

Abstract
Scientists, policymakers, and medical professionals alike have become increasingly worried about the rise of antibiotic resistance, and the growing number of infections due to bacteria like Clostridium difficile, which cause a significant number of deaths and are imposing increasing costs on our health care system. However, in the last few years, fecal microbiota transplantation (FMT), the transplantation of stool from a healthy donor into the bowel of a patient, has emerged as a startlingly effective means to treat recurrent C. difficile infections. At present, the FDA is proposing to regulate FMT as a biologic drug. However, this proposed classification is both underregulatory and overregulatory. The FDA's primary goal is to ensure that patients have access to safe, effective treatments—and as such they should regulate some aspects of FMT more stringently than they propose to, and others less so. This essay will examine the nature of the regulatory challenges the FDA will face in deciding to regulate FMT as a biologic drug, and will then evaluate available policy alternatives for the FDA to pursue, ultimately concluding that the FDA ought to consider adopting a hybrid regulatory model as it has done in the case of cord blood.
Keywords: antibiotic resistance, FDA, fecal transplantation, microbiome
Scientists, policymakers, and medical professionals alike have become increasingly worried about the rise of antibiotic resistance, particularly among hospital-acquired infections. The number of infections due to the most common nosocomial pathogen,1
Clostridium difficile, doubled between 2000 and 2005.2 This aggressive intestinal superbug causes more than 250,000 hospitalizations and 14,000 deaths in the USA each year,3 and it is estimated to cost the US health care system 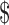 4.8 billion annually.4
4.8 billion annually.4
Although the first line of treatment for C. difficile remains antibiotic therapy, 25 per cent of patients experience a recurrence after their initial course, a number that has risen in the last decade.5 Failure rates after treatment with metronidazole, which is typically used to treat new cases of C. difficile, rose from 2.5 per cent to more than 18 per cent between 2000 and 2008.6 Failure rates after treatments with vancomycin or fidaxomicin, standard therapies for recurrences of C. difficile infection, have also risen, posing a significant challenge for patients with multiple recurrences.7 Should a patient experience a second recurrence, his or her chance of another relapse rises to 35 or 45 per cent,8 and reaches 60 per cent by the third recurrence.9 The limited effectiveness of antibiotic therapies for the treatment of recurrent C. difficile infection has prompted growing concern,10 as reflected in the White House's newly released National Action Plan to combat antibiotic resistant bacteria, and which prioritizes addressing the C. difficile problem.11
In the last few years, however, fecal microbiota transplantation (FMT) has emerged as an effective alternative means to treat recurrent C. difficile infections.12 FMT, also called fecal bacteriotherapy, is the transfer of stool from a healthy donor into the bowel of the patient. The cure rate for those who suffer two or more recurrences of C. difficile infection and receive a fecal transplant is 90 per cent—dramatically above the 30–40 per cent chance they face with antibiotic therapies.13
The US Food and Drug Administration (FDA) has responded to the emergence of this therapy by deciding that fecal microbiota meets the statutory definitions for both a drug and a biological product, and as such, warrants regulation by the agency as a drug.14 Drugs are defined in the Federal Food, Drug, and Cosmetic Act (FDCA) as ‘articles intended for use in the diagnosis, cure, mitigation, treatment, or prevention of disease, and articles (other than foods) that are intended to affect the structure and function of the body of man or other animals’.15 The Public Health Service (PHS) Act defines biological product as ‘a virus, therapeutic serum, toxin, antitoxin, vaccine, blood, blood component or derivative, allergenic product, protein, or analogous product applicable to the prevention, treatment, or cure of a disease or condition in human beings’.16 Biological products may be regulated as drugs under the FDCA, as biological products under the PHS Act, or under both laws.17
The FDA's decision to regulate fecal microbiota as a drug has both positive and negative consequences. Broadly speaking, there is a clear need for oversight of the use of fecal microbiota for transplantation, given the risks posed by stool-borne transmissible illnesses.18 The uniformity and expertise that the FDA can offer the industry are important institutional advantages both for stool providers navigating their legal responsibilities and for physicians seeking guidance on how to best help their patients.
However, the FDA's decision has proven problematic for scientific as well as legal and economic reasons. Not only does stool defy the typical scientific characterization that the FDA has long applied to small molecule and biologic drugs, but the potential do-it-yourself nature of the treatment poses particular concerns in the context of a regime involving periods of regulatory exclusivity. The goals of any regulation of FMT for the treatment of recurrent C. difficile infection should be to optimize access to the therapy while maintaining strict screening and reporting standards and oversight. This Essay will first examine how regulation under the drug paradigm fails to achieve these objectives, and will then evaluate available policy alternatives for the FDA to pursue, ultimately concluding that the FDA should pursue a hybrid regulatory model much like the one it has implemented in the case of cord blood and other human tissues.
I. AN INTRODUCTION TO FECAL MICROBIOTA TRANSPLANTATION
First reported in Western medical literature in 1958,19 FMT remained a fringe medical practice over the next 50 years. However, advances in molecular microbiology and the rising incidence and severity of C. difficile infection prompted a resurgence of interest in the therapy, especially as investigations consistently found that 90 per cent of recurrent C. difficile cases treated with FMT achieve clinical resolution.20
FMT refers to the practice of transplanting a filtered stool preparation from a healthy donor into the lower gastrointestinal tract, typically via colonoscopy or enema, or the upper gastrointestinal tract, for instance by nasojejunal tube. Published protocols guide the screening of stool donors for transmissible diseases including hepatitis and HIV,21 and the preparation of stool either for immediate use or for cryopreservation for later use.22 These protocols call for the stool to be mixed with a saline solution, filtered to remove fibrous material, and either administered to the patient immediately or mixed with glycerol and frozen until the time of administration. Protocols for encapsulating stool have also been validated and published.23
As mentioned above, systematic reviews and meta-analyses reviewing case series of FMT for recurrent C. difficile infection found that 90 per cent of patients were cured by the therapy.24 By comparison, as noted above, first-line antibiotics treatments cure C. difficile infection 82 per cent of the time,25 and after two or more recurrences, the cure rate of standard antibiotic therapies drops to below 40 per cent.26 In the first clinical trial evaluating the therapy, FMT proved so superior to standard antibiotics that the study's data and safety monitoring board stopped enrollment early, concluding that it was unethical to withhold the treatment from the members of the control group.27 Neither the systematic reviews and randomized trial nor a one- to two-year follow-up investigation found a link between FMT and any adverse events, though colonoscopy and upper routes of administration carry procedural risks.28 However, even though studies concluding a year or two post-FMT (a timeline which is typical of many clinical trials) have reported no adverse events, diseases that have been linked to the microbiome may surface years after FMT. As such, there remains a need for more investigation of the safety profile of FMT in the extreme long term.29
The mechanism by which the transplanted microbiota out-compete the C. difficile infection also requires further investigation. Analyses of microbiota communities before and after transplantation indicate that FMT addresses the lack of bacterial diversity in recurrent C. difficile patients and restores the composition of a normal microbiota community.30 It was even demonstrated that the presence of a single bacterial strain of C. scindens could enhance resistance to C. difficile infection in mice.31 These findings and others suggest the possibility of more targeted synthetic bacterial treatments of the infection, which companies including Seres Therapeutics are pursuing.32
While there is a paucity of data on the scale at which FMT is practiced today, OpenBiome, a public stool bank33 that supplies fecal microbiota preparations for clinical use, has provided material for more than 4,000 treatments of recurrent C. difficile infection to 300 clinical sites across the USA since it first began operations in 2013. OpenBiome is also sponsoring a multicenter study of the long-term safety profile of FMT in recurrent C. difficile patients, under an Investigational New Drug (IND) application.34
Beyond C. difficile, studies of the microbiome have revealed important relationships between intestinal bacteria and human health.35 FMT for the treatment of recurrent C. difficile infection represents a first foray into engineering the human microbiome to yield positive clinical results. Investigations into other applications of this therapy are underway, including testing FMT for the treatment of irritable bowel syndrome, inflammatory bowel disease, and a variety of metabolic diseases.36 Companies such as Vedanta Biosciences are also pursuing targeted synthetic bacterial treatments of subsets of these indications.37
II. THE DIFFICULTIES OF REGULATING FMT AS A DRUG
In May 2013, the FDA held a public workshop on FMT at which they first confirmed that fecal microbiota would be regulated as a drug.38 The FDA had written that while FMT ‘may be’ an effective therapy for the management of refractory C. difficile infection, its efficacy ‘ha[d] not yet been demonstrated in controlled clinical trials’.39 All uses of FMT would therefore need to be part of an IND application. In other words, patients who wished to be treated with a fecal transplant for recurrent C. difficile or other indications would need to participate in a clinical trial to do so.40
Physicians and scientists, including representatives of the Centers for Disease Control and members of professional medical societies, quickly responded with concern to the FDA's decision. They argued that the available evidence supporting FMT's effectiveness as a therapy for refractory C. difficile infection was too compelling for regulators to restrict its availability to the treatment groups of clinical trials.41 As such, just two months later, the FDA revised its decision and announced that it would exercise enforcement discretion when FMT was used to treat patients ‘with C. difficile infection not responding to standard therapies’.42 So long as the treating physician obtained adequate informed consent, the FDA would not require recurrent C. difficile patients to receive treatment through an FDA-reviewed clinical trial.
