

Abstract
Environmental and genetic factors represent key components in the establishment/maintenance of the intestinal microbiota. The aryl hydrocarbon receptor (AHR) is emerging as a pleiotropic factor, modulating pathways beyond its established role as a xenobiotic sensor. The AHR is known to regulate immune surveillance within the intestine through retention of intraepithelial lymphocytes, functional redistribution of Th17/Treg balance. Consequently, environmental/genetic manipulation of AHR activity likely influences host-microbe homeostasis. Utilizing C57BL6/J Ahr−/+ and Ahr−/− co-housed littermates followed by 18 days of genotypic segregation, we examined the influence of AHR expression upon intestinal microbe composition/functionality and host physiology. 16S sequencing/quantitative PCR (qPCR) revealed significant changes in phyla abundance, particularly Verrucomicrobia together with segmented filamentous bacteria, and an increase in species diversity in Ahr−/− mice following genotypic segregation. Metagenomics/metabolomics indicate microbial composition is associated with functional shifts in bacterial metabolism. Analysis identified Ahr−/−-dependent increases in ileal gene expression, indicating increased inflammatory tone. Transfer of Ahr−/− microbiota to wild-type germ-free mice recapitulated the increase Verrucomicrobia and inflammatory tone, indicating Ahr−/−-microbial dependence. These data suggest a role for the AHR in influencing the community structure of the intestinal microbiota.
The mammalian gastrointestinal tract represents an expansive and dynamic microbial ecosystem, comprising numerous phyla and a largely uncharacterized multitude of microbial species encompassing >1010 individuals. The collective microbiota (and its associated microbiome) interact cooperatively and competitively as a community through both intra- (microbe-microbe) and inter-kingdom (host-microbe) communication, to profoundly influence host homeostasis and physiology. Once established, typically shortly after birth, the high-level (phyla) composition of the intestinal microbiota may remain temporally stable. Despite the apparent stability, individual microbial species and their metabolic activity are in a constant state of flux, being highly sensitive to perturbation by environmental stimuli, including fluctuation in diet1,2,3,4, xenobiotics5, antibiotics6, stress7 and exposure to extrinsic commensal and/or pathogenic bacteria8. Additionally, intrinsic genetic factors impinge upon the composition of the microbiota. Indeed, for mammals, the maternal microbiota is the primary influence upon colonization and establishment of the nascent microbiota and subsequent programming of the immune system9. An increasing number of genetic loci are reported to impact the microbiota. Many of these loci are associated with immune function and bring in to focus the critical relationship between the immune system, microbiota and host physiology. Recently, the aryl hydrocarbon receptor (AHR) has gained considerable attention as a modulator of immune function, especially in the context of dietary AHR activation10. However, its role as a genetic determinant of microbial community structure has not been fully investigated.
Historically, AHR function was restricted to regulation of cytochrome P450-mediated detoxification and as a modulator of environmental pollutant-mediated toxicity (e.g. dioxin and polychlorobiphenyls); however, with the identification of endogenous, dietary and microbial ligands, the AHR is now increasingly recognized as a pleiotropic factor11,12. An increasing volume of evidence supports a pivotal role for the AHR as a modulator of immunological development, surveillance and function within barrier tissues such as the intestine13. Indeed, Ahr−/− mice are particularly susceptible to intestinal insults, such as exposure to dextran sodium sulfate. Conversely, expression of AHR in conjunction with ligand activation has been shown to be protective14. Of particular importance is the role of AHR and its activation by dietary or microbial-derived tryptophan derivatives such as indole-3 carbinol in facilitating gene expression associated with intestinal retention and function of group 3 innate lymphoid cells and intraepithelial lymphocytes15,16. Microbial stimulation of such cells, in combination with AHR transcriptional activity, drives the expression of IL22, which in turn stimulates epithelial cells to release anti-microbial factors including RegIII and defensins, thus contributing to epithelial barrier integrity and microbial homeostasis. Furthermore, AHR activity is a determinant in the differentiation of Th17 and Treg cells, the balance of which can profoundly influence the response to immunological stimuli at mucosal surfaces17.
The capacity of the AHR to direct immune functionality is therefore likely to impact host-microbe homeostasis, particularly within the intestine where microbial pressure is most intense. Here we provide evidence, using Ahr heterozygous and null C57BL6/J littermates, that the AHR contributes to the community structure of the microbiota and that genotypic segregation reinforces alterations in microbiota composition. Furthermore, we demonstrate that divergence of the microbiota between Ahr genotypes results in changes in metabolite abundance and host gene expression, which likely impacts overall host physiology.
Results
Genotypic segregation of Ahr −/+ and Ahr −/− littermates alters the community structure of the cecal microbiota
To examine the contribution of mouse AHR expression with regard to maintenance of the community structure of the intestinal microbiota, littermates (Ahr−/+ and Ahr−/−) from congenic female C57BL6/J (Ahr−/−) and male C57BL6.129-Ahrtm/Bra/J (Ahr−/+) matings were co-housed for 6 months prior to separation based upon genotype, as illustrated in the experimental overview (Supplemental Figure 1). We adopted this approach of heterozygous and null Ahr littermates in favor of homozygous and null animals to facilitate equivalent microbiota exposure at birth. Additionally, heterozygous Ahr mice exhibit similar physiological and transcriptional responses to homozygous Ahr counterparts18. Following 18 days of genotypic isolation under identical environmental and dietary conditions within the same vivarium, bacterial 16S rDNA gene profiling was performed with DNA isolated from cecal luminal contents (Fig. 1). Phyla level abundance analysis obtained from 16S rDNA gene sequencing (mean 180,000 reads/sample, n = 4/genotype) between each Ahr genotype revealed significant differences in the percentage abundance of 16S reads associated with Tenericutes (10-fold), TM7 (3-fold), Verrucomicrobia (3-fold), Proteobacteria (1.5-fold) and Actinobacteria (1.3-fold). No significant change in 16S reads was observed between Ahr genotypes with regard to Deferibacteria nor Bacteroidetes and Firmicutes, which together constitute the dominant (by percentage abundance) phyla in both mice and humans (Fig. 1a and Supplemental Figure 2). Although not significant, we observed opposing trends with 16S reads associated with Bacteroidetes and Firmicutes (increased and decreased, respectively) in Ahr−/− compared to Ahr−/+ counterparts. However, this was insufficient to significantly alter the Firmicutes/Bacteroidetes ratio (Fig. 1b). To validate these 16S rDNA gene sequencing observations we performed quantitative PCR analysis upon microbial DNA isolated from the cecal contents using selected phyla-specific primers (Supplementary Figure 3a). Such analysis of phyla and total bacterial abundance confirmed the lack of significant change with regard to Firmicutes or Bacteroidetes and established no overt influence of Ahr genotype upon total bacterial burden within the cecum.
Figure 1. Genotypic segregation of Ahr−/+ and Ahr−/− littermates alters the community structure of the cecal microbiota.

(a) Pie chart representation of the phyla level community structure of the cecal microbiota from genotypically segregated Ahr−/+ and Ahr−/− littermates as determined through 16S rDNA gene Illumina MiSeq sequencing. Data represent mean percentage abundance (n = 4/genotype, *p < 0.05, **p < 0.01 Student’s t-test). (b) Firmicutes/Bacteroidetes ratio based on phyla-specific 16S rDNA gene percentage abundance within cecal luminal contents.
