

Abstract
Advances in pharmacogenomic research and increasing industry interest in personalized medicine have important implications for the way that orphan drug policies are interpreted and applied. Concerns have been raised about the potential impact of pharmacogenomics and new genomic technologies on our understanding of how disease categories are delineated, and subsequently, how the concept of rare disease should be defined for the purposes of orphan drug policies. This article considers whether orphan drug legislation can be drafted in a way that will maximize benefits and minimize concerns relating to the impact of pharmacogenomics on orphan drug research and development. After reviewing the issues that may arise at the intersection of orphan drug policies and pharmacogenomics, this article will discuss the potential impact of pharmacogenomics at two critical points: orphan designation and approval of the drug product. At each of these points, the relevant aspects of current US orphan drug legislation are examined, focusing on the extent to which recent amendments may address concerns that have been raised previously. This analysis will then provide the foundation for a critical review and recommendations regarding the proposed new Canadian orphan drug framework.
Keywords: pharmaceutical policy, pharmacogenomics, orphan drug policy, rare diseases, Food and Drug Administration
INTRODUCTION
During the late 1970s and early 1980s, the lack of commercial development for drugs that treat rare diseases became an important political issue in the US.1 These so-called ‘orphan drugs’ were largely neglected by the pharmaceutical industry because they represented small markets that were unlikely to be profitable.2 The lobbying and public awareness efforts of a number of rare disease patient organizations in the US eventually culminated in the passage of the Orphan Drug Act of 19833 (ODA), which provides industry with support and incentives to develop orphan drugs, defined as drugs intended for use in treating a condition that affects less than 200,000 persons in the US. The substantial increase in the development of drugs for rare diseases over the past three decades is often directly attributed to the passage of the ODA4, and the Act is widely considered to be a success.5 Since the ODA was passed in 1983, more than 400 orphan drugs have been developed and marketed in the US,6 which suggests that the incentives are having an effect.7 Moreover, the last 10–15 years have been the most successful period of development for orphan drugs.8 According to the FDA, nearly 200 orphan drugs enter development each year and approximately one third of new drugs approved by the FDA are for the treatment of rare diseases.9 ODA incentives are also credited with contributing to ‘breakthrough innovation’ that provides advantages over previously available therapies.10
A concurrent trend that is contributing to the shift toward niche market development is recent advances in new genomic technologies that are now making ‘personalized medicine’ a reality. In 2003, the completion of the Human Genome Project—the international effort to map the entire human genome—‘laid the foundation for the development of new health care technologies and therapies, including genetic tests to assist in the diagnosis and prevention of disease and drug therapies that are tailored to the genetic characteristics of individual patients’.11 Pharmacogenomics, the study of the influence that genetic factors have on drug response,12 has emerged from genomics-related research and the development of new diagnostic approaches based on biomarkers.13 There is increasing interest in the pharmaceutical sector toward pairing pharmaceutical products with diagnostic tests that can stratify broader disease categories into rarer disease genotypes.14 Significantly, a growing number of products in clinical development now rely on a clinical biomarker,15 which suggests the mounting importance of pharmacogenomic-based drug development.
Advances in pharmacogenomic research and increasing industry interest in personalized medicine have important implications for the way that orphan drug policies are interpreted and applied. In the US, the Office of Orphan Products Development (OOPD) has indicated that pharmacogenomic products are treated the same as any other orphan drug submissions. However, concerns have been raised regarding orphan drug legislation generally and the impact of pharmacogenomics in this context in particular.16 Some express concern that orphan drug incentives are not adequately targeted to the diseases with greatest unmet medical needs and that the ODA has favored the development of treatments for ‘diseases that can, through ‘omics data and technologies, be recast as rare, or belong to the larger, more lucrative therapeutic class of oncology products’.17 Moreover, there are concerns that the ODA does not adequately distinguish between ‘true orphan drugs’ and ‘“Trojan” applicants that seek to co-opt the benefits for drugs that should not qualify as orphans’.18 However, in 2013, the FDA made a number of important amendments to the ODA Regulations that purport to address many of the challenges raised by the evolving drug development environment, including advances in pharmacogenomics.
The ODA has served as a model for legislation in a number of other jurisdictions, including Europe, Japan, and Australia. Although the legal framework implemented in these countries is similar, the definition of an orphan disease, the criteria which must be satisfied to obtain a designation of orphan status, the incentives provided to encourage the development of orphan drugs and the authorization process for orphan drugs, varies from jurisdiction to jurisdiction. Although for years the Canadian Government denied the need for the country to develop its own orphan drug policy, the government has recently reversed its policy and in December 2012, released a draft orphan drug policy for discussion. The release of the 2013 amendments to the ODA Regulations provides an opportunity to examine what lessons Canada can learn from the FDA's experience in drafting its own orphan drug policy, or indeed, whether a Canadian orphan drug policy is even necessary given the increasingly lucrative nature of niche markets.
This article considers whether orphan drug legislation can be drafted in a way that will maximize benefits and minimize concerns relating to the impact of pharmacogenomics on orphan drug research and development. After reviewing the issues that may arise at the intersection of orphan drug policies and pharmacogenomics, this article will discuss the potential impact of pharmacogenomics at two critical points: orphan designation and approval of the drug product. At each of these points, the relevant aspects of current US orphan drug legislation are examined, focusing on the extent to which recent amendments may address concerns that have been raised previously. This analysis will then provide the foundation for a critical review and recommendations regarding the proposed new Canadian orphan drug framework.
Issues at the Intersection of Orphan Drug Policies and Pharmacogenomics
In recent years, many concerns have been raised about the potential impact of pharmacogenomics and new genomic technologies on our understanding of how disease categories are delineated, and subsequently, how the concept of rare disease should be defined for the purposes of orphan drug policies. The connections between orphan drug development and pharmacogenomics have been widely recognized.19 It is estimated that over 80% of rare diseases are genetically-based,20 so it makes sense that pharmacogenomics could play an important role in the discovery and development of new treatments for rare disease. A 2013 report by Thomson Reuters suggests that the tremendous growth in orphan drug development over the past decade coincides with the increasing focus on personalized medicine, and that orphan disease markets will ‘propel the evolution of [personalized] medicine’.21 As argued by Haffner and colleagues, ‘[i]n an environment in which medicine is increasingly adapted to the needs of patients, the incentives of the Orphan Drug Act could arguably take on even greater importance’.22 The Obama Administration's announcement in January 2015 of a 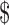 215 million investment to support ‘precision medicine’—another term often used to describe ‘personalized’ approaches to medicine that ‘take into account individual differences in people's genes, environments, and lifestyle’23—is yet another indication of the burgeoning importance of this area of research and development. Many pharmacogenomic drugs have already qualified for orphan drug status under the ODA,24 although at present these drugs represent only a small fraction of the total number of products that have received orphan drug designation.
215 million investment to support ‘precision medicine’—another term often used to describe ‘personalized’ approaches to medicine that ‘take into account individual differences in people's genes, environments, and lifestyle’23—is yet another indication of the burgeoning importance of this area of research and development. Many pharmacogenomic drugs have already qualified for orphan drug status under the ODA,24 although at present these drugs represent only a small fraction of the total number of products that have received orphan drug designation.
In certain cases, there may be a legitimate need to incentivize research into pharmacogenomic treatments that may only be effective in a particular subset of patients with a particular genetic biomarker. The Nuffield Council on Bioethics, for example, highlights that stratifying more common diseases into rarer disease genotypes may result in some patient subsets being so small that developing specific medicines targeting these groups may not be financially viable for drug developers.25 In such cases, orphan drug policies may provide the necessary incentives to encourage pharmaceutical companies to develop medicines for these narrow populations,26 perhaps even allowing drugs that would have otherwise failed to be targeted to a smaller subpopulation in which the drug is more likely to be safe and/or effective. On the other hand, there is concern that ‘[t]he nature of pharmacogenomics drugs may allow some pharmaceutical companies to game the system and abuse the ODA's built-in incentives for drug development’.27
The ODA provides incentives to manufacturers at two stages. First, in the development phase, sponsors can apply for an orphan drug designation, which gives them access to a variety of support and incentive measures, including a tax credit of 50% for the costs of clinical research, access to the OODP's clinical research grants program, a waiver of FDA user fees, and development and regulatory assistance.28 Second, if the drug is then approved, the manufacturer is granted a seven-year period of market exclusivity for the orphan indication(s) for which the drug is approved. This means that the FDA will not approve another drug for the same indication(s) during the exclusivity period, unless the holder of the exclusive license consents or cannot supply sufficient quantities of the drug,29 or the other product is shown to be clinically superior (and therefore not the ‘same’ drug).30 The exclusivity applies only to the approved orphan indication(s) and does not prevent the same drug from being designated or approved for a different use.31
Pharmacogenomics can impact the way that the ODA operates at a number of points in the product lifecycle. First, the use of pharmacogenomics to identify patient subsets can impact the way that disease categories are defined, and thus the size of the target population for the purposes of orphan designation. As the science advances, ‘the nomenclature and classification of disease is becoming increasingly complex’32 and consequently, the number of potential orphan diseases appears to be on the rise.33 Herder argues that ‘new insights from genomics and epigenomics are rendering the boundary between common and rare disease increasingly mutable, potentially exploding the scope of legislated definitions of orphan disease’.34 However, orphan designation is only the first step toward market approval for an orphan drug product, and many orphan designated drugs never reach the market or are not ultimately approved for the orphan indication.
Second, pharmacogenomics can have an impact on the way that orphan exclusive approval is granted within the rare disease or condition, or orphan subset, for which the designation was given. Pharmacogenomics can contribute to the narrowness of approved indications because stratification of the disease may increase the specificity with which the approved indication is defined, and may lead to multiple, narrow approvals within a single orphan designated disease or subset. Further, pharmacogenomics can assist in drug ‘repurposing’ efforts—that is, ‘the discovery of new useful activity in an older clinically used drug’35—allowing some drugs to achieve multiple orphan drug designations and approved indications. In these cases, orphan designation and exclusive approval may add to the profitability of a drug that has already been approved and widely marketed for other uses. The ODA thus provides an incentive for sponsors to invest in studying potential new uses of a drug for rare diseases.
PHARMACOGENOMICS AND ORPHAN DESIGNATION
One concern that has long plagued the ODA is the potential for drug developers to exploit the benefits of the Act by artificially subdividing diseases to create subgroups of patients that fall under the orphan drug prevalence threshold36—a practice referred to as ‘salami slicing’. A classic example of ‘salami slicing’ is the drug Epogen (epoetin alpha), which received an orphan designation from the FDA in 1986 for the treatment of anemia associated with end-stage renal disease.37 After the drug was approved by the FDA in 1989, the drug became widely prescribed for a wide variety of patients with anemia, not only anemia caused by end-stage renal failure. Consequently, through off-label use—prescribing a drug for indications not formally approved by drug regulators—in patients without end-stage renal disease, Epogen became a blockbuster drug and generated billions of dollars in revenue for its manufacturer.38
Loughnot argues that pharmacogenomics could potentially take ‘salami slicing to a new level’39 by allowing drug developers to ‘genetically subdivide diseases that affect a large portion of the population into groups small enough to qualify for orphan drug status’.40 The potential to stratify broader disease categories based on biomarker status significantly increases the potential number of orphan subsets that may be defined for the purpose of orphan designation.41 The FDA acknowledges that ‘what is considered a distinct “disease or condition” may change over time as scientific understanding evolves, which would affect prevalence determinations’.42 Maher and Haffner point out that ‘[w]hether such increasingly precise disease descriptions constitute separate diseases would normally appear to be an academic exercise, unless, as is now more often the case, a specific therapy targeting a specific mutation is developed’.43 That is, the very development of a new treatment can play an integral role in shaping the classification of a new rare disease or condition, or orphan subset.