The FDA's decision not to enforce the IND requirement for recurrent C. difficile infection has significantly expanded access to the therapy, in no small part by creating a window in which public stool banks like OpenBiome could operate.43 As a result, it has undoubtedly led to improved patient outcomes and reduced health care costs. A recent study of the cost-effectiveness of fecal transplantation has determined that it saves a conservative estimate of 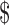 17,000 per patient.44 Public stool banks have developed protocols for screening and processing stool that are even more rigorous than previously published guidelines for directed donor fecal transplants,45 and that can represent even more cost savings by spreading the costs associated with stool screening and preparation over many treatments.46 However, the long-term regulatory outcome for FMT remains unclear, and as will be explained infra, if the FDA proceeds along its current pathway, the most likely outcome is one in which a single provider receives a license from the FDA to produce fecal microbiota for transplantation, at which time the FDA will likely no longer exercise its enforcement discretion.
17,000 per patient.44 Public stool banks have developed protocols for screening and processing stool that are even more rigorous than previously published guidelines for directed donor fecal transplants,45 and that can represent even more cost savings by spreading the costs associated with stool screening and preparation over many treatments.46 However, the long-term regulatory outcome for FMT remains unclear, and as will be explained infra, if the FDA proceeds along its current pathway, the most likely outcome is one in which a single provider receives a license from the FDA to produce fecal microbiota for transplantation, at which time the FDA will likely no longer exercise its enforcement discretion.
There are several problems with this outcome, each of which we will address in further detail in the sections to follow. First, to effectively regulate stool as a drug, regulators will need to address the challenge of characterizing active ingredients and guaranteeing safety and quality in spite of the variability of stool.47 The FDA has not yet publicly explained how it plans to address this challenge. Second, this outcome will likely impose significant burdens on the health care system, burdens that the FDA has not yet faced with other therapies. As an initial matter, it is probable that a license to produce fecal microbiota for transplantation will have the effect of granting market exclusivity to a single provider for the provision of ubiquitous human stool. Yet, in this case, like only a very few others, doing so would allow that provider to capture value from knowledge that already exists in the public domain, and is already being used by multiple providers to meet an urgent and growing public health need. This outcome would work against the public interest by inhibiting treatment choices for patients and raising prices across the health care system. When combined with the do-it-yourself potential of FMT, the decision to regulate fecal microbiota as a drug may impose safety risks that are unique to this therapy and do not arise in the context of other, more traditional drugs. Third, given the relative freedom with which clinicians may prescribe drugs off-label, regulating stool under the traditional drug paradigm may also discourage investment into explorations of fecal therapies for other indications, if off-label uses of fecal microbiota can meet demand from patients with those conditions to which researchers have made tentative links to microbiome health. This concern is not unique to FMT, but is worth attention because in this case the FDA has the ability to select an alternative regulatory paradigm that would mitigate, if not eliminate, this result.
A. Scientific complexities
From a scientific perspective, the regulation of stool as a drug is complicated by the material's complexity and inconsistency across samples. The microbial and metabolic contents of human stool are known to vary enormously across individuals and over time within individuals.48 Unless the active components are identified, purified, and tested, it will not be possible to guarantee that the product is consistent across batches.
This characteristic suggests that the regulation of stool should be tied to the process by which it is prepared for transplantation, rather than to the variable contents of the product.49 However, in the case of human stool, this ‘process’ not only includes preparation methods following stool collection; it is also driven by the complex and very specific life history of the individual donors, and might arguably be defined by the donors themselves.50 More practically, the ‘process’ associated with preparing fecal microbiota for transplantation could be narrowly defined by the very particular methods used to filter and prepare stool from donors who are selected in accordance with a consistent protocol.
Thus, under the drug regulation paradigm, given the characteristic variance of stool, the FDA would be underregulating stool from both a safety and an efficacy perspective should it simply provide traditional drug licenses to stool-based products. Stool preparations have distinct compositions and resultant therapeutic properties, and they also carry distinct pathogenic threats, and thus warrant ongoing safety monitoring and evaluation beyond that which typically accompanies an approved drug. Perhaps more importantly, it would be inappropriate to provide licenses that bear little relationship to the specific donor screening or stool preparation methods used by the stool provider. Instead, an approach that narrowly defines the process by which stool material is assessed and prepared for fecal transplantation would be more consistent with our understanding of the extreme complexity of the microbial system. However, the standard drug paradigm does not clearly allow the FDA to specify who may donate stool and which pathogens must be screened for. It is important that these complicated issues, many of which are still not well understood by science and medicine, be considered in a public regulatory context, rather than in the FDA's private negotiations with a single company.
B. Exclusivity problems
Other problems with the FDA's decision to regulate FMT as a drug stem from one of the FDA's often-overlooked powers: its statutory requirement to award periods of exclusivity for approved drugs under a variety of conditions. As one example, the Orphan Drug Act awards seven years of marketing exclusivity to FDA-designated orphan drugs,51 during which time the FDA may not approve a new or generic drug application for the same product and indication.52 The recently enacted Biologics Price Competition and Innovation Act goes further, providing 12 years of data exclusivity for reference biologics, during which the FDA may not approve an application for a biosimilar product that relies on the innovator company's clinical trial data.53
Although the appropriate lengths of exclusivity periods like these have been hotly contested,54 there is general agreement that in many cases an exclusivity period helps provide innovative drug manufacturers with sufficient incentives to carry new products through the long, expensive, development process.55 The Orphan Drug Act is the paradigmatic example: where the market for a drug is by definition small, a long exclusivity period helps assure pharmaceutical companies that they can recoup their investment into a drug for treating an orphan disease.56 Exclusivity then functions to raise the price of the drug above what it would be in an otherwise free market, but this is a social bargain that has been made for purposes of allowing companies to capture more of the value that they generate through innovation, incentivizing them to engage in innovative drug development activities that would not otherwise have occurred.
But sometimes this bargain breaks down. In some cases, the FDA has approved a drug when that very same drug was already widely, cheaply available on the market. The FDA's approval and grant of an exclusivity period in such cases may function to clear the market of existing therapies, permitting the new applicant to charge monopoly prices without fulfilling their end of the social bargain and bringing a truly new product to market. Scholars have been concerned about two such cases that have occurred recently, with disparate outcomes.
