

Abstract
The acetyl post-translational modification of chromatin at selected histone lysine residues is interpreted by an acetyl-lysine specific interaction with bromodomain reader modules. Here we report the discovery of the potent, acetyl-lysine competitive and cell active inhibitor PFI-3 that binds to certain Family VIII bromodomains while displaying significant, broader bromodomain family selectivity. The high specificity of PFI-3 for Family VIII was achieved through a novel bromodomain binding mode of a phenolic head group that led to the unusual displacement of water molecules that are generally retained by most other bromodomain inhibitors reported to date. The medicinal chemistry program that led to PFI-3 from an initial fragment screening hit is described in detail and additional analogues with differing Family VIII bromodomain selectivity profiles are also reported. We also describe the full pharmacological characterization of PFI-3 as a chemical probe, along with phenotypic data on adipocyte and myoblast cell differentiation assays.
TOC image
INTRODUCTION
Bromodomains are essential interaction motifs for the recruitment of transcriptional regulators to acetylated chromatin.1 They were first identified as a conserved sequence motif present in the Drosophila homologue of Brahma (BRM)2 and they constitute a family of 61 highly diverse interaction domains present in 46 proteins in humans.3 Bromodomains selectively recognize ε-N-lysine acetylation motifs, a key event in the reading process of these post-translational modifications that are important components of the so-called ‘epigenetic code’. The recent discovery of potent and highly specific inhibitors for the Bromodomain and extra-terminal domain (BET) family of bromodomains4 has stimulated intensive research activity particularly in oncology, where BET proteins regulate the expression of key oncogenes. The first wave of inhibitors from this BET bromodomain class has entered clinical testing and their progress has garnered significant attention.5 As a family of protein targets, bromodomains contain a central, deep and largely hydrophobic acetyl lysine binding pocket that is an attractive site for the development of selective and potent protein interaction inhibitors that mimic the endogenous ligands. The predicted favourable druggability of these acetyl lysine interaction domains6 suggests that other bromodomain members beyond the BET family may also be selectively targeted. Mammalian SWItch/Sucrose Non-Fermentable (SWI/SNF) complexes play a key role in cell differentiation and proliferation, and represent an essential component of the embryonic stem cell (ESC) core pluripotency transcriptional network.7 Brahma-related gene-1, (BRG1, SWI/SNF related, matrix associated, actin dependent regulator of chromatin, subfamily a 4 (SMARCA4)) and the related protein BRM (SMARCA2) are the central ATPase components of these multi-subunit complexes. In addition to an essential role in pluripotency and development, genetic lesions of SWI/SNF complexes have been strongly linked to cancer development and a synthetic lethal relationship between SMARCA4 and SMARCA2 has been reported.8 SMARCA2 and SMARCA4 are multi-domain proteins that contain both DNA and protein interaction modules within their structure. Importantly, these domains include a C-terminal bromodomain in each protein.9 The multi-domain PB1 protein contains six bromodomains classified to Family VIII of bromodomain containing proteins based on structural and sequence homology.3 Protein polybromo-1 (PB1) appears in the Polybromo, Brg1-Associated Factors (PBAF) complex (human analogue of SWI/SNF) alongside either SMARCA2 or SMARCA4. Inhibitors targeting the SMARCA2, SMARCA4 and PB1 bromodomains would be useful tools to study the modulation of these SWI/SNF mediated processes and any chemical probes identified could also offer starting points for drug like molecules of potential therapeutic utility.
RESULTS AND DISCUSSION
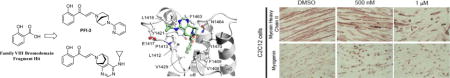
In the search for chemical starting points for Family VIII bromodomains, a fragment screen against the PB1(5) bromodomain using Differential Scanning Fluorimetry (DSF) successfully identified salicylic acid 1 as weak, but highly ligand efficient hit (Figure 1). This was confirmed as a direct interaction with the protein by Isothermal Titration Calorimetry (ITC). Of particular note was the high selectivity for Family VIII bromodomains over other bromodomain family members for what is a very small fragment molecule. This selectivity was explained by the identification of a rare binding mode in the field of bromodomain small molecule inhibitors where the phenolic functionality displaced water molecules typically retained in other bromodomain inhibitor complex binding modes.10 Very few other examples of such deep pocket binding have been reported in bromodomains.11 The crystal structure and characterization of the salicylic acid fragment hit 1 have been reported elsewhere.12 With a weak but ligand efficient fragment hit successfully crystallized in PB1(5), further molecular optimization was guided towards exploiting lipophilic interactions for larger ligands derived from the salicylic acid lead 1. These changes were designed to enhance potency levels closer to those set as an expectation of a quality chemical probe – around 100nM in a biochemical or biophysical assay.13 The PB1(5) small molecule X-ray structure points to asparagine N707 and tyrosine Y664 as key hydrogen bond interactions for the phenolic and carbonyl functional groups of 1 with the protein. The defined orientation of the carbonyl group, in accepting a hydrogen bond from N707, directs a potential substituent in more elaborated ligands out of the protein, through and over a lipophilic surface of amino acid side chains. Maintaining the phenolic and carbonyl substituents off the phenyl group as substructural requirements, structure activity relationships (SAR) were initially explored primarily through commercial purchase and file screening from the Pfizer chemical compound library. Enamides proved to be good surrogates for the previously stated key elements of the salicylic acid pharmacophore. Commercially available 2 was significantly more active than salicylic acid 1 in the primary DSF assay that was used to assess SAR, with compounds being tested at 10 μM (Table 1).
Figure 1.
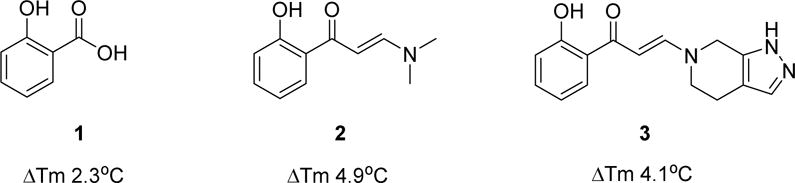
Fragment hit 1, a commercially sourced screening hit 2 and a novel, chemically synthesized analogue 3 with representative DSF data at 10 μM against the PB1(5) bromodomain.
Table 1.
DSF thermal shift (°C stabilization, 10 μM compound) SAR for Family VIII bromodomains. Values are derived from at least three determinations with standard deviations quoted.
