

Abstract
Background
GATA3 is pivotal for the development of T lymphocytes. While its effects in later stages of T cell differentiation are well recognized, the role of GATA3 in the generation of early T cell precursors (ETP) has only recently been explored. As aberrant GATA3 mRNA expression has been linked to cancerogenesis, we investigated the role of GATA3 in early T cell precursor acute lymphoblastic leukemia (ETP-ALL).
Methods
We analyzed GATA3 mRNA expression by RT-PCR (n = 182) in adult patients with T-ALL. Of these, we identified 70 of 182 patients with ETP-ALL by immunophenotyping. DNA methylation was assessed genome wide (Illumina Infinium® HumanMethylation450 BeadChip platform) in 12 patients and GATA3-specifically by pyrosequencing in 70 patients with ETP-ALL. The mutational landscape of ETP-ALL with respect to GATA3 expression was investigated in 18 patients and validated by Sanger sequencing in 65 patients with ETP-ALL. Gene expression profiles (Affymetrix Human genome U133 Plus 2.0) of an independent cohort of adult T-ALL (n = 83) were used to identify ETP-ALL and investigate GATA3low and GATA3high expressing T-ALL patients. In addition, the ETP-ALL cell line PER-117 was investigated for cytotoxicity, apoptosis, GATA3 mRNA expression, DNA methylation, and global gene expression before and after treatment with decitabine.
Results
In our cohort of 70 ETP-ALL patients, 33 % (23/70) lacked GATA3 expression and were thus defined as GATA3low. DNA methylation analysis revealed a high degree of GATA3 CpG island methylation in GATA3low compared with GATA3high ETP-ALL patients (mean 46 vs. 21 %, p < 0.0001). Genome-wide expression profiling of GATA3low ETP-ALL exhibited enrichment of myeloid/lymphoid progenitor (MLP) and granulocyte/monocyte progenitor (GMP) genes, while T cell-specific signatures were downregulated compared to GATA3high ETP-ALL. Among others, FLT3 expression was upregulated and mutational analyses demonstrated a high rate (79 %) of FLT3 mutations. Hypomethylating agents induced reversal of GATA3 silencing, and gene expression profiling revealed downregulation of hematopoietic stem cell genes and upregulation of T cell differentiation.
Conclusions
We propose GATA3low ETP-ALL as a novel stem cell-like leukemia with implications for the use of myeloid-derived therapies.
Electronic supplementary material
The online version of this article (doi:10.1186/s13045-016-0324-8) contains supplementary material, which is available to authorized users.
Keywords: GATA3, ETP-ALL, PER-117, Decitabine
Background
GATA3 is a transcription factor with a pivotal role in multiple developmental steps of T lymphopoiesis [1, 2], including the development of early T cell precursors (ETPs), a rare subpopulation of cells sharing characteristics with multipotent hematopoietic progenitors in the bone marrow [3]. ETPs are considered the most immature thymic cells with potential for complete T cell differentiation and retain plasticity for differentiation towards dendritic, NK, B, or myeloid cells [4]. In a murine model, GATA3 was required for the development of ETPs, whereas totipotent hematopoietic stem cells (HSCs) remained unaffected by in vivo manipulation of GATA3 expression levels. Indeed, in murine HSCs, GATA3 was silenced by DNMT3A-dependent DNA hypermethylation [5]. By losing repressive epigenetic marks during T lymphopoiesis, GATA3 functions as a key regulator of T cell differentiation through the interaction with a multitude of target genes that differ in a subpopulation specific manner [6]. For example, GATA3 was reported to restrain Notch activity, repress NK-cell fate and upregulate T cell lineage genes to facilitate T cell differentiation [7].
Lack of GATA3 has been linked to cancerogenesis, as absence of GATA3 expression was associated with poor prognosis and undifferentiated tumors in breast cancer [8]. Moreover, several other cancers exhibited aberrant GATA3 expression, including urothelial carcinoma [9], renal cell carcinoma [10], pancreatic cancer [11], cervical cancer [12], or Hodgkinʼs lymphoma [13]. In childhood B cell precursor acute lymphoblastic leukemia (BCP-ALL), specific germline variants of GATA3 were associated with a higher incidence of BCP-ALL and a higher risk of relapse [14, 15].
Given GATA3’s prominent role in both cancerogenesis and T cell development, we investigated GATA3 in ETP-ALL. ETP-ALL is a subtype of T-ALL characterized by a distinct gene expression profile (GEP) and a distinct immunophenotype with lack of CD1a and CD8, weak expression of CD5 and additional expression of more than 1 myeloid and/or stem cell marker [16]. ETP-ALL accounts for 11–15 % of cases with T-ALL [16–18] with similar distributions among pediatric and adult cohorts. We and others have characterized the mutational landscape of ETP-ALL with alterations in genes involved in cytokine and RAS signaling (e.g., NRAS, KRAS, FLT3, and JAK1), epigenetic regulation (e.g., EZH2, DNMT3A, and SUZ12), and hematopoietic development (e.g., ETV6, RUNX1, and IKZF1) [19, 20]. Notably, the incidence of activating NOTCH1 mutations is considerably lower in ETP-ALL (15 %) when compared to T-ALL (higher than 50 %) [17, 21]. GATA3 inactivating mutations were reported in 9 % of pediatric ETP-ALL patients predominantly affecting the DNA binding domain [19].
The prognostic relevance of ETP-ALL is controversially discussed. Comparing ETP-ALL with non-ETP-ALL, some reports indicate adverse prognosis in pediatric and adult patients with ETP-ALL with CR rates of 58–73 %, median event-free survival of 1.2 years, and 3-year overall survival of 30–60 % [16, 17, 22]. Other groups found similar outcome of ETP-ALL and non-ETP-ALL patients with 5-year overall survival rates of 67–93 and 77–92 %, respectively [23, 24].
Given the critical role that GATA3 plays in early lymphoid development, we investigated GATA3 in ETP-ALL, a stem cell-like leukemia blocked at the crossroads of lymphoid and myeloid differentiation. We hypothesized that aberrant GATA3 expression would divert ETP-ALL from the lymphoid fate and determine a novel biological subgroup of ETP-ALL.
Methods
Patient samples
Additional file 1: Figure S1 provides an overview over the sample cohorts and subsequent experiments.
Gene expression data (Affymetrix HG-U133 Plus 2.0 or A + B) were available for adult T-ALL (n = 83; including 30 patients with ETP-ALL and 53 patients with non-ETP-ALL, defined by gene expression profiling [16], GEO accession number GSE78132), BCP-ALL (n = 81, GSE13204) [25], normal controls (NC; n = 24, GSE13204) [25], and acute myeloid leukemia (AML; n = 130) [26, 27]. The T-ALL subgroup included consecutive patients with newly diagnosed ALL studied between 1999 and 2005 at two reference laboratories [25, 28].
Based on immunophenotyping of diagnostic samples at the central diagnostic reference laboratory of the German Multicenter Study Group for Acute Lymphoblastic Leukemia (GMALL) in Berlin, Germany, we identified additional 70 ETP-ALL samples [17]. Sufficient RNA for GATA3 mRNA expression analysis was available for all 70 samples, and sufficient genomic DNA (gDNA) for methylation assays was available for 69 samples of these adult ETP-ALL cases. As reference cohort, we used 112 non-ETP-ALL patients, of which 21 (19 %) had an immunophenotype of early T-ALL, 20 (18 %) of mature T-ALL, and 71 (63 %) of thymic T-ALL.
All patients, including the two independent cohorts of T-ALL, and normal controls gave written informed consent to participate in the study according to the Declaration of Helsinki. The studies were approved by the ethics board of the Johann Wolfgang von Goethe University, Frankfurt/Main, Germany.
Nucleic acid preparation and molecular characterization
Pretreatment bone marrow and peripheral blood samples from patients were used for gDNA and total RNA extraction using TRIzol (Life Technologies, Grand Island, NY, USA) according to the manufacturer’s protocol with minor modifications. Complementary DNA (cDNA) was synthesized using 500 ng of total RNA and avian myeloblastosis virus reverse transcriptase (RT-AMV; Roche, Mannheim, Germany) in the presence of RNase inhibitor (RNasin; Roche, Mannheim, Germany).
Samples of patients with ETP-ALL (n = 70) and non-ETP-ALL (n = 112) were investigated by comparative multiplex real-time PCR (RT-PCR) for expression of GATA3 (FWD: 5′-ACTACGGAAACTCGGTCAG-3′, REV: 5′-GTAGGGATCCATGAAGCAG-3′, Probe: 5′-CGGTGCAGAGGTACCCTCCG-3′) and glucose-6-phosphate isomerase (GPI) as a housekeeping gene. Relative GATA3 expression values of ETP-ALL (n = 70) and non-ETP-ALL (n = 112) were normalized to GATA3 expression in the human T-ALL cell line Jurkat. We identified a bimodal distribution of GATA3 mRNA expression levels by K-means clustering and defined a cutoff at an expression level of 0.2 relative to Jurkat and defined all samples with GATA3 expression below that cutoff as GATA3low and samples with higher expression as GATA3high (Fig. 1).
Fig. 1.