The story of colchicine, a drug used to treat acute gout flares, has likely been the most widely reported. Colchicine had been widely and cheaply available in generic form since the 1800s,57 long before the FDA began regulating the safety and efficacy of drugs. As such, even after the FDA acquired such authority in 1962,58 many drugs that were already on the market remained publicly available, although they were never formally evaluated by the FDA. A pharmaceutical company decided to invest in conducting a series of trials involving colchicine, to demonstrate its safety and its efficacy for gout as well as for an orphan disease known as familial Mediterranean fever.59
In 2009, the FDA approved the use of colchicine for these indications, and under the Orphan Drug Act the manufacturer received seven years of marketing exclusivity—for a product that was already on the market.60 The manufacturer and the FDA subsequently took legal action to force all other makers of colchicine to exit the market,61 and the manufacturer then raised the price per pill from about 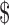 0.09 to
0.09 to 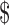 4.85. The grant of exclusivity resulted in significantly increased costs not only for patients but also for Medicare and Medicaid, common payers for the drug. Scholars have persuasively argued that the rewards granted to colchicine's now monopoly supplier vastly exceed the value of the information provided to the public, and that public health is likely to suffer as a result.62
4.85. The grant of exclusivity resulted in significantly increased costs not only for patients but also for Medicare and Medicaid, common payers for the drug. Scholars have persuasively argued that the rewards granted to colchicine's now monopoly supplier vastly exceed the value of the information provided to the public, and that public health is likely to suffer as a result.62
Another recent example involves a synthetic hormone used to reduce the risk of preterm births in pregnant women with a history of preterm births. The hormone had been available to women in compounded form for many years, and it was relatively inexpensive, at 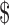 15 per injection or
15 per injection or 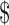 300 per pregnancy.63 But in February 2011, the FDA approved Makena, a branded form of the hormone, for this orphan indication. The marketing company first set a price roughly 100 times higher than that of the compounded therapy, for a total cost of almost
300 per pregnancy.63 But in February 2011, the FDA approved Makena, a branded form of the hormone, for this orphan indication. The marketing company first set a price roughly 100 times higher than that of the compounded therapy, for a total cost of almost 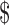 30,000 per pregnancy. Although they later cut that price in half, the cost still vastly exceeded the cost of the compounded form.64
30,000 per pregnancy. Although they later cut that price in half, the cost still vastly exceeded the cost of the compounded form.64
In this case, though, Makena's maker could not secure a monopoly over the distribution of the hormone. Although the FDA could not have approved another manufacturer's application under the Orphan Drug Act, the FDA stated that it would decline to take enforcement action against pharmacies that compounded the drug for patients unless safety problems were reported.65 This unusual decision was likely driven by several motivations. Historically, the FDA has had a complicated relationship with compounding pharmacies—its regulatory authority in this area is unclear, and the FDA's exercise of its enforcement discretion in this case avoided a legal battle with the compounding pharmacies. However, the FDA was also likely motivated by concerns about patient access, having observed the results of the colchicine case.
If the FDA continues in its efforts to regulate fecal microbiota as a drug, it may soon be required to make a similar choice in the case of FMT. Responding to an application from a company called Rebiotix, the FDA has recently designated ‘fecal microbiota’ as an orphan product for the treatment of recurrent C. difficile infection.66 Rebiotix is currently conducting clinical trials in an effort to gain FDA approval,67 and if it is successful, the Orphan Drug Act would prevent the FDA from approving another manufacturer's application to process and distribute whole stool material for FMT, barring a showing of clinical superiority.68
The question then becomes whether other stool banks would be able to stay in business, as in the case of Makena, or whether they would be forced out of the market, as in the case of colchicine. In our view, the FDA ought to continue exercising its enforcement discretion and permit other stool banks to remain as market participants, analogous to its behavior in the Makena case. However, the FDA certainly faces countervailing pressures. Perhaps most obviously, the FDA has prioritized the removal of unapproved drugs from the market.69 The FDA would also likely be sued for this behavior, as it was in the Makena case.70 Also, given the FDA's past attempts to curtail its use of enforcement discretion in the FMT context, it is difficult to think that it would be likely to continue exercising its enforcement discretion here.
Even if the FDA did continue to exercise its enforcement discretion after granting Orphan Drug exclusivity to one company, the award of exclusivity itself would likely spell trouble for other stool banking operations. In the case of colchicine, recall that before the FDA took action, the approved company independently brought suit against other colchicine manufacturers, seeking to force them to exit the market.71 Should the approved FMT company do the same, even if it would not be likely to win such a suit,72 the time and expense required by litigation would likely have an adverse effect on the ability of existing stool banks to continue providing care, especially given their non-profit approach.
Importantly, the possibility of having a single stool provider on the market in itself is not a problem. The survival of any particular FMT company is incidental to the primary goal: to ensure patients have access to safe, effective treatments. The problem, rather, is two-fold. First, to the extent that the prospect of granting exclusivity in the colchicine and Makena cases has troubled scholars because the reward received by companies would be disproportionate to the social contribution made, exclusivity for FMT raises the same concerns. But second, and even more troubling, is a factor unique to FMT: the potential for do-it-yourself treatments. Since monopoly power leads to monopoly prices, if Rebiotix chooses to sell patients stool for thousands or tens of thousands of dollars,73 depending on health insurance coverage,74 patients may resort to essentially free at-home transplantations, using friends or family members, screened at the patient's discretion, as donors. The problem is a simple one of trading off access, cost, and safety. At a low price, access can be assured. But at a high price, the prospect of a monopoly FMT system poses significant safety concerns.
C. Safety concerns
As we have explained, FMT as a therapy is unique for the difficulty of its characterization and the simplicity of its production, and each of these characteristics raises special safety concerns. First, the complexity of the microbial community in stool and the variability across stool samples makes it nearly impossible to guarantee the contents from batch to batch. As such, ongoing monitoring for the presence of possible pathogens is necessary for maintaining a safe product and should either be considered part of the approved manufacturing process or a condition imposed on manufacturers. Second, although there is little chance that patients will manufacture traditional small molecule therapies in their bathrooms, processing stool for transplantation at a basic level requires very little training or equipment. Instructional guides produced by non-professionals and posted on YouTube have received tens of thousands of views.75 Patient support groups and online forums include lengthy step-by-step instructions along with discussions of best practices for mixing stool in a low-cost blender and administering it via enema. The risk with any regulatory strategy that restricts access to this therapy, whether by raising barriers to clinical practice or by supporting monopolistic pricing, is that it will motivate desperate patients to pursue self-treatment.76
Unsupervised, do-it-yourself treatments carry considerable risk of the transmission of pathogens from improperly screened and handled stool. Few healthy individuals would be deemed eligible for donating stool for fecal transplantation. Only six per cent of prospective donors to OpenBiome pass the full screening process, which includes a 109-item clinical assessment administered by a nurse or physician, and 30 stool and blood screens.77 With a lack of long-term safety data on patient outcomes post-FMT, it is prudent to be overly cautious about screening for diseases that are potentially mediated by the microbiome; for instance, researchers have notably linked the microbiome to obesity,78 metabolic syndrome,79 and behavior.80 Similarly, it is just as important to collect longitudinal safety data to identify any conditions that may be transmitted via stool of which we are unaware.81 Thus, given the known and unknown risks that come with improper donor screening and inadequate patient follow-up, the ease with which patients may prepare and administer fecal transplants themselves without medical supervision, any regulatory outcome that results in restricted access by either limiting supply or significantly increasing the cost of therapy should be adopted extremely cautiously.82
III. TOWARD AN IMPROVED PARADIGM FOR FMT REGULATION
As we have explained, while FMT may fall within the broad statutory definition of ‘drug’,83 there are reasons to be concerned about regulating it wholesale under that paradigm. In the past, when similar concerns have arisen for other technologies involving human cellular or tissue-based products of various types, the FDA has adopted or developed alternative regulatory paradigms. It should also consider doing so here. There are several potential paradigms available, each with their own advantages and disadvantages.