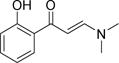
| Structure | PB1(2) | PB1(3) | PB1(4) | PB1(5) | SMARCA2A | SMARCA2B | SMARCA4 | |
|---|---|---|---|---|---|---|---|---|
| 2 |
|
2.0±0.2 | 0.5±0.1 | 0.7±0.2 | 4.9±0.6 | 4.8±0.4 | 2.0±0.2 | 3.3±0.2 |
| 3 |

|
4.1±0.3 | 1.7±0.1 | 1.2±0.2 | 7.8±0.7 | 7.8±0.1 | 4.8±0.2 | 7.0±0.3 |
| 4 |
|
3.2±1.9 | 1.8±0.5 | 1.0±0.2 | 6.9±0.7 | 8.2±0.7 | 3.9±0.2 | 6.3±0.6 |
| 5 |
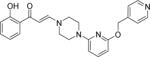
|
0.9±0.2 | 0.7±0.0 | 1.3±0.1 | 7.0±0.3 | 7.4±0.1 | 3.7±0.2 | 7.5±0.1 |
| 6 |

|
5.2±0.6 | 3.3±0.1 | 2.0±0.4 | 6.6±0.4 | 5.1±0.4 | 2.0±0.1 | 3.8±0.5 |
| 7 |
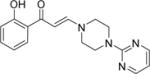
|
0.7±0.9 | 0.3±0.2 | 0.6±0.2 | 4.6±1.3 | 2.9±0.7 | 1.1±0.6 | 2.9±1.2 |
| 8 |
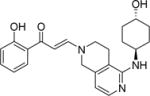
|
2.5±0.4 | 1.1±0.2 | 0.4±0.1 | 8.3 ±0.4 | 5.4±0.1 | 2.4±0.4 | 6.1±0.4 |
| 9 |
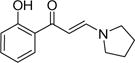
|
2.3±0.2 | 1.1±0.1 | 0.7±0.2 | 5.7±0.4 | 5.5±0.2 | 3.0±0.1 | 4.7±0.1 |
| 10 |
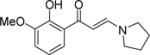
|
0.1±0.2 | 0.0±0.0 | 0.3±0.4 | 0.4±0.5 | 0.7±0.2 | −0.4±0.1 | 0.7±0.1 |
| 11 |
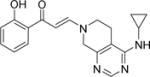
|
6.7±0.3 | 3.5±0.1 | 2.4±0.2 | 9.1±0.8 | 10.1±0.5 | 5.0±0.1 | 7.7±0.2 |
| 12 |
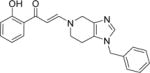
|
4.6±0.4 | 1.9±0.2 | 1.6±0.0 | 8.0±0.3 | 5.8±0.2 | 3.0±0.4 | 5.3±0.4 |
| 13 |
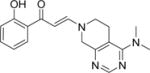
|
3.5±0.4 | 1.3±0.1 | 1.2±0.2 | 5.1±0.8 | 4.1±0.4 | 2.2±0.3 | 3.4±0.2 |
| 14 |
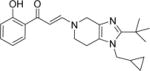
|
3.8±1.2 | 1.5±0.1 | 1.2±0.1 | 8.0±0.8 | 5.9±0.2 | 3.8±0.3 | 5.8±0.7 |
| 15 |
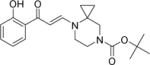
|
2.2±0.2 | 0.9±0.0 | 0.6±0.1 | 7.6±0.6 | 7.5±0.7 | 4.1±0.4 | 6.1±0.3 |
| 16 |
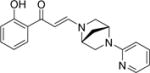
|
1.4±0.0 | 0.6±0.0 | 0.4±0.1 | 9.1±0.9 | 8.2±0.4 | 5.0±0.2 | 7.0±0.2 |
| 17 |
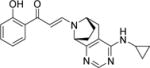
|
5.9±0.3 | 6.4±0.2 | 2.1±0.4 | 9.3±0.7 | 8.5±0.7 | NT | 7.4±0.7 |
| 18 |
|
NT | NT | NT | −0.4±0.3 | 0.7±0.3 | −0.4±0.2 | 0.7±0.3 |
The DSF thermal shift of 4.9°C for 2 in PB1(5) at 10 μM indicated significantly enhanced binding to the protein compared to salicylic acid 1 and that the acidic functional group was not essential. The predicted binding mode for 2 was confirmed by a crystal structure in PB1(5) showing the dimethylamino portion of the enamide emerging towards the protein surface (Figure 2A).
Figure 2.
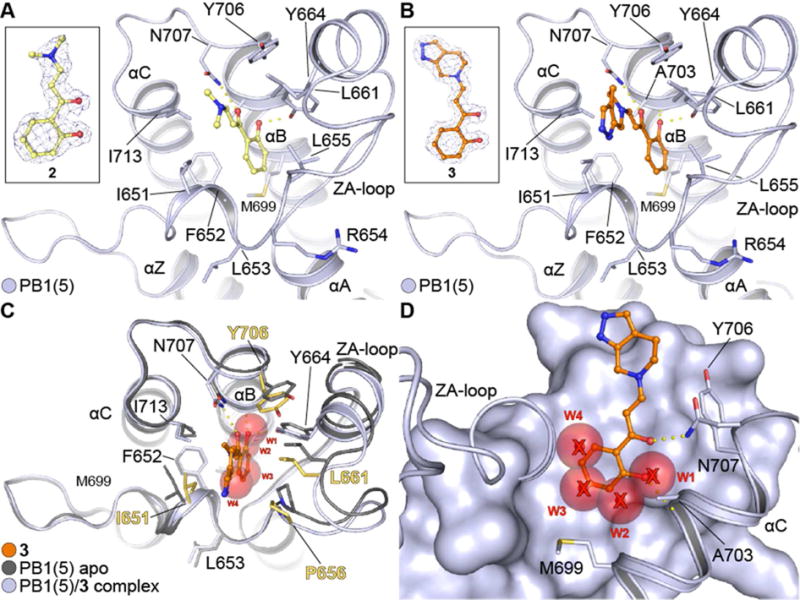
PB1 complexes with hydroxyphenyl-enamides. (A) Complex of PB1(5) with 2. The ligand occupies the acetyl-lysine cavity of the bromodomain directly coordinating to the conserved asparagine (N707) and Y664. Key residues are annotated and secondary structure elements are displayed. The ligand’s 2FcFo map contoured at 2σ is displayed in the inset. (B) Complex of PB1(5) with 3. As in (A) the compound binds in an acetyl-lysine mimetic fashion, directly engaging the conserved asparagine (N707) and Y664 as well as A703 via a backbone interaction. Key residues and secondary structure elements are annotated. The ligand’s 2FcFo map contoured at 2σ is displayed in the inset. (C) Comparison of apo PB1(5) (PDB ID: 3G0J) and PB1(5)-3 complex. The ligand inserts deep into the hydrophobic pocket of the protein resulting in distortion of the plasticity and an outwards motion of the ZA-loop. Key residues that change orientation are highlighted in yellow. The four conserved water molecules found in most bromodomain structures (w1-4) have been displaced in the complex. (D) Side view of 3 bound onto PB1(5) overlaid with the four conserved water molecules found on the apo-PB1(5) structure. The ligand inserts deep into the cavity displacing the four water molecules (w1-4) and engaging the protein via N707 and A703 while stacking over M699.
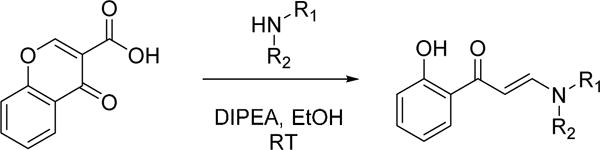
A number of commercially available enamides showed similar levels of activity. The selection of enamides available was further enhanced by a simple one step, chemical synthesis of analogs in which both primary and secondary amines could be functionalized with chromone-3-carboxylic acid through a decarboxylative ring opening (Scheme 1).14
Scheme 1.
General method for library chemistry amenable synthesis of Family VIII bromodomain binders
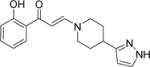
The protocol was sufficiently robust to be run using parallel synthetic chemistry methods with a high success rate for the amine monomer set selected as the library variable. The first library run yielded 130 compounds at a success rate of 73% and a number of pyrrolidine, piperidine and piperazine based compounds possessed significant activity against PB1(5), SMARCA2 and SMARCA4 as judged by DSF (Compounds 3–8, Table 1). For compounds such as 3, Tm shift values increased by over 2°C against PB1(5), SMARCA2 (both isoforms) and SMARCA4 when compared to the simple dimethylamine derivative 2. A crystal structure in PB1(5) was secured for compound 3 which exemplifies the key features of the interaction mode (Figure 2B–D). Compounds derived from primary amine monomers showed poor binding. A small number of functionalized chromone-3-carboxylic acid templates were run in the library (as a negative SAR control), and as the crystal structure of the initial fragment hit 1 suggested, room deep in the bromodomain pocket was restricted and a specific substitution pattern on the phenyl group was required for activity. As an example, the closely related pyrrolidines 9 and 10 show that the addition of a methoxy group ortho to the phenol eliminated Family VIII activity completely. In general, the first round of compound synthesis yielded analogs with a relatively limited interaction with PB1(2) as determined by DSF, indicating the potential for intra-family VIII selectivity for compounds derived from pyrrolidines, piperidines and piperazines. This was not easily explained by the crystal structures available or protein sequence differences, but his contrasts with earlier examples from the BET family where all chemotypes inhibit all members of this bromodomain sub-family and selectivity between BRD2, 3, 4 and T has yet to be established through conventional modes of inhibition.15
A second round of library chemistry looked at a greater number of fused, bicyclic amines based on compound 3. This compound had stood out in the first round as showing notable levels of PB1(2) activity in the DSF assay compared to other analogs. These bicyclic amines were primarily sourced, being slightly more complex in structure, from the Pfizer compound collection of secondary amines and not from commercial suppliers. 320 compounds were delivered at a 77% synthetic success rate using the same parallel synthesis protocol. Physicochemical properties were constrained such that they would not compromise potential cell penetration for compounds. Targets were designed within cLogP lower and upper limits of 1 and 4. Once again, compounds with significant PB1(5) activity were identified by DSF with Tm shifts of >8°C (Compounds 11–15, Table 1). These fused bicyclic compounds presented a different Family VIII selectivity profile compared to the first library, with PB1(2) activity now observed alongside PB1(5), SMARCA2 and SMARCA4.