The potential for ‘salami slicing’ is not a new concern, having been previously considered in earlier revisions of the US orphan drug legislation. In discussing the criteria for orphan drug designation, the 1991 notice of proposed rulemaking stated that a subset of a common disease or condition ‘would qualify for designation only if the subset is medically plausible’ and that ‘arbitrary’ subsets would be unacceptable.44 However, the 1992 regulations offered little guidance on the meaning of this rather ambiguous phrase, providing only that the concept of ‘medically plausible’ is interpreted flexibly depending on the specific facts of each case.45 Moreover, a request to further define the term ‘arbitrary’ in this context was rejected on the basis that ‘every FDA decision on arbitrariness would necessarily be highly fact dependent’.46 According to Herder, until recently, the addition in 1992 of the requirement that the disease in question be considered ‘medically plausible’ was the only relevant constraint that has been adopted by the FDA to distinguish between ‘rare diseases that have long been identified as such’ and ‘those which have been reclassified as rare by virtue of new scientific insights’.47
The question of orphan subsets was addressed again, in more detail, in the most recent regulatory amendments. In the 2011 notice of proposed rulemaking, the FDA acknowledged that ‘[b]ecause the term “medically plausible” has not been further clarified through regulations or guidance, it has been misinterpreted to mean any medically recognizable or any clinically distinguishable subset of persons with a particular disease or condition’, and that inappropriate application of the ‘medically plausible’ concept could result in artificially narrow subsets.48 Although it can certainly be beneficial for manufacturers to develop more effective treatments for some subsets of common diseases and conditions, it could be said that this departs from the original intent of the ODA to target very rare diseases that currently lack any effective treatments.49 Indeed, the FDA believes such an interpretation would frustrate the intent of the ODA and divert resources away from research and development of true orphan drugs by allowing ‘a non-rare disease or condition to be artificially subdivided into smaller groups for establishing subsets that are under the prevalence limit for designation’.50 The FDA therefore proposed to amend the regulation ‘to remove the term “medically plausible”… and instead provide a description of how an appropriate subset may be identified for the purpose of orphan drug designation’.51 The Final Rule adopted in 2013 added a definition of ‘orphan subset’ to clarify that an appropriate subset may exist where ‘use of the drug in a subset of persons with a non-rare disease or condition may be appropriate but use of the drug outside of that subset (in the remaining persons with the non-rare disease or condition) would be inappropriate owing to some property(ies) of the drug’.52
The FDA insists that the 2013 amendments are consistent with the agency's longstanding approach to identifying ‘medically plausible’ subsets.53 Some may continue to question how effectively the FDA can distinguish between ‘true’ and ‘artificial’ subsets of disease, particularly since evaluation of orphan subsets is complex and is often based on uncertain evidence.54 Herder further notes that ‘[p]olicing the artificial creation of orphan diseases may be difficult given the potential information asymmetries between the FDA and the companies it regulates’.55 If there were significant concerns about the FDA's previous approach, the fact that it maintains that the new language is consistent with this approach may undermine confidence in the potential for the new language to better distinguish between ‘real’ and ‘artificial’ groups. However, an analysis of the revised wording, along with some examples of subsets that have been accepted or rejected by the FDA, suggests that these concerns may have been mitigated somewhat.
Defining Orphan Subsets in the Pharmacogenomic Context
In the 2013 Final Rule, although the FDA accepted that ‘biomarker-based and other targeted treatments’ could be used to define subsets, it expressly rejected the proposition ‘that an orphan subset can exist whenever there is a basis for using the drug in the subset of interest, regardless of whether the drug can also be used in the remaining persons with the disease or condition’.56 At the core of the analysis is consideration of the ‘property or properties of the drug that preclude its use in the remaining persons with the non-rare disease or condition, outside of the orphan subset [emphasis added]’.57 According to the stated approach of the FDA, sponsors have to not only demonstrate ‘why this subset … should be targeted for an orphan drug treatment’,58 but also why the drug cannot also be used outside the subset. The 2013 amendments provide specific guidance as to what factors may or may not inform whether an appropriate orphan subset exists.
Many aspects of this guidance are directly relevant to the pharmacogenomic context. This is likely due to the fact that that much of the new wave of concern around ‘salami slicing’ has arisen from the impact of new genomic technologies on the classification of rare diseases and conditions. First, the 2013 Final Rule provides that where a drug's mechanism of action suggests that the drug would not have significant activity outside of a subset of patients with a particular type of tumor or biomarker, this may establish an orphan subset.59 This is directly relevant to pharmacogenomic therapies which target specific biomarkers or tumor mutations. Second, the Final Rule indicates that where previous clinical experience with the drug indicates that the drug does not demonstrate significant activity in a particular subset of patients, this may inform whether an acceptable orphan subset exists.60 Pharmacogenomic research can assist in the identification of patient subsets that are more likely to respond to drug therapy. Third, the Final Rule lists the drug's toxicity profile as a relevant factor in defining orphan subsets. For example, patients with a particular non-rare disease or condition who ‘are refractory to, or intolerant of, other less toxic drugs’ could be a subset for the purposes of a more toxic drug, whereas other patients with the same disease or condition would not be appropriate candidates for that drug.61 Pharmacogenomic research is often used to identify those patients who may be at a higher risk of adverse drug reactions due to their genetic profile.62
The FDA may grant multiple orphan designations for a particular disease or orphan subset—indeed, this has become common practice.63 However, it is worth noting that the FDA's acceptance of an orphan subset for one drug does not mean that the same subset will be accepted for subsequent applicants. The FDA states that the prevalence estimate may be narrowed owing to one or more properties of the drug that allow for the existence of an orphan subset [emphasis added]’.64 That is, although a particular orphan subset may be designated for a given drug product, this same orphan subset may be rejected for another drug because the appropriateness of the subset is evaluated separately based on the specifics of each drug.
Similarly, the factors that are not sufficient to define an orphan subset are informative in the pharmacogenomic context. The 2013 Final Rule also notes that clinical trial eligibility, a sponsor's plans to study the drug only for a ‘select indication’, the particular grade or stage of a disease, or a low likelihood of use in a broader population are not sufficient in themselves to establish an orphan subset.65 With pharmacogenomics, biomarkers can be used to prospectively select patient subpopulations—a strategy known as enrichment of the study population—that are more likely to respond to a given drug therapy so that the treatment effect is more likely to be detected.66 In such cases, later-stage studies may only be conducted in patient groups with a particular biomarker status. According to the FDA, restricting clinical trial eligibility to biomarker-positive patients, or only choosing to study the drug in a particular patient subset, is not sufficient to establish an orphan subset. Again, the sponsor is required to show not just that the drug is more effective or less likely to cause adverse effects within the subset population, but also that the difference between the subset and the larger group is sufficient to prevent the drug from being a viable treatment option for patients in the larger group. As such, this approach does show some potential for limiting what could be seen as abuse, particularly since the sponsor bears the burden of showing that drug is inappropriate for use outside of the orphan subset.67
Loughnot suggested in 2005 that ‘the increased precision that pharmacogenomics brings to pharmacology might eventually help provide the FDA with the ability to define “medically plausible”’.68 Ultimately, as the science advances, pharmacogenomics will likely increase not only the number of potential rare diseases and orphan subsets, but also the precision with which these diseases and subsets may be delineated. As noted by Haffner and colleagues, ‘advances in genomics and proteomics have led to increasingly precise disease definitions’.69 As advances in pharmacogenomics increase the ability of researchers to measure non-response, this may raise the bar for sponsors who are trying to establish an orphan subset. Maher and Haffner note that ‘a clearer understanding of drug non-response is often revealed when response is stratified’. Consequently, as the lines between response and non-response are more clearly defined, orphan subsets will hopefully become less prone to manipulation.
Current Evidence of Subdivision based on Pharmacogenomics
Concerns about the potential for pharmacogenomics to increase the practice of ‘salami slicing would primarily arise in circumstances where the prevalence of the broader disease category is over 200,000 cases in the US, but the prevalence of the ‘medically plausible’ subtype is less than 200,000 cases. There are a few cases where a pharmacogenomic-based biomarker was likely a determinative factor in bringing an orphan subset below the prevalence threshold. For example, although non-small cell lung cancer accounts for 85%70 of the approximately 400,000 cases of lung cancer in the US71—thus placing the disease well above the orphan designation threshold—in 2010, Xalkori (crizotinib) received an orphan designation for the ‘treatment of ALK-positive, MET-positive, or ROS-positive72 non-small cell lung cancer’. In this case, the biomarker status of the non-small cell lung cancer (ie ALK-positive, MET-positive, or ROS-positive) appears to have been the factor that brought the indication under the prevalence threshold to qualify for orphan drug status.73 As another example, in 2011, there were some 960,000 Americans living with melanoma,74 but in 2010, Zelboraf (vemurafenib) received an orphan designation for the ‘treatment of patients with IIb to Stage IV melanoma positive for the BRAF (v600) mutation’.75 If, as the 2013 amendments suggest, the stage of disease alone is usually not sufficient to define an appropriate orphan subset (see below), then the biomarker selection for the BRAF(v600) mutation appears to be important to bringing the target population below the 200,000 patient prevalence threshold.
In clarifying their longstanding approach to eligibility for orphan subsets, the guidance provided by the FDA in the 2013 amendments may help to decipher the reasons behind their decisions to deny orphan drug designations to some pharmacogenomic drugs in the past. For example, before receiving approval for the breast cancer (specifically, HER2-positive metastatic breast cancer) drug Herceptin (trastuzumab) in September 1998, its manufacturer, Genentech, applied for orphan drug status with the FDA, but the designation was denied. At the time, there was an estimated 165,000 metastatic breast cancer patient in the US, of whom approximately 30%, or 49,500 people, had HER2 overexpressing tumors76—well below the 200,000 cut-off for orphan drug designation. However, the FDA denied Herceptin orphan drug status.77 While the exact reason for the denial was unclear, the OOPD indicated that the most common reasons for refusal is disagreement between drug sponsors and regulatory authorities over how the target population is defined. 78 In 2002, Shah suggested that Herceptin was most likely not approved for orphan designation ‘because the size of the population of HER2 overexpressers was underestimated, as it is overexpressed in cancers other than that of the breast’.79 In particular, it was clear from the success of clinical trials that Herceptin could potentially also be used in the treatment of a range of other possible cancers including bladder, pancreatic, ovarian, colorectal, and prostate.80 However, it seems unlikely that the possibility of treating multiple types of cancer with Herceptin was in fact the reason for the denial since in the 2013 Final Rule, the FDA explicitly states that ‘[a] drug that shows promise in multiple, different rare diseases or conditions may be eligible for multiple designations, one for each disease or condition, because FDA considers the prevalence within each disease or condition’.81 Rather, it is more likely that the reason for the denial was that the stage of the disease (metastatic, or Stage IV breast cancer) was not an acceptable subset to limit the target population since ‘FDA currently considers Stage I breast cancer to be the same “disease or condition” as Stage IV breast cancer when evaluating orphan drug designation requests for products that treat breast cancer’.82 Since breast cancer was estimated to affect nearly three million women in the US in 2011,83 the sponsor presumably failed to demonstrate that the drug would not be effective in a broader subset of patients (ie in HER2-positive breast cancer in other stages).