A. Blood
Although blood, like FMT, facially appears to qualify as a ‘drug’ under the FDCA, the FDA has promulgated regulations establishing an entirely separate system for regulating blood and blood products as biologic products under the PHS Act. This regulatory system is largely focused on ensuring the safety of the blood supply, with the FDA restricting the possible donor pool84 and requiring extensive testing and quarantine of donated blood before it can be transfused.85 The FDA also regulates blood banks themselves, with highly specified registration and facility requirements.86 Yet, generic licenses are issued to banks meeting requirements, rather than being awarded exclusively to one or a small number of institutions. In many ways, the FDA regulates blood more like a commodity, rather than a therapeutic compound.87
The FDA's regulation of whole blood and blood products is in some ways an artifact of history, however. Blood transfusions were routinely performed in the 1800s, and the discovery of blood typing in the early 1900s greatly improved the safety and efficacy of the treatment.88 By the time the FDA officially acquired the authority to regulate blood in 1970,89 the safety and efficacy of transfusions was beyond question. A far greater concern, in the FDA's view, was the potential for scarcity. Unlike most traditional drugs, production of blood cannot easily be scaled up. It relies on donors, and the possibility of granting exclusive licenses to certain banks would be unthinkable, in light of the potential human cost. The FDA's commitment of resources to create blood's existing regulatory structure must be understood in the context of this history.
Blood and fecal microbiota share many scientific characteristics that distinguish them both from traditional drugs. Unlike traditional drugs, blood and fecal microbiota pose safety concerns involving transmissible diseases. Further, in both cases we ought to be concerned about the potential for scarcity, due to the need at present to procure both blood and stool from human donors. And just as the efficacy of blood and blood products was known and appreciated prior to the FDA's involvement in this area, FMT's efficacy for at least one indication—recurrent C. difficile infection—has already been demonstrated.
Yet, blood and fecal microbiota are distinct in at least one important way: unlike blood, scientists believe that FMT may be useful for a wide range of potential indications that implicate the human microbiome. Some of these, such as ulcerative colitis or Crohn's Disease, may naturally be suspected due to FMT's locus of action. Yet others, like Parkinson's disease, multiple sclerosis, and childhood regressive autism,90 are also potential indications. Research into these indications but also into the microbiome more broadly is desperately needed, and the current paradigm for regulating blood is not set up to oversee such clinical examination.
Instead, then, we might look to another regulatory paradigm whose history more closely resembles that of FMT. Specifically, FMT's development quite strongly resembles the history of the use of cord blood, whose potential to treat a range of diseases was not discovered until the 1980s.91 Cord blood, which is regulated by the FDA under its related oversight system for human cells and tissues, is a second potential model for FMT.
B. Tissue
Blood is not the only therapeutic that the FDA has carved out of its system for regulating traditional drugs. The FDA has also issued special regulations for human cells, tissues, and cellular and tissue-based products or HCT/Ps, which are ‘human cells or tissues that are intended for implantation, transplantation, infusion, or transfer into a human recipient’,92 a group that includes bones, ligaments, skin, and cord blood.93 Like blood, these human tissues are regulated as biological products under the PHS Act, in which the safety of the specimen is paramount.94 But for HCT/Ps and specifically for cord blood, the FDA has chosen to regulate different uses in different ways.
Specifically, where banked cord blood is intended for use by its donor or that donor's close relatives, that cord blood is regulated like general whole blood, as a biological product, in a way that prioritizes the safety of the process. Accordingly, private cord blood banks must be registered and licensed much like typical blood banks, and they must comply with all tissue regulations. Cord blood stored for personal use may be used for any indication. Where cord blood is intended for use by a patient unrelated to the donor, however, it is regulated both as a biological product and as a drug.95 This means that cord blood when sourced from an unrelated donor must be approved for use under an IND or BLA for the requested indication, a process that might involve extensive clinical trials. To put it succinctly, for cord blood, FDA regulation is concerned with both the intended recipient and the intended use, toggling the type and level of regulation along both dimensions.
A key virtue of adopting the HCT/P approach in the FMT context is that it would permit the FDA to regulate different uses of FMT differently. There is already strong evidence to suggest that FMT can be safe and effective for the treatment of recurrent C. difficile infection.96 However, whether FMT can be safe and effective for the treatment of other indications is not known. As such, the FDA might use their hybrid approach to regulating HCT/Ps like cord blood as a guide. In effect, choosing to regulate FMT as a biological tissue product would allow the FDA to permit its use in treating recurrent C. difficile infection, while still requiring additional proof before it could be prescribed for other indications.
Adopting the HCT/P regulatory paradigm wholesale would, however, permit individuals to bank and use their own stool or stool from any first- or second-degree relatives for any purpose. As such, regulators ought to be concerned about the potential for this carve-out to reduce the benefits of adopting the HCT/P approach in terms of regulating different indications in different ways. Arguably, though, it is preferable for individuals who are determined to use FMT for unapproved indications to do so through a process that at the very least minimizes the safety concerns associated with the therapy. If no such exemption existed, the reality is that patients would undergo FMT in an unregulated, do-it-yourself fashion, not that they would be unable to undergo FMT at all. The FDA has faced this same tradeoff in the context of semen, which has similar potential for at-home administration. The FDA therefore imposes fewer and less strict requirements on directed sperm donors than on anonymous donors.97
In order to regulate the safety of FMT along the same lines as either blood or tissue, though, the FDA would need to either interpret or amend its existing regulations. At present, although fecal material qualifies as a biological product, the FDA's Tissue Reference Group has recommended that it does not meet the statutory definition of an HCT/P and therefore could not at present be subsumed within the relevant regulations.98 The Reference Group did not publicly explain their reasons, but a close reading of the regulations suggests two primary reasons the Reference Group may have reached this conclusion, each of which can be surmounted.
First, the Group members may have reasoned that although fecal microbiota preparations are at least partly composed of ‘human cells or tissues that are intended for implantation, transplantation, infusion, or transfer into a human recipient’, fecal microbiota falls within the explicit regulatory carve-out for ‘secreted or extracted human products’, which include milk and collagen.99 These products are not considered to be HCT/Ps, and therefore are not regulated under the tissue paradigm—their collection and storage is not subject to the same rigorous safety requirements that apply to cord blood products. Importantly, the FDA has made exceptions to this exception before—the carve-out for secreted products goes onto state that ‘semen is considered an HCT/P’, suggesting that otherwise semen would be considered a secreted product as well.100
If this limitation on what constitutes an HCT/P is the basis for the Reference Group's conclusion, the FDA would likely need to conduct an official notice-and-comment rulemaking to clarify that fecal microbiota is also considered an HCT/P.101 A simpler approach would be to attempt to issue an interpretive rule or guidance document indicating that fecal matter simply does not qualify as a ‘secreted or extracted human product’.102 However, this approach might be viewed by courts to ‘purport[] to impose legally binding obligations or prohibitions on regulated parties’,103 and therefore might be rejected as an effort to circumvent the Administrative Procedure Act.
Second, the Reference Group may have reasoned that FMT does not require the transplantation of ‘human cells or tissue’, but that it rather requires the transplantation of microbial cells or tissue.104 This concern is easily surmounted through guidance documents. The simplest option would be for the FDA to clarify that FMT as it is currently practiced simply does fall into the category of ‘articles containing … human cells or tissues’. Although it is possible that in the future the material for FMT will not need to be sourced from humans and therefore will not necessarily contain human cells,105 at present the raw material for FMT is inexorably sourced from humans and inevitably contains human cells.
Yet, either of these approaches would require a significant expenditure of resources, which the FDA might not be willing to allocate without political pressure of the kind they faced in the cord blood context.106 Given the potential for publicly reported adverse events that may be attributed to the underregulation of processing and storage of stool samples, the FDA might be willing to take such actions, but it is by no means clear at this time. If they are not willing to devote the resources, a third option—simple continued use of enforcement discretion—might be even more attractive.