Through using the DSF assay, identification of both moderately selective and ‘pan’ profiles for Family VIII bromodomain binders from the same chemotype offered the chance to design chemical tools that could be used in conjunction with one another to establish the roles of these bromodomain containing proteins in SWI/SNF complexes, where both PB1(2) and PB1(5) bromodomain containing proteins can be present alongside either SMARCA2 or SMARCA4. In total, some 400 compounds were synthesized in these two rounds of library chemistry and the most potent compounds (initially judged by Tm shift in DSF assays) were progressed to Isothermal Titration Calorimetry (ITC) for absolute binding affinity determination. Compound 11 met the pharmacological probe criteria with particularly potent binding, as determined by ITC, to PB1(2), PB1(5) and SMARCA4 at 152nM, 28nM and 49nM respectively.
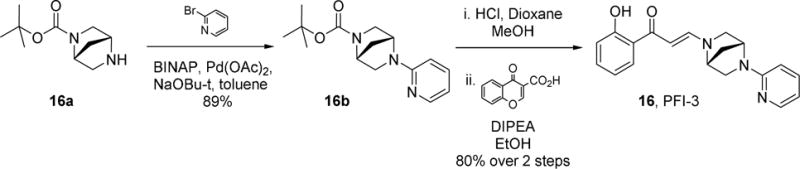
Despite this promising profile and the relative ease with which SAR could be explored through the one step chemical synthesis of analogs, the utility of the chemotype remained in question given the potential for chemical instability of the embedded enamide within the structure. This functional group could be hydrolytically unstable as a masked 1,3-dicarbonyl and importantly, render the compounds unsuitable for their primary intended use as chemical probes for cell biology. Chemical stability assessment of compound 11 confirmed the expected potential for hydrolysis and the rate of decomposition was considered unsuitable for use in extended cellular assays. The next round of compound design focused on combining strategies for chemical stability with the potent binding affinity for Family VIII bromodomains. Factors influencing the mechanism for hydrolysis such as leaving group ability of the amine, steric hindrance of the hydrolysed centre and likelihood for forming an intermediate iminium ion were considered. Compound 15 had indicated that substitution next to the hydrolytically displaced nitrogen atom of the departing amine was tolerated. The bridged piperazine proposed, in what was finally identified as the nominated chemical probe 1612 (PFI-3, Scheme 2), conferred significantly enhanced chemical stability over the earlier prototype 11.
Scheme 2.
Synthetic route to compound 16 (PFI-3, PF-6687252)
The improved chemical stability over a range of conditions relevant to cellular biology studies, confirmed compound 16 as a first in class chemical probe for PB1(5), SMARCA2 and SMARCA4 while offering considerable selectivity over PB1(2). Compound 16 had a half-life of >250 hours in PBS, pH7.4 at 20°C. In contrast, compound 11 had a half-life of 20 hours under the same conditions. Greater than 90% of the compound 16 was recovered after one week in cell media at 37°C (Supplementary data, Table T1). Strong Tm shift data for 16 (with the exception of PB1(2), was confirmed by <100nM ITC KD determinations for PB1(5) and SMARCA4.12 Compound 16 showed complete selectivity over all non-family VIII bromodomains as judged by both a Tm shift panel of 40 bromodomain proteins and the Bromoscan screen from DiscoveRx.16,17
With just three bromodomains targeted by compound 16, the attraction of a less selective Family VIII profile, particularly a compound with PB1(2) activity, was sought in order to offer tools of orthogonal selectivity combinations for cellular biology experiments. The chemical synthesis libraries initially performed had identified compound 11 as a potent binder of PB1(2) in addition to its SMARCA2, SMARCA4 and PB1(5) activity and but for its insufficient chemical stability, would have been nominated as a chemical probe. Having established strategies for conferring chemical stability in the more selective and synthetically expedient piperazine system that yielded compound 16, a similar bicylization approach was applied to compound 11. This required a considerably lengthier and complex synthetic route that ultimately yielded compound 17 (Supplemental Scheme S1). Compound 17 (Figure 3A) is a potent binder to most of the Family VIII bromodomains as determined by ITC (Figure 3B) and is not active against the remaining bromodomain proteins as judged by a Tm shift panel of forty bromodomains and also BROMOscan® profiling from DiscoveRx (Supplementary Figure F3 and Figure F4). A protein co-crystal structure for compound 17 was solved in SMARCA 2 (Figure 3C and D) and this was also used to infer the absolute stereochemistry of the compound that was prepared as a single enantiomer, having used chiral Supercritical Fluid Chromatography (SFC) to separate a racemic amine intermediate during the synthesis (Supplementary Scheme S1). A small molecule X-Ray structure of a heavy atom containing amide derivative of amine 17f (Supplementary Scheme S1) was also secured and this provided definitive confirmation of the absolute stereochemistry of compound 17 (Supplemental Figure F1)
Figure 3.
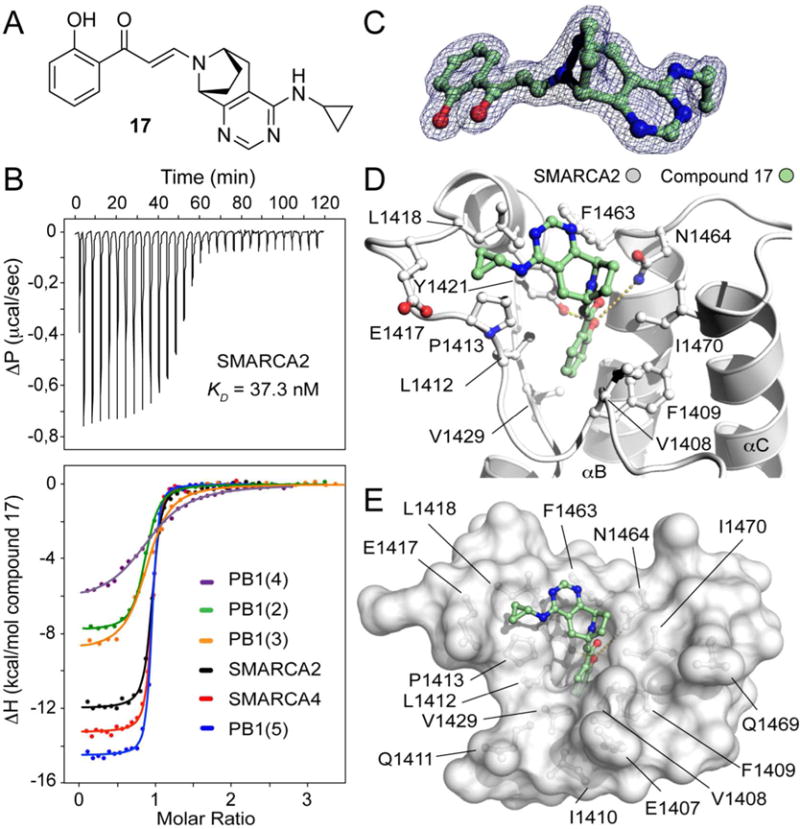
Compound 17 is a potent pan-inhibitor of SMARCA2/SMARCA4 and PB1 families. (A) Chemical structure of inhibitor 17. (B) Isothermal titration data of the interaction of compound 17. Top panel shows the raw binding exotherms for each SMARCA2 injection into the cell containing the inhibitor solution. Bottom panel displays normalized binding enthalpies and the corresponding non-linear least squares fits (Insert) for the two families SMARCA2/SMARCA4 and PB1 bromodomains. (C) 2Fo-Fc electron density map contoured at 1.5 σ around the inhibitor at 1.7 Å resolution. (D) Complex of SMARCA2 with compound 17. The hydroxyphenyl propenone core scaffold adopts the same conformation of previous crystallized analogues in the acetyl-lysine binding site. The cyclopropanamine group is pointing towards the ZA-loop stabilized by hydrophobic interactions with the lipophilic side chains of L1418, P1413 and E1417. (E) Surface representation of the SMARCA2 acetyl-lysine binding site in complex with compound 17.