In most cases, stratification based on genetic biomarkers does not appear to be necessary to bring the target population below the 200,000 patient threshold; pharmacogenomic data is often used to stratify diseases that are already rare enough to be eligible for orphan designation. Indeed, Greenbaum notes that ‘the vast majority of pharmacogenomics drugs fall within the literal definition of an orphan drug’.84 That is, most orphan designations that have been granted for pharmacogenomic drug products are for diseases where the broader disease category already falls below the 200,000 person threshold, such as chronic myelogenous leukemia85, pancreatic cancer86, or acute lymphoblastic leukemia.87 Moreover, biomarker status is only one of many different factors that can be used to subdivide disease categories. Most rare diseases and orphan subsets granted orphan designation are already subdivided based on a wide range of factors such as chronic or acute state, age of the target population (eg adult vs. pediatric), or the underlying cause of disease, just to name a few.
Only once a larger body of examples is available to examine will it be possible to fully assess to what extent the ODA remains open to abuse through ‘salami slicing’. The clarification provided in the 2013 Final Rule does seem to reduce the potential for abuse, and experience to date seem to suggest that instances in which subsets are artificially created to bring products below the orphan drug prevalence threshold will be fairly rare; in most cases, subdivision based on biomarkers may be entirely legitimate. While this is an issue that should continue to be monitored, other jurisdictions, like Canada, can learn from the FDA's recent efforts to define the orphan subset concept in a way that minimizes the potential for abuse.
PHARMACOGENOMICS AND ORPHAN DRUG APPROVAL
Concerns about the potential misappropriation of orphan designation through salami slicing may be tempered by the fact that obtaining an orphan designation is only the first step toward market approval. While many of the benefits under the ODA accrue as soon as an orphan designation is received (namely assistance in clinical trials, the 50% tax credit for clinical trial costs, and access to federal grants), these benefits will ultimately be of little value if they do not lead to market authorization for the indication (or a subset thereof) for which the orphan designation was granted. The seven-year market exclusivity, also known as orphan exclusive approval, is arguably the most important incentive for drug developers seeking orphan designation.88
The bar for obtaining orphan designation is significantly lower than that for obtaining market approval and only a small fraction of orphan designated drugs ever reach the US market.89 As noted by Maher and Haffner, ‘[e]valuation of [a request to consider a subset of a prevalent disease for orphan designation] frequently rests on both incomplete knowledge of disease etiology and uncertain therapeutic mechanism of action, and is both difficult and complex’.90 For example, as of 2012, there had been 2661 successful orphan product designations granted by the FDA, which had led to 408 approved orphan products (representing about 15% of orphan drug designations).91 As with other orphan drugs, only a portion of pharmacogenomic drugs that receive an orphan designation are approved for the US market, though the proportion of designated pharmacogenomic-based drugs ultimately approved appears to be somewhat higher than for other classes of drugs.92
Once a drug that has been granted orphan designation is approved for a rare disease or condition, the market exclusivity provisions in the ODA prevent the FDA from approving ‘such drug for such disease or condition’ for a period of seven years from the date of approval, unless the holder of the first approval consents or cannot supply sufficient quantities of the drug.93 The regulations specify that once a designated drug receives exclusive approval, ‘no approval will be given to a subsequent sponsor of the same drug for the same use or indication for 7 years’ unless one of the exceptions applies.94 The phrase ‘same drug’ is defined by regulation to mean a drug that contains the same active moiety or principal molecular features as a previously approved drug and is intended for the same use.95 However, the regulations specify that a subsequent drug is not the ‘same drug’ if it ‘can be shown to be clinically superior to the first drug’.96 The regulations define a ‘clinically superior’ drug as one that ‘is shown to provide a significant therapeutic advantage over and above that provided by an approved drug’, through greater safety, efficacy, or other ‘major contribution to patient care’.97
A sponsor can obtain an orphan designation for a previously approved drug for the same rare disease or condition if it ‘can present a plausible hypothesis that its drug may be clinically superior to the first drug’,98 and then can receive its own exclusive orphan drug approval if it can demonstrate this clinical superiority.99 The ‘plausible hypothesis’ of clinical superiority standard for orphan designation is easier to establish than the ‘clinical superiority’ standard required for orphan exclusive approval. According to the FDA, ‘development of improved versions of existing drugs … is achieved through liberally granting designation based on a plausible hypothesis of clinical superiority, allowing drugs to benefit from development incentives that flow from designation’.100 Maher and Haffner note that ‘unlike the FDA market approval review, the orphan designation review takes place at an earlier stage of product development, sometimes even prior to any clinical studies having been performed’.101 It is also worth noting that demonstration of clinical superiority at the approval stage need not be on the same basis as the hypothesis presented at the designation stage.102
The current approach to requiring proof of clinical superiority at the approval stage was called into question by the September 2014 decision of the US District Court for the District of Columbia in the case of Depomed Inc. v. US Department of Health and Human Services et al. The case arose from a 2012 complaint launched by the pharmaceutical firm Depomed challenging the FDA's decision to deny orphan drug exclusivity for the drug Gralise.103 Other drugs with the same active ingredient (gabapentin) had previously been approved and marketed for the same indication (post-herpetic neuralgia). Having provided a plausible hypothesis of clinical superiority over these earlier products, Gralise was granted orphan drug designation. However, FDA refused to grant exclusive approval because the sponsor had not proved the clinical superiority of its product. None of the previously approved products had received orphan drug designation, however, so Depomed argued that the clinical superiority requirement should not apply. Looking at the relevant statutory provisions, the District Court found that a plain-language reading of the ODA mandates the FDA to ‘recognize exclusivity for any drug that the FDA has designated [as an orphan drug] and granted marketing approval’.104 It was therefore not open to the FDA to impose the additional requirement of proving clinical superiority as a condition of granting exclusivity. As a result, the FDA was ordered by the District Court to grant orphan drug exclusivity for Gralise ‘without requiring proof of clinical superiority or imposing any additional conditions on Depomed’.105
It is important to note that the District Court's decision expressly acknowledges that FDA can still impose conditions for orphan drug designation because it has been granted the authority in the ODA to make regulations on this issue.106 Therefore, the FDA can still use clinical superiority to determine whether a drug for which orphan drug designation is sought is the same drug as one previously designated and approved. In the Court's view, this should allay FDA's concerns that removing the clinical superiority requirement for exclusive approvals could lead to sponsors ‘evergreening’ or obtaining ‘serial exclusivity’ for their products (an issue discussed in more detail in the next section), contrary to the policy goals of the ODA.107 A sponsor can only obtain orphan drug exclusivity for a product that has been designated as an orphan drug, and the FDA can deny this designation to the sponsor of a drug that is the same as (ie not clinically superior to) a previously approved drug.
Following this decision, in December 2014, the FDA issued a ‘clarification of policy’ in which the Agency stated that the District Court decision was limited to the specific case of Gralise and that as such, the Agency will continue to apply its existing regulations which ‘require the sponsor of a designated drug that is the “same” as a previously approved drug to demonstrate that its drug is “clinically superior” to that drug upon approval in order for the subsequently approved drug to be eligible for orphan drug exclusivity’.108 This seems to be a fairly aggressive position, considering that the District Court's decision questioned the FDA's authority to impose these conditions on exclusivity, and has led to considerable speculation about its implications.109 The issue is unlikely to be definitively resolved unless and until these issues are relitigated in further court challenges or appeals. While considerable uncertainty remains, the specific implications for the issues discussed in this article may be limited, given that FDA's authority to set conditions for orphan drug designation remains undisturbed, and the specific factual context of the Depomed case—where the same drug had been previously approved but not designated as an orphan drug—are quite unusual.110
Incentivizing Reformulation and Repurposing
The pharmaceutical sector is fiercely competitive and brand name drug companies are always seeking ways to squeeze more profits out of drug products, particularly those that are approaching patent expiry or that are no longer under patent protection. A common tactic is to seek new periods of patent protection or market exclusivity by discovering new uses or new target populations for existing drug products—a strategy often referred to as ‘repurposing’. The National Institutes of Health in the US have described repurposing ‘as a key initiative to fight against stagnation in drug development’.111 In particular, repurposing is an important strategy in the development of therapies for rare diseases: ‘The sheer number of unmet medical needs to be found among orphan and rare disease suggests that drug repurposing among existing clinically used drugs may be a major solution to this societal medical need’.112 In addition, drug developers may seek to reformulate or improve upon existing drug products in order to qualify for a new period of market exclusivity. It is worth noting that once the patent and market exclusivity periods have expired on a pharmaceutical product, anyone may seek an orphan designation and orphan exclusive approval for that product.113
Evergreening, Clinical Superiority and Multiple Approvals
A common means of extending the lifecycle of a patented drug product is through drug reformulation where a drug company modifies the characteristics of an existing drug product enough to qualify for a new patent or period of data exclusivity.114 Pharmaceutical companies are often accused of ‘evergreening’ their products by making trivial and needless modifications to patented medicines in order to extend the term of patent protection or exclusivity. The FDA has acknowledged that one of the potential concerns with allowing new periods of orphan exclusivity for an already approved drug is that it ‘could permit inappropriate “evergreening” of exclusive approval periods’ by allowing a sponsor to apply for a new designation (and then exclusive approval) near the end of a previous exclusivity period.115 As noted by the FDA ‘“evergreening” would allow orphan exclusivity to be extended indefinitely for the same drug for the same use without any meaningful benefit to patients, a result at odds with the seven-year exclusivity period provided by the statute’.116 However, reformulation is not considered to be inappropriate if the sponsor can demonstrate clinical superiority;117 a sponsor that improves its own previously approved drug can be eligible for a new exclusivity period if clinical superiority is shown.118 As the FDA notes, the requirement of clinical superiority is intended to encourage ‘the development of potentially safer and more effective orphan drugs—rather than encouraging minor modifications to already approved drugs that confer no meaningful benefit to patients.’119 Indeed, if the intent is to create incentives to improve treatment options, then arguably it shouldn't matter who develops the clinically superior alternative, whether the original sponsor or a competitor. In this way, the ODA may incentivize research into improved versions or new applications of existing drug products for a rare disease or condition.