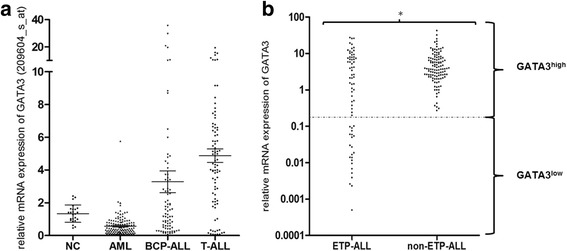
GATA3 mRNA expression in patient samples. a Affymetrix-based mean differential expression of GATA3 based on normalized expression values in normal controls (NC) and selected hematological disorders. Horizontal lines indicate mean GATA3 expression s.e. Note the segmented y-axis. b RT-PCR-based analyses of GATA3 mRNA expression relative to Jurkat on a logarithmic scale revealed lower GATA3 expression in ETP-ALL (n = 70) than in non-ETP-ALL (n = 112) (4.82 vs. 6.29, p = 0.0005 indicated by asterisk). We identified a bimodal distribution of GATA3 expression by K-means clustering with a cutoff at a relative expression of 0.2 (indicated by the dotted line). The GATA3low cohort contained only cases with ETP-ALL (i.e., GATA3low ETP-ALL)
Western blot
GATA3 protein levels were measured using standard western blotting techniques using the GATA3 antibody HG3-35 (Santa Cruz Biotechnology Inc., Dallas, TX, USA).
Gene expression profiling
A GATA3-associated GEP was generated from data of 83 adult T-ALL patients (including 30 ETP-ALL and 53 non-ETP-ALL samples defined by hierarchical clustering using a list of genes reported as differentially expressed in pediatric ETP-ALL [16], GEO accession number GSE78132). For analysis, common probe sets between HG-U133 Plus 2.0 and HG-U133 A + B (Affymetrix, Santa Clara, CA, USA) were identified and quantile normalized. For ANOVA analysis, the type of chip was integrated as a random effect to take the batch effect into account. GATA3 expression was calculated from signals obtained from probe sets 209602_s_at, 209603_at, and 209604_s_at, respectively. As for the RT-PCR expression levels, we identified a bimodal distribution of GATA3-normalized expression values and defined a cutoff at 8.2 on a logarithmic scale to categorize patients below that cutoff as GATA3low and with higher expression as GATA3high. A GATA3-dependent GEP was generated by the comparison of the expression profiles from GATA3low (n = 11) and GATA3high (n = 72) samples. Lists of genes with at least 1.5-fold under- or overexpression comparing GATA3low and GATA3high were generated (Additional file 2: Table S1), and statistical significance was calculated by ANOVA with a FDR ≤0.05. Data analysis was carried out with Partek Genomic Suite v6.6 Software (Partek Inc., St. Louis, MO, USA).
Lists of up- and downregulated genes (Additional file 2: Table S1) were uploaded to the DAVID Bioinformatics server (http://david.abcc.ncifcrf.gov) to define enriched KEGG pathway annotations [29].
Additionally, GEP was performed on the Affymetrix® HG-U133 Plus 2.0 platform with ETP-ALL cell lines (PER-117, Loucy, in triplicates) and non-ETP-ALL cell lines (BE13, Molt4, Jurkat, RPMI8402; in duplicates) (GEO accession number GSE78166).
For gene set enrichment analysis (GSEA), GATA3-supervised GEPs were examined for enrichment of curated gene sets representing ETP-ALL [19], HSC [30], T cell differentiation [30], granulocyte/macrophage progenitors (GMP) [30], and myeloid/lymphoid progenitors (MLP) [31] comparing GATA3low ETP-ALL (n = 11) and GATA3high ETP-ALL (n = 19) cases. Additionally, we used decitabine-induced changes of GEP as a discriminator to analyze enrichment of these curated gene lists in PER-117 cells. Data analyses were carried out with the GSEA desktop application version 2.0.12 [32, 33] from the Broad Institute (http://www.broadinstitute.org/gsea).
Methylation analysis
We assessed global DNA methylation analyses in 12 ETP-ALL and 14 BCP-ALL samples by the Illumina Infinium® HumanMethylation450 BeadChip platform. Hybridization was performed according to the manufacturer’s protocol. The signals generated for unmethylated and methylated cytosine nucleotides by single-nucleotide extension of locus-specific methylation probes were transformed into β values ranging from 0 to 1 (representing 0 to 100 %) for each of the 450,000 interrogated CpG residues. We assumed differential methylation, if more than three differentially methylated sites (DMS) with a p value <0.05 were present for each gene and the absolute difference of the corresponding β values was greater than 0.17. Data analysis was carried out with Partek Genomic Suite v6.6 Software (Partek Inc., St. Louis, MO, USA).
Sufficient amounts of gDNA for bisulfite conversion was available for 69 ETP-ALL and 48 AML samples, which was carried out using the EpiTect Bisulfite Kit (QIAGEN, Hilden, Germany) according to the manufacturer’s instructions. For validation of the differentially methylated region of GATA3 detected by global methylation analysis, primers were designed for amplification and pyrosequencing based on the bisulfite converted sequence of GATA3 (genomic location: GRCh37: chr10:8097750-8098004) and used in the Pyrosequencing Assay Design Software v1.0 (Biotage, Uppsala, Sweden) for assay design. Amplification of a 255-bp sequence was carried out in all 69 bisulfite converted ETP-ALL samples using a 5′-GGAGGAGGTGGATGTGTTTTTTAAT-3′ forward and a 3′-AACCCCAATTTTTTTATAAATAAACCA-5′reverse biotinylated primer. Additionally, 13 representative samples of the non-ETP-ALL cohort were selected for analysis by pyrosequencing; 100 ng of bisulfite-converted gDNA was used per reaction with Taq-DNA-polymerase (Hot Start Mix S, peqlab, Erlangen, Germany). Samples were analyzed for specificity and correct size by 2 % agarose gel electrophoresis.
For pyrosequencing, a 5′-GTTACGGTGTAGAGGTATTTT-3′ sequencing primer was used. The percentage of CpG site methylation was calculated through the ratio of the relative content of thymine (i.e., unmethylated cytosine) and the relative content of cytosine (i.e., methylated cytosine) using the Pyro Q-CpG Software version 1.0.9 (Biotage, Uppsala, Sweden). Four of 12 CpG sites covered by the sequencing primer failed quality control due to the reference sequence pattern at the end of the amplicon. The remaining eight CpG sites were included to calculate the mean percentage of methylation for each sample.
Cell culture and chemicals
The immature T-ALL cell line PER-117 [34] was grown in RPMI media with 10–20 % fetal bovine serum and cultured at 37 °C in a 5 % CO2 humidified chamber. PER-117 exhibited an immature phenotype resembling ETP-ALL (CD7+CD5−CD1a−cyCD3+CD33+TdT−CD10−CD34−CD117−), and gene expression profiling based on microarray analysis revealed an ETP-ALL phenotype (Additional file 3: Figure S2) including high expression of GATA2, CEBPα, or NFE2 and low expression of LEF1 and GATA3 (GATA3low ETP-ALL).
Additionally, the ETP-ALL cell line Loucy (with high GATA3 expression, GATA3high ETP-ALL) and the non-ETP-ALL cell lines, Jurkat, Molt4, BE13, and RPMI8402 were obtained from the German Resource Center for Biological Material, DSMZ (Braunschweig, Germany) and previously characterized on a molecular level [35]. 5-Azacytidine and 5-aza-deoxycytidine were purchased from Sigma-Aldrich (St. Louis, MO, USA).
Cell proliferation assay
Cell proliferation was measured with the WST-1 reagent according to the manufacturer’s instructions (Roche Diagnostics GmbH, Germany). Cell lines were treated with various concentrations of 5-azacytidine (Sigma-Aldrich, St. Louis, USA) and 5-aza-deoxycytidine (Sigma-Aldrich, St. Louis, USA), and absorbance was measured after 48, 72, and 96 h by optical density absorption analyses at a wavelength of 450 nm using an ELISA multiplate reader.
Apoptosis assay
Apoptosis was measured using Annexin V Apoptosis Detection Kit (BD Pharmingen, Heidelberg, Germany). Cells were labeled with Annexin V and 7-amino-actinomycin D (7-AAD) after treatment with 5-azacytidine and 5-aza-deoxycytidine. Analyses were performed by FACS Calibur (Becton-Dickinson) to determine the percentage of apoptotic cells from combined 7-AAD incorporation and Annexin V binding.
Statistical analysis
The statistical difference of gene expression between two independent groups was tested by the non-parametric Mann-Whitney U test. For non-parametric correlation of mRNA expression and DNA methylation, Spearman’s rank correlation coefficient was calculated. Fisher’s exact test was used to test for the association between two kinds of classifications (e.g., 2 × 2 contingency table).
For all tests, a p value <0.05 (two-sided) was considered to indicate a significant difference. All calculations were performed using the SPSS software version 19 (SPSS Inc., Chicago, IL, USA), GraphPad Prism® software version 5 (GraphPad Software Inc., La Jolla, CA, USA), and Partek Genomic Suite v6.6 Software (Partek Inc., St. Louis, MO, USA).
Results
Lack of GATA3 expression in ETP-ALL
We first assessed GATA3 mRNA expression by microarray analysis and found that mean expression of GATA3 was higher in T-ALL (4.88 ± 0.41, mean ± s.e., n = 83) than in other cohorts (NC 1.33 ± 0.11, n = 24; AML 0.57 ± 0.05, n = 130; and BCP-ALL 3.28 ± 0.66, n = 81; all values are mean ± s.e., p < 0.001) (Fig. 1a).
To further explore GATA3 expression in T-ALL, we analyzed GATA3 mRNA expression by quantitative RT-PCR in larger cohorts of ETP-ALL (n = 70) and non-ETP-ALL (n = 112). The mean relative expression of GATA3 was lower in ETP-ALL than in non-ETP-ALL (4.82 ± 0.78 vs. 6.29 ± 0.60, mean ± s.e., p = 0.0005). Interestingly, we found a bimodal distribution of GATA3 expression with one third of ETP-ALL patients lacking GATA3 expression (23/70, 33 %, GATA3low ETP-ALL). In contrast, none of 112 non-ETP-ALL samples lacked GATA3 expression, which consisted of 71 thymic, 21 early, and 20 mature T-ALL patient samples (Fig. 1b). In agreement with this, the non-ETP-ALL cell lines Molt4, Jurkat, RPMI8402, and BE13 all expressed GATA3, while PER-117 [34], a cell line with an ETP-ALL immunophenotype and GEP (Additional file 3: Figure S2) lacked GATA3 expression. Western blotting revealed that differential GATA3 mRNA expression translated into differential protein expression levels (Additional file 4: Figure S3).