C. Enforcement discretion
As discussed above, the FDA has currently chosen to use its enforcement discretion to de facto permit physicians to provide FMT for the treatment of recurrent C. difficile infections. If the FDA continued to exercise this discretion, it would be able to achieve a similar outcome as it could achieve in the HCT/P context. That is, physicians would be free to provide FMT to treat C. difficile infections not responding to standard therapies, but a company hoping to market FMT for other indications would still need to complete clinical trials.
Importantly, if the FDA chose to continue exercising its enforcement discretion for FMT for recurrent C. difficile, health care providers would not be without expert advice on the subject. Professional organizations like the American Gastroenterological Association have issued best practice guidelines for FMT, including restrictions on donor selection, sample processing, and facilities management.107 The FDA's regulation of blood products provides a helpful example. In that context, private standard-setting organizations like the American Association of Blood Banks had already begun to regulate the blood supply before the FDA was involved in the process, a role which the professional association continues to fulfill today. Although these guidelines lack the legal force of the FDA's regulatory scheme, they are not toothless. Physicians subject to malpractice actions who have failed to adhere to guidelines that represent the standard of care may be subject to professional discipline. By extension, stool banks would face pressure to demonstrate compliance with best practice guidelines from the clinical sites and insurers using their services.
A key virtue of this approach is that it permits the FDA to postpone the off-label prescription problems that might arise if it does approve Rebiotix's FMT application. Essentially, although manufacturers are not permitted to advertise their products for off-label uses, doctors are free to prescribe therapies and devices for off-label indications, with one study suggesting that about 20 per cent of all drugs are prescribed off label.108 Too often there is no scientific support for off-label prescriptions, and with a grant of exclusivity, the manufacturer has little financial incentive to conduct further validating studies. Because approving FMT for the treatment of recurrent C. difficile infections would permit physicians to prescribe it for all other unproven indications, it would perversely delay the FDA's acquisition of the very information it wants most—data on the use of FMT for other, non-C. difficile indications—by dampening the financial incentive to conduct those additional trials.109
This approach also has benefits from a regulatory efficiency standpoint. When compared with the possibility of constructing an entirely new regulatory system, as the FDA has done in the context of blood, or even completing a notice-and-comment rulemaking to bring FMT within the scope of the HCT/P regulatory scheme, the possibility that the FDA could achieve a somewhat similar result by simply doing nothing is undoubtedly attractive. Further, it is useful to recall that the use of stool for FMT is only a bridge technology, at least in the C. difficile context. Companies seeking to understand the microbiome and use it directly to treat the disease110 will (perhaps soon) obviate the need to use donated stool for these purposes. As such, any regulatory action taken by the FDA may be obsolete within a decade.
This approach is certainly not a perfect one. The possibility that enforcement discretion may be withdrawn at any time places stool processing companies in a precarious, uncertain position regarding their legality. It also does not avoid the off-label prescribing problem, but simply delays it until such time as the FDA approves FMT for a different indication. Still, the virtues of the status quo ought to be considered carefully.
D. Feasibility in light of path-dependency
The FDA cannot pursue this option on a blank slate. The possibilities above must be considered in light of decisions that the agency has already made in this area that may inhibit its ability to adopt a more flexible paradigm in the near term. To briefly summarize what has been described above, in 2012, the FDA's Tissue Reference Group recommended that fecal microbiota did not meet the statutory definition of an HCT/P.111 In 2013, the FDA explained that it considered fecal microbiota for transplantation a drug, and would regulate it as such. A few months later, the agency permitted Rebiotix to pursue licensure for its fecal microbiota preparation under the approval program for orphan drugs and subsequently awarded it an Orphan Drug designation for treating recurrent C. difficile infection.112 In light of the scientific, legal, and public health arguments against regulating fecal microbiota preparations for C. difficile therapies as drugs, what options do the rest of these decisions leave the FDA?
Above, we considered the option of surmounting the Reference Group's recommendation through regulation, and indeed, the FDA could conceivably regulate FMT as an HCT/P in addition to regulating it as a drug, though this route may prove costly. We also considered the FDA's ability to continue exercising its enforcement discretion in an attempt to promote access to FMT for recurrent C. difficile, but this option would indeed be jeopardized in light of its prior decisions in this area. Beyond regulation, though, there are other ways in which the FDA could use various legal tools to minimize the social harms that granting an exclusive license to provide stool for transplantation would cause.
One option would be for the FDA to narrowly construe its Orphan Drug designation, defining the drug itself narrowly by the very specific processing methodology to which it is subject. As a result, other companies seeking to provide stool for transplantation would still need to complete clinical trials, but companies would be able to invent around Rebiotix's method. Or alternatively, the FDA might approve another company's FMT preparation on the grounds of clinical superiority.113 As a third option, since the existing designation is limited to recurrent C. difficile infections, a product could be tested and approved for initial occurrences of C. difficile.114 However, these would be unusual outcomes.
There are still other more promising ways for the FDA to regulate this new and rapidly evolving field with flexibility. Perhaps the most novel would be to carefully monitor any approved company with exclusivity for the presence of a drug shortage. Defined broadly as a situation in which ‘the demand or projected demand for the drug within the United States exceeds the supply of the drug’,115 in such an event the FDA is empowered to take a range of actions, including obtaining the product from other sources. And here it is again critical to remember the ways in which FMT is not a typical drug because a company cannot simply ‘scale up’ production of stool by building more factories or increasing production efficiency. It must instead recruit additional healthy donors and collect the product from them. OpenBiome, with which one of the authors of this Essay is affiliated, has curated the raw material for just 7400 treatments in its two years of existence. The FDA might reasonably decide that in the event of a shortage, whether produced by an unanticipated surge in demand or barrier to supply, it would take any single corporation too long to recruit enough healthy donors and de-risk them through multiple rounds of screens for it to provide coverage alone. Indeed, any shortage, whether now or in the future, would be made worse if a single company held a monopoly over stool procurement and processing.
IV. CONCLUSION
The question of how the FDA should regulate FMT to prioritize the safety of the procedure while at the same time ensuring its effectiveness is a complicated one. As we have explained, trying to shoehorn FMT into the traditional drug regulatory paradigm is problematic, with the potential to both underregulate safety and overregulate access. Regulating FMT as a tissue would more fully address our concerns, since so much is still unknown about FMT's potential efficacy across a wide range of indications.
A model much like the one the FDA has adopted in the case of HCT/Ps like cord blood would seem to be the most appropriate solution. Under this scenario, the FDA would enforce a suite of regulations that control for the safety of fecal microbiota preparations so long as the material is only being used to treat recurrent C. difficile infection. For all other uses, the FDA would require demonstrations of safety and efficacy following the path for investigational new drugs. This paradigm would grant C. difficile patients safe access to ubiquitous human stool, the data for which is available in the public domain, while at the same time encouraging scientists to study new applications for the therapy.
FMT presents the FDA with a new regulatory challenge, and it provides the agency with a chance for thinking critically and creatively about the most appropriate ways to oversee this new technology. The FDA should seize the opportunity, as other emerging technologies like 3-D printing and mobile health may soon pose similar regulatory challenges for allowing access under medical supervision while restricting that access to evidence-based practices.116 We hope the considerations presented in this Essay will help the FDA in its endeavors to do so.
Footnotes
Rachel E. Sachs, JD, MPH, is an Academic Fellow at the Petrie-Flom Center for Health Law Policy, Biotechnology, and Bioethics at Harvard Law School.
Carolyn A. Edelstein, MPA, is the Director of Global Partnerships at OpenBiome, a public, non-profit stool bank.
Shelley S. Magill et al., Multistate Point-Prevalence Survey of Health Care-Associated Infections, 370 New Eng. J. Med. 1198, 1198 (2014).