It is important to note that the scope of the market exclusivity is determined by the approved indications, not by the orphan designation. The FDA generally grants orphan-drug designation for use of a drug in all patients with a rare disease or condition and expects sponsors to seek approval on this basis, but sometimes the approval will be narrower if the data submitted only supports use in a subset of patients or indications.120 In the 2013 amendments, the FDA set out to clarify the scope of market exclusivity by replacing the term ‘subset [of uses]’ with ‘select indication(s) or use(s)’. The regulations now provide that if orphan exclusive approval ‘is limited to only particular indication(s) or uses(s) within the rare disease or condition for which the drug was designated, FDA may later approve the drug for additional indication(s) or uses(s) within the rare disease or condition not protected by the exclusive approval’.121 That is, the sponsor may obtain multiple periods of seven-year market exclusivity for each approved indication or use that falls within the orphan designation. Each new period of market exclusivity will begin to run from the date of approval for the new (ie not previously approved) indication or use122 thus staggering the market exclusivity based on the approval date of each new orphan indication.123
Loughnot expresses concern that pharmacogenomics could contribute to evergreening tactics by ‘help[ing] identify patients who are susceptible to adverse drug reactions’, allowing a sponsor to ‘create significantly better clinical trial results without altering a drug at all’ by including only patients who are less likely to have adverse reactions.124 This tactic of ‘enriching’ clinical trial populations with patients who are most likely to benefit from the drug under study has become common practice in the development of pharmacogenomic products, as well as in other areas of drug development. As noted above, the 2013 amendments make clear that for the purposes of orphan designation, clinical trial eligibility and the decision to only study the drug in a particular patient population are insufficient to establish an acceptable orphan subset.125 Subsequently, at the approval stage, if the issue is the sponsor attempting to ‘evergreen’ an existing orphan exclusive approval on its own product, simply retargeting a drug at a narrower patient population within an already approved orphan indication would likely not be sufficient to obtain a new period of market exclusivity. An already approved drug could be targeted toward an unapproved indication within the orphan designation, since the ODA clearly permits multiple approvals within a designation. However, this would not be ‘evergreening’ per se since the approval would be for a new indication.
It is common for an orphan designated drug to be ultimately approved for a narrower indication than was set out in the designation. For example, the pharmacogenomic-based drug Xalkori (crizotinib) initially received orphan designation for the treatment of ALK-positive, MET-positive, or ROS-positive (three different types of biomarkers) non-small cell lung cancer but has so far only received FDA approval for treatment of ALK-positive non-small cell lung cancer.126 A broader orphan designation widens the potential scope of orphan exclusive approvals that may be obtained under a single designation; if the scope of the orphan designation is too narrow, additional designation will likely need to be obtained.
As noted above in the context of orphan designation, pharmacogenomics may both multiply the number of potential rare diseases and orphan subsets that may be designated and increase the precision with which these diseases and subsets may be defined; ‘[w]ith therapies becoming increasingly guided by a more complete understanding of both genomics and proteomics, the number of potential orphan diseases should be expected to increase’.127 Similarly, at the approval stage, pharmacogenomics may narrow the scope of approved indications within a designated rare disease or subset since the approval may specify increasingly precise conditions of use or target populations. Accordingly, pharmacogenomics may increase the trend toward having multiple orphan approvals within a single orphan designation since pharmacogenomics increases the stratification of disease and the specificity with which disease subtypes and/or subpopulations may be defined. Nonetheless, as discussed in the next section, the potential for off-label prescribing may erode the distinction between approved and unapproved indications within an orphan designation.
Pharmacogenomics and Repurposing
Through subgroup analyses, pharmacogenomics can assist in drug repurposing efforts by identifying new targets for treatment or pinpointing patient subpopulations in which an existing drug may be more effective. As Greenbaum notes, ‘[n]ew technological tools are now being used to determine if there are additional targets of current drugs on the market, or so called “off-targets”’.128 Such repurposing is an example of ‘retrospective’ pharmacogenomic drug development, where sponsors can use data generated in previous clinical trials to identify potential new indications.129 In addition, pharmacogenomic research may be able to ‘rescue’ drugs that may have failed all comer clinical trials by defining a more appropriate patient population and conducting enriched clinical trials; traditional randomized clinical trials may mask treatment efficacy by including participants for whom the drug has poor efficacy.130 Further, pharmacogenomics enables genetic profiling of subpopulations at an increased risk of adverse drug events. The FDA, for example, has acknowledged that if ‘new science enables us to determine that the adverse events are restricted to a small, identifiable segment of the population, public health could be improved by making the drug available to others who could benefit without undue risk’.131 Consequently, pharmacogenomics may even allow drugs that have been withdrawn from the market due to rare but serious adverse events to be reintroduced for a specific subpopulation under more restricted terms of authorization.132
A previously approved drug may receive an orphan designation for an unapproved use, regardless of whether the previous approval was for a rare or non-rare disease or condition.133 Even where a drug was previously approved for a common indication, it can receive orphan designation for a different indication that qualifies as an orphan disease, and manufacturers can be granted market exclusivity for an off-patent drug for orphan indications. The support and incentives provided under the ODA may result in ‘previously discarded treatments being revived’134 or in new orphan applications for already successful drug products. Haffner and colleagues note that orphan exclusive approval under the ODA is important for the development of older drugs135—namely, drugs that are no longer covered by patent protection. Sponsors may be able to maximize the sales potential for existing drugs by obtaining new orphan designations. The FDA has explicitly stated that ‘[a] drug that shows promise in multiple, different rare diseases or conditions may be eligible for multiple designations, one for each disease or condition, because FDA considers the prevalence within each disease or condition’.136 Gleevec (imatinib), for example, has received seven separate orphan designations and orphan exclusive approvals.137 According to a report by Thomson Reuters, about 15% of orphan drugs analyzed in one study had subsequent launches for additional rare diseases.138
The FDA openly encourages drug developers to pursue orphan indications for drugs that have already been approved for more common conditions. The OOPD has created a database of products that have received both orphan status designation for a rare disease and a market authorization for the treatment of more common diseases. This database ‘offers sponsors a useful tool for finding special opportunities to develop niche therapies that are already well-advanced through development’ and thus ‘represent a far “easier lift” to drug developers than beginning with an untested new therapy compound’. 139 Even drugs that have achieved blockbuster sales in broad patient markets may be eligible for orphan designation where that same drug can also be used to treat an orphan condition. Indeed, the FDA has granted orphan designation to over 100 drugs with existing approvals for more common diseases,140 including to some highly successful blockbuster drugs such as Prozac (fluoxetine),141 Viagra (sildenafil citrate),142 and Neurontin (gabapentin).143 However, of these only gabapentin has received market authorization for an orphan indication.
It is problematic when incentives are used in situations where they are not intended or needed—which could be the case where orphan drug policies—intended to encourage sponsors to develop products that would not otherwise be financially viable—are used simply to enhance the profitability of products that would already viable without any incentives. In these situations, concerns of abuse or exploitation may be raised. The ODA does not consider the previous profitability of the drug in determining whether a new orphan application should receive an orphan designation, and subsequently, orphan exclusive approval.144 Rather, the ODA is aimed at encouraging the development of orphan treatments that would otherwise not be developed. Even if a drug had already achieved blockbuster sales for another indication, this does not necessarily translate into making it financially viable to pursue new orphan indications for the drug product since this additional research and development can entail significant cost. Thus, the incentive to investigate and test the drug's potential for a particular indication could be necessary and useful, even if the drug is already profitable. Therefore, on balance, it is legitimate to allow orphan designations for drugs previously approved for other indications, including common diseases. It is quite fair to say that ‘[f]rom the perspective of the patient with a rare disease, whether a drug is also effective in treating a more prevalent disorder is irrelevant’.145 From this perspective, anything that could encourage a sponsor to identify and test the drug as a potential therapy for the patient's condition might be beneficial.
Finally, as noted above, the potential for off-label prescribing may erode the value of the orphan drug exclusivity: although manufacturers are prohibited from marketing drugs for off-label uses, physicians are free to prescribe drugs off-label,146 potentially allowing generic drugs to be prescribed for indications that are protected by orphan exclusivity. Typically, generic drugs must have the same labeling as the innovative drug product to which they are compared in their application for market approval.147 However, where the innovator has patent or exclusivity protection for a particular use or condition, if the generic copies these protected elements in the innovator's labeling, they risk an infringement action.148 However, the generic may seek permission from the FDA to ‘carve out’ the protected language, which would theoretically limit the generic's market to conditions or uses that are not covered by patent or exclusivity rights.149 While a detailed discussion of the carve-out policy is beyond the scope of this article, it is worth noting that these provisions could potentially undermine an innovator's incentive to pursue repurposed orphan applications for existing drug products. As noted by Mahn, innovative drug companies are ‘concerned that the “carve out” rule, coupled with the practice of prescribing and substituting generic drugs “off label”, threatens their ability to recover the large investments needed to discover new uses or to improve the safety or efficacy profiles for old drugs’.150
Proposals for a Canadian Orphan Drug Framework
In the past, suggestions that Canada consider a US-style orphan drug framework had been rejected by Health Canada on the basis that sufficient flexibility and incentives already existed in the Canadian legislation.151 In a 1997 policy statement, Health Canada cited several mechanisms which could be applied to orphan drugs, including tax incentives, fee reductions for drugs with small market potential, and access to unapproved drugs through the Special Access Program. Health Canada concluded that these mechanisms were sufficient because approximately 60% of US-approved orphan drugs were available in Canada.152 They further noted that ‘the number of persons with rare diseases in Canada may not be sufficient to support substantial clinical trial research and development in the area of Orphan Drugs’.153 Currently, Canadians may access orphan drugs through the Health Canada's Special Access Program, by participating in clinical trials, or where the drug has been approved through the regular drug approval process.154
Interestingly, the 1997 policy statement by Health Canada reported that ‘[t]here has not been significant pressure from industry or special interest groups in Canada to develop an Orphan Drug policy’.155 Recently, however, there have been renewed calls for an orphan drug policy in Canada. The Canadian Organization for Rare Disorders, for example, has argued that without an orphan drug policy, manufacturers have no motivation to seek market approval for orphan drugs in Canada and consequently, ‘Canadians with rare disorders run the risk of being among the last in the developed countries to gain access to new medicines, if at all’.156 BIOTECanada, the national association representing the biotechnology industry, also strongly supports the development of a Canadian orphan drug framework, stating that the initiative ‘will help Canada to compete in attracting investment to nurture [orphan drug] products into the marketplace, and see new Canadian solutions for unmet medical needs developed in Canada’.157 Orphan drug policies have also been framed as a means to promote personalized medicine: a decade ago, in a report to the Canadian Government, the External Advisory Committee on Smart Regulation noted that ‘[w]ith the recent developments in pharmacogenomics, and the resulting ability to target treatments for sub-groups of the population, it may be timely to consider implementing a legislative framework to facilitate access to these drugs’.158 According to the Canadian Organization for Rare Disorders, Health Canada's proposed orphan drug framework would be ‘the first step towards Canada taking a leadership position in personalized medicine’.159
Despite its earlier position that there was no need for an orphan drug policy in Canada, in October 2012, the federal government announced its plans to develop an orphan drug framework for the designation, authorization and monitoring of orphan drugs. Soon after in December 2012, Health Canada released an Initial Draft Discussion Document for A Canadian Orphan Drug Framework, which proposed a regulatory framework for orphan drugs, including provisions relating to orphan drug designation, scientific and clinical protocol advice, special market authorization for orphan drugs, and post-market assessment and management. The criteria for orphan designation outlined in the proposed Canadian framework mirror those of the European legislation, with the exception that they do not consider the economic viability of the drug. In particular, the Canadian framework adopts the same prevalence criteria as the European Union legislation in the definition of an ‘orphan drug’: a drug that ‘is intended for the diagnosis, treatment, mitigation or prevention of a life-threatening, seriously debilitating, or serious and chronic disease or condition affecting not more than five in 10 thousand persons in Canada’.160 As is the case in other jurisdictions, Health Canada's proposed legislation contains a series of incentives including priority review for marketing authorization,161 fee reductions for small to medium enterprises, and scientific and clinical protocol advice.162
Currently, the Canadian orphan drug framework is still in the draft discussion phase, and Health Canada has provided only a high-level description of the elements of the proposed framework. As such, it is too early to assess how pharmacogenomics products will be handled under the Canadian orphan drug policy. As the proposed framework is developed and implemented, Canada can learn from the US experience with the ODA and the debate surrounding how rare diseases are categorized and understood in order to maximize the benefits and minimize the potential for abuse of an orphan drug regime. In particular, Health Canada will have to consider how orphan subsets will be defined for the purposes of orphan designation and the terms and conditions for the approval and market exclusivity for orphan drugs.