GATA3 silencing is mediated by aberrant DNA methylation
To explore the regulation of GATA3 expression, we investigated global DNA methylation on the Illumina HumanMethylation 450 k platform in 12 ETP-ALL samples (Fig. 2), which were selected according to GATA3 mRNA expression (GATA3low vs. GATA3high) and mutational status of DNMT3A. The genomic locus of GATA3 (NC_000010.10) was represented by 72 CpG sites.
Fig. 2.
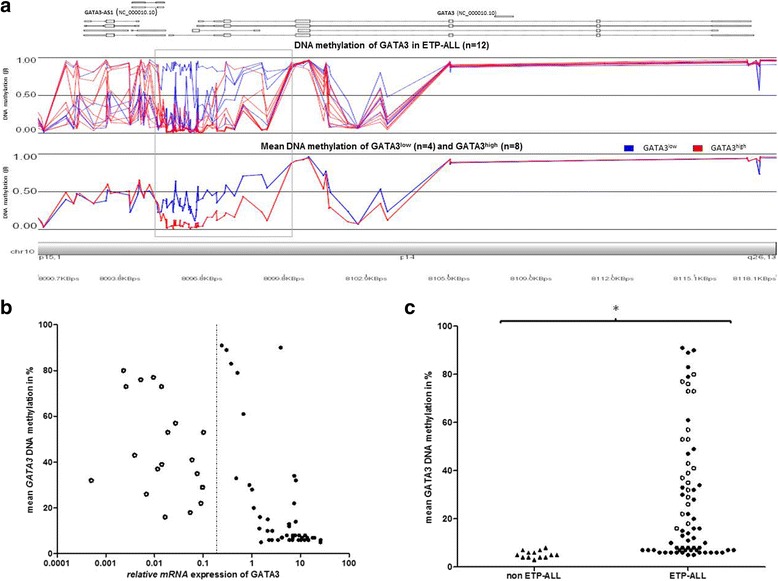
GATA3 silencing is regulated by DNA methylation in ETP-ALL. a 10p15 chromosome plot depicting DNA methylation of 12 ETP-ALL patient samples as assessed by Illumina Infinium® HumanMethylation450 BeadChip. β values representing DNA methylation for each patient (upper panel) and mean DNA methylation (lower panel) of GATA3low ETP-ALL (n = 4, blue) and GATA3high ETP-ALL (n = 8, red). Comparing GATA3low ETP-ALL (n = 4, blue) with GATA3high ETP-ALL (n = 8, red), 35 differentially methylated sites were located within a 6-kb segment of GATA3 (indicated by the gray box), including the CpGs that were analyzed by pyrosequencing in a larger cohort of patients. b GATA3 DNA methylation as assessed by pyrosequencing (of CpGs within the gray box in a) was negatively correlated to GATA3 mRNA expression in ETP-ALL (n = 64, r = −0.73, p < 0.0001). The dotted line indicates the cutoff to distinguish GATA3low (empty dots) and GATA3high (solid dots) samples. c Pyrosequencing revealed higher GATA3 DNA methylation in ETP-ALL (n = 69) than in non-ETP-ALL (n = 13) (28 vs. 5 %, p < 0.0001 indicated by asterisk). Empty and solid dots indicate GATA3low and GATA3high ETP-ALL, while triangles indicate non-ETP-ALL
Four of 12 ETP-ALL patients were defined as GATA3low (mean GATA3 expression ± s.e.: 0.08 ± 0.05), while the remaining eight patients were GATA3high (mean GATA3 expression ± s.e.: 6.4 ± 1.5). GATA3 DNA methylation of all 72 GATA3 CpG sites was higher in GATA3low ETP-ALL compared to GATA3high ETP-ALL (mean 45 vs. 23 %, p < 0.0001). We detected 35 of 72 GATA3 CpG sites with a significantly higher degree of methylation in GATA3low ETP-ALL compared to GATA3high ETP-ALL (mean 46 vs. 19 %, p < 0.0001). All 35 DMS clustered in a 6-kb region (genomic location: GRCh37: chr10:8095478-8101513; indicated by the grey box in Fig. 2a), which mapped to a GATA3 CpG island (GRCh37: chr10:8091375-8098329) that was previously reported to be differentially methylated in cancer [10, 36].
Comparing DNMT3A-mutated (n = 6) and DNMT3A wild-type (n = 6) ETP-ALL, we found lower GATA3 methylation in DNMT3A-mutated versus DNMT3A wild-type samples (16 vs. 35 %, p < 0.0001) at the GATA3 CpG island (GRCh37: chr10:8091375-8098329), but GATA3 expression, as determined by RT-PCR, was not different between the DNMT3A mutated (n = 6) and wild-type (n = 6) ETP-ALL cases (4.4 vs. 3.8, p = 0.84). Notably, all 16 DMS within the GATA3 CpG island clustered in a 3.3-kb region (GRCh37: chr10:8092037-8095363) just upstream of the 6-kb region that correlated to GATA3 gene mRNA expression, but remarkably without overlap.
To validate these findings in a larger sample set, DNA methylation was analyzed by pyrosequencing in 69 ETP-ALL samples; 11 of 69 samples were also investigated by the Illumina Human Methylation assay. Capturing a segment of the GATA3 CpG island (GRCh37: chr10:8097750-8098004) (Additional file 5: Figure S4), we assessed 64 samples of which both GATA3 mRNA expression and GATA3 DNA methylation were available. We confirmed a high degree of concordance between pyrosequencing and the Illumina Human Methylation assay in samples analyzed in parallel on both platforms (n = 11, R2 = .94). By pyrosequencing, we confirmed a higher degree of DNA methylation in GATA3low ETP-ALL (n = 19) compared to GATA3high ETP-ALL (n = 45) (mean 46 vs. 21 %, p < 0.0001). GATA3 expression and DNA methylation were inversely correlated (r = −0.73, p < 0.0001) (Fig. 2b). When we compared ETP-ALL to non-ETP-ALL, DNA methylation was lower in non-ETP-ALL (5 %, range 3–8 %, n = 13) than in ETP-ALL (28 %, range 5–91 %, n = 69; p < 0.0001) (Fig. 2c).
GATA3low ETP-ALL is associated with FLT3 mutations
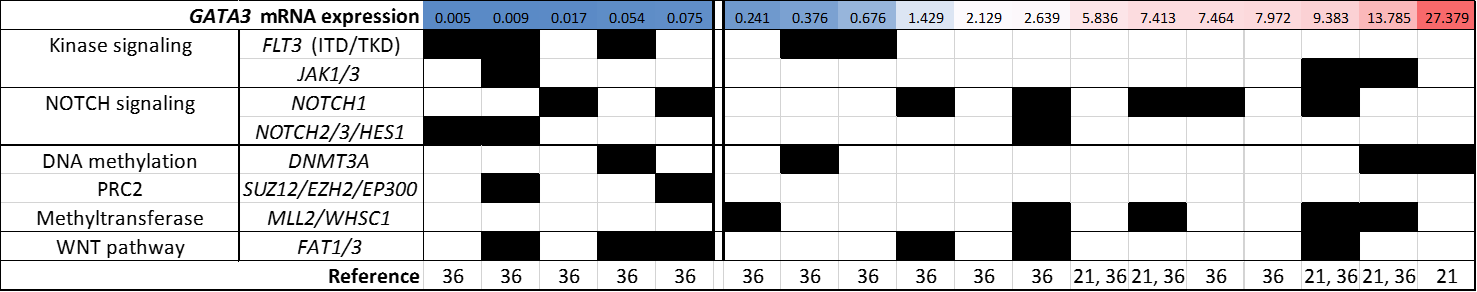
Our group previously assessed the mutational landscape of ETP-ALL by whole exome sequencing [20] and targeted NGS re-sequencing [37]. Within this cohort of ETP-ALL, we have investigated the mutational pattern with respect to GATA3 expression (Additional file 6: Table S2). In contrast to pediatric cohorts, we found no GATA3 mutations in this cohort, including a screen for hotspot mutations of exon 4 in an additional expansion cohort of 70 samples of adult ETP-ALL.
Comparing GATA3low and GATA3high ETP-ALL, we found FLT3 mutations (including both internal tandem duplications and mutations in the tyrosine kinase domain) in 79 % of GATA3low ETP-ALL (15/19), while only 15 % of GATA3high ETP-ALL (7/46) were FLT3 mutated (p < 0.001). Comparing FLT3+- and FLT3−-ETP-ALL, we found lower GATA3 expression (0.5 vs. 7.0, p < 0.0001) and higher GATA3 methylation as detected by pyrosequencing (51 vs. 8 %, p < 0.0001) in FLT3+-ETP-ALL. NOTCH1 mutations were generally infrequent in our cohort of ETP-ALL (11/65, 17 %) and less frequent still in GATA3low ETP-ALL than in GATA3high ETP-ALL (2/19, 11 % vs. 9/46, 20 %). Notably, we detected mutations in genes of the PRC2 complex in only a small number of adult ETP-ALL patients (5/70, 7 %) with marginal differences between GATA3low and GATA3high ETP-ALL cases (2/23, 9 vs. 3/47, 6 %). DNMT3A, EZH2, SUZ12, and EP300 mutations were similarly distributed among GATA3low and GATA3high samples as well as all other investigated genes.
