients and rats treated with 3-nitropropionic acid. Int. J. Dev. Neurosci. 26, 665–671 [DOI] [PubMed] [Google Scholar]
- 43. Okuda M., Horn H. F., Tarapore P., Tokuyama Y., Smulian A. G., Chan P. K., Knudsen E. S., Hofmann I. A., Snyder J. D., Bove K. E., and Fukasawa K. (2000) Nucleophosmin/B23 is a target of CDK2/cyclin E in centrosome duplication. Cell 103, 127–140 [DOI] [PubMed] [Google Scholar]
- 44. Okuwaki M., Tsujimoto M., and Nagata K. (2002) The RNA binding activity of a ribosome biogenesis factor, nucleophosmin/B23, is modulated by phosphorylation with a cell cycle-dependent kinase and by association with its subtype. Mol. Biol. Cell. 13, 2016–2030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Prinos P., Lacoste M. C., Wong J., Bonneau A. M., and Georges E. (2011) Mutation of cysteine 21 inhibits nucleophosmin/B23 oligomerization and chaperone activity. Int. J. Biochem. Mol. Biol. 2, 24–30 [PMC free article] [PubMed] [Google Scholar]
- 46. Herrera J. E., Correia J. J., Jones A. E., and Olson M. O. (1996) Sedimentation analyses of the salt- and divalent metal ion-induced oligomerization of nucleolar protein B23. Biochemistry 35, 2668–2673 [DOI] [PubMed] [Google Scholar]
- 47. Zhou B. P., Liao Y., Xia W., Spohn B., Lee M. H., and Hung M. C. (2001) Cytoplasmic localization of p21Cip1/WAF1 by Akt-induced phosphorylation in HER-2/neu-overexpressing cells. Nat. Cell Biol. 3, 245–252 [DOI] [PubMed] [Google Scholar]
- 48. van den Heuvel S., and Harlow E. (1993) Distinct roles for cyclin-dependent kinases in cell cycle control. Science 262, 2050–2054 [DOI] [PubMed] [Google Scholar]
- 49. D'Mello S. R., Galli C., Ciotti T., and Calissano P. (1993) Induction of apoptosis in cerebellar granule neurons by low potassium: inhibition of death by insulin-like growth factor I and cAMP. Proc. Natl. Acad. Sci. U.S.A. 90, 10989–10993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Yalcin A., Koulich E., Mohamed S., Liu L., and D'Mello S. R. (2003) Apoptosis in cerebellar granule neurons is associated with reduced interaction between CREB-binding protein and NF-κB. J. Neurochem. 84, 397–408 [DOI] [PubMed] [Google Scholar]
- 51. Koulich E., Nguyen T., Johnson K., Giardina C., and D'mello S. (2001) NF-κB is involved in the survival of cerebellar granule neurons: association of IκBβ [correction of Iκβ] phosphorylation with cell survival. J. Neurochem. 76, 1188–1198 [DOI] [PubMed] [Google Scholar]
- 52. Dastidar S. G., Landrieu P. M., and D'Mello S. R. (2011) FoxG1 promotes the survival of postmitotic neurons. J. Neurosci. 31, 402–413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Pfister J. A., Ma C., Morrison B. E., and D'Mello S. R. (2008) Opposing effects of sirtuins on neuronal survival: SIRT1-mediated neuroprotection is independent of its deacetylase activity. PLoS One 3, e4090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Bardai F. H., Verma P., Smith C., Rawat V., Wang L., and D'Mello S. R. (2013) Disassociation of histone deacetylase-3 from normal huntingtin underlies mutant huntingtin neurotoxicity. J. Neurosci. 33, 11833–11838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Bardai F. H., Price V., Zaayman M., Wang L., and D'Mello S. R. (2012) Histone deacetylase-1 (HDAC1) is a molecular switch between neuronal survival and death. J. Biol. Chem. 287, 35444–35453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Dastidar S. G., Bardai F. H., Ma C., Price V., Rawat V., Verma P., Narayanan V., and D'Mello S. R. (2012) Isoform-specific toxicity of Mecp2 in postmitotic neurons: suppression of neurotoxicity by FoxG1. J. Neurosci. 32, 2846–2855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Murphy T. H., Schnaar R. L., and Coyle J. T. (1990) Immature cortical neurons are uniquely sensitive to glutamate toxicity by inhibition of cystine uptake. FASEB J. 4, 1624–1633 [PubMed] [Google Scholar]
- 58. Ratan R. R., Murphy T. H., and Baraban J. M. (1994) Macromolecular-synthesis inhibitors prevent oxidative stress-induced apoptosis in embryonic cortical-neurons by shunting cysteine from protein-synthesis to glutathione. J. Neurosci. 14, 4385–4392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Moffat J., Grueneberg D. A., Yang X., Kim S. Y., Kloepfer A. M., Hinkle G., Piqani B., Eisenhaure T. M., Luo B., Grenier J. K., Carpenter A. E., Foo S. Y., Stewart S. A., Stockwell B. R., Hacohen N., et al. (2006) A lentiviral RNAi library for human and mouse genes applied to an arrayed viral high-content screen. Cell 124, 1283–1298 [DOI] [PubMed] [Google Scholar]
- 60. Neef D. W., Turski M. L., and Thiele D. J. (2010) Modulation of heat shock transcription factor 1 as a therapeutic target for small molecule intervention in neurodegenerative disease. PLoS Biol. 8, e1000291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Verma P., Pfister J. A., Mallick S., and D'Mello S. R. (2014) HSF1 protects neurons through a novel trimerization- and HSP-independent mechanism. J. Neurosci. 34, 1599–1612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Chen H.-M., Wang L., and D'Mello S. R. (2008) A chemical compound commonly used to inhibit PKR, {8-(imidazol-4-ylmethylene)-6H-azolidino[5,4-g] benzothiazol-7-one}, protects neurons by inhibiting cyclin-dependent kinase. Eur. J. Neurosci. 28, 2003–2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.