Quantitative PCR analysis of individual taxa or species, based on amplification of microbial DNA, is typically normalized to total bacterial abundance, in essence representing bacteria normalized to bacteria and thus lacks independent normalization. Additionally, a significant fraction of isolated microbial DNA may be derived from non-viable or metabolically inactive bacterial cells and therefore biologically or functionally irrelevant at the moment of isolation19. Furthermore, reports have demonstrated that 16S gene sequencing is prone to taxa-specific bias resulting in misrepresentation of abundance20. Therefore, to establish that these 16S rDNA gene sequencing observations accurately reflect quantification of viable, functionally relevant microbial taxa and provide an independent normalization parameter, we utilized an RNA-based quantitative PCR approach. Total RNA isolated from intact ceca (cecum and luminal microbial contents) was used as a template for quantitative reverse transcriptase PCR analysis and normalized to bacteria-independent, eukaryotic Rpl13a expressed by cecal tissue (Supplementary Figure 3b). Quantitative analyses of phyla abundance based on RNA isolated from intact ceca corroborated both DNA-based PCR and 16S rDNA gene sequencing data demonstrating no significant change in overall bacterial burden, Firmicutes or Bacteroidetes but confirmed that the enrichment of Verrucomicrobia likely represented a 3-fold increase in viable bacteria. These data indicate that AHR expression influences the high-level community structure of the microbiota within the cecal lumen albeit through adjustment of lower abundance phyla including Verrucomicrobia.
Expanded sequence analysis identified numerous significant differences in 16S read abundance between Ahr−/+ and Ahr−/− mice at the taxonomic class, order, family, and genus levels (Fig. 2). The trend towards depletion of 16S reads associated with Firmicutes in Ahr−/− mice correlated with a reduction in the class Clostridia, principally unclassified genera belonging to the family Lachnospiraceae (r = 0.9802, p < 0.05). Conversely, the trend towards enrichment of 16S reads associated with Bacteroidetes in Ahr−/− mice correlated with an increase in the class Bacteroidia, principally unclassified genera belonging to the order Bacteroidales (r = 0.9981, p < 0.01). The observed Ahr−/− mice significant enrichment of Proteobacteria-associated 16S reads correlated with an increase in unclassified genera belonging to the class Alphaproteobacteria (r = 0.9862, p < 0.05). The significant enrichment of Actinobacteria-associated 16S reads correlated with an increase in unclassified genera belonging to the family Coriobacteriaceae (r = 0.9994, p < 0.001). The significant enrichment of 16S reads associated with Tenericutes in Ahr−/− mice was attributable to an increase in the genus Anaeroplasma. The trend towards depletion of Deferribacteres observed in Ahr−/− mice was solely attributable to a decrease in the genus Mucisprillium. The significant enrichment of 16S reads associated with Verrucomicrobia in Ahr−/− mice appeared to be solely attributable to an increase in the genus Akkermansia.
Figure 2. Analysis of microbial taxonomic population changes in ceca of Ahr−/+ and Ahr−/− mice following genotypic segregation.

Heat map representation of individual percentage 16S read abundance at taxonomic (phyla, class, order, family and genus) levels of cecal microbiota from genotypically segregated Ahr−/+ and Ahr−/− littermates. Data are represented as Log2 transformed 16S read percentage abundance for each individual (n = 4/genotype) with high to low abundance indicated by red to blue, respectively.
To examine potential relationships between the cecal microbiota within and between each of the Ahr genotypes, intra-taxa level correlations were determined (Supplemental Figure 4 and Supplementary file 1). Analysis of phyla-phyla interactions within Ahr−/+ mice indicated that Firmicutes abundance is positively correlated with Tenericutes but inversely correlated with Bacteroidetes. Despite retaining the inverse relationship with Bacteroidetes in Ahr−/− mice, Firmicutes failed to exhibit the correlation with Tenericutes within this genotype. A number of discordant class-class relationships were observed between the Ahr genotypes. The most significant relationship in Ahr−/+ mice was exhibited by the positive correlation between Mollicutes and Alphaproteobacteria. Mollicutes abundance in Ahr−/+ mice also correlated positively with Clostridia, and Erysipelotrichia but negatively with Bacteroidia. However, these relationships were not apparent in Ahr−/− mice. At the level of order-order interactions, Ahr−/+ mice exhibited significant positive correlations between Erysipelotrichales, Clostridales and Anaeroplasmatales. A significant positive correlation was also observed between Verrucomicrobiales and Bdellovibrionales in Ahr−/+ mice. None of these relationships were evident with Ahr−/− mice. Conversely, Ahr−/− mice exhibited a significant negative correlation that was absent in Ahr−/+ mice between Lactobacillales and Burkholderiales. Significant Ahr−/+-associated family-family level interactions included positive correlations between Bdellovibrionaceae and Streptococcaceae, Verrucomicrobiaceae; Anaeroplasmataceae and Eubacteriaceae, Erysipelotrichaceae. Within Ahr−/− animals, the positive correlation between Bdellovibrionaceae and Streptococcaceae was reversed and the positive association with Verrucomicrobiaceae lost and replaced with a negative correlation with Sutterellaceae. Ahr−/− mice also exhibited a loss of association between Anaeroplasmataceae and Eubacteriaceae, Erysipelotrichaceae but acquired a positive correlation with Prevotellaceae. Additionally, significant positive correlations observed in Ahr−/+ animals between Clostridiaceae and Prevotellaceae; Eubacteriaceae and Porphyromonadaceae; and a negative association between Rikenellaceae and Prevotellaceae were not exhibited by Ahr−/− mice. Numerous Genus-Genus level differences were evident between Ahr genotypes, including positive associations between Akkermansia and Vampirovibrio, Ruminococcus, Allobaculum; Clostridium XVa and Oscillibacter and Odorbacter; and Lactobacillus and Bilophila, Hydrogenoanerobacterium and Psuedoflavonifracter in Ahr−/+ animals that were not exhibited by Ahr−/− mice. Combined, these data indicate differences in the steady-state cecal microbial landscape of AHR expressing and non-expressing mice, suggesting a role for AHR in maintaining the bacterial ecosystem of the intestine.
Segregation of cecal Akkermansia muciniphila abundance between Ahr genotypes
16S rDNA gene sequencing analyses identified Verrucomicrobia as a prominent phyla-level divergence between genotypically segregated Ahr−/+ and Ahr−/− mice (Fig. 1). Using primers specific for Akkermansia muciniphila (A. muciniphila), the only currently cultured species of Verrucomicrobia known to colonize rodents and humans, quantitative PCR analysis performed on RNA isolated from intact ceca further established that the increase in 16S reads associated with Verrucomicrobia observed in Ahr−/− animals is likely attributable to increased colonization by viable A. muciniphila (Supplemental Figure 3). To further examine this change in A. muciniphila representation further, fecal abundance was assessed from the point of genotypic segregation (Fig. 3a). Quantitative PCR performed on fecal DNA revealed an increased A. muciniphila load in Ahr−/− mice when compared to Ahr−/+ animals. Following segregation, Ahr−/− mice exhibited an increase in A. muciniphila, which was significantly different from than that observed in Ahr−/+ animals. An examination of body weight revealed a significant degree of weight loss by Ahr−/− mice upon segregation, which was not evident in Ahr−/+ littermates (Fig. 3b). Examination of food intake revealed a mean intake of ~3 g/mouse/day by both genotypes thus indicating that the weight loss exhibited by Ahr−/− animals could not be due to diminished appetite (Fig. 3c). Analysis of blood glucose levels did not reveal any significant difference between the Ahr genotypes over the time course of segregation, suggesting that the observed increase in A. muciniphila does not impact serum glucose levels (Fig. 3d).