Orphan Designation
Under the draft orphan drug framework, to qualify for orphan designation, the drug must either not currently be authorized for the Canadian market, or if already approved, must ‘provide a potentially substantial benefit for the patient distinguishable from the existing therapy’.163 The proposed framework also permits a sponsor to submit an application for orphan designation on the basis of a designation from a recognized country if the proposed orphan drug and indication in Canada is the same to that under which the foreign designation was issued.164 As in the US, a single drug may be eligible for multiple orphan designations for different rare diseases.165
While the draft Canadian framework contains many of the same types of incentives that are currently offered under the US system, there are some important incentives that are not included in the draft framework or that are offered under more restricted terms. First, the financial incentives under the Canadian framework are more restricted. Perhaps most significantly, the ODA offers a tax credit for 50% of the cost of clinical trials, whereas the draft Canadian orphan drug framework does not currently propose any type of specific tax credit for orphan designated drugs.166 In addition, under the US system, drugs that have been granted an orphan designation are exempt from the application fee that sponsors must normally pay to the FDA when making a regulatory submission;167 these application fees can be over two million dollars per product depending on the type of application.168 In contrast, although few details are provided at this stage, the Canadian draft framework proposes only a fee reduction169 rather than a fee exemption, and further this reduction is aimed at small and medium enterprises, 170 whereas the US exemption is available to any applicant. As a result, the financial incentives available upon orphan designation under the proposed Canadian orphan drug framework are arguably less enticing than those offered under the ODA and consequently, may be less likely to encourage the creation of artificial subsets. Finally, as noted above, concerns about the potential misappropriation of orphan designation through salami slicing should be tempered by the fact that designation is only the first step toward market approval, and only a fraction of orphan drugs will ultimately be approved.
Under the Canadian draft framework, the government will be ‘required to maintain and make available to the public a list of orphan drug designations’.171 This is in line with the Orphan Drug Product designation database maintained by the FDA and the Register of Designated Orphan Medicinal Products maintained by the European Medicines Agency (EMA). However, it is worth noting that while the FDA does not make public any information about the drug products that have been denied orphan designation,172 in Europe, the EMA also maintains a register of drug products that have been refused orphan drug designation,173 which includes links to various documents giving the basis for the decision. Making public information on which sponsors have applied for and been refused orphan designation could potentially add a level of public scrutiny to the activities of pharmaceutical companies that apply for orphan drug designation, hopefully discouraging companies from making questionable applications for designation. This type of transparency measure is important for ensuring the accountability and responsiveness of the orphan drug regime, and Health Canada should aim to make public information about both successful and unsuccessful applications for orphan designation.
As the orphan drug framework develops, Health Canada must establish criteria for defining acceptable orphan subsets, particularly given the controversy that had arisen over this subject south of the border. Although designation will ultimately be assessed on a case-by-case basis, the long running uncertainty in the US around the definition of a ‘medically plausible’ subset, and the subsequent need for clarifications under the 2013 amendments demonstrate the need for regulatory authorities to provide more detailed guidelines around what factors may and may not inform the existence of an acceptable orphan subset—particularly in light of advances in new genomic technologies and evolution in our understanding of disease categories. The US approach of requiring the sponsor to demonstrate why patients outside of the orphan subset are not good candidates for the drug appears to be a good approach—particularly as it puts the onus on the sponsor to justify their framing of the target population. Further, given that the current Canadian draft framework proposes to ‘allow a sponsor to submit an application on the basis of designation of a recognized country’,174 the policies adopted by the FDA in respect of orphan subsets may very well lead to a corresponding orphan designation in Canada if the US orphan designation is used as the basis for the application.
Orphan Exclusivity and Approval
At the approval stage, the Canadian orphan drug framework does not propose any new category of exclusivity for orphan drug products. Instead, the framework proposes linkage to the existing data exclusivity provisions. These provide eight years of exclusivity for an innovative drug, defined as ‘a drug that contains a medicinal ingredient not previously approved in a drug … and that is not a variation of a previously approved medicinal ingredient’.175 The exclusivity operates by preventing a competitor from obtaining market approval for another drug based on a direct or indirect comparison with the innovative drug.176 An additional six months of exclusivity is provided for innovative drugs that have been the subject of studies relating to use of the drug in pediatric populations.177 Similar provisions for ‘new chemical entity’178 and ‘pediatric’179 exclusivity exist in the US but are distinct from orphan drug exclusivity.
By proposing linkage to the innovative drug and pediatric exclusivity periods,180 Health Canada's draft orphan drug framework appears to indicate that up to an eight-and-a-half year exclusivity period would be available for orphan drugs. This period is longer than the seven-year period of market exclusivity granted for orphan drugs in the US, though shorter than the ten years provided by the European orphan drug legislation. However, there is an important distinction between how the existing data exclusivity offered by Health Canada operates in comparison with the orphan exclusivity offered under the US orphan drug regime: in the US, drugs that have been previously approved are still eligible for the seven-year orphan exclusivity for each new approved orphan indication, whereas under the proposed Canadian system, a previously approved drug would not be eligible for a new period of exclusivity if the sponsor subsequently sought approval for an orphan indication.181 That is, the sponsor would not be granted a fresh period of eight-year exclusivity for a new orphan indication if the drug has already been approved for another indication because the exclusivity is only available for innovative drugs. At most, perhaps, an extension (similar to the six-month pediatric extension) might lengthen a previously granted period of exclusivity. Unless there are plans to alter the existing exclusivity to allow an orphan drug to receive multiple periods of exclusivity, the draft Canadian orphan drug framework is unlikely to provide much incentive for pharmaceutical companies to pursue research into new orphan applications for existing drugs that have already been approved, or that are no longer under patent protection.
While only granting a single period of data exclusivity for each drug product has the advantage of preventing abuse of the orphan drug incentives through evergreening strategies, the disadvantage is that this approach will do little to encourage the repurposing of existing drug products for orphan indications, or the development of clinically superior reformulations of existing treatments for orphan diseases. Health Canada will need to do more than simply apply the existing eight- or eight-and-a-half-year exclusivity to the orphan drug context if it wants to provide these incentives. Rather, as demonstrated by the controversy around the assessment of clinical superiority under the US orphan drug policy, Health Canada will have to clearly articulate the circumstances under which orphan drug products will be eligible for market exclusivity. As the Canadian framework takes shape, the orphan exclusivity provisions should encourage the development of alternative treatments that offer genuine and significant benefits for patients by setting strict standards for what constitutes a clinically superior product that entitles a sponsor to a new exclusive approval for what would otherwise be the ‘same drug’. The difficulty is that it is currently unclear how the Canadian exclusive approval would work, and therefore, how the issue would play out under the new framework.
Finally, it is worth noting that Health Canada specifically acknowledges that ‘greater uncertainties may exist for orphan drugs given the complexities of the diseases, the small and vulnerable populations and the treatment environment itself’,182 and that consequently, ‘greater abilities to plan for and resolve those uncertainties are needed once the drug is on the market’. 183 Accordingly, the draft Canadian orphan drug framework includes expanded transparency measures, including the publication of the key information on when an application for market authorization for an orphan drug has denied. Health Canada notes that ‘[t]he publication of negative decisions and the basis for these decisions is important for orphan drugs in circumstances when an existing marketed re-purposed drug could continue to be used off-label’.184 Unfortunately, the FDA does not currently make publicly accessible, in their online orphan drug database or otherwise, any specific information about when an application for market authorization for an orphan-designated drug has been unsuccessful.
CONCLUSION
Given the rapid evolution of the field of pharmacogenomics, its impact on orphan drug policies will need to be continually reassessed to ensure that policies remain responsive to the current drug development paradigm. The recent revision of the ODA Regulations in 2013 is encouraging because the FDA has at least demonstrated that it is alive to the concerns raised by the increasing detail with which potential orphan subsets can be defined. Although questions remain about whether the amendments will be sufficient to prevent abuse of the provisions, it appears likely that some of the concerns have been mitigated. The US experience can provide lessons for other jurisdictions, including Canada as it develops its new orphan drug policy.
A central dilemma in addressing the issue of salami slicing in orphan designation is that it implies judgments about what should be regarded as ‘true’ versus ‘artificial’ subdivision of disease categories—an assessment that must be made with reference to the purposes of the orphan legislation. However, it is important to note that this does not necessarily imply that developing treatments for ‘artificial’ groups that don't qualify as orphan subsets is not valuable. As noted by Herder, ‘[i]n the abstract, it is not obvious that orphan disease policy should prioritize diseases that have long been understood as rare over diseases like breast cancer that may soon be subdivided into multiple rare diseases’ since either may be in need of ‘new and improved’ treatment options.185 Rather, the more important question is whether the objectives of the ODA are fulfilled by incentivizing development of such treatments.
In pharmaceutical development, there is a general desire to encourage the development of more and better treatment options for patients with rare diseases. While this is a worthy goal, we should bear in mind that the more specific objective of orphan drug legislation is to target incentives toward the development of drugs that would not otherwise be developed due to a lack of financial viability. Some argue that orphan drug legislation is overinclusive because it provides incentives where they are not really needed. The marked increase in applications for orphan designations in recent years has led to suggestions that the ODA ‘risks becoming a victim of its own prodigious success’,186 and tends to confirm suspicions that the orphan drug market is one that the pharmaceutical industry now views as potentially lucrative. There are ‘orphan’ drugs that are actually very profitable due to the very high prices that can be charged for rare disease therapies187 or to extensive off-label use for more common conditions188—both of which are in themselves sources of significant controversy. The outstanding question is whether it is better to err on the side of offering orphan incentives more liberally, even if this leaves the policy open to abuses such as salami slicing, or to be more restrictive in granting incentives but risk of dissuading the development of some orphan drugs, such as those that target biomarker-based orphan subsets.