Distinct transcriptional program of GATA3low ETP-ALL
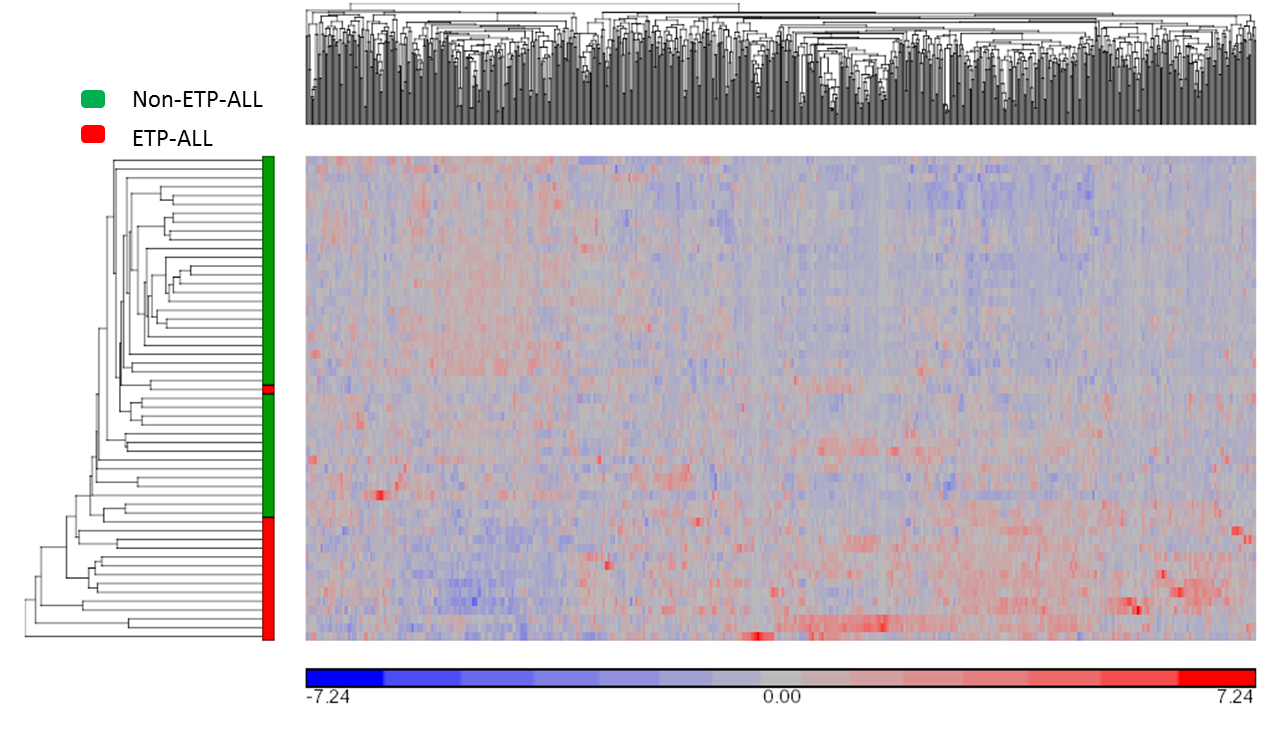
To explore differences of the transcriptional program, microarray expression data of 83 T-ALL patients were available; 11 of 83 patients were defined as GATA3low, while the remaining 72 patients were classified as GATA3high. Including probe sets with at least 1.5-fold overexpression, we detected 1435 differentially expressed probes sets in GATA3low compared to GATA3high T-ALL cases (Additional file 2: Table S1). Hierarchical clustering with this gene list revealed a GATA3low-derived gene expression signature (Fig. 3a). Importantly, this GATA3low GEP identified all but one case of ETP-ALL in an independent cohort of pediatric T-ALL (Additional file 7: Figure S5) [16]. Annotation of the top 267 DEG (i.e., genes with fold change ≥3×) using the KEGG pathway database demonstrated significant enrichment of upregulated genes associated to cancer and, notably, AML, while genes associated to T cell signaling were downregulated in GATA3low samples (Additional file 8: Table S3).
Fig. 3.
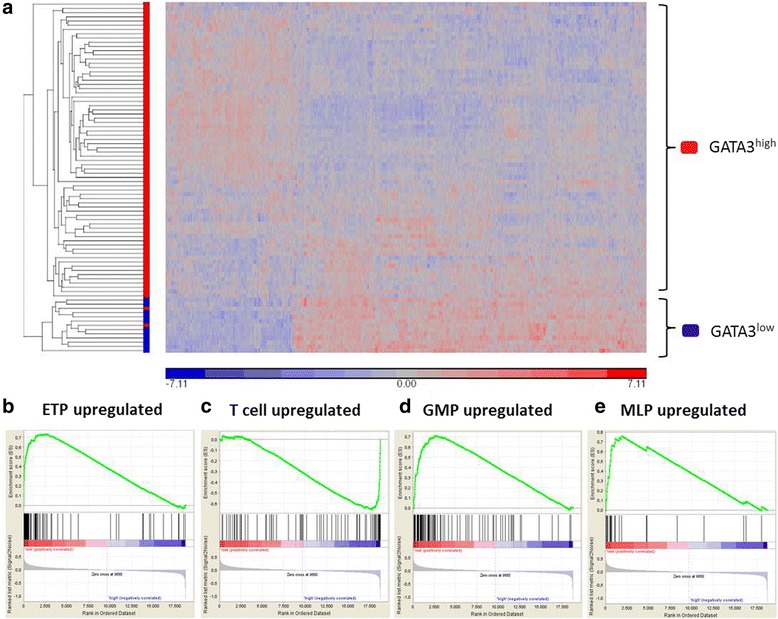
GATA3low ETP-ALL exhibits a distinct transcriptional program. a Heat map of 1.5-fold DEG comparing GATA3low (n = 11) with GATA3high (n = 72) in patients with T-ALL (n = 83), including 30 patients with ETP-ALL and 53 patients with non-ETP-ALL. b–e GSEA restricted to ETP-ALL (n = 30) comparing GATA3low (n = 11) and GATA3high ETP-ALL (n = 19) revealed enrichment of ETP-ALL genes (NES = 1.5, p < 0.01) (b), depletion of T cell differentiation genes (NES = 1.6, p < 0.008) (c), enrichment of GMP-based genes (NES = 1.5, p = 0.06) (d), and enrichment of MLP-based genes (NES = 1.5, p = 0.04) in GATA3low ETP-ALL (e)
Next, we intended to dissect molecular differences by comparing GATA3low and GATA3high in cases with ETP-ALL only. In a cohort of 30 adult patients with ETP-ALL, we identified 11 patient samples as GATA3low and 19 as GATA3high (Additional file 9: Figure S6). Applying GSEA, we found significant enrichment of ETP-ALL-associated genes (NES = 1.51, p = 0.01, FDR = 0.05) (Fig. 3b) and depletion of genes involved in T cell differentiation (NES = −1.6, p = 0.008, FDR = 0.01) in GATA3low ETP-ALL cases (Fig. 3c). Moreover, we also found enrichment for GMP-based genes (NES = 1.5, p = 0.06, FDR = 0.1, Fig. 3d) and MLP-based genes (NES = 1.5, p = 0.04, FDR = 0.14, Fig. 3e) in the GATA3low group.
Decitabine restores GATA3 expression in PER-117 cells
Given the high degree of GATA3 DNA methylation, we studied whether hypomethylating agents (HMA) could convert methylation-induced GATA3 silencing. We used PER-117 as a model for GATA3low ETP-ALL with high GATA3 DNA methylation (mean DNA methylation 92 % 1 %), low GATA3 mRNA expression (relative expression to Jurkat 0.002), and an ETP-like immunophenotype and GEP (Additional file 3: Figure S2). We evaluated GATA3 DNA methylation by pyrosequencing and GATA3 expression by RT-PCR after treatment with decitabine.
Treatment of PER-117 with decitabine (48 h, 5 μM) increased GATA3 mRNA expression by 2.2-fold (n = 4, p < 0.001) while lowering GATA3 DNA methylation from 91 to 78 % (n = 4, p < 0.05) (Fig. 4a). In contrast, another ETP-ALL cell line, Loucy, exhibited higher GATA3 expression (GATA3high ETP-ALL) than PER-117 and treatment with decitabine failed to induce GATA3 expression. In PER-117, decitabine induced 50 % growth inhibition at a concentration (IC50) of 4 μM (n = 9, p < 0.05) and enhanced apoptosis at the IC50 from 10 to 29 % after 48 h (n = 5, p < 0.05) (Fig. 4b).
Fig. 4.