Figure 3. Segregation of Akkermansia muciniphila abundance between Ahr genotypes.

(a) Quantitative real time PCR analysis of fecal Akkermansia muciniphila abundance prior to and following genotypic segregation. Data represent min-max and median fecal Akkermansia muciniphila 16S rDNA gene abundance normalized to Eubacteria from Ahr−/+ (blue) and Ahr−/− (red) (n=4/genotype). Analyses were performed using Student’s t-test *p < 0.05. (b) Quantification of percentage weight change during the course of genotypic segregation. Mice were weighed every 48 h at 10–11 am and percentage weight change relative to initial weight calculated. Data represent mean percentage weight change +/− SD in Ahr−/+ (blue circle) and Ahr−/− (red triangle) (n = 4/genotype). Analyses were performed using Student’s t-test *p < 0.05. (c) Quantification of food intake during genotypic segregation. Data represent cumulative food intake expressed as g/mouse (n = 4/genotype) from Ahr−/+ (blue circle) and Ahr−/− (red circle). (d) Analysis of blood glucose during genotypic segregation. Blood glucose levels were determined every 48 h at 10–11 am through tail clip bleed and a hand-held glucose monitor. Data represent mean glucose concentration (mg/dL) +/− SD from Ahr−/+ (blue circle) and Ahr−/− (red circle).
Cecal microbiota from Ahr −/+ and Ahr −/− mice exhibit divergent metagenomic metabolic pathway profiles
Having established that genotypically segregated Ahr−/+ and Ahr−/− littermates harbor divergent cecal microbial populations without an overall change in total bacterial burden, we utilized whole genome metagenomic pathway analysis, adopting the HMP Unified Metabolic Analysis Network 2 (HUMAnN2) pipeline, to assess the potential differences in metabolic pathway representation within the differing microbial community structures. HUMAnN2 analyses allows for identification and assessment of those pathways that are represented with greater than 50% pathway coverage. Such analyses were performed and identified 1144 MetaCyc pathways or superpathways that were represented in either or both Ahr genotypes. Of these represented pathways, 468 (~40%) exhibited a significant difference in abundance between Ahr−/+ and Ahr−/− animals, indicating the potential for metabolic consequences within the host (Fig. 4a). The 70 most prominent differences were observed with pathways associated with amino acid biosynthesis and degradation; purine/pyrimidine biosynthesis/salvage; folate transformations; glycolysis and anaerobic respiration, all of which exhibited significantly enhanced representation in cecal microbiota from Ahr−/− mice. Despite these changes in pathway abundance, analysis of the Shannon diversity index associated with the cohort of metagenomic pathways in each genotype failed to identify a significant separation (Fig. 4b). Linear discriminate analysis (LDA) was performed to identify those pathways that discriminate between Ahr genotypes (Fig. 4c). In Particular, LDA analysis identified UMP, lysine and methionine, UDP N-acetyl glucosamine, mycolate biosynthetic pathways, gluconeogenesis and fatty acid elongation processes as the most discriminatory factors within the metagenome of Ahr−/− associated cecal microbiota. Whereas the Ahr−/+ cecal metagenome was most associated with pyrimidine salvage, chorismate, glutamate and glutamine, isoleucine, histidine, valine and chorismate from 3-dehydroquinate biosynthetic pathways, as well as pyruvate to acetate processes. These results support 16S rDNA gene sequencing data demonstrating a functionally altered community structure.
Figure 4. HUMAnN2 metagenomic metabolic pathway analysis.

(a) Metagenomic pathways exhibiting significant changes in representation. Microbial DNA from cecal contents was sequenced and pathway coverage determined using the HUMAnN2 pipeline. The pathway coverage output representing the number of gene families associated with each pathway were processed to include only those pathways with >50% coverage. Data represent those pathways exhibiting a difference in abundance below a significance threshold of p < 0.001 (Student’s t-test) expressed as a z-score. (b) Shannon diversity index associated with metagenomic pathways. Diversity indices were calculated and presented as min-max and median H values (n = 4/genotype). (c) Linear discriminant analysis (LDA) of metagenomic pathways. Pathway abundance data were applied to the LDA algorithm with a α = 0.05 threshold for Kruskal-Wallis and pairwise Wilcoxon tests combined with a 2.0 logarithmic LDA cut-off to identify pathway components which most significantly discriminate between Ahr genotypes. Data represent Log10 LDA score for indicated pathways within Ahr−/+ (blue) and Ahr−/− (red) mice.
Alteration of microbial community structure is associated with changes in cecal metabolite signatures in genotypically segregated Ahr −/+ and Ahr −/− littermates
Our observation that genotypic segregation of Ahr−/+ and Ahr−/− littermates facilitates changes in the representation of selected microbial metabolic pathways prompted an examination of the metabolite profiles associated with the cecal luminal contents (Fig. 5a). Relative quantification through 1H-NMR peak integration analyses identified significant decreases in the short-chain fatty acids (SCFAs) butyrate and propionate within the luminal contents of Ahr−/− mice, indicative of reduced microbial fermentation. Ahr−/− mice exhibited a significant increase in lactate that inversely correlated with glucose levels, suggesting an elevated glycolytic flux, which is supported by metagenomic pathway analyses indicating greater representation of the microbial glycolytic pathway in Ahr−/− animals. Consistent with our pathway analyses demonstrating enrichment of various amino acid biosynthetic pathways in microbiota from Ahr−/− mice, we observed significant increases in the relative abundance of branched-chain amino acids (valine, leucine and isoleucine), alanine, lysine, glutamate, tyrosine and phenylalanine within cecal luminal contents.
Figure 5. Genotypically segregated Ahr mice exhibit differential cecal metabolite profiles.

(a) Cecal contents from genotypically segregated Ahr−/− and Ahr−/+ mice. Data represent relative min-max and median peak integration for the indicated metabolite. (b) Heat map representations of correlative taxa-level interactions between cecal microbiota and metabolites from genotypically segregated Ahr−/+ and Ahr−/− littermates. Data represent Pearson correlation (r) coefficients with positive to negative correlation indicated by red to blue, respectively. Correlation coefficients and p values are detailed in Supplementary file 2.
To examine potential associations between the observed changes in cecal metabolites and the altered microbial landscape in Ahr−/− mice, Pearson correlation coefficients were determined (Fig. 5b and Supplemental file 2). The reduction in the SCFA butyrate failed to associate at the phylum or class level but exhibited a significant positive correlation with the order Burkholderiales (r = 0.9794, p < 0.05) and the genera Oscillibacter and Parabacteroides (r = 0.998, p < 0.01). In contrast, cecal butyrate levels in Ahr−/+ mice exhibited positive correlations with the phylum Bacteroidetes (r = 0.9595, p < 0.05) and the Sutterellaceae (r = 0.9946, p < 0.01) family, combined with negative associations at family and genus levels with Erysipelotrichiaceae (r = −0.9748, p < 0.05) and Eubacterium (r = −0.9596, p < 0.05) respectively. Within Ahr−/− animals, the reduction in the SCFA propionate negatively correlated with the Mollicutes class (r = −0.9665, p < 0.05). Within Ahr−/+ mice, propionate abundance negatively correlated with Anaerotruncus (r = −0.9183, p < 0.05), members of Firmicutes. Although no significant difference in the SCFA acetate was observed between Ahr genotypes, its abundance exhibited divergent microbiota associations, being negatively correlated with Muscispirillum (r = −0.969, p < 0.05) and positively with Oscillibacter (r = 0.9579, p < 0.05) in Ahr−/+ mice but not Ahr−/− animals. The significant increase in cecal lactate observed in Ahr−/− mice negatively associated with Clostridium XVb (r = −0.9684, p < 0.05) and positively with Allobaculum (r = 0.9988, p < 0.05) but negatively with Clostridales (r = −0.9543, p < 0.05) in Ahr−/+ animals. Cecal glucose significantly positively correlated with Ruminococcaceae (r = 0.9637, p < 0.05) in Ahr−/− mice but with Lachnospiraceae (r = 0.9683, p < 0.05) in Ahr−/+ animals. Cecal oligosaccharide content exhibited a significant negative correlation with Coriobacteriaceae (r = −0.9870, p < 0.05) in Ahr−/− mice. In contrast, within Ahr−/+ animals oligosaccharide levels were positively associated with Lactobacilliaceae (r = 0.9757, p < 0.05). These data provide supporting evidence that the altered microbiota together with concomitant changes in metabolic pathway representation observed within Ahr−/− mice leads to quantifiable metabolite differences.