The cellular activity of compound 17 for SMARCA2 was assessed using a fluorescence recovery after photo-bleaching (FRAP) assay (Figure 4)18 U2OS were treated with the Histone deacetylase (HDAC) inhibitor suberoylanilide hydroxamic acid (SAHA) to induce hyperacetylation resulting in a better assay window. Cells were transfected with a Green Fluorescent Protein (GFP)-SMARCA2 construct or as control a GFP-SMARCA2 construct, in which the conserved asparagine (N1464F) essential for recognizing the acetylated lysine had been mutated. Compound 17 accelerated FRAP half-recovery time (t1/2) significantly in a dose dependent manner to the level of the N1464 mutant indicating that compound 17 was able to bind to the SMARCA2 bromodomain and displace full length SMARCA2 from chromatin (Fig. 4A and B). We next tested if compound 17 was able to also displace the GFP-linked PB1 from chromatin. Again, SAHA was used to increase the assay window. The assay did show a larger variability than the SMARCA2 assay, possibly due to the presence of six bromodomains acting as chromatin binding and scaffolding domains.19 Mutation of the conserved asparagine in the second (N263A) and fifth (N707F) bromodomain of PB1 respectively, indicated that a significant difference in half-recovery time could only be achieved by preventing the second bromodomain of PB1 from binding to chromatin. Compound 17 was able to displace PB1 from chromatin at 1 μM concentration, but not at a lower concentration of 100 nM as observed in the SMARCA2 FRAP assay. This data is in good agreement with the mutant data indicating that the second bromodomain, for which 17 has a lower potency, is the relevant domain for chromatin binding (Fig. 4C and D). As expected, the inactive compound 18 had no effect on the half-recovery time of GFP-PB1.
Figure 4.
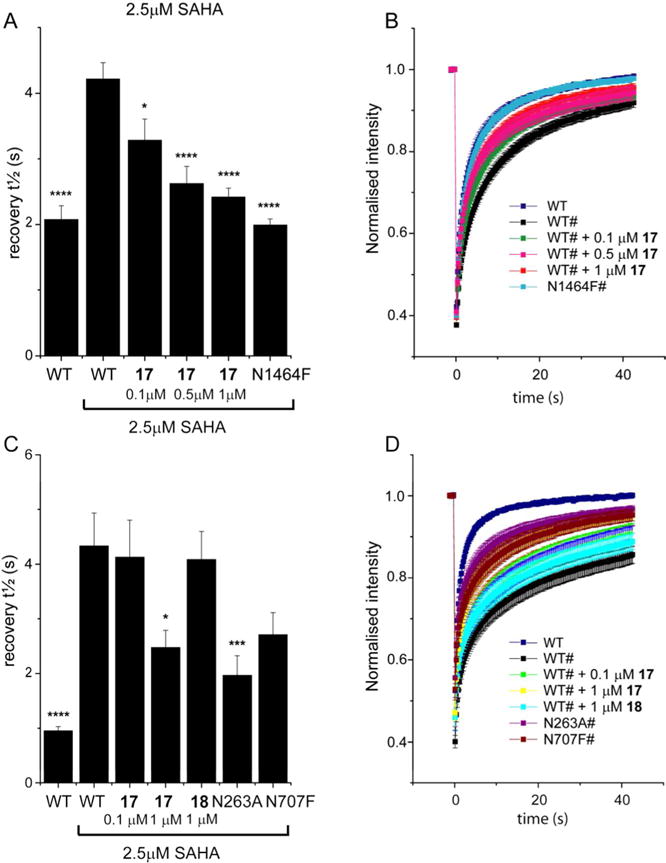
SMARCA2 and PB1 FRAP Assays. (A): Half-life of fluorescence recovery (t½) for transfected cells expressing GFP-tagged SMARCA2 (WT) treated with compound 17 at different doses or mutant SMARCA2 (N1464F). Shown are bars representing the mean t½ calculated from individual recovery curves of at least 20 cells per group and error bars depict the standard error of the mean (SEM). (B): Raw data fluorescent recovery curves corresponding to the fluorescence recovery shown in panel (A). (C): Half recovery times of fluorescence recovery (t½) for transfected cells expressing GFP-tagged PB1 (WT) treated with compound 17 at different doses or mutant PB1 (N263A or N707F). Shown are bars representing the mean t½ calculated from individual recovery curves of at least 20 cells per group and error bars depict the standard error of the mean (SEM). (D): Raw data fluorescent recovery curves corresponding to the fluorescence recovery shown in panel (C). * indicates a p-value compared to WT treated with 2.5 μM SAHA **** p<0.0001; *** p<0.001; * p<0.03.
With novel tools for Family VIII bromodomains in hand, known to be cell penetrant through target engagement assays, phenotypes for the molecules were sought. The BioMAP® primary human cell platform from DiscoveRx has been shown to successfully identify potential phenotypes for bromodomain chemical probes.20 The early and strong phenotype derived from binding BET bromodomains by small molecules is very clearly identified by BioMAP systems with broad effects at multiple concentrations detected across a panel of cell-based assays modeling various aspects of human disease biology. With nothing known about potential phenotypes derived from small molecule binding to Family VIII bromodomains, compound 16 (PFI-3) was profiled in BioMAP by DiscoveRx. Under the standardized conditions run in the BioMAP assay platform protocol, compound 16 had a largely ‘silent’ profile based on statistically significant effects outside the historical control envelope (Figure 2B). The compound showed no inhibitory effects against a large number (148) of cellular end points in twelve primary human cell-based systems. This is in stark contrast to our previously disclosed BET inhibitor PFI-1 profiled in the same platform which exhibited strong inhibition of inflammation, immune activation, tissue/matrix remodeling and hemastasis-related biomarkers, illustrating that not all bromodomain containing proteins will present obvious phenotypes when bound to potent small molecule inhibitors.
With no obvious phenotype found in the BioMAP system, the potential for PB1 mediated cellular efficacy of 16 and 17 was tested using two clear cell renal cell carcinoma (ccRCC) cell lines (786-O and Rcc4), the former having a wild type PB1 expression and the latter having PB1 downregulation.21 Based on this role for PB1, compound treatment of a cell line with a wild type expression should result in an increase in proliferation.21 However, treatment with either compound 16 or 17 (that bind to the PB1 bromodomains) did not show any effect on cell line proliferation (Supplementary Figure F5).
The potential importance of Family VIII bromodomain containing proteins in the chromatin remodeling SWI/SNF complexes and their role in cancer has however been highlighted through many recent publications.22 In combination with knock down experiments, compound 16 showed no role for bromodomain inhibition in the cancer phenotypes highlighted. The Adenosine Triphosphate (ATP)ase domain of SMARCA2 and SMARCA4 proved to be the functionally important domain with respect to cancer cell line efficacy.23 Cellular activity in FRAP assay systems and a Stem cell phenotype have been disclosed elsewhere for compound 16,12 but additional, non-oncology phenotypes for compounds 16 and 17 are now reported here.