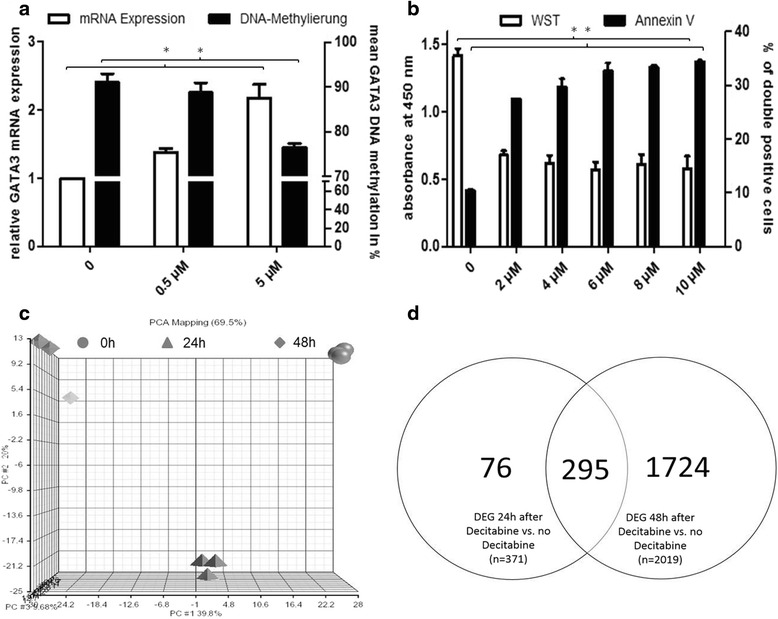
Decitabine reverses GATA3 silencing in PER-117 cells, an in vitro model for GATA3low ETP-ALL. a Treatment with decitabine (5 μM) increased GATA3 mRNA expression (n = 4, p < 0.001 indicated by the asterisk, all values are mean ± s.d.) and decreased GATA3 DNA methylation after 48 h (n = 4, p < 0.05 indicated by the asterisk, all values are mean ± s.d.) as detected by pyrosequencing. Note the two segments of the right y-axis for improved visualization. b Treatment with decitabine impaired proliferation as detected by WST assay with an IC50 of 4 μM and induced apoptosis as detected by flow cytometry of incorporation of 7-AAD and Annexin V binding after 48 h (n ≥ 5, p < 0.05 indicated by the asterisk, all values are mean ± s.d). c Principal component analysis of global gene expression profiles of PER-117 cells before and after treatment with decitabine (5 μM). d Venn diagram indicating the number of differentially expressed probe sets in PER-117 cells after 24 and 48 h and at both time points compared with no treatment
We analyzed global gene expression of PER-117 cells by Affymetrix microarrays before and after treatment with decitabine at a final concentration of 5 μM at three time points (0, 24, and 48 h). At both 24 and 48 h after decitabine treatment, we detected significant changes in global gene expression compared to untreated cells (Fig. 4c) with 2019 differentially expressed probe sets (fold change of ≥1.5 and FDR <0.05) after 48 h of exposure to decitabine (Fig. 4d). Principal component analysis revealed differential changes of global gene expression after 24 and 48 h: GEP changes represented by the first principal component expanded up until 48 h, while GEP changes subsumed by the second and third principal components were nearly completely reversible after 48 h (Fig. 4c).
Pathway analysis of all DEG after 48 h of decitabine treatment identified significant upregulation of p53 signaling genes (e.g., FAS, CDKN1a, or MDM2), while genes involved in cell cycling (e.g., CDK6, RB1, GSK3B, or E2f5) were downregulated upon treatment with decitabine. Moreover, we found downregulation of cancer-associated genes such as BCL2, FLT3LG, or PIK3r5 in cells treated with decitabine (Additional file 10: Table S4). Importantly, GATA3 ranked among the top upregulated genes (fold change of 2.2, p < 0.0001) confirming doubled GATA3 mRNA expression levels determined by RT-PCR.
To further characterize the transcriptional changes upon decitabine treatment, we performed GSEA comparing untreated with treated PER-117 cells. In cells treated with decitabine, we found downregulation of HSC genes (NES = 1.28, p < 0.001, FDR = 0.16) and, in line with increased GATA3 expression, upregulation of T cell differentiation (NES = 1.16, p = 0.06, FDR = 0.22).
Discussion
Here, we discovered a novel, molecularly distinct subgroup of T-ALL patients lacking GATA3 expression (GATA3low). All GATA3low T-ALL patients exhibited an immunophenotype of ETP-ALL, while GATA3high T-ALL patients were of thymic, early, or mature subtypes. The subgroup of GATA3low ETP-ALL is molecularly and clinically relevant as it lacks T lineage commitment in favor of a sustained myeloid gene expression signaling and a high rate of FLT3 mutations.
Clustering analysis revealed a third of our cohort’s ETP-ALL samples to be GATA3low. To study mechanisms of silenced GATA3 mRNA expression, we investigated DNA methylation. We identified a CpG island of GATA3 with consistently higher GATA3 DNA methylation in GATA3low ETP-ALL compared to GATA3high ETP-ALL including more than 30 DMS. This GATA3 CpG island was differentially methylated in renal cell carcinoma [10] and thyroid adenocarcinoma. In fact, cg01255894, a hypermethylated CpG site in ETP-ALL, was among the top 25 methylation probes that were most negatively correlated with gene expression [36]. Notably, GATA3 DNA hypermethylation was absent in non-ETP-ALL indicating that GATA3 silencing was a distinct mechanism in ETP-ALL. It is tempting to relate this finding to reports of murine DNMT3A-deficient mice, where GATA3 silencing was associated with DNMT3A-dependent DNA hypermethylation in HSC [5]. Indeed, when we compared DNMT3A mutated and DNMT3A wild-type ETP-ALL, we found lower GATA3 DNA methylation in samples with mutated DNMT3A, but GATA3 mRNA expression was not different between DNMT3A wild-type and mutated ETP-ALL. Thus, DNMT3A contributes to GATA3 DNA methylation; however, redundant mechanisms are likely required for GATA3 silencing in GATA3low ETP-ALL. Importantly, hypermethylation of GATA3 was found only in the subset of GATA3low ETP-ALL, but not in other leukemic subtypes such as typical T-ALL or BCP-ALL. Notably, in 49 samples from patients with AML, GATA3 expression was similarly low as in GATA3low ETP-ALL (mean 0.2 vs. 0.03), but DNA hypermethylation was absent in AML (17 vs. 46 %). Thus, GATA3low ETP-ALL may reflect the transformed precursor stage of yet non-committed ETP that physiologically harbor GATA3 DNA hypermethylation.
In order to explore the cell of origin of GATA3low ETP-ALL, we identified a GATA3low-specific GEP in a cohort of T-ALL, including ETP-ALL and non-ETP-ALL patient samples. GATA3low and GATA3high samples generated distinct gene expression clusters in a supervised analysis. The biological significance of this observation was underscored when we validated our GATA3low signature by identifying cases with ETP-ALL in an independent cohort of pediatric patients with T-ALL [16] by unsupervised hierarchical clustering. Moreover, pathway annotation of DEG indicated upregulation of myeloid genes and downregulation of T cell differentiation. Perhaps unsurprisingly, we found depletion of T cell signaling and enrichment of myeloid signaling when we performed GSEA comparing GATA3low ETP-ALL with T-ALL. By restricting the analysis to ETP-ALL only, we confirmed enrichment of GMP and MLP signatures and depletion of T cell differentiation in GATA3low samples compared to GATA3high ETP-ALL, which pointed at a specific molecular bracket within ETP-ALL. The specificity of GATA3 in this regard was further underscored when we analyzed other relevant transcription factors involved in T cell differentiation. Other transcription factors, such as MEF2C, PU.1, BCL11B, LMO1-3, HOXA1, TCF-1, or LYL1 failed to identify subsets with meaningful gene set enrichment in neither “typical” T-ALL nor ETP-ALL. Only the transcription factor LEF1 segregated cases into subgroups with similar gene set enrichment patterns as GATA3 subgroups, albeit with significant overlap of GATA3low/LEF1low cases. LEF1 is an important effector of WNT signaling and, like GATA3, known to be essential for early stages of T cell development. In T cell malignancy, LEF1 was implicated in transforming T cells in the absence of TCF1 [38].
The observation of a myeloid gene expression signature was further supported by the high frequency of FLT3 mutations in GATA3low ETP-ALL. It is important to note that neither of the investigated cases fulfilled the diagnostic criteria for leukemia of ambiguous lineage or acute myeloid leukemia. Therefore, these findings point to T-lymphoblastic precursors with multilineage potential as cells of origin of GATA3low ETP-ALL. Indeed, enrichment of ETP-ALL genes in GATA3low compared with GATA3high ETP-ALL reinforced this assumption as ETP-ALL by itself is characterized by upregulation of stem cell genes and myeloid-derived gene expression [19].
Ultimately, the significance of GATA3low ETP-ALL as a subgroup of ETP-ALL will depend on the implementation of distinct therapeutic interventions. In our ETP-ALL cohort, we found no significant outcome differences comparing GATA3low and GATA3high ETP-ALL (1-year OS 75 vs. 79 %) in a retrospective analysis of 52 patients. In general, the clinical outcome of ETP-ALL remains controversial, as reports of adverse risk in pediatric and adult ETP-ALL [16, 22, 39] have been challenged by reports indicating no outcome differences between ETP-ALL and non-ETP-ALL patient cohorts [23, 24]. This controversy might be in part due to the definition of ETP-ALL by GEP or flow cytometry as well as differences in treatment intensities, especially MRD-directed approaches to treatment intensification [16, 19, 23, 24, 40].
In any case, the mutational and transcriptional profile of GATA3low ETP-ALL provides rationale for implementing targeted therapies in patients with failure of lymphoid-directed therapies. Low incidence of NOTCH1 mutations in GATA3low ETP-ALL will likely render NOTCH-targeted therapies (e.g., γ-secretase inhibitors) ineffective. On the other hand, FLT3 mutations were detected in more than 75 % of GATA3low ETP-ALL and our group has previously shown in vitro efficacy of FLT3 inhibitors in human T-ALL cell lines [17]. Importantly, GATA3 DNA hypermethylation implicates epigenetic therapies in GATA3low ETP-ALL, such as decitabine, a hypomethylating agent approved for the treatment of myelodysplastic syndrome, chronic myelomonocytic leukemia, and AML. Our data demonstrate that decitabine induced apoptosis in PER-117 cells while lowering GATA3 DNA methylation. Subsequent induction of GATA3 expression may function as a surrogate of T cell differentiation which we also observed in PER-117 cells upon decitabine treatment. This is in line with murine breast cancer, where lack of GATA3 is associated with undifferentiated tumors [8]. The IC50 of decitabine in our experiments was comparable to AML cell lines [41, 42], and current dosing of decitabine in AML results in a similar range of steady state plasma levels [41]. Although further experiments are necessary to evaluate in vivo efficacy of HMA in T-ALL, it is likely that similar doses of decitabine will be required in T-ALL and AML. It is important to note that forced GATA3 overexpression alone failed to induce relevant changes in proliferation, apoptosis, or differentiation in PER-117 cells, which we attributed to GATA3 dose sensitivity. Instead, subtle changes of GATA3 expression are needed to divert aberrant DNA hypermethylation towards an equilibrium of optimum methylation in T lymphoblasts [43], for which GATA3 induction upon treatment with decitabine serves as yet another example.