Segregation of ileal segmented filamentous bacteria abundance between Ahr genotypes
Previous studies have highlighted the significance of ileum-specific segmented filamentous bacteria (SFB) (Candidatus savagella or arthromitus) in determining host-microbe responses21. We therefore examined the degree of ileal colonization of this Clostrida species within genotypically segregated Ahr mice. Quantitative PCR analysis of terminal ileum RNA revealed a marked and significant increase in viable SFB abundance in Ahr−/− mice when compared to their heterozygous littermates (Fig. 6a). PCR quantification of total bacterial burden within the ileum of these mice also indicated a significantly increased bacterial load. The similar magnitude of the increases associated with SFB and total bacteria suggested that the elevated bacterial burden within the terminal ileum of Ahr−/− mice might be largely attributable to SFB expansion. To further examine the Ahr−/−-associated increase in SFB, we visually examined the terminal ilea of these mice through scanning electron microscopy (Fig. 6b). Visualization of SFB revealed marginal to non-existent colonization in Ahr−/+ animals but identified large numbers of Ahr−/− epithelia-associated SFB, confirming the quantitative expansion observed in these mice. The established influence of SFB in determining host immunological responses to the microbiota and other antigenic sources suggests that the distinct difference in colonization is likely to influence the physiology of these mice16.
Figure 6. Segregation of ileal Segmented filamentous bacteria abundance between Ahr genotypes.

(a) Real time PCR quantification of ileal SFB abundance together with total bacterial load normalized to eukaryotic Rpl13a using specific primers upon cDNA generated from total RNA isolated from intact ileal tissue (ileum and luminal contents) from Ahr−/+ (blue) and Ahr−/− (red) mice. Data represent mean (n = 4/genotype) 16S rRNA gene abundance −/+ SD. Analyses were performed using Student’s t-test *p < 0.05, **p < 0.01. (b) Scanning electron microscopy of terminal ileum excised from Ahr−/+ and Ahr−/− mice demonstrating increased SFB colonization. Scale bar indicates 60 μm. Data are representative of images from multiple mice of each Ahr genotype.
Genotypically segregated Ahr −/− mice display an enhanced inflammatory tone within the intestine
Our experimental approach using Ahr−/+ and Ahr−/− littermates cohoused for 6 months allowed for initial exposure to the same maternal microbiota, yet analysis identifies microbial divergence following subsequent genotypic segregation, thus implicating host influences. The ileum is regarded as a major site of host-microbiota interaction, surveillance and education through modulation of host immunity. We therefore examined gene expression within the terminal ilea of genotypically segregated Ahr mice to determine if it may account for the observed microbial divergence. Quantitative PCR and NanoString™ expression analysis were performed upon ileal samples and identified significant differences in gene targets previously reported to be influenced by or modulate the microbiota (Fig. 7). Ileal expression of the acute phase reactants Saa1, 2 & 3, which are induced in response to commensal bacteria, particularly segmented filamentous bacteria (SFB), displayed Ahr genotype-dependent expression with significantly elevated levels of Saa1 and Saa3 but not Saa2 in Ahr−/− mice (Fig. 7a). Associated with the increase in Saa1/3, we also observed a corresponding significant increase in Il17a expression, which is consistent with the expansion of SFB and Th17 development in these mice21. However, ELISA-based quantification of ileal IL22, a contributor to microbiota-dependent epithelial Saa induction, exhibited decreased abundance in Ahr−/− animals (Fig. 7b). These mice also displayed enhanced expression of the siderophore Lcn2 and the LPS and pathogen-associated molecular pattern co-receptor Cd14. In conjunction with this increase in pro-inflammatory markers, NanoString™ quantification revealed a significant reduction in ileal expression of the anti-microbial defensin Def24a. Significantly reduced ileal expression of c-kit (Cd117) and Ereg were also observed in Ahr−/− mice (Fig. 7c). Conversely, a significant increase in tight junction-associated Cldn1 was evident in Ahr−/− animals. Additionally, Ahr−/− mice exhibited a trend towards enhanced expression of the cytokines Il1b, Il6, Cxcl5 and Il10, each suggestive of an inflammatory response, although these increases failed to achieve statistical significance (data not shown). Despite the observed changes in gene expression suggestive of heightened inflammatory signaling and diminished anti-microbial expression, histological examination of ileal tissue failed to identify any overt evidence of gross inflammation in Ahr−/− animals whilst maintained under a specific pathogen-free environment (Fig. 7d).
Figure 7. Genotypically segregated Ahr mice exhibit differential ileal gene expression profiles.

(a) Real time PCR quantification of ileal gene expression normalized to eukaryotic Rpl13a using specific primers upon cDNA generated from total RNA isolated from intact ileal tissue (ileum and luminal contents) from Ahr−/+ (blue) and Ahr−/− (red) mice. Data represent mean (n = 4/genotype) relative gene expression −/+ SD. Analyses were performed using Student’s t-test *p < 0.05, **p < 0.01, ***p < 0.001. (b) ELISA quantification of ileal IL22 from Ahr−/+ (blue) and Ahr−/− (red) mice. Data represent min-max and median IL22 (n = 4/genotype) level from Ahr−/+ (blue) and Ahr−/− (red) mice expressed as pg/mL. Analyses were performed using Student’s t-test *p < 0.05. (c) NanoString™ quantification of ileal gene expression. Total RNA isolated from ilea was processed through the NanoString nCounter system. Data were normalized to positive control probes (assay and house-keeping genes) and represent mean relative expression level +/− SD. Analyses were performed using Student’s t-test *p < 0.05, **p < 0.01, ***p < 0.001. (d) Histological sections of terminal ilea of Ahr−/− and Ahr−/+ mice. Terminal ilea were excised, formalin fixed, sectioned (5 μm) and stained with hematoxylin & eosin. Data are representative of 10× and 60× magnification images of multiple sections from each genotype (n = 4/genotype). (e) Cecal microbiota profile from genotypically segregated Ahr mice is transmissible to wild type germ free animals. Pooled cecal contents isolated from genotypically segregated mice (n = 4/genotype) were used to orally inoculate wild type germ free animals. Cecal abundance of Eubacteria, A. muciniphila and SFB were determined by qPCR performed on total RNA following 5 days of germ free animals. Data represent min-max and median abundance of indicated species following normalization to eukaryotic Rpl13a in the ceca of formerly germ free animals (n = 4/treatment; *p < 0.05, **p < 0.01 and ***p < 0.001). (f) Ileal gene expression in wild type germ free animals inoculated with cecal contents from genotypically segregated Ahr mice. Ileal expression of indicated targets were determined by qPCR on RNA isolated following 5 days of colonization. Data represent mean expression normalized to Rpl13a (n = 4/treatment; *p < 0.05, **p < 0.01).