There are now 11.2 cases of C. difficile infection per 10,000 inpatient hospitalizations. Marya D. Zilberberg, Andrew F. Shorr & Marin H. Kollef, Increase in Adult Clostridium Difficile–Related Hospitalizations and Case-Fatality Rate, United States, 2000–2005, 14 Emerg. Infect. Dis. 929, 929 (2008).
The White House, National Action Plan for Combating Antibiotic-Resistant Bacteria, 60, Mar. 2015, https://www.whitehouse.gov/sites/default/files/docs/national_action_plan_for_combating_antibotic-resistant_bacteria.pdf (accessed June 18, 2015).
Erik R. Dubberke & Margaret A. Olsen, Burden of Clostridium Difficile on the Healthcare System, 55 Clin. Infect. Dis. S88, S88 (2012).
Ciaran P. Kelly & J. Thomas LaMont, Clostridium Difficile—More Difficult Than Ever, 359 New Eng. J. Med. 1932, 1936 (2008).
Id. at 1935.
Laurica A. Petrella et al., Decreased Cure and Increased Recurrence Rates for Clostridium Difficile Infection Caused by the Epidemic C. Difficile BI Strain, 55 Clin. Infect. Dis. 351, 352 (2012).
Kelly & LaMont, supra note 5, at 1936.
Id.; see also Gauree G. Konijeti et al., Cost-Effectiveness of Competing Strategies for Management of Recurrent Clostridium Difficile Infection: A Decision Analysis, 58 Clin. Infect. Dis. 1507, 1507 (2014).
See eg J. C. O'Horo et al., Treatment of Recurrent Clostridium Difficile Infection: A Systematic Review, 42 Infection 43, 43 (2014).
The White House, supra note 3, at 5, 60.
See generally eg Zain Kassam, Christine H. Lee & Richard H. Hunt, Review of the Emerging Treatment of Clostridium difficile Infection with Fecal Miocrobiota Transplantation and Insights into Future Challenges, 34 Clin. Lab. Med. 787 (2014). Although there is no scientific reason to believe that FMT would not be efficacious in treating first occurrences of Clostridium difficile, it has not yet been tested in such populations, and given the unknown long-term safety profile of FMT, it may not be suitable for such purposes on balance.
Konijeti et al., supra note 9, at 1511.
Food & Drug Administration, Fecal Microbiota for Transplantation: Scientific and Regulatory Issues 309, May 2, 2013, http://www.fda.gov/downloads/BiologicsBloodVaccines/NewsEvents/WorkshopsMeetingsConferences/UCM352903.pdf (accessed June 18, 2015).
21 U.S.C. § 321(g)(1) (2012).
42 U.S.C. § 262(i)(1) (2012).
Some therapies that might be colloquially referred to as ‘drugs’—for instance, some oncology treatments—are also considered to be biological products and regulated through Biologics License Applications, rather than New Drug Applications. For the purposes of this Article, however, we are concerned with the group of ‘biological products’ that includes substances that are generally viewed as parts of or sourced from the human body—substances like blood, bone, and even semen.
See Matthew J. Hamilton et al., Standardized Frozen Preparation for Transplantation of Fecal Microbiota for Recurrent Clostridium difficile Infection, 107 Am. J. Gastroenterol 761, supplementary appendix 1 (2012).
See generally Ben Eiseman et al., Fecal Enema as an Adjunct in the Treatment of Pseudomembranous Enterocolitis, 44 Surgery 854 (1958).
Zain Kassam et al., Fecal Microbiota for Clostridium difficile Infection: Systematic Review and Meta-Analysis, 108 Am. J. Gastroenterol 500, 508 (2013); Sumei Sha et al., Systematic Review: Faecal Microbiota Transplantation Therapy for Digestive and Nondigestive Disorders in Adults and Children, 39 Aliment. Pharmacol. Ther. 1003, 1004 (2014).
Hamilton, supra note 18, at supplementary appendix 1.
See generally id.
See generally id.
See Kassam, supra note 20, at 504–5; Sha, supra note 20, at 1003.
See Kelly & LaMont, supra note 5, at 1935.
Id. at 1936.
Els van Nood et al., Duodenal Infusion of Donor Feces for Recurrent Clostridium difficile, 368 New Eng. J. Med. 407, 409 (2013).
See Kassam, supra note 20, at 505; Lawrence J. Brandt, et al., Long-Term Follow-up of Colonoscopic Fecal Microbiota Transplant for Recurrent Clostridium difficile Infection, 107 Am. J. Gastroenterol. 1079, 1079 (2012) (following patients for a mean of 17 months post-FMT).
See Kassam, supra note 21 at 506.
See eg Matthew J. Hamilton et al., High-Throughput DNA Sequence Analysis Reveals Stable Engraftment of Gut Microbiota Following Transplantation of Previously Frozen Fecal Bacteria, 4 Gut Microbes 125 (2013).
See generally Charlie G. Buffie et al., Precision Microbiome Reconstitution Restores Bile Acid Mediated Resistance to Clostridium difficile, 517 Nature 205 (2015).
Seres Therapeutics, Seres Health Presents Final Data for Study of SER-109 in Recurrent Clostridium difficile Infection at ICAAC 2014 Conference, Sept. 10, 2014, http://serestherapeutics.com/news/newsroom/seres_health_presents_final_data_for_study_of_ser-109_in_recurrent_clostridium_difficile_infection_at_icaac_2014_conference/ (accessed June 18, 2015)
In addition to OpenBiome, some hospitals and clinics also manage internal stool banking programs. In late 2014, BloodSource, a California-based blood bank, opened AdvancingBio, a stool bank that has processed and banked 47 fecal microbiota preparations as of Mar. 2015. Sammy Caiola, Sacramento Nonprofit Center's Collection Helps Patients with Intestinal Disease, Sacramento Bee, Mar. 13, 2015, http://www.sacbee.com/news/local/health-and-medicine/healthy-choices/article14072969.html (acces-sed June 18, 2015).
OpenBiome, OpenBiome Receives Grant to Support FMT Patient Safety Study, Aug. 7, 2014, http://www.openbiome.org/press-releases/2014/8/8/openbiome-receives-grant-to-support-fmt-patient-safety-study (accessed June 18, 2015).
Sha, supra note 20, at 1004.
See eg Paul Moayyedi et al., Canadian Association of Gastroenterology Position Statement: Fecal Microbiota Transplant Therapy, 28, Can. J. Gastroenterol. Hepatol. 66, 67 (2014).
See Alex Lash, With Vedanta Deal, J&J Marks Big-Pharma Milestone in the Microbiome, XConomy, Jan. 13, 2015, http://www.xconomy.com/boston/2015/01/13/with-vedanta-deal-jj-marks-big-pharma-milestone-in-the-microbiome/ (accessed June 18, 2015).
See supra note 14, at 309.
Food & Drug Administration, Public Workshop: Fecal Microbiota for Transplantation, Announcement of May 2013 Workshop, http://www.fda.gov/BiologicsBloodVaccines/NewsEvents/WorkshopsMeetingsConferences/ucm341643.htm (accessed June 18, 2015).
Patients would likely also be able to access FMT through an expanded access protocol.
Guidance for industry: enforcement policy regarding investigational new drug requirements for use of fecal microbiota for transplantation to treat Clostridium difficile Infection not responsive to standard therapies; availability, 78 Fed. Reg. 42,965, 42,965 (July 18, 2013).
Id.
Mark B. Smith, Colleen Kelly & Eric J. Alm, How to Regulate Faecal Transplants, 506 Nature 290, 290 (2014).
Konijeti et al., supra note 9, at 1511.
Mark B. Smith et al., A Scalable Workflow for Screening, Processing and Characterizing Donor Stool for Use in Fecal Microbiota Transplantation; Poster presentation, 2014 James W. Freston Conference, 2014 Am. Gastroenterological Ass'n (Aug. 16, 2014).