Another area that should be of interest to Canada as it develops its orphan drug framework is the potential to encourage the use of pharmacogenomics research to repurpose drugs for use in treating rare disorders. In the US, the possibility of a new period of orphan drug exclusivity provides an incentive for manufacturers to invest in repurposing, and there is even talk of adding a new type of extension to exclusivity or patent terms to further stimulate this activity.189 As discussed above, the exclusivity proposed in the Canadian draft framework for orphan drugs would need to be modified to provide an effective incentive for repurposing approved drugs for new orphan indications.
Finally, the impact of pharmacogenomics on orphan drug development makes it all the more important to ensure that orphan drug policies be implemented with a high level of transparency and coordination. In introducing the draft orphan drug framework, the Canadian Government stated that ‘a key focus of this new approach will be on international information-sharing and collaboration for the development and regulation of orphan drugs’.190 Specifically, the proposed Canadian orphan drug regulations would ‘allow Health Canada to operationally align and participate in well-established activities of the US and the European Union including designation, scientific/protocol advice and pre- and post-market information sharing’.191 Transparency is also addressed through plans to publish the basis for decisions on marketing authorization. As the science of drug development becomes more and more complex, measures to minimize abuse of incentives and to enhance the safety of off-label use by sharing information among regulatory agencies, manufacturers, and the public are essential.
Acknowledgments
We would like to acknowledge Emily Harris (JD 2015) and Priscila Padilla (JD 2015) at the University of Saskatchewan and Nicholas Reist (JD 2016) at the University of Toronto for their assistance with background research and references.
Footnotes
Abbey S. Meyers, History of the American Orphan Drug Act, National Organization for Rare Disorders, Feb. 18, 2000, at 1, https://www.rarediseases.org/docs/policy/HistoryoftheAmericanOrphanDrugAct.pdf (accessed Dec. 10, 2014).
Id. at 1.
21 U.S.C. § 360bb(a)(2).
Timoty R. Coté, Kui Xu & Anne R. Pariser, Accelerating Orphan Drug Development, 9 Nat. Rev. Drug Disc. 901, 901 (2010).
Paul D. Maher & Marlene Haffner, Orphan Drug Designation and Pharmacogenomics: Options and Opportunities, 20 BioDrugs 71, 72 (2006).
US Food and Drug Administration, Developing Products for Rare Diseases & Conditions, http://www.fda.gov/forindustry/DevelopingProductsforrareDiseasesConditions/default.htm (accessed Dec. 10, 2014). In contrast, between 1973 and 1983, only 10 orphan drugs made it to market.
Olivier Wellman-Labadie & Youwen Zhou, The US Orphan Drug Act: Rare Disease Research Stimulator or Commercial Opportunity?, 95 Health Pol'y 216, 221 (2010); Maher & Haffner, supra note 5, at 71.
Reuters, The Economic Power of Orphan Drugs, 2012, at 4, http://thomsonreuters.com/business-unit/science/subsector/pdf/the-economic-power-of-orphan-drugs.pdf (accessed Dec. 10, 2014)
Jonathan D. Rockoff, Drug Makers See Profit Potential in Rare Diseases, Wall Street Journal, Jan. 30, 2013, http://www.wsj.com/articles/SB10001424127887323926104578273900197322758 (accessed Apr. 2, 2015).
Christopher-Paul Milne & Joyce Tait, Evolution Along the Government-Governance Continuum: FDA's Orphan Products and Fast Track Programs as Exemplars of “What Works” for Innovation and Regulation, 64 Food Drug Law J. 733, 742–43 (2009).
Ian Kerr, Jennifer Chandler & Timothy Caulfield, Emerging Health Technologies, in 501 Canadian Health Law and Policy, (Jocelyn Downie et al. eds, 4th ed. 2011), at 504.
National Human Genome Research Institute, Frequently Asked Questions About Pharmacogenomics, http://www.genome.gov/27530645 (accessed Dec. 10, 2014).
Id.
Roger Collier, Bye, Bye Blockbusters, Hello Niche Busters, 183 Can. Med. Ass'ns J. e697 (2011).
Personalized Medicines Coalition, Personalized Medicine by the Numbers, 2014, http://www.personalizedmedicinecoalition.org/Userfiles/PMC-Corporate/file/pmc_personalized_medicine_by_the_numbers.pdf (accessed Dec. 10, 2014).
David Loughnot, Potential Interactions of the Orphan Drug Act and Pharmacogenomics: A Flood of Orphan Drugs and Abuses? 31 Am. J. L. & Med. 365 (2005); Wellman-Labadie & Zhou, supra note 7; Matthew Herder, When Everyone is an Orphan: Against Adopting a U.S.-styled Orphan Drug Policy in Canada, 20 Acct. Res. 227 (2013).
Herder, supra note 16, at 57.
Loughnot, supra note 16, at 365.
Maher & Haffner, supra note 5; Marlene E. Haffner, Josep Torrent-Farnell & Paul D. Maher, Does Orphan Drug Legislation Really Answer the Needs of Patients?, 371 Lancet 2041, 2043 (2008); Loughnot, supra note 16; Dov Greenbaum, Incentivizing Pharmacogenomic Drug Development: How the FDA Can Overcome Early Missteps in Regulating Personalized Medicine, 40 Rutgers L. J. 97 (2008).
Health Canada, Office of Legislative and Regulatory Modernization, Initial Draft Discussion Document for a Canadian Orphan Drug Regulatory Framework, Dec. 13, 2012, at 4, http://www.orpha.net/national/data/CA-EN/www/uploads/Initial-Draft-Discussion-Document-for-A-Canadian-Orphan-Drug--Regulatory-Framework.doc (accessed Dec. 11, 2014). [hereinafter Health Canada Draft Orphan Drug Framework].
Reuters, supra note 8, at 4.
Haffner, Torrent-Farnell & Maher, supra note 19, at 2043.
The White House, FACT SHEET: President Obama's Precisions Medicine Initiative, Jan. 30, 2015, http://www.whitehouse.gov/the-press-office/2015/01/30/fact-sheet-president-obama-s-precision-medicine-initiative (accessed Feb. 27, 2015).
Valerie G. Koch, Incentivizing Utilization of Pharmacogenomics in Drug Development, 15 J. Health Care L. & Pol'y 263 (2012).
Nuffield Council on Bioethics, Regulation and Public Policy, in Pharmacogenetics: ethical issues 50 (2003).
Id. at 50.
Greenbaum, supra note 19, at 101.
Loughnot, supra note 16, at 368, 370.
21 U.S.C. § 360cc; 21 C.F.R § 316.31.
21 C.F.R. § 316.3((b)(14); § 316.35(c). However, the market exclusivity period may be shortened if the orphan designation is withdrawn or revoked by the FDA, the marketing approval is withdrawn, the sponsor agrees to the withdrawal, or the sponsor is unable to provide sufficient quantity of the drug: 21 C.F.R. § 316.31.
21 C.F.R. §§ 316.20, 316.23, 316.31(b).
Maher & Haffner, supra note 5, at 77.
Id. at 78.
Herder, supra note 16.
Christopher A. Lipinski, Drug Repurposing, 8 Drug Disc. Today 57, 57 (2011).
Loughnot, supra note 16, at 370, 371.
Id. at 370.
Id. at 371.
Id. at 374.
Id. at 366
Maher & Haffner, supra note 5, at 78.
Orphan Drug Regulations (Final Rule), 78 Fed. Reg. 35,117, 35,120–1 (June 12, 2013) [hereinafter Final Rule 2013].
Maher & Haffner, supra note 5, at 77.
Orphan Drug Regulations (Proposed Rule), 56 Fed. Reg. 3338, 3339 (Jan. 29, 1991).
Id.
Orphan Drug Regulations (Final Rule), 57 Fed. Reg. 62,076, 62,077 (Dec. 29, 1992) [hereinafter Final Rule 1992].
Herder, supra note 16, at 236.
Orphan Drug Regulations (Proposed Rule), 76 Fed. Reg. 64,868, 64,869 (Oct. 19, 2011) [hereinafter Proposed Rule 2011].
For example, see Sara Reardon, Regulators Adopt More Orphan Drugs, 508 Nature 16, 17 (2014).
Final Rule 2013, supra note 42, at 35,119. See also Proposed Rule 2011 supra note 48 at 64,869.
Id. at 64,869 and §§ 316.3(b)(13), 316.20(b)(6) .
Final Rule 2013, supra note 42, at 35,119.
Id. at 35,119, 35,121.
Maher & Haffner, supra note 5, at 77.
Herder, supra note 16, at 246.
Final Rule 2013, supra note 42, at 35,121.
Id. at 35,119.
Herder, supra note 16, at 246.
Final Rule 2013, supra note 42, at 35,120.
Id.
Id.
For example, genetic modeling has been used to study and identify the role of cytochrome P450 in the metabolism of codeine, which may identify patients at risk of adverse reactions to the painkiller. See Thomas Eissing, Jörg Lippert & Stefen Willmann, Pharmacogenomics of Codeine, Morphine, and Morphine-6-glucuronide: Model-based Analysis of the Influence of CYP2D6 Activity, UGT2B7 Activity, Renal Impairment, and CYP3A4 Inhibition, 16 Mol. Diagn. Ther. 43 (2012).
For example, there have been 20 separate drugs granted orphan designation for the treatment of various types of chronic myelogenous leukemia between 1986 and 2011. Indeed, several pharmacogenomic products—imatinib, nilotinib and dasatinib—have been granted orphan designations for this particular disease. See US Food and Drug Administration, Orphan Drug Product Designation Database, http://www.accessdata.fda.gov/scripts/opdlisting/oopd/index.cfm (accessed Dec. 11, 2014).
Final Rule 2013, supra note 42, at 35,126. This point was made in response to a comment that the FDA should ‘make public its finding on the acceptability of specific prevalence data to reduce uncertainty about designation requirements’. In rejecting the idea of publicly disclosing prevalence data, the FDA also noted that ‘such an approach would unfairly allow subsequent sponsors to get a “free ride” in designation requests’.64
Final Rule 2013, supra note 42, at 35,120.
US Food and Drug Administration, Enrichment Strategies for Clinical Trials to Support Approval of Human Drugs and Biological Products (Draft Guidance), 2012, http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM332181.pdf (accessed May 6, 2014).
Final Rule 2013, supra note 42, at 35,120. During consultations, the FDA received many comments expressing the concern that ‘in order to establish an orphan subset, sponsors would have to prove a negative: That the drug would not potentially benefit other subsets of persons with the non-rare disease or condition’.
Loughnot, supra note 16, at 380.
Haffner, Torrent-Farnell & Maher, supra note 19, at 2041.
Julian R. Molina et al., Non-Small Cell Lung Cancer: Epidemiology, Risk Factors, Treatment, and Survivorship, 83 Mayo Clin. Proc. 582 (2009).
American Lung Association, Lung Cancer Fact Sheet, http://www.lung.org/lung-disease/lung-cancer/resources/facts-figures/lung-cancer-fact-sheet.html (accessed Dec. 11, 2014).
ALK, MET and ROS are all biomarkers that are clinically relevant to lung cancer. See, for example, Grzegorz J. Korpanty et al., Biomarkers That Currently Affect Clinical Practice in Lung Cancer: EGFR, ALK, MET, ROS-1, and KRAS, 4 Front. Oncol. 204 (2014).