Inflammatory gene expression associated with the microbiota of Ahr −/− mice is transmissible to germ-free animals
To assess whether the microbiota associated with Ahr−/− animals is a causative agent behind the enhanced inflammatory gene expression observed in these mice, we examined the effect of cecal microbiota transfer from genotypically segregated Ahr mice into germ free wild type animals (Fig. 7e). Germ free wild type mice were administered with a single oral inoculum of pooled cecal contents from Ahr−/− or Ahr−/+ animals and maintained under a germ free environment for 5 days prior to analysis of cecal bacterial abundance and host ileal gene expression. This colonization timeframe was adopted to maintain the original inoculate community structure and limit microbial drift arising from the Ahr+/+ status of the germ free recipients. Quantitative PCR analysis of bacterial abundance performed on RNA isolated from ceca confirmed microbial colonization had occurred in formerly germ free animals. Inoculation from Ahr−/− or Ahr−/+ mice resulted in similar levels of eubacteria colonization (Fig. 7e). Consistent with the increased abundance in donor animals, germ free animals inoculated with cecal contents from Ahr−/− mice exhibited a greater degree of colonization by A. muciniphila than that obtained with Ahr−/+ animals. Similarly, SFB colonization of germ free animals was established to a greater degree following inoculation with Ahr−/− animal-derived cecal contents. Analysis of host gene expression identified significantly increased expression of Saa3, Lcn2, Cd14 and Il17a within the ilea of germ free animals exposed to Ahr−/−-derived cecal microbiota when compared to those inoculated with cecal contents from Ahr−/+ mice and control non-inoculated germ free animals (Fig. 7f).
Discussion
A number of genetic and toxicological studies demonstrate the involvement of the AHR and its ligands in the establishment, maintenance and regulation of immune function5,10,15,22. Given the intimate association between host immunity and the microbiota we have therefore examined the impact of genetic AHR ablation upon the composition of the intestinal microbiota. Using a mouse model of co-housed Ahr−/+ and Ahr−/− littermates, subsequently segregated based upon genotype we demonstrate significant differences with regard to microbial community structure between these genotypes. This demonstrates that despite the opportunity for acquisition of a common microbiota within littermates the AHR genotype contributed to a selective pressure on the microbiota of the offspring. Despite the importance of AHR in facilitating the development and function of many immunological cell types e.g. Th17, Treg, B cells and mast cells, microbial overgrowth was not evident within the ceca of Ahr−/− mice. The dominant (by percentage abundance) phyla, Firmicutes and Bacteroidetes, were not overtly influenced by genetic AHR ablation; consequently, no change in Firmicutes/Bacteroidetes ratio was observed indicating that a major phyla level shift in community structure had not occurred. However, numerous significant differences between Ahr genotypes were observed at sub-phyla taxonomic levels, indicating that Ahr−/− mice harbor similar microbiota that undergoes a significant divergence upon segregation. Furthermore, metagenomic and metabolomic analyses indicate that redistribution of the microbiota in Ahr−/− animals was associated with significant changes in the cecal microbiome and metabolites and is thus likely to contribute to host physiology.
The most prominent microbial differences witnessed between the Ahr genotypes were marked Ahr−/−-associated expansions of Akkermansia muciniphila (A. muciniphila) and segmented filamentous bacteria (SFB) within the cecum and ileum respectively. Interestingly, both species are widely studied and reported to exert profound effects upon rodent and human physiology arising from their proximity to the intestinal epithelium21,23. In humans and rodents, A. muciniphila typically accounts for <1–5% of the total microbiota23,24. Occupying a niche within the luminal surface of the mucus layer, A. muciniphila degrades host mucin oligosaccharides liberating nutrients for other microbes to utilize25,26. Importantly, increased A. muciniphila abundance is suggested to confer a number of beneficial effects with regard to maintenance of epithelial barrier integrity, and protection from inflammatory intestinal conditions, colorectal cancer, insulin resistance, dyslipidemia and obesity, although the mechanisms have not yet been established27,28,29,30. Our observation of A. muciniphila enrichment in Ahr−/− mice is intriguing and suggests that these mice may exhibit some of the health benefits associated with A. muciniphila expansion. Indeed, Ahr−/− mice are reported to be resistant to diet-induced obesity and protected from insulin insensitivity, although this was not examined in the context of the microbiota31. Our data, which demonstrates a significant weight loss without a concomitant reduction in food intake, which tracked with increased fecal A. muciniphila abundance in Ahr−/− animals, is supportive for a role of A. muciniphila in reducing adiposity. However, the association of weight loss and A. muciniphila in segregated Ahr−/− mice could not be attributed to SCFAs, which are reported to exhibit anti-adipogenic activity32. Indeed, with the exception of phenylalanine in Ahr−/+ mice, we failed to identify any correlation between A. muciniphila and cecal metabolites. We did not observe any overt inflammation within Ahr−/− mice under pathogen-free conditions which is consistent with other reports15. AHR activity has previously been shown to modulate local and systemic expression of inflammatory mediators, cytokines and acute phase reactants33,34,35. In addition, previous studies have demonstrated that ablation of AHR expression renders increased sensitivity to a range of intestinal insults including models of inflammatory bowel disease and pathogenic infection although these observations however, were not specifically examined in the context of the microbiota14,36,37. The elevation of inflammation-associated markers, such as Saa, Lcn2 and Cd14 within the ileum of Ahr−/− animals suggests that these mice may be subject to low-grade inflammatory stress38,39.
The etiology of enhanced inflammatory tone and the change in the microbial landscape exhibited by Ahr−/− mice is likely to be multi-factorial and interrelated. SCFA levels, which are diminished in Ahr−/− animals as a likely consequence of a decrease in Clostridia species, has previously been shown to enhance mucosal barrier function, thus bacterial translocation may be more permissive in these mice and would be consistent with the observed increase in expression of ileal Lcn2 and Cd14. Additionally, loss of AHR expression is demonstrated to disrupt the immunological milieu of the intestine allowing for decreased microbial surveillance that may in part account for the expansion of A. muciniphila. Reduced colonization of intra-epithelial lymphocytes reported in Ahr−/− mice is likely to exert a profound effect upon inflammatory signaling and the microbial ecosystem15. Indeed, we observed significantly decreased IL22 levels that were associated with decreased expression of the anti-microbial Def24a. It is conceivable that reduced IL22-mediated defensin production may have contributed to the preferential expansion of A. muciniphila due to their proximity to the normally anti-microbial peptide rich mucus. Similarly, T and B cell-deficient Rag1−/− mice also exhibit expansion of A. muciniphila. Additionally, the ilea of Ahr−/− mice demonstrated a robust expansion of SFB, which penetrate the mucus layer to physically attach to the epithelial layer40. Therefore reduced anti-microbial peptide activity is likely to facilitate SFB expansion. Importantly, these studies were performed using co-housed Ahr−/+ and Ahr−/− littermates, indicating Ahr−/− animals are acutely receptive to SFB colonization and expansion when compared to Ahr−/+ littermates, despite an equivalent initial colonization environment. Consistent with previous studies, SFB expansion in Ahr−/− mice was accompanied by significant increase in Il17a expression within the ileum despite studies indicating the apparent requirement for AHR signaling to generate a Th17 polarizing environment15,16. A heightened IL17A response is likely to contribute to the manifestation of inflammation and increased sensitivity to insults observed within the Ahr−/− mice.