See Smith, supra note 42, at 291.
Id.
See generally eg J. Gregory Caporaso et al., Moving Pictures of the Human Microbiome, 12 Genome Biol. R50 (2011); Lawrence A. David et al., Host Lifestyle Affects Human Microbiota on Daily Timescales, 15 Genome Biol. R89 (2014); Human Microbiome Project Consortium, Structure, Function, and Diversity of the Healthy Human Microbiome, 486 Nature 207 (2012).
The FDA currently regulates many biologic drugs in this fashion, regulating not simply the compound itself but the process by which the drug is produced.
David, supra note 48, at 1–2.
Orphan drugs are those which treat a ‘rare disease or condition’, 21 U.S.C.A. § 360bb(a) (2006), which is defined primarily by the number of people in the United States afflicted with the disease. Id. at § 360cc(a)(2).
21 U.S.C.A. § 360cc(a) (2006). The FDA may, however, approve an application of a different drug for the same indication. Joseph A. Levitt & John V. Kelsey, The Orphan Drug Regulations and Related Issues, 48 Food & Drug L.J. 525, 526–28 (1993). As such, the way in which the FDA defines the drug is key to the strength of the Act's exclusivity periods. Id.
42 U.S.C.A. § 262(k)(7)(A) (2011).
Compare eg Federal Trade Commission, Follow-on Biologic Drug Competition 5, 2009 (suggesting that having no exclusivity period would be sufficient to promote innovation), with Biotechnology Industry Organization, A Follow-On Biologics Regime Without Strong Data Exclusivity Will Stifle the Development of New Medicines 1, 4 2007 (arguing for a minimum of 14 years of exclusivity). See also Yaniv Heled, Patents vs. Statutory Exclusivities in Biological Pharmaceuticals–Do We Really Need Both?, 18 Mich. Telecomm. & Tech. L. Rev. 419, 423 n.5 (2012).
See eg Rebecca S. Eisenberg, The Shifting Functional Balance of Patents and Drug Regulation, 19 Health Aff. 119, 122 (2001); Arti K. Rai, The Information Revolution Reaches Pharmaceuticals: Balancing Innovation Incentives, Cost, and Access in the Post-Genomics Era, 2001 U. Ill. L. Rev. 173, 183 (2001).
See Richard A. Merrill, The Architecture of Government Regulation of Medical Products, 82 Va. L. Rev. 1753, 1790–91 (1996).
Aaron S. Kesselheim & Daniel H. Solomon, Incentives for Drug Development—The Curious Case of Colchicine, 362 New Eng. J. Med. 2045, 2045 (2010).
Kefauver-Harris Amendment to the Food, Drug & Cosmetic Act, Pub. L. No. 87–781, 76 Stat. 780 (1962).
Kesselheim & Solomon, supra note 57, at 2045.
Under the Hatch-Waxman Act, the manufacturer also received three years of exclusivity for the use of colchicine to treat gout, its historical use, id., but since the periods run concurrently, the total exclusivity time is just seven years.
Id. at 2046.
Id.
Yesha Patel & Martha M. Rumore, Hydroxyprogesterone Caproate Injection (Makena) One Year Later, 37 P & T 405, 406 (2012).
Id.
Food & Drug Administration, Questions and Answers on Updated FDA Statement on Compounded Versions of Hydroxyprogesterone Caproate, 2012, http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm310215.htm (accessed June 18, 2015).
Food & Drug Administration, Orphan Drug Designation for Fecal Microbiota, Mar. 10, 2014, http://www.accessdata.fda.gov/scripts/opdlisting/oopd/OOPD_Results_2.cfm?Index_Number=421013 (accessed June 18, 2015).
See eg Rebiotix, Rebiotix Announces First Ever Randomized Study of a Microbiota-Based Drug for Recurrent Clostridium difficile Infection Under an FDA IND, Dec. 16, 2014, http://www.rebiotix.com/index.php/first-randomized-clinical-trial-microbiota-drug-clostridium-difficile-infection (accessed June 18, 2015). The product, RBX2660, is a fecal microbiota preparation for enema administration, derived from human donors. As such, it is not compositionally consistent. See Alex Park, Should We Regulate Poop as a Drug?, Mother Jones, Aug. 18, 2014, http://www.motherjones.com/environment/2014/08/fecal-transplant-fda-openbiome-rebiotix (accessed June 18, 2015).
Depomed, Inc. v. United States Dep't of Health & Human Servs., No. 12-cv-1592, 2014 WL 4457225, at 14 (D.D.C. Sept. 5, 2014).
Food & Drug Administration, Unapproved Drugs Initiative, 2014, http://www.fda.gov/Drugs/Guidance-ComplianceRegulatoryInformation/EnforcementActivitiesbyFDA/SelectedEnforcementActionson-UnapprovedDrugs/ucm118990.htm (accessed June 18, 2015). Although it might be supposed that the preparation of stool for FMT is more akin to compounding than it is to the production of colchicine, and therefore that stool banks might be able to evade the exclusivity requirements in the way that compounding pharmacies have, this is unlikely. Unlike compounding pharmacies, no stool banks have argued that their activities lie entirely outside the FDA's regulatory authority. Our argument here is simply that the FDA has selected a suboptimal regulatory paradigm.
Makena's maker argued that the FDA had violated its own rules by not exercising its enforcement authority over the compounding pharmacies. But before the courts could officially decide the case, the parties settled. Kurt R. Karst, KV Lawsuit Involving Makena and Compounded 17P Concludes in Sopranos Style, FDA Law Blog, July 7, 2014, http://www.fdalawblog.net/fda_law_blog_hyman_phelps/2014/07/kv-lawsuit-involving-makena-and-compounded-17p-concludes-in-sopranos-style.html (accessed June 18, 2015).
See generally Complaint, Mutual Pharmaceutical Co., Inc. v. Watson Pharmaceuticals, Inc., No. 2:09-cv-05700-PA-RZ (C.D. Cal. Aug. 4, 2009).
Cf. Denial of Motion for a Preliminary Injunction (Doc. 139), Mutual Pharmaceutical Co., Inc. v. Watson Pharmaceuticals, Inc., No. 2:09-cv-05700-PA-RZ (C.D. Cal. Oct. 19, 2009).
Rebiotix might benchmark the price of its therapy against the price of vancomycin, which at its least expensive (short-course and low-dose) costs between $1000 and $1500. Andrew Pollack, The Hidden Price of Drugs, New York Times, May 31, 2011, http://prescriptions.blogs.nytimes.com/2011/05/31/the-hidden-price-of-drugs/ (accessed June 18, 2015). Fidaxomicin, another common antibiotic, costs twice as much.
Today, to our knowledge no insurer covers the use of frozen, ready-to-use fecal microbiota preparations.
See eg HomeFMT, Fecal Transplant (FMT), http://www.youtube.com/watch?v=xLIndT7fuGo (accessed June 18, 2015).
Semen is another substance with the potential for at-home administration. Perhaps responding to precisely these kinds of do-it-yourself concerns, the FDA imposes fewer and less strict requirements on directed sperm donors (where the recipient and donor know each other) than on anonymous donors. See eg 21 C.F.R. § 1271.90(a)(2) (2001); Food & Drug Administration, Donor Eligibility Final Rule and Guidance Questions and Answers, 2009, http://www.fda.gov/BiologicsBloodVaccines/TissueTissueProducts/QuestionsaboutTissues/ucm102842.htm (accessed June 18, 2015).
Many (55%) are excluded through the clinical interview, while a further 65% fail the stool screens. The most common reason for failure at the stool screen stage is for asymptomatic rotavirus (20%), which has traditionally not been included in screening protocols. Blood screen failure is extremely rare. Laura Burns et al., Donor Recruitment and Eligibility for Fecal Microbiota Transplantation: Results from an International Public Stool Bank, Digestive Diseases Week 2015 (May 19, 2015) (poster abstract).