In an FDA presentation in March 2014, non-small cell lung cancer was given as an example of a common disease, while non-small cell lung cancer with EGFR mutation was listed as an example of an acceptable orphan subset. See James H. Reese, FDA Orphan Drug Designation 101, at 23, http://www.ema.europa.eu/docs/en_GB/document_library/Presentation/2014/03/WC500164160.pdf (accessed Jan. 13, 2015).
National Cancer Institute, SEER Stat Fact Sheets: Melanoma of the Skin, http://seer.cancer.gov/statfacts/html/melan.html (accessed Dec. 18, 2014).
US Food and Drug Administration, supra note 63.
Adam Hedgecoe, The Politics of Personalised Medicine: Pharmacogenetics in the Clinic 119 (2004).
Jai Shah, Economic and Regulatory Considerations in Pharmacogenomics for Drug Licensing and Healthcare, 21 Nat. Biotechnol. 747, 749 (2002).
Id.
Id.
Hedgecoe, supra note 76, at 119. However, although Herceptin was denied orphan drug status for the treatment of patients with metastatic breast cancer, the FDA subsequently granted orphan designation to Herceptin for the treatment of pancreatic cancer in 1999 (though marketing approval has not been granted for this indication) and for the treatment of HER2-overexpressing advanced adenocarcinoma of the stomach in Oct. 2009 (for which market approval has been granted)—see Orphan Drug Product designation database, supra note 63.
The FDA provides the following example of when the same drug may be eligible for three separate orphan designations: ‘One for the treatment of ovarian cancer, one for the treatment of multiple myeloma, and one for the treatment of Kaposi's sarcoma, even if the cumulative prevalence of all three diseases or conditions would exceed 200,000’. See Final Rule 2013, supra note 42, at 35,120.
Id.
National Cancer Institute, SEER Stat Fact Sheets: Breast Cancer, http://seer.cancer.gov/statfacts/html/breast.html (accessed Dec. 18, 2014).
Greenbaum, supra note 19, at 101.
In 2010, there were an estimated 31,566 cases of chronic myeloid leukemia in the US. See Leukemia and Lymphoma Society, Facts, 2014, at 8, https://www.lls.org/content/nationalcontent/resourcecenter/freeeducationmaterials/generalcancer/pdf/facts.pdf (accessed Dec. 18, 2014).
In 2011, there were an estimated 43,538 cases of pancreatic cancer in the US. See National Cancer Institute, SEER Stat Fact Sheets: Pancreas Cancer, http://seer.cancer.gov/statfacts/html/pancreas.html (accessed Dec. 18, 2014).
In 2010, there were an estimated 66,030 cases of acute lymphoblastic leukemia in the US. See Leukemia and Lymphoma Society, supra note 85, at 8.
Loughnot, supra note 16, at 369; Greenbaum, supra note 19, at 134, 135; Gary A. Pulsinelli, The Orphan Drug Act: What's Right with It, 15 Santa Clara High Tech. L. J 299, 310 (1999).
Health Canada Draft Orphan Drug Framework, supra note 20, at 4.
Maher & Haffner, supra note 5, at 77.
Health Canada Draft Orphan Drug Framework, supra note 20, at 6.
Some drugs have had mixed success, obtaining orphan drug status for multiple indications, but receiving market authorization for only a portion of these orphan indications. For example, Xalkori (crizotinib), Herceptin (trastuzamab), Erbitux (cetuximab) and Tasigna (nilotinib) have received multiple orphan designations, but received FDA approval for only one orphan indication. Other drugs have received orphan drug status but been unsuccessful in achieving any market approval for an orphan indication. Both Tarceva (erlotinib) and Tykerb (lapatinib) have orphan designations without any corresponding market authorization for an orphan indication. Conversely, Dasatinib (Sprycel) has received both orphan designation and market approval for two separate orphan indications. US Food and Drug Administration, supra note 63.
21 U.S.C. § 360cc.
21 C.F.R. § 316.3(b)(12).
21 C.F.R. § 316.3(b)(14).
Id.
Id. at § 316.3(b)(3). The 2013 amendments to the ODA noted that ‘[t]he following factors, when applicable to severe or life-threatening diseases, may in appropriate cases be taken into consideration when determining whether a drug makes a major contribution to patient care: convenient treatment location; duration of treatment; patient comfort; reduced treatment burden; advances in ease and comfort of drug administration; longer periods between doses; and potential for self-administration’. See Final Rule 2013, supra note 42, at 35,125. However, most of these factors were previously set in the preamble to the Final Rule 1992, supra note 42, at 62079.
21 C.F.R. § 316.20(a).
Id. at § 316.34(c).
Final Rule 2013, supra note 42, at 35,122.
Maher & Haffner, supra note 5, at 71.
Final Rule 2013, supra note 42, at 35,128.
Complaint for Declaratory, Injunctive and Other Relief, Depomed Inc. v. US Department of Health and Human Services et al., No. 1:12-cv-01592-RLW (Sept. 25, 2012).
Id.at 33.
Id.
Id at 7.
Id at 29, 30.
See US Food and Drug Administration, Policy on Orphan Drug Exclusivity; Clarification, 79 Fed. Reg. 76,888 (Dec. 23, 2014). Although FDA originally filed a notice of appeal in Nov. 2014, the appeal was withdrawn only days later. See Kurt R. Karst & Michelle L. Butler, FDA Issues Post-Depomed Policy Statement; Agency Doubles Down on Clinical Superiority Requirement, FDA Law Blog, Dec. 22, 2014, http://www.fdalawblog.net/fda_law_blog_hyman_phelps/2014/12/fda-issues-post-depomed-policy-statement-agency-doubles-down-on-clinical-superiority-requirement.html (accessed Dec. 23, 2014).
Grossman, for example, suggests that it is possible that FDA ‘calculated how rarely this case is likely to come up and decided that for another plaintiff to emerge with a similar situation and a deep enough set of pockets to want to sue [is unlikely]’. Steven Grossman, President of health policy consultancy HPS Group, quoted in Fiona Barry, FDA Defiant on Orphan Exclusivity Rules after Court Judgement, In-Pharma, Jan. 7, 2015, http://www.in-pharmatechnologist.com/Regulatory-Safety/FDA-defiant-on-orphan-exclusivity-rules-even-after-court-judgement (accessed Apr. 2, 2015). Alternatively, Karst and Butler suggest that ‘[p]erhaps this out-of-left-field-strategy is FDA's way of drawing out another lawsuit from an affected sponsor so that the Agency can have another crack to relitigate the issue in court’. Id.
Depomed, supra note 104 at 14.
Tal Burt & Savita Dhillon, Pharmacogenomics in Early-phase Clinical Development, 12 Pharmacogenomics 1085, 1090 (2013).
Lipinski, supra note 35, at 57.
As an example, the growth hormone somatropin has received orphan designations from Novo Nordisk, EMD Serono, and Eli Lilly and has multiple approved rare disease indications. US Food and Drug Administration, supra note 63.
Rebecca S. Yoshitani & Ellen S. Cooper, Pharmaceutical Reformulation: The Growth of Lifecycle Management, 7 Hous. J. Health L. & Pol'y 379, 380 (2007).
Proposed Rule 2011, supra note 48, at 64,870.
Final Rule 2013, supra note 42, at 35,126.
Id. at 35,123.
Id.
Id.
Id. This is distinct from the idea of ‘orphan subset’ discussed above because in this case we are looking at specific uses or indications within a rare disease or condition, whereas an orphan subset is a low-prevalence subset within a non-rare disease or condition—Proposed Rule 2011, supra note 48, at 64,871.
21 C.F.R. § 316.31(b).
Id. at § 316.31(b). Specifically, § 316.31(b) states that ‘FDA will recognize a new orphan drug exclusive approval for these new (not previously approved) indication(s) or use(s) from the date of approval of the drug for such new indication(s) or use(s)’.
For example, in 2006, Kalydeco (ivacaftor) received an orphan designation for the treatment of cystic fibrosis. According to the Cystic Fibrosis Foundation, approximately 30,000 people in the US have cystic fibrosis—well below the 200,000 person threshold required to qualify for orphan designation under the ODA—Cystic Fibrosis Foundation, Frequently Asked Questions, http://www.cff.org/aboutcf/faqs (accessed Dec. 18, 2014). Cystic fibrosis is a complex disease that may be caused by over 1800 different genetic mutations of a particular gene. Kalydeco is currently only approved for very specific indications within the cystic fibrosis designation: in 2012; Kalydeco was approved for a limited group of patients with a particular mutation of the gene that causes cystic fibrosis (G551D mutation). Subsequently, in Feb. 2014, Kalydeco was approved for the treatment of patients with eight additional cystic fibrosis mutations (G551D, G178R, S549N, S549R, G551S, G1244E, S1251N, S1255P, and G1349D)—Cystic Fibrosis Foundation, FDA Approves Expanded Use of Kalydeco™ for Eight Additional Mutations that Cause CF, Feb. 21, 2014, http://www.cff.org/aboutCFFoundation/NewsEvents/2--21-FDA-Approves-Expanded-Use-of-Kalydeco-for-CF.cfm (accessed Dec. 18, 2014).
Loughnot, supra note 16, at 376.
Final Rule 2013, supra note 42, at 35,120.
Some orphan approvals are even more specific. For example, nilotinib (Tasigna) initially received an orphan designation for the ‘treatment of chronic myelogenous leukemia’, but ultimately received market approval for ‘chronic phase (CP) and accelerated phase (AP) Philadelphia chromosome positive chronic myelogenous leukemia (CML) in adult patients resistant to or intolerant to prior therapy that included imatinib’. US Food and Drug Administration, supra note 63.
Maher & Haffner, supra note 5 at 78.
Greenbaum, supra note 19, at 120.
Id. at 118.
Felix W. Frueh, Back to the Future: Why Randomized Controlled Trials Cannot Be the Answer to Pharmacogenomics and Personalized Medicine, 10 Pharmacogenomics 1077 (2009).
US Food and Drug Administration, The Future of Drug Safety — Promoting and Protecting the Health of the Public (FDA's Response to the Institute of Medicine's 2006 Report), Jan. 2007, at 3, http://www.fda.gov/downloads/Drugs/DrugSafety/PostmarketDrugSafetyInformationforPatientsandProviders/UCM171627.pdf (accessed Dec. 18, 2014).
Burt & Dhillon, supra note 111, at 1090.
Final Rule 2013, supra note 42, at § 316.23(b).
Maher & Haffner, supra note 5, at 76.
Haffner, Torrent-Farnell & Maher, supra note 19, at 2042.
Final Rule 2013, supra note 42, at 35,120.
Novartis received seven separate orphan designations for imatinib for the treatment of: chronic myelogenous leukemia (Jan. 2001), gastrointestinal stromal tumors (Nov. 2001), idiopathic hypereosinophilic syndrome (Aug. 2005), systemic mastocytosis (Sep. 2005), myeloproliferative disorders/myelodysplastic syndromes (Oct. 2005), Philadelphia-positive acute lymphoblastic leukemia (Oct. 2005) and dermatofibrosarcoma protuberans (Dec. 2005). US Food and Drug Administration, supra note 63.
Reuters, supra note 8, at 7.