Transfer of Ahr−/− animal-derived microbiota into formerly germ free wild type animals and a subsequent increase in inflammation-associated gene expression within these animals suggests that a microbial component is causative of the change in inflammatory tone in both Ahr−/− and formerly germ free wild type animals. Whether colonization of germ free animals with Ahr−/−-derived microbiota directly facilitated the increase in inflammatory markers or inoculation with microbial metabolites, such as LPS stimulated expression is unclear. However, considering gene expression was assessed after five days argues against such metabolite-mediated changes. Furthermore, the enhanced expression however of genes known to be sensitive to SFB colonization, e.g. Saa1/3 and Il17a, suggests a direct consequence of microbial interaction with the epithelium and education of intestinal immune cells.
The data presented herein suggest that absence of AHR expression has a marked effect upon the microbiota, particularly those that are in close proximity to the epithelium, and provides further evidence that host genotype exerts a significant influence. It is unclear whether colonization or altered abundance of species, such as A. muciniphila or SFB, is causative or a marker of the overall redistribution and change in diversity of the microbiota in Ahr−/−. However, the influence of SFB in determining the immunological status of the host, together with the capacity of A. muciniphila to provide nutrients for the benefit of other species, which in turn cooperate and compete in a complex network, may point to important roles for these bacteria in shaping the intestinal microbiota and host physiology.
The use of global Ahr−/− animals in this study did not allow the relative contribution of AHR expression within different intestinal cell types to be addressed. However, studies utilizing conditional intestinal epithelia-specific Ahr−/− mice may highlight roles for AHR in non-immune cells in determining the intestinal microbial ecosystem. Furthermore, this and future studies may provide a framework to assess the influence of dietary AHR ligands upon the composition of the intestinal microbiota, microbiome and host physiology. Importantly, the observed differences in microbiota composition demonstrated here and the growing evidence supporting the importance of the microbiota in shaping host physiology suggest that studies utilizing Ahr deficient mice and/or AHR ligands may require assessment of the influence of the microbiota for a comprehensive interpretation of results.
Methods
Animals and husbandry
All animal studies were performed with approval and under the auspices of the Institutional Animal Care and Use Committee (IACUC, The Pennsylvania State University). All animal experiments were carried out in accordance to IACUC guidelines. C57BL6/J mice were originally purchased from Jackson Laboratories (Bar Harbor, ME.) and subsequently bred in-house. Ahr null mice (B6.129-Ahrtm1Bra/J) were obtained as a gift (Dr. Christopher Bradfield, University of Wisconsin, Madison, WI.) and subsequently bred in-house. Animals were housed in autoclaved polypropylene cages with corncob bedding under a specific pathogen-free environment within the same vivarium under a standard 12 h light/dark cycle (light 0600–1800 h eastern standard time) with ad libitum access to autoclaved standard animal chow and water.
Ahr genotyping
Female Ahr−/+ and Ahr−/− littermates were genotyped using genomic DNA isolated from tail clips as previously described (www.Jax.org B6;129-AhrtmBra/J strain # 0027272).
Bacterial DNA isolation
Bacterial genomic DNA was isolated from 100 mg cecal contents and fecal pellets using Powersoil DNA isolation kit (Mobio, Carlsbad, CA.) according to the manufacturer instructions. For metagenomic studies, cecal bacterial DNA was extracted using EZNA stool DNA kit (Omega Bio-tek, Norcross, GA) following the manufacturer recommended instructions. Following extraction using either method, DNA concentration was normalized to 20 ng/µl and stored at −80 °C.
16S rDNA gene Illumina MiSeq sequencing
Bacterial DNA extracted using a Powersoil DNA isolation kit and 20 ng template DNA were amplified across the V4/V4 region of the 16S rDNA gene utilizing the primers (515F: TCGTCGGCAGCGTCAGATGTGTATAAGAGACAGGTGCCAGCMGCCGCGGTAA and 806R: GTCTCGTGGGCTCGGAGATGTGTATAAGAGACAGGGACTACHVGGGTWTCTAAT) in combination with FastStart high-fidelity amplification kit (Roche, Indianapolis, IN.) and the following cycling conditions (94 °C, 3 min; 94 °C, 15 sec, 55 °C, 45 sec; 72 °C, 1 min for 30 cycles; 72 °C, 8 min). Verification of product amplification was demonstrated through agarose gel electrophoresis and the visualization of a single product of 359 bp. Amplified V4 16S rDNA gene products were supplied to the Genomics Core Facility (The Pennsylvania State University) for 16S sequencing using the Illumina MiSeq platform. De-multiplexed sequencing reads were processed through the Mothur software package41. Reads were trimmed at 320 bp and subsequently aligned to the Silva reference genome and chimaeras removed with the Uchime software package42. Reads were classified using the Ribosome Database Project classifier set to 75% and used to compile a phylogenetic tree on Mothur with clearcut. Classified reads were normalized to total reads to yield relative taxonomic abundance.
Whole genome pathway metagenomic analysis
Microbial DNA was isolated from cecal contents using ENZA stool DNA kit. Whole genome sequencing was performed using the Illumina HiSeq 2500 platform within the Pennsylvania State University Genomics core facility. Raw sequencing reads, averaging 18 × 106 reads/sample, were processed through the HUMAnN2 v0.99 (http://huttenhower.sph.harvard.edu/humaan2/manual#markdown-header-workflows)43 pipeline using default parameters to generate gene family, pathway abundance and pathway coverage outputs. Pathway abundance and coverage outputs were merged to identify pathways with greater than/equal to 50% gene family coverage within a given pathway. Pathways with greater than/equal to 50% coverage were further processed through linear discriminate analysis (LDA) effective size bio-discovery algorithm (LEfSe) (http://huttenhower.org/galaxy). Shannon diversity indices were calculated using the formula 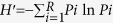 , where Pi represents the abundance of ith pathway as a fraction of the sum of all pathway abundance.
, where Pi represents the abundance of ith pathway as a fraction of the sum of all pathway abundance.
Scanning electron microscopy
Scanning electron microscopy was performed using a JSM 5400 SEM (JEOL) microscope (The Pennsylvania State University). Briefly, ilea were excised, flushed with sterile PBS and cut longitudinally. 5 mm2 sections were mounted on needles and fixed in 2% gluteraldehyde overnight at 4 °C. Samples were washed three times with 0.1 M sodium cacodylate buffer, pH 7.4, and then post fixed with 1% osmium tetroxide in 0.1 M sodium cacodylate buffer. The samples were washed and dehydrated through a graded ethanol series, then critical point dried, mounted, and coated with 10 nm gold palladium alloy. Images were acquired on a JSM 5400 SEM (JEOL).