The SWI/SNF complex mediates a broad spectrum of biological processes including cell proliferation, differentiation, DNA replication and repair. Mouse knockout models demonstrate that SMARCA4 is necessary for early embryogenesis, whereas SMARCA2 knockout mice are viable without overt visible defects.24 Previous studies have established the role of SMARCA4 in regulating the transcriptional cascade of gene expression during muscle cell and adipocyte differentiation.25 Furthermore, it has been shown that the C-terminal region of SMARCA4, that includes the bromodomain, is subjected to post-translational modification that regulates SMARCA4 activity in myogenesis.26 These observations prompted us to dissect the function of the SMARCA4 bromodomain in the cell differentiation process using the small molecule tools 16 and 17 instead of gene mutagenesis.
Cell viability was measured by standard 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay and neither compound showed significant cytotoxic effect on cells up to 50 μM dose (Figure 6A). SMARCA4 in cell target engagement was confirmed for compounds 16 and 17 through a SMARCA4 FRAP assay (Supplementary Figure F6) analogous to the SMARCA2 method shown in Figure 4. Both compounds blocked C2C12 myoblast differentiation in cells grown in the presence of either compound 16 or compound 17. Defects in cell fusion, evident through shortened myotubes containing fewer nuclei than controls, as well as failure to induce MyHC expression were observed in cells treated with 1 μM of compound 16 or 5 μM of compound 17 (Figure 6B). Compounds 16 and 17 also attenuated adipocyte differentiation and lipid accumulation was decreased dramatically. Significant effects were seen at 1 μM for compound 16 and 5 μM for compound 17. Near complete inhibition of adipogenesis was achieved at 20 μM of compound 16 (Figure 6C).
Figure 6.
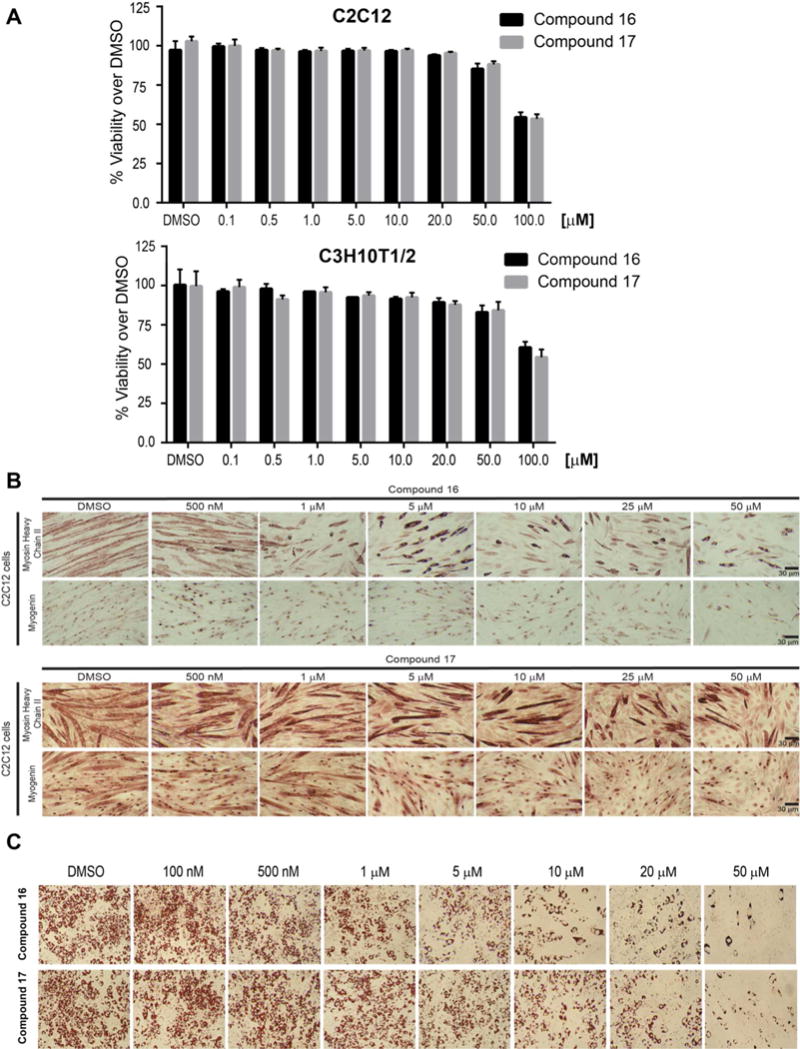
(A) Cytotoxicity tests for the compounds were conducted in C2C12 and C3H10T1/2 cells which were treated with increasing doses of vehicle (DMSO), compound 16 or compound 17 for 72 hours. Cell viability was measured by standard MTT assay. The compounds did not have significant cytotoxic effect on cells up to 50 μM dose. Cells treated with the highest dose of 100 μM caused 50% cell death. (B) Compounds 16 and 17 block differentiation in C2C12 myoblasts normally seen within 48 hours. This is assessed by monitoring the expression of the early marker of differentiation, myogenin, and by monitoring cells undergoing fusion to become myotubes that express Heavy Chain (MyHC). Blockage of Myogenin expression required significantly higher concentrations of both compounds. (C) Compound 16 and 17 attenuated adipocyte differentiation. On addition of an adipogenic cocktail, C3H10T1/2 mesenchymal cells can be induced to differentiate to adipocytes that are manifested by the accumulation of lipid droplets visualized by Oil Red O staining. In the presence of increasing doses of both compounds, there were fewer cells differentiated and lipid accumulation was decreased dramatically. Compound 16 was more effective than compound 17 in blocking adipogenesis.
Compound 16 (PFI-3) is a first in class chemical probe for Family VIII bromodomains and was discovered through optimization of the fragment hit salicylic acid 1. Using two rounds of library synthesis and optimization for chemical stability, all guided by structure based design strategies, compound 16 was nominated as a chemical probe and has been used to identify novel examples of small molecule mediated phenotypes in myoblasts and adipocytes. The identification of compound 17 offers a cell permeable, Family VIII bromodomain tool compound with additional PB1(2) activity over and above that of PFI-3 (16). Although the use of compounds 16 and 17 alongside one another did not offer any significant differentiation in phenotype in the cellular systems studied in this work, they stand as well characterized tool compounds to further study hypotheses surrounding the role of certain bromodomain containing proteins within the SWI/SNF complex, in cells.
SELECTED EXPERIMENTAL PROCEDURES
Chemistry Methods and Compound Characterization
Proton (1H NMR), carbon (13C NMR), and fluorine (19F NMR) magnetic resonance spectra where obtained in DMSO-d6 at 400, 100, 376 MHz and respectively unless otherwise noted. The following abbreviations were utilized to describe peak patterns when appropriate: br = broad, s = singlet, d = doublet, and m = multiplet. High-resolution mass measurements were obtained on an Agilent ToF mass spectrometer. All air and moisture sensitive reactions were carried out under an atmosphere of dry nitrogen using heat-dried glassware and standard syringe techniques. Tetrahydrofuran (THF) and acetonitrile were purchased from EMD anhydrous and were used without further drying. Flash chromatography was performed using an Analogix Intelliflash 280 or Biotage SP1 purification system with Sepra Si 50 silica gel using ethyl acetate/heptane mixtures as solvent unless otherwise indicated. HPLC was carried out on an Agella Venusil ASB C18 column (21.2 × 150 mm, 5 Wm). A flow rate of 0.5–150 mL/min was used with mobile phase A: water + 0.1% modifier (v/v) and B: acetonitrile + 0.1% modifier (v/v). The modifier was formic acid, trifluoroacetate, ammonia acetate, or hydrochloric acid. Quality control (QC) analysis was performed using a LCMS method. Acidic runs were carried out on a Shimadzu XB-C18 (2.1 × 30 mm, 5 Wm), X-Bridge (50 × 4.6 mm, 5 Wm), Gemini NX C18 (50 ×4.6, 3 μm), or Gemini NX C18 (50 × 4.6, 5 Wm). A flow rate of 1.0–1.2 mL/min was used with mobile phase A: water + 0.1% modifier (v/v) and B: acetonitrile + 0.1% modifier (v/v). For acidic runs the modifier was trifluoroacetic acid. A Shimadzu 20AB pump ran a gradient elution from 0% to 98% B over 2 min followed by a 1 min hold at 95% B. Detection was achieved using a Shimadzu 10A detector set at 220 or 260 nm followed in series by a Shimadzu MS2010EV or Applied Biosystem API 2000 mass spectrometer in parallel.