For example, the FDA notes that ‘these drugs have already been subjected to pre-clinical (e.g., pharmacokinetic and toxicologic) testing and are already deemed to be pharmacologically active, effective and safe in some clinical context’. US Food and Drug Administration, A Valuable Resource for Drug Developers: The Rare Disease Repurposing Database (RDRD), http://www.fda.gov/ForIndustry/DevelopingProductsforRareDiseasesConditions/HowtoapplyforOrphanProductDesignation/ucm216147.htm (accessed Dec. 19, 2014).
Based on the list of drugs included in the table ‘Orphan-designated products with at least one marketing approval for a common disease indication’ prepared by the FDA, see id.
The FDA granted two orphan drug designations for fluoxetine: one in Apr. 1999 for the treatment of autism and the other in Apr. 2004 for the treatment of body dysmorphic disorder in children and adolescents. However, fluoxetine never received FDA approval for either of these orphan indications. US Food and Drug Administration, supra note 63.
Sildenafil citrate is most commonly known under the brand-name Viagra and is used for the treatment of erectile dysfunction. In July 2011, sildenafil citrate received an orphan designation under the trade name Revatio from the FDA for the treatment of pediatric pulmonary arterial hypertension but has not yet received FDA approval for this indication. Id.
The FDA granted gabapentin orphan drug status for the treatment of amyotrophic lateral sclerosis in July 1995, but market approval was never granted for this indication. Subsequently, in Nov. 2010, gabapentin was granted orphan status for the management of post-herpetic neuralgia under the trade name Gralise, for which marketing approval was granted in Jan. 2011. Id.
Interestingly, when the ODA was passed in 1983, demonstrating that a particular orphan drug would be unprofitable was a prerequisite to obtaining an orphan designation, and sponsors thus had to provide financial information to support their application irrespective of the size of the target population. However, in 1984 the ODA was amended to introduce the 200,000 person prevalence threshold for the definition of a rare disease. Following the amendments, ‘a sponsor could still seek orphan drug designation by demonstrating that the financial criteria of the law were applicable but was not required to do so if the target patient population was less than 200,000’. See M. Angeles Villarreal, Orphan Drug Act: Background and Proposed Legislation in the 107th Congress (CRS Report for Congress), July 25, 2001, at 2, http://www.law.umaryland.edu/marshall/crsreports/crsdocuments/RS20971.pdf (accessed Dec. 19, 2014).
Haffner, Torrent-Farnell & Maher, supra note 19, at 2041.
Christopher M. Wittich, Christopher M. Burkle, & William L. Lanier, Ten Common Questions (and Their Answers) About Off-label Drug Use, 87 Mayo Clin. Proc. 982 (2012).
Generic drugs are approved by the FDA based on the submission of an Abbreviated New Drug Application. ‘Generic drug applications are termed “abbreviated” because they are generally not required to include preclinical (animal) and clinical (human) data to establish safety and effectiveness. Instead, generic applicants must scientifically demonstrate that their product is bioequivalent (i.e., performs in the same manner as the innovator drug).’ See Food and Drug Administration, Abbreviated New Drug Application (ANDA): Generics, updated Sept. 18, 2014, http://www.fda.gov/Drugs/DevelopmentApprovalProcess/HowDrugsareDevelopedandApproved/ApprovalApplications/AbbreviatedNewDrugApplicationANDAGenerics/ (accessed Mar. 9, 2015).
Terry Mahn, Protecting New Investments in Old Drugs,Fish and Richardson PC, updated Mar./Apr. 2009, http://www.fr.com/files/Uploads/bios/Mahn_Terry/Protecting_New_Investments.pdf (accessed Apr. 2, 2015).
Id.
Id at 39.
Durhane Wong-Reiger, Canada's Long Journey toward an Orphan Drug Framework, Advocate, 2013, at 20.
Id. at 20.
Health Canada, Orphan Drug Policy (letter sent to associations, file no: 96–037419), Jan. 16, 1997, https://web.archive.org/web/20130722055233/, http://www.hc-sc.gc.ca/dhp-mps/prodpharma/applic-demande/pol/orph_pol-eng.php (accessed July 2013) [hereinafter Health Canada 1997 Policy Statement].
Health Canada Draft Orphan Drug Framework, supra note 20, at 5.
Health Canada 1997 Policy Statement, supra note 153, at 8.
Canadian Organization for Rare Disorders, Canada's Orphan Drug Policy, July 6, 2005, at 4, http://paperzz.com/download/1749313#pdf (accessed Dec. 19, 2014).
BIOTECanada, Helping Canadians with Rare Diseases: Government of Canada Rare Disease Strategy Offers Hope and Solutions for Patients, http://www.biotech.ca/en/policy-matters/health-bio/rare.aspx (accessed Dec. 19, 2014).
External Advisory Committee on Smart Regulation, Smart Regulation: A Regulatory Strategy for Canada (Report to the Government of Canada), Sep. 2004, at 93, http://publications.gc.ca/collections/Collection/CP22-78-2004E.pdf (accessed Dec. 19, 2014).
Canadian Organization for Rare Disorders, Current Issues, http://www.raredisorders.ca/currentIssues.html (accessed Dec. 19, 2014).
Health Canada Draft Orphan Drug Framework, supra note 20, at 10.
Orphan drugs often treat serious or life-threatening diseases for which few or no alternative treatments exist, which can increase pressure on regulators to speed the approval of these drugs, even in the face of uncertain safety and efficacy: Shannon Gibson & Trudo Lemmens, Niche Markets and Evidence Assessment in Transition: A Critical Review of Proposed Drug Reforms 22 Med. Law Rev. 200 (2014). Priority review avenues can be a strong incentive for drug developers since the sooner a drug hits the market, the sooner the drug—which is typically running under the patent clock—can begin to generate revenue.
Protocol assistance during clinical development is an important incentive for drug developers: due to the difficulties of conducting clinical trials in small patient populations, advice from regulators on how to design clinical trials that can save the sponsor a significant amount of time and money: Herder, supra note 16, at 238.
Health Canada Draft Orphan Drug Framework, supra note 20, at 10.
Id. at 6.
Id. at 7.
A Scientific Research and Experimental Development tax credit as well as provincial and territorial research and development tax credits are available, and Canada has been described as having ‘one of the most generous R&D tax-credit programs among major industrial countries’: Dirk Czarnitzki, Petr Hanel & Julio Miguel Rosa, Evaluating the Impact of R&D Tax Credits on Innovation: A Microeconometric Study on Canadian Firms 40 Res. Pol'y 217, 218 (2011). However, there has been no indication that a tax credit specific to orphan drug development would be added in Canada.
The Prescription Drug User Fee Act of 1992 authorized the FDA to assess user fees for the regulatory review of human drug applications. The orphan exemption is only one of many reductions, waivers and exemptions that may be granted under the act. Any sponsor who has been granted an orphan designation for a drug product may claim this exemption as long as the drug application is limited to an indication for a rare disease or condition. See US Food and Drug Administration, User Fee Waivers, Reductions, and Refunds for Drug and Biological Products (Guidance for Industry), Sep., 2011, at 11, http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/ucm079298.pdf (accessed Dec. 19, 2014).
US Food and Drug Administration, Prescription Drug User Fee Rates for Fiscal Year 2015, 79 Fed. Reg. 44,807, 44,810 (Aug. 1, 2014).
In recognition of the fact that drug review fees can be substantial and may be prohibitive to start-up companies and non-profit organizations, Health Canada offers fee reductions to certain sponsors. Successful applicants can have their drug review fees ‘capped at 10% of the drug's anticipated sales revenue in Canada for its first three years on the market’: Health Canada, Health Canada's Proposal to Parliament for User Fees and Service Standards for Human Drugs and Medical Devices Programs, Apr., 2010, at Table 1A, http://www.hc-sc.gc.ca/dhp-mps/finance/costs-couts/fee-propo-frais-eng.php pdf (accessed Dec. 19, 2014). If the fee reductions under the Canadian orphan drug framework are similarly limited to small and medium enterprises, then this incentive would not be available to the major players in the pharmaceutical industry.
Health Canada Draft Orphan Drug Framework, supra note 20, at 7.
Id. at 12.
However, the FDA does have a database of ‘Post-market Requirements and Commitments’ that includes information on post-market studies and clinical trials that sponsors are required or have agreed to conduct. Numerous pharmacogenomic drug products are included in this database, including Herceptin, Gleevec and Erbitux. See US Food and Drug Administration, Postmarketing Requirements and Commitments: Introduction, http://www.fda.gov/Drugs/GuidanceComplianceRegulatoryInformation/Post-marketingPhaseIVCommitments/default.htm (accessed Dec. 20, 2014).
European Commission, Orphan Refusals, http://ec.europa.eu/health/documents/community-register/html/orh_others.htm (accessed Dec. 20, 2014).
Health Canada Draft Orphan Drug Framework, supra note 20, at 6.
Canada Food and Drug Regulations, C.R.C., c. 870, s. C.08.004.1(1).
Id. at s. C.08.004.1(3). Most commonly, this prevents a generic manufacturer from using the innovative drug product as the Canadian reference product for the purposes of generic approval. See Health Canada, Guidance Document: Data Protection under C.08.004.1 of theFood and Drug Regulations (Guidance document), http://www.hc-sc.gc.ca/dhp-mps/prodpharma/applic-demande/guide-ld/data_donnees_protection-eng.php (accessed Dec. 20, 2014).
Id. at s. C.08.004.1(4).
21 U.S.C. § 355(c)(3)(E)(ii).
Id. at § 355(a).
Health Canada Draft Orphan Drug Framework, supra note 20, at 7.
Health Canada, Data Protection under C.08.004.1, supra note 176, at 2.
Health Canada Draft Orphan Drug Framework, supra note 20, at 6.
Id.
Id. at 7.
Herder, supra note 16, at 245.
Reardon, supra note 49, at 16.
The market exclusivity granted to orphan drugs is sometimes viewed as contributing to the high price of orphan drugs—id. at 16. However, other commentators counter that these high prices merely reflect the rising costs of developing advanced biotechnology drugs and the lack of economies of scale in drug development for rare diseases: Haffner, Torrent-Farnell & Maher, supra note 19, at 2042, 2043.
Id. at 2041.
Kurt R. Karst, Exclusivity Creep: OPEN ACT Would Open Up Hatch-Waxman and the BPCIA to Add a 6-Month Extension to Existing Exclusivities, FDA Law Blog, Dec. 4, 2014, http://www.fdalawblog.net/fda_law_blog_hyman_phelps/2014/12/exclusivity-creep-open-act-would-open-up-hatch-waxman-and-the-bpcia-to-add-a-6-month-extension-to-ex.html (accessed Dec. 20, 2014).
Health Canada, An Orphan Drug Framework for Canada, http://www.hc-sc.gc.ca/ahc-asc/media/nr-cp/_2012/2012-147a-eng.php (accessed Dec. 20, 2014).
Health Canada Draft Orphan Drug Framework, supra note 20, at 5.
FUNDING
This work was funded by the Government of Canada through Genome Canada and the Ontario Genomics Institute (OGI-064: Enhanced CARE for RARE Genetic Diseases in Canada), and through a second Genome Canada grant (Ethical and Legal Issues of Cancer Initiating Stem Cell Research).
CONFLICT OF INTEREST
None declared.