Quantitative PCR expression analysis
Mice were euthanized through carbon dioxide asphyxiation and terminal ilea (1 cm distal to cecal junction) or whole cecum excised and immediately frozen in liquid nitrogen and stored at −80 °C. Tissues were homogenized in 1 ml Tri Reagent (Sigma, St. Louis, MO.) together with 10–20 1 mm zirconia/silica beads (BioSpec Products, Bartletsville, OK) using a Bertin Precellys 24 homogenizer (VWR, Radnor, PA.) and total RNA isolated following manufacturers directions. 2 μg RNA were utilized as template for cDNA synthesis using the High Capacity cDNA Archive kit (Applied Biosystems, Foster City, CA.) following manufacturers directions. cDNA were diluted 1:10 in nuclease-free water and 6 μl used as a template for quantitative PCR. 20 μl reactions comprised template, 9 μl PerfeCTa SYBR mastermix (Quanta Biosciences, Gaithersburg, MD), 2 μl 3 nM forward primer, 2 μl 3 nM reverse primer and 1 μl 10 mg/ml BSA. Reactions were performed using the cycling conditions (95 °C, 3 min; 95 °C, 30 sec, target-specific annealing °C, 72 °C, 45 sec for 40 cycles; 72 °C, 5 min) on a CFX Connect platform (Biorad). Sequences of primers are described in Supplemental Material. Melt curve analysis of amplicons identified single melt peaks.
ELISA
Mice were euthanized through carbon dioxide asphyxiation and terminal ilea (1 cm distal to cecal junction) excised and immediately frozen in liquid nitrogen and stored (−80 °C). Tissues were homogenized in PBS supplemented with complete mini protease inhibitor cocktail (Roche), followed by centrifugation (10,000 × g for 10 min at 4 °C). Supernatants were transferred to fresh tubes, normalized to 400 μg/ml protein and stored at −80 °C. Samples were shipped and analyzed (Eve Technologies, Calgary, Canada).
1H-NMR cecal metabolite analysis
1H-NMR analyses were performed as previously described5. Briefly, cecal content samples (~50 mg) were subjected to three consecutive freeze-thaws and directly extracted by precooled phosphate buffer with homogenization using the Precellys Tissue Homogenizer. After extraction, 550 μL of each extract was centrifuged and then transferred to 5 mm NMR tubes. Cecal extracts were recorded at 298 K on a Bruker Avance III 600 MHz spectrometer (Bruker Biospin, Germany) operating at 600.08 MHz for 1H, equipped with a Bruker inverse cryogenic probe.
Cecal microbiota transfer
Female Ahr−/+ and Ahr−/− littermates were derived from C57BL6/J × (B6.129-Ahrtm1Bra/J) matings and cohoused for 8 weeks prior to genotypic segregation. Following 14 days of segregation mice were euthanized and cecal contents extracted directly into anaerobic liquid dental medium (Anaerobe Systems, Morgan Hill, CA.). Cecal contents from 4 mice were pooled for each genotype to eliminate inter-animal variation in microbial composition. Pooled cecal contents were administered by gavage to 8 week-old germ-free C57BL6/J mice housed in isolation chambers within the Pennsylvania State University germ free facility. Mice were maintained within the germ free facility for 5 days under a standard 12 h light/dark cycle (light 0600–1800 h eastern standard time) with ad libitum access to sterile standard animal chow and water prior to euthanasia and sample collection.
Statistical analysis
Where indicated, two-tailed unpaired parametric Students’ t-test was performed. Correlation analyses were performed using Pearson’s correlation coefficient (r) with a confidence interval of 95% and a two-tailed significance test. Significance thresholds of *p < 0.05, **p < 0.01 and ***p < 0.001 were applied. Statistical analyses and graphing was performed using Graphpad Prism v6.
Additional Information
How to cite this article: Murray, I. A. et al. Expression of the aryl hydrocarbon receptor contributes to the establishment of intestinal microbial community structure in mice. Sci. Rep. 6, 33969; doi: 10.1038/srep33969 (2016).
Supplementary Material
Acknowledgments
The authors would like to thank the following: Dr. Christopher Bradfield (University of Wisconsin, Madison, WI.) for providing Ahr−/− mice, Pennsylvania State University Genomics Core Facility for assistance with 16S rDNA gene sequencing and metagenomics, Microscopy facility for assistance with scanning electron microscopy the Metabolomics Core Facility for NMR analysis, which are part of the Pennsylvania State University Huck Intstitutes of the Life Sciences. We also thank Marcia Perdew for editorial assistance. This project was supported by Agriculture and Food Research Initiative Competitive Grant # 2014-06624 from the USDA National Institute of Food and Agriculture [G.H.P.]. This work was also supported by the National Institutes of Health and National Institute of Environmental Health Sciences [Grant ES004869, ES019964] [G.H.P.]. Additional support was provided by the National Institutes of Health and National Institute of Environmental Health Sciences [Grant ES022186] [A.D.P.].
Footnotes
Author Contributions I.A.M., R.G.N., L.Z., A.D.P. and G.H.P. designed research; I.A.M., R.G.N., L.Z. performed the research; I.A.M., L.Z. and A.D.P. contributed new reagents/analytic tools; I.A.M., R.G.N., L.Z., A.D.P. and G.H.P. analyzed data; I.A.M. and G.H.P. prepared the manuscript.
References
- Carmody R. N. et al. Diet dominates host genotype in shaping the murine gut microbiota. Cell host & microbe 17, 72–84, doi: 10.1016/j.chom.2014.11.010 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- David L. A. et al. Diet rapidly and reproducibly alters the human gut microbiome. Nature 505, 559–563, doi: 10.1038/nature12820 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin U. H. et al. Microbiome-derived tryptophan metabolites and their aryl hydrocarbon receptor-dependent agonist and antagonist activities. Molecular pharmacology 85, 777–788, doi: 10.1124/mol.113.091165 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonnenburg E. D. et al. Diet-induced extinctions in the gut microbiota compound over generations. Nature 529, 212–215, doi: 10.1038/nature16504 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L. et al. Persistent Organic Pollutants Modify Gut Microbiota-Host Metabolic Homeostasis in Mice Through Aryl Hydrocarbon Receptor Activation. Environmental health perspectives 123, 679–688, doi: 10.1289/ehp.1409055 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lichtman J. S. et al. Host-Microbiota Interactions in the Pathogenesis of Antibiotic-Associated Diseases. Cell reports 14, 1049–1061, doi: 10.1016/j.celrep.2016.01.009 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Mahony S. M. et al. Early life stress alters behavior, immunity, and microbiota in rats: implications for irritable bowel syndrome and psychiatric illnesses. Biological psychiatry 65, 263–267, doi: 10.1016/j.biopsych.2008.06.026 (2009). [DOI] [PubMed] [Google Scholar]
- Flores G. E. et al. Temporal variability is a personalized feature of the human microbiome. Genome biology 15, 531, doi: 10.1186/s13059-014-0531-y (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez de Aguero M. et al. The maternal microbiota drives early postnatal innate immune development. Science 351, 1296–1302, doi: 10.1126/science.aad2571 (2016). [DOI] [PubMed] [Google Scholar]
- Kiss E. A. et al. Natural aryl hydrocarbon receptor ligands control organogenesis of intestinal lymphoid follicles. Science 334, 1561–1565, doi: 10.1126/science.1214914 (2011). [DOI] [PubMed] [Google Scholar]
- Hubbard T. D. et al. Adaptation of the human aryl hydrocarbon receptor to sense microbiota-derived indoles. Scientific reports 5, 12689, doi: 10.1038/srep12689 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubbard T. D., Murray I. A. & Perdew G. H. Indole and Tryptophan Metabolism: Endogenous and Dietary Routes to Ah Receptor Activation. Drug metabolism and disposition: the biological fate of chemicals 43, 1522–1535, doi: 10.1124/dmd.115.064246 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esser C. & Rannug A. The aryl hydrocarbon receptor in barrier organ physiology, immunology, and toxicology. Pharmacological reviews 67, 259–279, doi: 10.1124/pr.114.009001 (2015). [DOI] [PubMed] [Google Scholar]