(E)-3-(4,5-dihydro-1H-pyrazolo[3,4-c]pyridin-6(7H)-yl)-1-(2-hydroxyphenyl)prop-2-en-1-one (3)
To a mixture of 4,5,6,7-tetrahydro-1H-pyrazolo[3,4-c]pyridine (203 mg, 1.65 mmol) in EtOH (5 mL) and DMF (0.8 mL) was added DIPEA (303 mg, 3 mmol) followed by chromone-3-carboxylic acid (285 mg, 1.5 mmol). The mixture was stirred at 35°C for 16 h. TLC (DCM/MeOH = 10/1) showed the starting material was consumed. The volatiles were evaporated off and the residue was treated with DCM. Insoluble material was removed via filtration. The filtrate was concentrated and the residue was purified by flash column (EA in PE: 0–62%) to give 3 (90 mg, 20%) as a yellow solid. 1H NMR (400 MHz, Methanol-d4) δ 8.11 (d, J = 12.3 Hz, 1H), 7.90 (dd, J = 8.1, 1.6 Hz, 1H), 7.49 (s, 1H), 7.44 – 7.28 (m, 1H), 6.96 – 6.80 (m, 2H), 3.79 (t, J = 5.8 Hz, 2H), 3.33 (p, J = 1.6 Hz, 2H), 2.82 (t, J = 5.8 Hz, 2H);); 13C NMR (100 MHz, Methanol-d4) δ 163.7, 135.2, 130.0, 121.7, 119.6, 118.8, 113.9, 21.9; HRMS [M+H] for C15H15N3O2, calc., 270.1237, found, 270.1232; LCMS = 95.7%, m/z = 270.1 (M+H). HPLC = 97%, t = 3.02 min; HRMS [M+H] for C15H16N3O2, calc., 270.1237, found, 271.236.
(1R,4R)-tert-butyl 5-(pyridin-2-yl)-2,5-diazabicyclo[2.2.1]heptane-2-carboxylate (16b)
To a stirred mixture of (1R,4R)-tert-butyl 2,5-diazabicyclo[2.2.1]heptane-2-carboxylate 16a (500 mg, 2.52 mmol, [α ]20 = 49.8° (c= 0.79 EtOH)) in toluene (~12.6 mL) was added 2-bromopyridine (0.481 mL,5.04 mmol), potassium, tert-butoxide (1 M in THF, 2.52 mL, 2.52 mmol), palladium acetate (28 mg, 0.126 mmoll) and BINAP (78 mg, 0.126 mmol) at room temperature, then stirred for another 16 hours at 110°C. TLC (DCM: MeOH = 10:1) showed the reaction was complete. Water (3 mL) was added and the mixture was extracted with EA (10×3), the organic layer was dried by anhydrous Na2SO4, purified by column chromatography to give 16b as a yellow solid (620 mg, 89%). 1H NMR (400 MHz, DMSO-d6) δ = 1.31 – 1.46 (m, 9 H), 1.90 (d, J=11.32 Hz, 1 H), 3.18 (d, J=9.76 Hz, 1 H), 3.25 (d, J=9.37 Hz, 1 H), 3.29 – 3.40 (m, 1 H), 3.49 (t, J=8.39 Hz, 1 H), 4.39 – 4.54 (m, 1 H), 4.78 (d, J=6.63 Hz, 1 H), 6.49 – 6.63 (m, 1 H), 7.46 – 7.56 (m, 1 H), 8.07 (d, J=5.07 Hz, 1 H).
(E)-1-(2-hydroxyphenyl)-3-((1R,4R)-5-(pyridin-2-yl)-2,5-diazabicyclo[2.2.1]heptan-2-yl)prop-2-en-1-one (16, PFI-3, PF-06687252)
To a solution of the starting material 16b (620 mg, 2.25 mmol, 1.0 eq) in methanol (1 mL) was added 4 N HCl in dioxane (5 mL, 20 mmol, 9 equiv). The reaction was homogenous and slightly yellow in color. The reaction was stirred for 30 mins and LCMS/TLC indicated the consumption of the starting material and a new peak with the correct M+H (=176). The reaction was concentrated to a yellow solid and used without further purification as the HCl salt. LCMS = 90%, t = 0.16 m/z = 176.0. To a solution of the crude amine (477 mg, 2.25 mmol, 1.0 equiv) in ethanol (11.3 mL) was added diisopropylethylamine (1.98 ml, 11.3 mmol, 5.0 equiv). The reaction was stirred and was homogenous. To the reaction was added chromone-3-carboxylic acid (428 mg, 2.25, 1.0 equiv). The reaction was stirred at room temperature and followed by LCMS and TLC. The reaction was judged complete after 3 hours and the reaction was concentrated to an oil residue. The residue was purified via silica column (80g, 20% ethyl acetate in heptane to 100% over 10 column volumes; desired material started eluting at ~80% ethyl acetate and peaked at ~85% ethyl acetate) to provide the desired material 16 as a yellow solid. 1H NMR (400 MHz, DMSO-d6) δ = 2.02 – 2.12 (m, 2 H), 3.34 – 3.41 (m, 2 H), 3.53 (d, J=11.32 Hz, 1 H), 3.62 (d, J=8.98 Hz, 1 H), 4.80 (s, 1 H), 4.98 (s, 1 H), 5.65 – 5.90 (m, 2 H), 6.50 – 6.66 (m, 2 H), 6.76 – 6.83 (m, 2 H), 7.35 (t, J=7.61 Hz, 1 H), 7.49 – 7.59 (m, 1 H), 7.83 – 7.90 (m, 1 H), 8.10 (d, J=4.68 Hz, 1 H), 8.26 (d, J=12.10 Hz, 1 H); 1H NMR (400 MHz, CHLOROFORM-d) δ = 2.04 – 2.20 (m, 2 H), 3.35 – 3.55 (m, 3 H), 3.69 (d, J=8.53 Hz, 1 H), 4.44 (br. s., 1 H), 5.08 (br. s., 1 H), 5.71 (d, J=12.05 Hz, 1 H), 6.36 (d, J=8.53 Hz, 1 H), 6.62 – 6.67 (m, 1 H), 6.79 (t, J=7.78 Hz, 1 H), 6.92 (d, J=7.53 Hz, 1 H), 7.34 (t, J=7.03 Hz, 1 H), 7.48 (t, J=8.28 Hz, 1 H), 7.61 (d, J=9.03 Hz, 1 H), 8.09 – 8.20 (m, 2 H), 13.85 (s, 1 H); 13C NMR (DMSO-d6) δ = 36.9, 54.2, 55.6, 55.9, 63.9, 89.8, 107.2, 112,4, 117.4, 117.9, 119.9, 128.8, 133.8, 137.3, 147.9, 150.4, 156.9, 162.4, 189.9; LCMS = 100%, t = 0.60 m/z = 322.1; HRMS [M+H] for C19H20N3O2, calc., 322.1550, found, 322.1551; Optical rotation 4.4 mgs in 2 mL MeOH cell length 1 dm; measured rotation (degrees) = +0.2652; [a]=120.5.
Supplementary Material
Figure 5.