Conclusions
GATA3 silencing occurs in about one third of adult ETP-ALL patients and is associated with GATA3 DNA hypermethylation. Lack of GATA3 engages a transcriptional program that is characterized by enrichment of myeloid signatures and loss of T cell differentiation against a background of both T-ALL and ETP-ALL. We propose a novel stem cell-like leukemia termed GATA3low ETP-ALL with a high frequency of FLT3 mutations as a distinct molecular entity with sensitivity to hypomethylating agents.
Acknowledgements
This work was supported by a research grant from the Deutsche Krebshilfe (Mildred Scheel Professur) and the Wilhelm Sander-Stiftung to CD Baldus.
Authors’ contributions
LF, MN, and CDB designed and supervised the research. LF and MN led the data analysis. UK, SH (Heesch), and MM led the methods development. SS, TH, KS, and NG acquired the data and performed the data analysis. CS, JOT, SH (Heller), LHM, KI, and LB performed the experimental validation. LF, MN, and CDB prepared the figures. LF and CDB wrote the manuscript. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Additional files
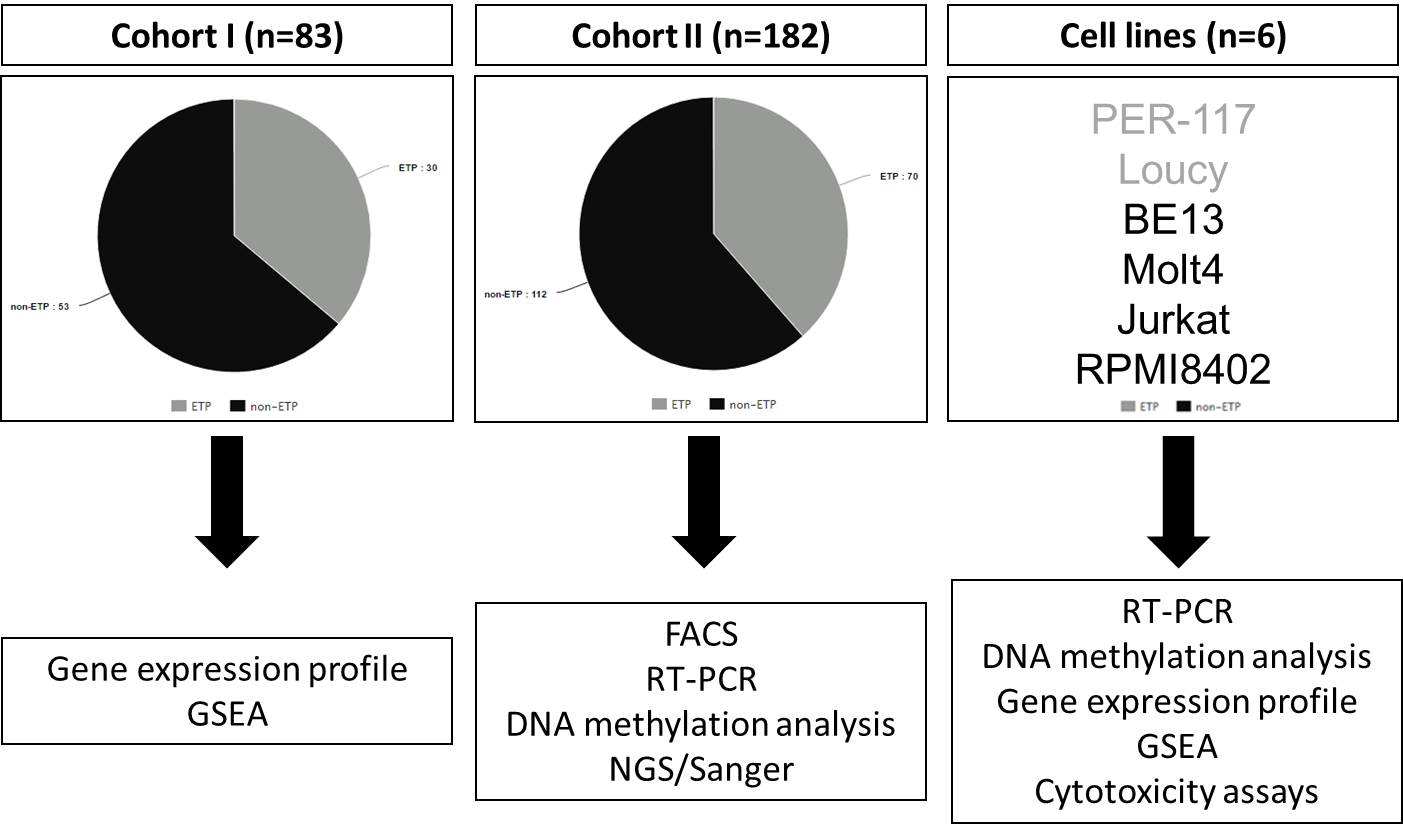{kind=link}
Flowchart depicting the patient sample cohorts. Strategy to define ETP-ALL is provided as well as platforms for subsequent analyses. (JPG 93 kb)
List of DEG comparing patients with low and high GATA3 mRNA expression. Microarray analyses of diagnostic bone marrow samples of 83 adult patients with T-ALL were used to generate a list of DEG comparing GATA3low and GATA3high. Only probe sets with at least 1.5-fold up- or downregulation with respect to GATA3 expression and a FDR <0.05 were included. (XLSX 102 kb)
{kind=link}
PER-117 and Loucy are ETP-ALL cell lines. Hierarchical clustering of ETP-ALL and non-ETP-ALL cell lines, using a set of genes differentially expressed in pediatric patients with ETP-ALL compared with non-ETP-ALL [20]. (PNG 63 kb)
{kind=link}
GATA3 expression in human leukemia cell lines. (A) RT-PCR-based analysis of GATA3 mRNA expression relative to Jurkat indicates differential GATA3 mRNA expression. (B) Western blot analysis revealed differential GATA3 protein expression (predicted molecular weight of 47 kDa). (JPG 35 kb)
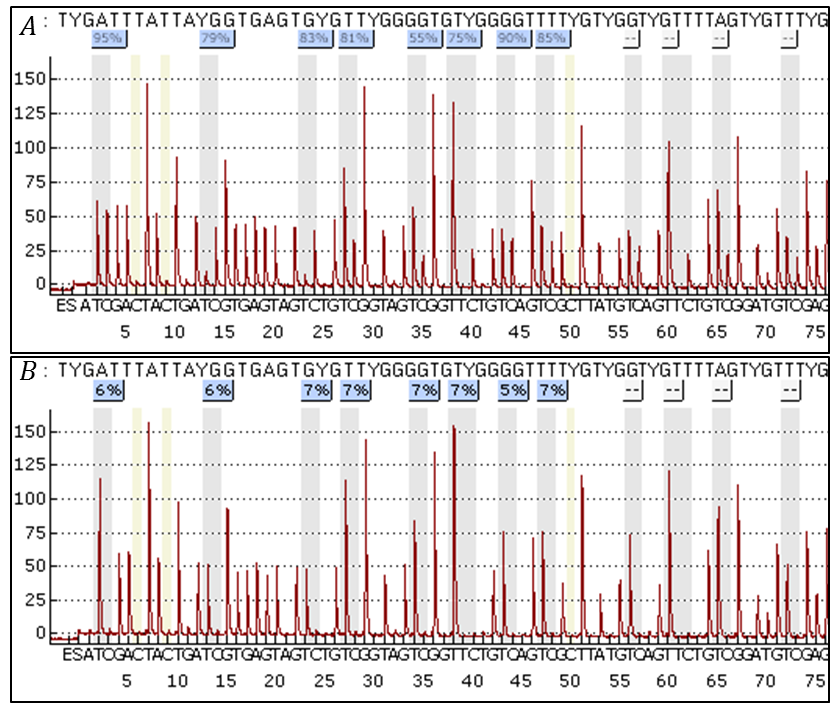{kind=link}
Pyrograms showing GATA3 DNA methylation in ETP-ALL in eight consecutive CpG sites across 64 bases. The sequence of the GATA3 amplicon is indicated at the top, while the sequence at the bottom of each panel indicates the dispensation order. The grey boxes highlight peaks resulting from sequential dispensations of C and T from which DNA methylation was calculated (blue boxes). (A) Representative ETP-ALL sample exhibiting high GATA3 DNA methylation. (B) Representative ETP-ALL sample exhibiting low GATA3 DNA methylation. (PNG 226 kb)
{kind=link}
Next generation sequencing comparing the mutational pattern of GATA3low (n = 5) and GATA3high ETP-ALL (n = 13). Sanger Sequencing was performed for selected genes in an extended cohort (n = 70) confirming a high rate of FLT3 mutations in GATA3low ETP-ALL. (PNG 47 kb)
{kind=link}
The GATA3low gene expression profile identified cases with ETP-ALL in an independent cohort of T-ALL. Unsupervised hierarchical cluster analysis of diagnostic bone marrow samples from 55 pediatric patients with T-ALL, using a set of genes defined by low GATA3 expression in adult T-ALL. Classification of ETP-ALL and non-ETP-ALL was performed according to the ETP gene expression signature published along with this dataset. (PNG 484 kb)
{kind=link}
KEGG pathway annotation of DEG in GATA3low versus GATA3high T-ALL patients. Lists of threefold over- and underexpressed genes of GATA3low patients were uploaded to DAVID Bioinformatics Resources v6.7. Listed are selected KEGG annotated pathway elements significantly enriched in GATA3low up- or downregulated DEGs. (PNG 34 kb)
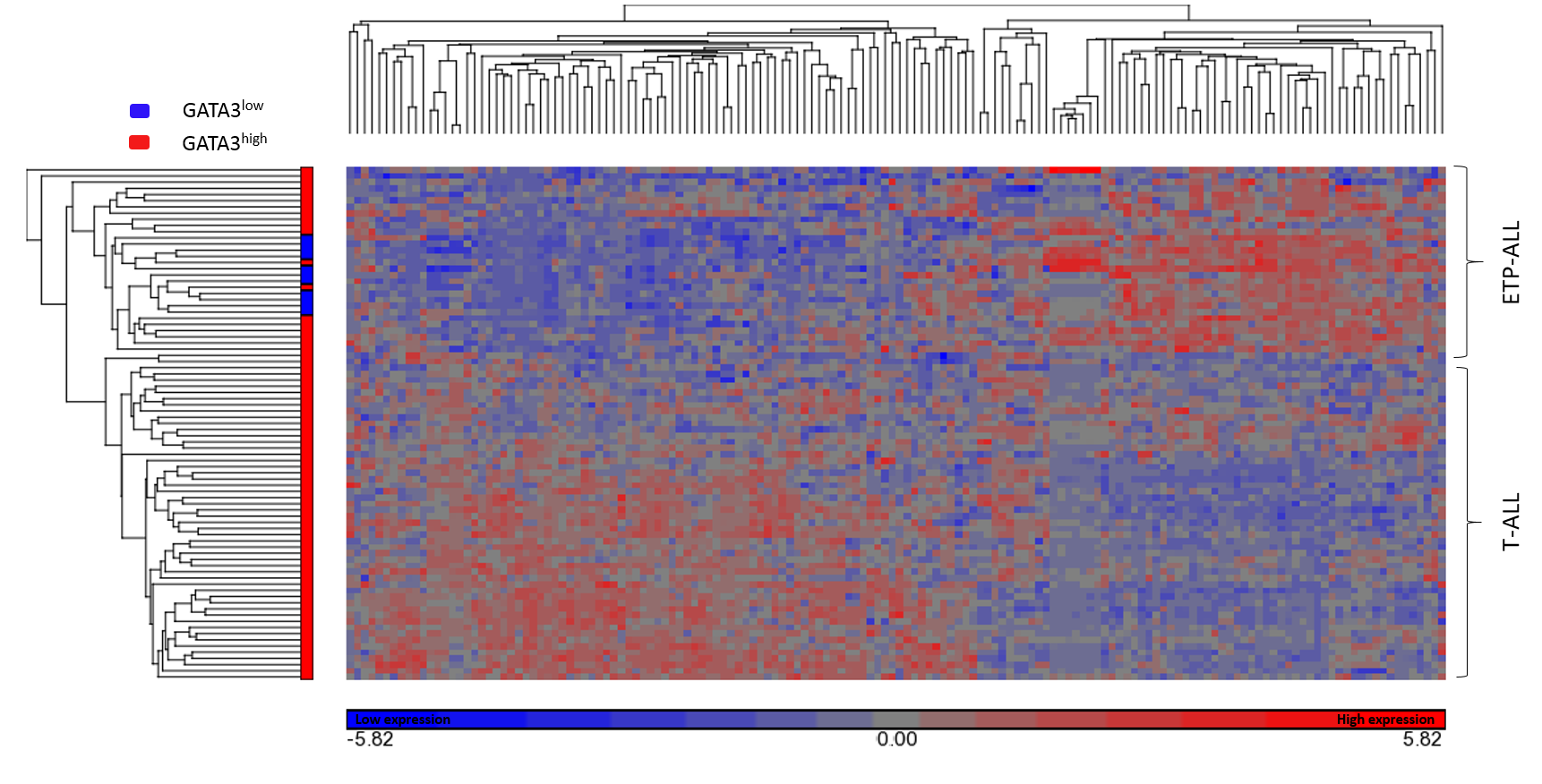{kind=link}
Validation of the GATA3low ETP-ALL cluster in adult patients with T-ALL using the ETP signature derived from pediatric T-ALL. Unsupervised hierarchical cluster analysis of diagnostic bone marrow samples from 83 adult patients with T-ALL, using a set of genes defined in pediatric T-ALL [20]. Blue = GATA3low ETP-ALL (n = 11), red = GATA3high (n = 72). Patients assigned to ETP-ALL and “typical” T-ALL are indicated. (PNG 748 kb)
{kind=link}
KEGG pathway annotation of DEG in PER-117 cells before versus after treatment with decitabine. Lists of 1.5-fold over- and underexpressed genes of cells treated with decitabine (5 μM) were uploaded to DAVID Bioinformatics Resources v6.7. Listed are selected KEGG annotated pathway elements significantly enriched in cells treated with decitabine. (PNG 61 kb)
References
- 1.Pai SY, Truitt ML, Ho IC. GATA-3 deficiency abrogates the development and maintenance of T helper type 2 cells. Proc Natl Acad Sci U S A. 2004;101:1993–1998. doi: 10.1073/pnas.0308697100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ho IC, Vorhees P, Marin N, Oakley BK, Tsai SF, Orkin SH, Leiden JM. Human GATA-3: a lineage-restricted transcription factor that regulates the expression of the T cell receptor alpha gene. EMBO J. 1991;10:1187–1192. doi: 10.1002/j.1460-2075.1991.tb08059.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hosoya T, Kuroha T, Moriguchi T, Cummings D, Maillard I, Lim KC, Engel JD. GATA-3 is required for early T lineage progenitor development. J Exp Med. 2009;206:2987–3000. doi: 10.1084/jem.20090934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bell JJ, Bhandoola A. The earliest thymic progenitors for T cells possess myeloid lineage potential. Nature. 2008;452:764–767. doi: 10.1038/nature06840. [DOI] [PubMed] [Google Scholar]