- Monteleone I. et al. Aryl hydrocarbon receptor-induced signals up-regulate IL-22 production and inhibit inflammation in the gastrointestinal tract. Gastroenterology 141, 237–248, 248 e231, doi: 10.1053/j.gastro.2011.04.007 (2011). [DOI] [PubMed] [Google Scholar]
- Li Y. et al. Exogenous stimuli maintain intraepithelial lymphocytes via aryl hydrocarbon receptor activation. Cell 147, 629–640, doi: 10.1016/j.cell.2011.09.025 (2011). [DOI] [PubMed] [Google Scholar]
- Qiu J. et al. Group 3 innate lymphoid cells inhibit T-cell-mediated intestinal inflammation through aryl hydrocarbon receptor signaling and regulation of microflora. Immunity 39, 386–399, doi: 10.1016/j.immuni.2013.08.002 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quintana F. J. et al. Control of T(reg) and T(H)17 cell differentiation by the aryl hydrocarbon receptor. Nature 453, 65–71, doi: 10.1038/nature06880 (2008). [DOI] [PubMed] [Google Scholar]
- Schmidt J. V., Su G. H., Reddy J. K., Simon M. C. & Bradfield C. A. Characterization of a murine Ahr null allele: involvement of the Ah receptor in hepatic growth and development. Proceedings of the National Academy of Sciences of the United States of America 93, 6731–6736 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maurice C. F., Haiser H. J. & Turnbaugh P. J. Xenobiotics shape the physiology and gene expression of the active human gut microbiome. Cell 152, 39–50, doi: 10.1016/j.cell.2012.10.052 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schloss P. D., Gevers D. & Westcott S. L. Reducing the effects of PCR amplification and sequencing artifacts on 16S rRNA-based studies. PloS one 6, e27310, doi: 10.1371/journal.pone.0027310 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanov I. I. et al. Induction of intestinal Th17 cells by segmented filamentous bacteria. Cell 139, 485–498, doi: 10.1016/j.cell.2009.09.033 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stockinger B., Hirota K., Duarte J. & Veldhoen M. External influences on the immune system via activation of the aryl hydrocarbon receptor. Seminars in immunology 23, 99–105, doi: 10.1016/j.smim.2011.01.008 (2011). [DOI] [PubMed] [Google Scholar]
- Derrien M., Vaughan E. E., Plugge C. M. & de Vos W. M. Akkermansia muciniphila gen. nov., sp. nov., a human intestinal mucin-degrading bacterium. International journal of systematic and evolutionary microbiology 54, 1469–1476, doi: 10.1099/ijs.0.02873-0 (2004). [DOI] [PubMed] [Google Scholar]
- Derrien M., Collado M. C., Ben-Amor K., Salminen S. & de Vos W. M. The Mucin degrader Akkermansia muciniphila is an abundant resident of the human intestinal tract. Applied and environmental microbiology 74, 1646–1648, doi: 10.1128/AEM.01226-07 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caputo A. et al. Whole-genome assembly of Akkermansia muciniphila sequenced directly from human stool. Biology direct 10, 5, doi: 10.1186/s13062-015-0041-1 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Passel M. W. et al. The genome of Akkermansia muciniphila, a dedicated intestinal mucin degrader, and its use in exploring intestinal metagenomes. PloS one 6, e16876, doi: 10.1371/journal.pone.0016876 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dao M. C. et al. Akkermansia muciniphila and improved metabolic health during a dietary intervention in obesity: relationship with gut microbiome richness and ecology. Gut 65, 426–436, doi: 10.1136/gutjnl-2014-308778 (2016). [DOI] [PubMed] [Google Scholar]
- Derrien M., Belzer C. & de Vos W. M. Akkermansia muciniphila and its role in regulating host functions. Microbial pathogenesis, doi: 10.1016/j.micpath.2016.02.005 (2016). [DOI] [PubMed] [Google Scholar]
- Dingemanse C. et al. Akkermansia muciniphila and Helicobacter typhlonius modulate intestinal tumor development in mice. Carcinogenesis 36, 1388–1396, doi: 10.1093/carcin/bgv120 (2015). [DOI] [PubMed] [Google Scholar]
- Roopchand D. E. et al. Dietary Polyphenols Promote Growth of the Gut Bacterium Akkermansia muciniphila and Attenuate High-Fat Diet-Induced Metabolic Syndrome. Diabetes 64, 2847–2858, doi: 10.2337/db14-1916 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu C. X. et al. Aryl hydrocarbon receptor deficiency protects mice from diet-induced adiposity and metabolic disorders through increased energy expenditure. International journal of obesity 39, 1300–1309, doi: 10.1038/ijo.2015.63 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canfora E. E., Jocken J. W. & Blaak E. E. Short-chain fatty acids in control of body weight and insulin sensitivity. Nature reviews. Endocrinology 11, 577–591, doi: 10.1038/nrendo.2015.128 (2015). [DOI] [PubMed] [Google Scholar]
- DiNatale B. C. et al. Kynurenic acid is a potent endogenous aryl hydrocarbon receptor ligand that synergistically induces interleukin-6 in the presence of inflammatory signaling. Toxicological sciences: an official journal of the Society of Toxicology 115, 89–97, doi: 10.1093/toxsci/kfq024 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lahoti T. S. et al. Aryl Hydrocarbon Receptor Activation Synergistically Induces Lipopolysaccharide-Mediated Expression of Proinflammatory Chemokine (c-c motif) Ligand 20. Toxicological sciences: an official journal of the Society of Toxicology 148, 229–240, doi: 10.1093/toxsci/kfv178 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lahoti T. S. et al. Aryl hydrocarbon receptor antagonism mitigates cytokine-mediated inflammatory signalling in primary human fibroblast-like synoviocytes. Annals of the rheumatic diseases 72, 1708–1716, doi: 10.1136/annrheumdis-2012-202639 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh N. P. et al. Activation of aryl hydrocarbon receptor (AhR) leads to reciprocal epigenetic regulation of FoxP3 and IL-17 expression and amelioration of experimental colitis. PloS one 6, e23522, doi: 10.1371/journal.pone.0023522 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takamura T. et al. Lactobacillus bulgaricus OLL1181 activates the aryl hydrocarbon receptor pathway and inhibits colitis. Immunology and cell biology 89, 817–822, doi: 10.1038/icb.2010.165 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reigstad C. S., Lunden G. O., Felin J. & Backhed F. Regulation of serum amyloid A3 (SAA3) in mouse colonic epithelium and adipose tissue by the intestinal microbiota. PloS one 4, e5842, doi: 10.1371/journal.pone.0005842 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deatherage Kaiser B. L. et al. A Multi-Omic View of Host-Pathogen-Commensal Interplay in -Mediated Intestinal Infection. PloS one 8, e67155, doi: 10.1371/journal.pone.0067155 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atarashi K. et al. Th17 Cell Induction by Adhesion of Microbes to Intestinal Epithelial Cells. Cell 163, 367–380, doi: 10.1016/j.cell.2015.08.058 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schloss P. D. et al. Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Applied and environmental microbiology 75, 7537–7541, doi: 10.1128/AEM.01541-09 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar R. C., Haas B. J., Clemente J. C., Quince C. & Knight R. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 27, 2194–2200, doi: 10.1093/bioinformatics/btr381 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abubucker S. et al. Metabolic reconstruction for metagenomic data and its application to the human microbiome. PLoS computational biology 8, e1002358, doi: 10.1371/journal.pcbi.1002358 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.