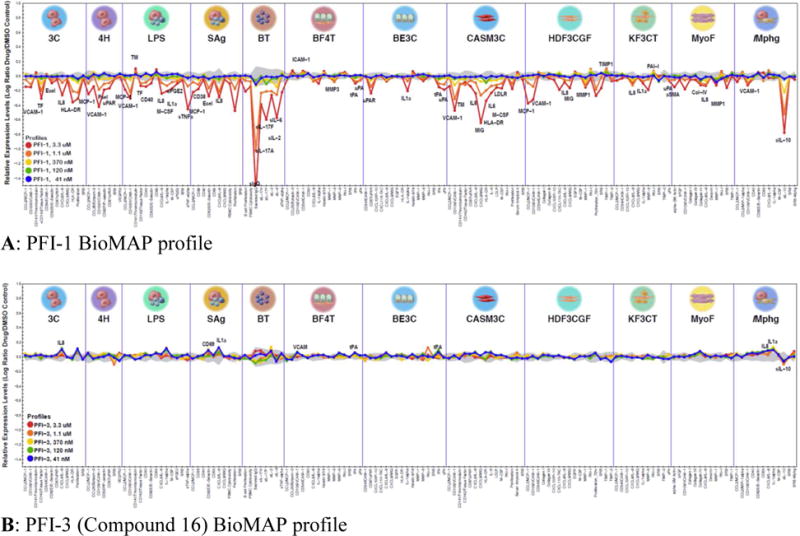
Contrast in BioMAP profile of the (A) BET bromodomain inhibitor PFI-1 and the (B) Family VIII bromodomain inhibitor PFI-3 (16) in 12 human primary cell-based systems against a number of cellular endpoints. Compounds were tested over 5 concentrations as indicated and effects were normalized to vehicle and expressed as log ratio. The grey shading is the 95% vehicle control envelope for historical control data. Values that fall outside of this envelope at two or more consecutive concentrations and with an effect size >20% ((|log10 ratio|> 0.1) are denoted as statistically significant and are annotated. The protein-based parameters tested in each system are listed along the bottom of the graph.
Acknowledgments
The Structural Genomics Consortium is a registered charity (number 1097737) that receives funds from AbbVie, Bayer, Boehringer Ingelheim, the Canadian Institutes for Health Research, the Canada Foundation for Innovation, Genome Canada, GlaxoSmithKline, Janssen, Lilly Canada, Merck, the Novartis Research Foundation, the Ontario Ministry of Economic Development and Innovation, Pfizer, Takeda and the Wellcome Trust. Aspects of this work were supported by NIH grants CA185926, CA82834 and GM56244.
ABBREVIATIONS USED
- BET
bromodomain and extra C-terminal domain
- SWI/SNF
SWItch/Sucrose Non-Fermentable
- BRG1
brahma-releated gene 1
- SMARCA
SWI/SNF related, matrix associated, actin dependent regulator of chromatin, subfamily a
- PB1
protein polybromo
- DSF
differential scanning fluorimetry
- ITC
isothermal titration calorimetry
- SAR
structure activity relationship
- NT
not tested
- DIPEA
diisopropylethylamine
- EtOH
ethanol
- RT
room temperature
- BINAP
2,2′-bis(diphenylphosphino)-1,1′-binaphthyl
- OAc
acetate
- NaOBu-t
sodium tertiarybutoxide
- MeOH
methanol
- FRAP
fluorescence recovery after photobleaching
- HDAC
histone deacetylase
- SAHA
suberoylanilide hydroxamic acid
- GFP
green fluorescent protein
- ccRCC
cell renal cell carcinoma
- MTT
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
Footnotes
ASSOCIATED CONTENT: Supporting Information: Supplementary biological methods and chemistry experimental procedures; Cellular activity; X-ray statistics. This material is available free of charge via the Internet at http://pubs.acs.org. Accession codes: Coordinates have been deposited with the Protein Data Bank (Compound 2 5E7D, compound 3 4Q0N, compound 17 5KDH).
PFI-3 is commercially available and is also known as PF-6687252
References
- 1.Filippakopoulos P, Knapp S. Targeting bromodomains: epigenetic readers of lysine acetylation. Nat Rev Drug Discov. 2014;13:337–56. doi: 10.1038/nrd4286. [DOI] [PubMed] [Google Scholar]
- 2.Tamkun JW, Deuring R, Scott MP, Kissinger M, Pattatucci AM, Kaufman TC, Kennison JA. Brahma: A regulator of Drosophila homeotic genes structurally related to the yeast transcriptional activator SNF2/SWI2. Cell. 1992;68:561–572. doi: 10.1016/0092-8674(92)90191-e. [DOI] [PubMed] [Google Scholar]
- 3.Filippakopoulos P, Picaud S, Mangos M, Keates T, Lambert JP, Barsyte-Lovejoy D, Felletar I, Volkmer R, Müller S, Pawson T, Gingras AC, Arrowsmith CH, Knapp S. Histone recognition and large-scale structural analysis of the human bromodomain family. Cell. 2012;149:214–231. doi: 10.1016/j.cell.2012.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.(a) Brand M, Measures AM, Wilson BG, Cortopassi WA, Alexander R, Hoss M, Hewings DS, Rooney TPC, Paton RS, Conway SJ. Small molecule inhibitors of bromodomain-acetyl-lysine interactions. ACS Chem Biol. 2015;10:22–39. doi: 10.1021/cb500996u. [DOI] [PubMed] [Google Scholar]; (b) Garnier JM, Sharp PP, Burns CJ. BET bromodomain inhibitors: a patent review. Exp Opin Ther Pat. 2014;24:185–99. doi: 10.1517/13543776.2014.859244. [DOI] [PubMed] [Google Scholar]; (c) Chung CW. Small molecule bromodomain inhibitors: extending the druggable genome. Progress in Medicinal Chemistry. 2012;51:1–55. doi: 10.1016/B978-0-12-396493-9.00001-7. [DOI] [PubMed] [Google Scholar]
- 5.Jung M, Gelato KA, Fernandez-Montalvan A, Siegel S, Haendler B. Targeting BET bromodomains for cancer treatment. Epigenomics. 2015;7:487–501. doi: 10.2217/epi.14.91. [DOI] [PubMed] [Google Scholar]
- 6.Vidler LR, Brown N, Knapp S, Hoelder S. Druggability analysis and structural classification of bromodomain acetyl-lysine binding sites. J Med Chem. 2012;55(17):7346–7359. doi: 10.1021/jm300346w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ho L, Crabtree GR. Chromatin remodelling during development. Nature. 2010;463:474–784. doi: 10.1038/nature08911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Masliah-Planchon J, Bieche I, Guinebretiere JM, Bourdeaut F, Delattre O. SWI/SNF chromatin remodeling and human malignancies. Ann Rev Path – Mech. 2015;10:145–171. doi: 10.1146/annurev-pathol-012414-040445. [DOI] [PubMed] [Google Scholar]
- 9.Singh M, Popowicz GM, Krajewski M, Holak TA. Structural ramification for acetyl-lysine recognition by the bromodomain of human BRG1 protein, a central ATPase of the SWI/SNF remodeling complex. Chembiochem. 2007;8:1308–1316. doi: 10.1002/cbic.200600562. [DOI] [PubMed] [Google Scholar]
- 10.Gallenkamp D, Gelato KA, Haendler B, Weinmann H. Bromodomains and their pharmacological inhibitors. ChemMedChem. 2014;9:438–464. doi: 10.1002/cmdc.201300434. [DOI] [PubMed] [Google Scholar]