- 5.Challen GA, Sun D, Jeong M, Luo M, Jelinek J, Berg JS, Bock C, Vasanthakumar A, Gu H, Xi Y, et al. Dnmt3a is essential for hematopoietic stem cell differentiation. Nat Genet. 2012;44:23–31. doi: 10.1038/ng.1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wei G, Abraham BJ, Yagi R, Jothi R, Cui K, Sharma S, Narlikar L, Northrup DL, Tang Q, Paul WE, et al. Genome-wide analyses of transcription factor GATA3-mediated gene regulation in distinct T cell types. Immunity. 2011;35:299–311. doi: 10.1016/j.immuni.2011.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Van de Walle I, Dolens AC, Durinck K, De Mulder K, Van Loocke W, Damle S, Waegemans E, De Medts J, Velghe I, De Smedt M, et al. GATA3 induces human T-cell commitment by restraining Notch activity and repressing NK-cell fate. Nat Commun. 2016;7:11171. doi: 10.1038/ncomms11171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chou J, Lin JH, Brenot A, Kim JW, Provot S, Werb Z. GATA3 suppresses metastasis and modulates the tumour microenvironment by regulating microRNA-29b expression. Nat Cell Biol. 2013;15:201–213. doi: 10.1038/ncb2672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Higgins JP, Kaygusuz G, Wang L, Montgomery K, Mason V, Zhu SX, Marinelli RJ, Presti JC, Jr, van de Rijn M, Brooks JD. Placental S100 (S100P) and GATA3: markers for transitional epithelium and urothelial carcinoma discovered by complementary DNA microarray. Am J Surg Pathol. 2007;31:673–680. doi: 10.1097/01.pas.0000213438.01278.5f. [DOI] [PubMed] [Google Scholar]
- 10.Cooper SJ, Zou H, Legrand SN, Marlow LA, von Roemeling CA, Radisky DC, Wu KJ, Hempel N, Margulis V, Tun HW, et al. Loss of type III transforming growth factor-beta receptor expression is due to methylation silencing of the transcription factor GATA3 in renal cell carcinoma. Oncogene. 2010;29:2905–2915. doi: 10.1038/onc.2010.64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gulbinas A, Berberat PO, Dambrauskas Z, Giese T, Giese N, Autschbach F, Kleeff J, Meuer S, Buchler MW, Friess H. Aberrant gata-3 expression in human pancreatic cancer. J Histochem Cytochem. 2006;54:161–169. doi: 10.1369/jhc.5A6626.2005. [DOI] [PubMed] [Google Scholar]
- 12.Steenbergen RD, OudeEngberink VE, Kramer D, Schrijnemakers HF, Verheijen RH, Meijer CJ, Snijders PJ. Down-regulation of GATA-3 expression during human papillomavirus-mediated immortalization and cervical carcinogenesis. Am J Pathol. 2002;160:1945–1951. doi: 10.1016/S0002-9440(10)61143-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Stanelle J, Doring C, Hansmann ML, Kuppers R. Mechanisms of aberrant GATA3 expression in classical Hodgkin lymphoma and its consequences for the cytokine profile of Hodgkin and Reed/Sternberg cells. Blood. 2010;116:4202–4211. doi: 10.1182/blood-2010-01-265827. [DOI] [PubMed] [Google Scholar]
- 14.Perez-Andreu V, Roberts KG, Harvey RC, Yang W, Cheng C, Pei D, Xu H, Gastier-Foster JES, Lim JY, et al. Inherited GATA3 variants are associated with Ph-like childhood acute lymphoblastic leukemia and risk of relapse. Nat Genet. 2013;45:1494–1498. doi: 10.1038/ng.2803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Migliorini G, Fiege B, Hosking FJ, Ma Y, Kumar R, Sherborne AL, da Silva Filho MI, Vijayakrishnan J, Koehler R, Thomsen H, et al. Variation at 10p12.2 and 10p14 influences risk of childhood B-cell acute lymphoblastic leukemia and phenotype. Blood. 2013;122:3298–3307. doi: 10.1182/blood-2013-03-491316. [DOI] [PubMed] [Google Scholar]
- 16.Coustan-Smith E, Mullighan CG, Onciu M, Behm FG, Raimondi SC, Pei D, Cheng C, Su X, Rubnitz JE, Basso G, et al. Early T-cell precursor leukaemia: a subtype of very high-risk acute lymphoblastic leukaemia. Lancet Oncol. 2009;10:147–156. doi: 10.1016/S1470-2045(08)70314-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Neumann M, Coskun E, Fransecky L, Mochmann LH, Bartram I, Sartangi NF, Heesch S, Gokbuget N, Schwartz S, Brandts C, et al. FLT3 mutations in early T-cell precursor ALL characterize a stem cell like leukemia and imply the clinical use of tyrosine kinase inhibitors. PLoS One. 2013;8:e53190. doi: 10.1371/journal.pone.0053190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Allen A, Sireci A, Colovai A, Pinkney K, Sulis M, Bhagat G, Alobeid B. Early T-cell precursor leukemia/lymphoma in adults and children. Leuk Res. 2013;37:1027–1034. doi: 10.1016/j.leukres.2013.06.010. [DOI] [PubMed] [Google Scholar]
- 19.Zhang J, Ding L, Holmfeldt L, Wu G, Heatley SL, Payne-Turner D, Easton J, Chen X, Wang J, Rusch M, et al. The genetic basis of early T-cell precursor acute lymphoblastic leukaemia. Nature. 2012;481:157–163. doi: 10.1038/nature10725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Neumann M, Heesch S, Schlee C, Schwartz S, Gokbuget N, Hoelzer D, Konstandin NP, Ksienzyk B, Vosberg S, Graf A, et al. Whole-exome sequencing in adult ETP-ALL reveals a high rate of DNMT3A mutations. Blood. 2013;121:4749–4752. doi: 10.1182/blood-2012-11-465138. [DOI] [PubMed] [Google Scholar]
- 21.Paganin M, Ferrando A. Molecular pathogenesis and targeted therapies for NOTCH1-induced T-cell acute lymphoblastic leukemia. Blood Rev. 2011;25:83–90. doi: 10.1016/j.blre.2010.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jain N, Lamb AV, O'Brien S, Ravandi F, Konopleva M, Jabbour E, Zuo Z, Jorgensen J, Lin P, Pierce S, et al. Early T-cell precursor acute lymphoblastic leukemia/lymphoma (ETP-ALL/LBL) in adolescents and adults: a high-risk subtype. Blood. 2016;127:1863–1869. doi: 10.1182/blood-2015-08-661702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wood B, Winter SS, Dunsmore KP, Devidas M, Chen S, Asselin B, Esiashvili N, Loh ML, Winick NJ, Carroll WL, Raetz EA, Hunger SP. ASH Annual Meeting and Exposition. San Francisco, CA: ASH; 2014. T-lymphoblastic leukemia (T-ALL) shows excellent outcome, lack of significance of the early thymic precursor (ETP) immunophenotype, and validation of the prognostic value of end-induction minimal residual disease (MRD) in Children’s Oncology Group (COG) study AALL0434. [Google Scholar]