Peter J. Turnbough et al., An Obesity-Associated Gut Microbiome with Increased Capacity for Energy Harvest, 444 Nature 1027, 1027 (2006).
Anne Vrieze et al., Transfer of Intestinal Microbiota From Lean Donors Increases Insulin Sensitivity in Individuals With Metabolic Syndrome, 143 Gastroenterology 913, 913 (2012).
Stephen M. Collins et al., The Adoptive Transfer of Behavioral Phenotype Via the Intestinal Microbiota: Experimental Evidence and Clinical Implications, 16 Curr. Op. Microbiol. 240, 240 (2013).
Smith, supra note 43, at 290.
Some at-home experimentation with fecal transplantation is unavoidable given the hype that has surrounded the potential for this treatment to help patients with chronic, difficult-to-treat conditions such as ulcerative colitis or Crohn's disease. See eg Emily Eakin, The Excrement Experiment, New Yorker, Dec. 1, 2014, at 64, 64. Beyond its use for treating recurrent C. difficile, FMT's efficacy remains largely unknown, and as such, it is necessary to require that patients seeking FMT under medical supervision for all other indications enroll in a clinical trial, despite concerns associated with do-it-yourself treatments. More must be known about the risks and benefits of FMT to drive effective medical practice, even though it will result in patients who cannot participate in trials resorting to self-administered variations.
21 U.S.C. § 321(g)(1) (2012).
21 C.F.R. § 640.3 (2015); Food & Drug Administration, Keeping Blood Transfusions Safe: FDA's Multi-Layered Protections for Donated Blood, 2013, http://www.fda.gov/BiologicsBloodVaccines/SafetyAvailability/BloodSafety/ucm095522.htm (accessed June 18, 2015).
21 C.F.R. § 640.5 (2015); Food & Drug Administration, Keeping Blood Transfusions Safe: FDA's Multi-Layered Protections for Donated Blood 2013, http://www.fda.gov/BiologicsBloodVaccines/SafetyAvailability/BloodSafety/ucm095522.htm (accessed June 18, 2015).
21 C.F.R. § 607 et seq. (2015).
Peter Barton Hutt, Richard A. Merrill & Lewis A. Grossman, Food and Drug Law: Cases and Materials 1154 (4th ed., Foundation Press 2014).
Kara W. Swanson, Banking on the Body 24, 30 (2014).
Pub. L. No. 91–515, 84 Stat. 1297, 1308 (1970) [codified at 42 U.S.C. § 262(a) (2012)]; Hutt, Merrill & Grossman, supra note 87, at 1153.
See Sha, supra note 20, at 1028.
Eliane Gluckman et al., Hematopoietic Reconstitution in a Patient with Fanconi's Anemia by Means of Umbilical-Cord Blood from an HLA-Identical Sibling, 321 New Eng. J. Med. 1174, 1174 (1989); see also Jennifer Kulynych, Blood As A Biological ‘Drug’: Scientific, Legal, and Policy Issues in the Regulation of Placental and Umbilical Cord Stem Cell Transplantation, 32 U. Rich. L. Rev. 407, 408 (1998).
21 C.F.R. § 1271.3(d) (2014).
Id.
The FDA only began to regulate many of these products in earnest in the late 1970s, once the potential for disease transmission became known. Richard A. Merrill, Human Tissues and Reproductive Cloning: New Technologies Challenge FDA, 3 Hous. J. Health L. & Pol'y 1, 10 (2002).
Food & Drug Administration, Cord Blood Banking: Information for Consumers, 2012, http://www.fda.gov/BiologicsBloodVaccines/ResourcesforYou/Consumers/ucm236044.htm (accessed June 18, 2015).
See generally van Nood, supra note 27.
See eg 21 C.F.R. § 1271.90(a)(2) (2001).
Food & Drug Administration, Tissue Reference Group: FY 2012 Update, 2012, http://www.fda.gov/biologicsbloodvaccines/tissuetissueproducts/regulationoftissues/ucm152857.htm (accessed June 18, 2015).
21 C.F.R. § 1271.3(d)(3) (2014).
This is not precisely accurate because semen is also explicitly enumerated as an HCT/P, but in common parlance semen would otherwise be considered as something which is ‘secreted’ by the body.
5 U.S.C. § 553; 5 U.S.C. § 551(4).
5 U.S.C. § 553(b)(3)(A), (d)(2).
Nat'l Min. Ass'n v. McCarthy, 758 F.3d 243, 251 (D.C. Cir. 2014).
This is not the most natural reading of the regulation, which requires only that the article ‘contain[]’ human cells or tissues, and in fact many scientists have begun to refer to the microbiome as a human organ. See eg Alexander Khoruts et al., Development of Fecal Microbiota Transplantation Suitable for Mainstream Medicine, 13 Clin. Gastroenterol. & Hepatol. 246, 247 (2015). However, in a May 2013 public meeting, Rebiotix stated that it had received a letter from the FDA reaching this interpretation. Food & Drug Administration, Fecal Microbiota for Transplantation: Scientific and Regulatory Issues 229, May 2, 2013, http://www.fda.gov/downloads/BiologicsBloodVaccines/NewsEvents/WorkshopsMeetingsConferences/UCM352902.pdf (accessed June 18, 2015). As such, we must take this argument seriously.
See infra text accompanying note 110.
See eg Kulynych, supra note 91, at 409, 410 (discussing various statutory efforts in this area).
Letter from the Presidents of the Infectious Disease Society of America, the American Gastroenterological Association, the American Society for Gastrointestinal Endoscopy et al., Current Consensus Guidance on Donor Screening and Stool Testing for FMT, July 5, 2013, http://www.gastro.org/research/Joint_Society_FMT_Guidance.pdf (accessed June 18, 2015).
David C. Radley et al., Off-label Prescribing Among Office-Based Physicians, 166 Arch. Intern. Med. 1021, 1021 (2006).
Although these concerns technically apply to all standard drugs, we might both be particularly worried about off-label uses of FMT and particularly concerned about incentives for companies to perform clinical trials here. Off-label uses of FMT might be particularly concerning because we are just beginning to understand the relationship between the human microbiome and disease. Scientists may have a relatively complete picture of how a given small molecule drug interacts with the body, but the potential health effects of microbiome manipulation require much greater study than they have received to date. Widespread off-label prescribing has the potential to hamper efforts to do so. And given the difficulty of protecting technologies like FMT by traditional intellectual property means, companies will lack other incentives to pursue such testing. See generally Amy Kapczynski & Talha Syed, The Continuum of Excludability and the Limits of Patents, 122 Yale L. J. 1900 (2013).
See eg Seres Therapeutics, About Seres Therapeutics, 2015, http://serestherapeutics.com/about/about_seres_therapeutics/ (accessed June 18, 2015).
Food & Drug Administration, Tissue Reference Group: FY 2012 Update, 2012, http://www.fda.gov/biologicsbloodvaccines/tissuetissueproducts/regulationoftissues/ucm152857.htm (accessed June 18, 2015).
See supra note 66.
21 C.F.R. § 316.20(a) (2014).
See Levitt & Kelsey, supra note 51, at 526, 528.
Food & Drug Administration, Drug Shortage Management, 12, 2014, http://www.fda.gov/downloads/AboutFDA/CentersOffices/OfficeofMedicalProductsandTobacco/CDER/ManualofPoliciesProcedures/UCM079936.pdf (accessed June 18, 2015).
The FDA has undoubtedly faced numerous new challenges throughout its history, and it has had to consider how it might adapt old statutes to new problems. Merrill, supra note 94, at 2, 3; see also generally Jody Freeman & David B. Spence, Old Statutes, New Problems, 163 U. Pa. L. Rev. 1 (2014). But the do-it-yourself nature of FMT will soon be echoed by 3-D printing technology and mobile health developments.