- 11.Harner MJ, Chauder BA, Phan J, Fesik SW. Fragment-based screening of the bromodomain of ATAD2. J Med Chem. 2014;57:9687–9692. doi: 10.1021/jm501035j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fedorov O, Castex J, Tallant C, Owen DR, Martin S, Aldeghi M, Monteiro O, Filippakopoulos P, Picaud S, Trzupek JD, Gerstenberger BS, Bountra C, Willmann D, Wells C, Philpott M, Rogers C, Biggin PE, Brennan P, Bunnage ME, Schuele R, Günther T, Knapp S, Müller S. Selective targeting of the BRG/PB1 bromodomains impairs embryonic and trophoblast stem cells maintenance. Sci Adv. 2015;1:e1500723. doi: 10.1126/sciadv.1500723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.(a) Frye SV. The art of the chemical probe. Nat Chem Biol. 2010;6:159–161. doi: 10.1038/nchembio.296. [DOI] [PubMed] [Google Scholar]; (b) Bunnage ME, Chekler EL Piatnitski, Jones LH. Target validation using chemical probes. Nat Chem Biol. 2013;9:195–199. doi: 10.1038/nchembio.1197. [DOI] [PubMed] [Google Scholar]; (c) Arrowsmith CH, Audia JE, Austin C, Baell J, Bennett J, Blagg J, Bountra C, Brennan PE, Brown PJ, Bunnage ME, Buser-Doepner C, Campbell RM, Carter AJ, Cohen P, Copeland RA, Cravatt B, Dahlin JL, Dhanak D, Edwards AM, Frye SV, Gray N, Grimshaw CE, Hepworth D, Howe T, Huber KVM, Jin J, Knapp S, Kotz JD, Kruger RG, Lowe D, Mader MM, Marsden B, Mueller-Fahrnow A, Müller S, O’Hagan RC, Overington JP, Owen DR, Rosenberg SH, Roth B, Ross R, Schapira M, Schreiber SL, Shoichet B, Sundstrom M, Superti-Furga G, Taunton J, Toledo-Sherman L, Walpole C, Walters MA, Willson TM, Workman P, Young RN, Zuercher WJ. The promise and peril of chemical probes. Nat Chem Biol. 2015;11:536–541. doi: 10.1038/nchembio.1867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ghosh CK, Khan S. Heterocyclic systems; 10. Defunctionalization of 4-oxo-4H-[1]-benzopyran-3-carboxylic acids and -3-carboxaldehydes. Synthesis. 1981;9:719–721. [Google Scholar]
- 15.Müller S, Knapp S. Discovery of BET bromodomain inhibitors and their role in target validation. MedChemComm. 2014;53:288–296. [Google Scholar]
- 16.Philpott M, Yang J, Tumber T, Fedorov O, Uttarkar S, Filippakopoulos P, Picaud S, Keates T, Felletar I, Ciulli A, Knapp S, Heightman TD. Mol BioSyst. 2011;7:2899–2908. doi: 10.1039/c1mb05099k. [DOI] [PubMed] [Google Scholar]
- 17.Ciceri P, Müller S, O’Mahony A, Fedorov O, Filippakopoulos P, Hunt JP, Lasater EA, Pallares G, Picaud S, Wells C, Martin S, Wodicka LM, Shah NP, Treiber DK, Knapp S. Dual kinase-bromodomain inhibitors for rationally designed polypharmacology. Nat Chem Biol. 2014;10:305–312. doi: 10.1038/nchembio.1471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Philpott M, Rogers CM, Yapp C, Wells C, Lambert JP, Strain-Damerell C, Burgess-Brown NA, Gingras AC, Knapp S, Müller S. Assessing cellular efficacy of bromodomain inhibitors using fluorescence recovery after photobleaching. Epigenetics & Chromatin. 2014;7:14. doi: 10.1186/1756-8935-7-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Thompson M. Polybromo-1: the chromatin targeting subunit of the PBAF complex. Biochimie. 2009;913:309–319. doi: 10.1016/j.biochi.2008.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hammitzsch A, Tallant C, Fedorov O, O’Mahony A, Brennan PE, Hay DA, Martinez FO, Al-Mossawi MH, de Wit J, Vecellio M, Wells C, Wordsworth P, Müller S, Knapp S, Bowness P. CBP30, a selective CBP/p300 bromodomain inhibitor, suppresses human Th17 responses. Proc Nat Acad Sci USA. 2015;112:10768–10773. doi: 10.1073/pnas.1501956112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pawłowski R, Mühl SM, Sulser T, Krek W, Moch H, Schraml P. Loss of PBRM1 expression is associated with renal cell carcinoma progression. Int J Cancer. 2012;132:E11–E17. doi: 10.1002/ijc.27822. [DOI] [PubMed] [Google Scholar]
- 22.(a) Helming KC, Wang X, Roberts CWM. Cancer vulnerabilities of mutant SWI/SNF complexes in cancer. Cell. 2014;26:309–317. doi: 10.1016/j.ccr.2014.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Hoffman GR, Rahal R, Buxton F, Xiang K, McAllister G, Frias E, Bagdasarian L, Huber J, Lindeman A, Chen D, Romero R, Ramadan N, Phadke T, Haas K, Jaskelioff M, Wilson BG, Meyer MJ, Saenz-Vash V, Zhai H, Myer VE, Porter JA, Keen N, McLaughlin ME, Mickanin C, Roberts CWM, Stegmeier F, Jagani Z. Functional epigenetics approach identifies BRM/SMARCA2 as a critical synthetic lethal target in BRG1-deficient cancers Proc. Nat Acad Sci USA. 2014;111:3128–3133. doi: 10.1073/pnas.1316793111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vangamudi B, Paul TA, Shah PK, Kost-Alimova M, Nottebaum L, Shi X, Zhan Y, Leo E, Mahadeshwar HS, Protopopov A, Futreal A, Tieu TN, Peoples M, Heffernan TP, Marszalek JR, Toniatti C, Petrocchi A, Verhelle D, Owen DR, Draetta G, Jones P, Palmer WS, Sharma S, Andersen JK. The SMARCA2/4 ATPase domain surpasses the bromodomain as a drug target in SWI/SNF-mutant cancers: Insights from cDNA rescue and PFI-3 inhibitor studies. Cancer Res. 2015;75:3865–3878. doi: 10.1158/0008-5472.CAN-14-3798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.(a) Bultman S, Gebuhr T, Yee D, La Mantia C, Nicholson J, Gilliam A, Randazzo F, Metzger D, Chambon P, Crabtree G, Magnuson TA. Brg1 null mutation in the mouse reveals functional differences among mammalian SWI/SNF complexes. Mol Cell. 2000;6:1287–1295. doi: 10.1016/s1097-2765(00)00127-1. [DOI] [PubMed] [Google Scholar]; (b) Reyes JC, Barra J, Muchardt C, Camus A, Babinet C, Yaniv M. Altered control of cellular proliferation in the absence of mammalian brahma (SNF2alpha) EMBO J. 1998;17:6979–6991. doi: 10.1093/emboj/17.23.6979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.(a) Ohkawa Y, Yoshimura S, Higashi C, Marfella CG, Dacwag CS, Tachibana T, Imbalzano AN. Myogenin and the SWI/SNF ATPase Brg1 maintain myogenic gene expression at different stages of skeletal myogenesis. J Biol Chem. 2007;282:6564–6570. doi: 10.1074/jbc.M608898200. [DOI] [PubMed] [Google Scholar]; (b) Ohkawa Y, Marfella CG, Imbalzano AN. Skeletal muscle specification by myogenin and Mef2D via the SWI/SNF ATPase Brg1. EMBO J. 2006;25:490–501. doi: 10.1038/sj.emboj.7600943. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) de la Serna IL, Carlson KA, Imbalzano AN. Mammalian SWI/SNF complexes promote MyoD-mediated muscle differentiation. Nat Genet. 2001;27:187–190. doi: 10.1038/84826. [DOI] [PubMed] [Google Scholar]; (d) Salma N, Xiao H, Mueller E, Imbalzano AN. Temporal recruitment of transcription factors and SWI/SNF chromatinremodeling enzymes during adipogenic induction of the peroxisome proliferator-activated receptor gamma nuclear hormone receptor. Mol Cell Biol. 2004;24:4651–4663. doi: 10.1128/MCB.24.11.4651-4663.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nasipak BT, Padilla-Benavides T, Green KM, Leszyk JD, Mao W, Konda S, Sif S, Shaffer SA, Ohkawa Y, Imbalzano AN. Opposing calcium-dependent signaling pathways control skeletal muscle differentiation by regulating a chromatin remodelling enzyme. Nat Commun. 2015;17:7441. doi: 10.1038/ncomms8441. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.