- 24.Conter V, Valsecchi MG, Buldini B, Parasole R, Locatelli F, Colombini A, Arico M, Rizzari C, Putti MC, Barisone E, Nigro LL, Santoro N, Ziino O, Pession A, Testi AM, Micalizzi C, Casale F, Silvestri D, Cazzaniga G, Biondi A, Basso G. 56th ASH Annual Meeting and Exposition. San Francisco, CA: ASH; 2014. Outcome of early T-cell precursor acute lymphoblastic leukemia in AIEOP patients treated with the AIEOP-BFM ALL 2000 study. [Google Scholar]
- 25.Haferlach T, Kohlmann A, Wieczorek L, Basso G, Kronnie GT, Bene MC, De Vos J, Hernandez JM, Hofmann WK, Mills KI, et al. Clinical utility of microarray-based gene expression profiling in the diagnosis and subclassification of leukemia: report from the International Microarray Innovations in Leukemia Study Group. J Clin Oncol. 2010;28:2529–2537. doi: 10.1200/JCO.2009.23.4732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Marcucci G, Baldus CD, Ruppert AS, Radmacher MD, Mrozek K, Whitman SP, Kolitz JE, Edwards CG, Vardiman JW, Powell BL, et al. Overexpression of the ETS-related gene, ERG, predicts a worse outcome in acute myeloid leukemia with normal karyotype: a Cancer and Leukemia Group B study. J Clin Oncol. 2005;23:9234–9242. doi: 10.1200/JCO.2005.03.6137. [DOI] [PubMed] [Google Scholar]
- 27.Kuhnl A, Kaiser M, Neumann M, Fransecky L, Heesch S, Radmacher M, Marcucci G, Bloomfield CD, Hofmann WK, Thiel E, Baldus CD. High expression of IGFBP2 is associated with chemoresistance in adult acute myeloid leukemia. Leuk Res. 2011;35:1585–1590. doi: 10.1016/j.leukres.2011.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Haferlach T, Kohlmann A, Schnittger S, Dugas M, Hiddemann W, Kern W, Schoch C. Global approach to the diagnosis of leukemia using gene expression profiling. Blood. 2005;106:1189–1198. doi: 10.1182/blood-2004-12-4938. [DOI] [PubMed] [Google Scholar]
- 29.da Huang W, Sherman BT, Lempicki RA. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009;37:1–13. doi: 10.1093/nar/gkn923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Novershtern N, Subramanian A, Lawton LN, Mak RH, Haining WN, McConkey ME, Habib N, Yosef N, Chang CY, Shay T, et al. Densely interconnected transcriptional circuits control cell states in human hematopoiesis. Cell. 2011;144:296–309. doi: 10.1016/j.cell.2011.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Doulatov S, Notta F, Eppert K, Nguyen LT, Ohashi PS, Dick JE. Revised map of the human progenitor hierarchy shows the origin of macrophages and dendritic cells in early lymphoid development. Nat Immunol. 2010;11:585–593. doi: 10.1038/ni.1889. [DOI] [PubMed] [Google Scholar]
- 32.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, Mesirov JP. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mootha VK, Lindgren CM, Eriksson KF, Subramanian A, Sihag S, Lehar J, Puigserver P, Carlsson E, Ridderstrale M, Laurila E, et al. PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat Genet. 2003;34:267–273. doi: 10.1038/ng1180. [DOI] [PubMed] [Google Scholar]
- 34.Kees UR, Ford J, Price PJ, Meyer BF, Herrmann RP. PER-117: a new human ALL cell line with an immature thymic phenotype. Leuk Res. 1987;11:489–498. doi: 10.1016/0145-2126(87)90082-8. [DOI] [PubMed] [Google Scholar]
- 35.Kalender Atak Z, De Keersmaecker K, Gianfelici V, Geerdens E, Vandepoel R, Pauwels D, Porcu M, Lahortiga I, Brys V, Dirks WG, et al. High accuracy mutation detection in leukemia on a selected panel of cancer genes. PLoS One. 2012;7:e38463. doi: 10.1371/journal.pone.0038463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Park R. Thyroid adenocarcinoma: correlation between mRNA expression and DNA methylation. 2013. [Google Scholar]
- 37.Neumann M, Vosberg S, Schlee C, Heesch S, Schwartz S, Gökbuget N, Hoelzer D, Graf A, Krebs S, Bartram I, Blum H, Brüggemann M, Hecht J, Bohlander SK, Greif PA, Baldus CD. Mutational spectrum of adult T-ALL. Oncotarget. 2014. [DOI] [PMC free article] [PubMed]
- 38.Yu S, Zhou X, Steinke FC, Liu C, Chen SC, Zagorodna O, Jing X, Yokota Y, Meyerholz DK, Mullighan CG, et al. The TCF-1 and LEF-1 transcription factors have cooperative and opposing roles in T cell development and malignancy. Immunity. 2012;37:813–826. doi: 10.1016/j.immuni.2012.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Neumann M, Heesch S, Gokbuget N, Schwartz S, Schlee C, Benlasfer O, Farhadi-Sartangi N, Thibaut J, Burmeister T, Hoelzer D, et al. Clinical and molecular characterization of early T-cell precursor leukemia: a high-risk subgroup in adult T-ALL with a high frequency of FLT3 mutations. Blood Cancer J. 2012;2:e55. doi: 10.1038/bcj.2011.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Homminga I, Pieters R, Langerak AW, de Rooi JJ, Stubbs A, Verstegen M, Vuerhard M, Buijs-Gladdines J, Kooi C, Klous P, et al. Integrated transcript and genome analyses reveal NKX2-1 and MEF2C as potential oncogenes in T cell acute lymphoblastic leukemia. Cancer Cell. 2011;19:484–497. doi: 10.1016/j.ccr.2011.02.008. [DOI] [PubMed] [Google Scholar]
- 41.Karahoca M, Momparler RL. Pharmacokinetic and pharmacodynamic analysis of 5-aza-2'-deoxycytidine (decitabine) in the design of its dose-schedule for cancer therapy. Clin Epigenetics. 2013;5:3. doi: 10.1186/1868-7083-5-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Berg T, Guo Y, Abdelkarim M, Fliegauf M, Lubbert M. Reversal of p15/INK4b hypermethylation in AML1/ETO-positive and -negative myeloid leukemia cell lines. Leuk Res. 2007;31:497–506. doi: 10.1016/j.leukres.2006.08.008. [DOI] [PubMed] [Google Scholar]
- 43.Schoofs T, Berdel WE, Muller-Tidow C. Origins of aberrant DNA methylation in acute myeloid leukemia. Leukemia. 2014;28:1–14. doi: 10.1038/leu.2013.242. [DOI] [PubMed] [Google Scholar]