

Abstract
Host prion (PrPC) genotype is a major determinant for the susceptibility to prion diseases. The Q/K222-PrPC polymorphic variant provides goats and mice with high resistance against classical scrapie and bovine spongiform encephalopathy (BSE); yet its effect against atypical scrapie is unknown. Here, transgenic mice expressing the goat wild-type (wt) or the K222-PrPC variant were intracerebrally inoculated with several natural cases of atypical scrapie from sheep and goat and their susceptibility to the prion disease was determined. Goat wt and K222-PrPC transgenic mice were 100% susceptible to all the atypical scrapie isolates, showing similar survival times and almost identical disease phenotypes. The capacity of the K222-PrPC variant to replicate specifically the atypical scrapie strain as efficiently as the goat wt PrPC, but not the classical scrapie or cattle-BSE as previously reported, further suggests the involvement of concrete areas of the host PrPC in the strain-dependent replication of prions.
Introduction
Scrapie is a fatal neurodegenerative disease of sheep and goats caused by the conversion of the host cellular prion protein (PrPC) into a pathological misfolded form (PrPSc). Scrapie occurs in a variety of phenotypes within two forms—classical and atypical scrapie forms—which differ in their incubation periods, clinical signs, neuropathological lesion profiles and/or PrPSc biochemical properties. The zoonotic potential of classical scrapie has been recently proposed from transmission studies of a panel of classical scrapie isolates in non-human primates [1] and in humanized transgenic mice [2], highlighting the necessity of deciphering the factors involved in prion transmission.
Susceptibility of sheep and goats to scrapie is strongly determined by the PrPc encoding gene (Prnp) and the prion strain. Strategies to promote breeding for the Prnp allele linked to classical scrapie resistance, A136R154R171, in sheep herds were implemented in some European countries and in the USA, resulting in rapid decline of classical scrapie outbreaks [3]. Unfortunately, A136R154R171 genotype does not provide resistance towards atypical scrapie [4].
We have previously demonstrated the high resistance of the goat K222-PrPC variant to a wide variety of classical scrapie isolates using transgenic (Tg) mice [5]. These results were confirmed by oral and intracerebral inoculations of a natural classical scrapie isolate in goats expressing this polymorphism [6] and we therefore proposed the K222-PrPC variant as a good candidate for developing selective breeding programs in goat herds. These studies are very valuable to control the spread of prion diseases in animals, but also provide crucial information about the role of the host genotype in the prion pathobiology. In the present study, we analyze the role of K222-PrPC variant on the resistance/susceptibility of goats to atypical scrapie using goat-PrP Tg mice.
Materials and methods
Ethics statements
Animal experiments were carried out in strict accordance with the recommendations of the Code for Methods and Welfare Considerations in Behavioral Research with Animals (Directive 86/609EC) and approved by the Committee on the Ethics of Animal Experiments (CEEA) of the Spanish Instituto Nacional de Investigación y Tecnología Agraria y Alimentaria (INIA); Permit Number: CEEA2011/050.
Transmission studies
Groups of 5–6 Tg mice (6–7 weeks old) expressing either the goat wild type (wt) PrPC (Q222-Tg501) or the K222-PrPC variant (K222-Tg516) at two fold the PrPC levels in the goat brain [5] were intracerebrally inoculated into the right parietal lobe with 20 μL of 10% brain homogenate (w/v) from sheep and goats naturally infected with atypical scrapie (see Table 1 for isolate information). Mice were monitored daily and their neurological status assessed twice a week. Mice were humanely euthanized when the progression of the disease was evident or at the end of their life span [≈650 days post-inoculation (dpi)]. During necropsy, brains were sliced sagittally and one hemibrain was fixed in 10% buffered formalin for histopathological analysis. The remaining hemibrain was homogenized as 10% (w/v) for the detection of proteinase K-resistant PrP (PrPres) by Western blot. Survival time was calculated as the mean number of survival dpi for all the mice positive for PrPres in the brain, with the standard error included, while attack rate was expressed as the proportion of PrPres-positive mice among the mice inoculated.
Table 1.
Description of natural atypical scrapie isolates
| Isolate | PrP genotype | Description and references | Supplier |
|---|---|---|---|
| Sheep-Sc M45 | wta | Cerebellum from a natural atypical scrapie case in sheep | NEIKERc |
| Goat-Sc I15 | wtb | Brain from a natural atypical scrapie case in goat | IZSTOd |
| Goat-Sc 12-09106 | wt; H/H154 | Cerebellum from a natural atypical scrapie case in goat | INIAVe |
aWild type (wt) sheep prion protein genotype: A136R154Q171/A136R154Q171.
bWild type (wt) goat prion protein genotype: I142R154R211Q222S240/I142R154R211Q222S240.
cInstituto Vasco de Investigación y Desarrollo Agrario (NEIKER), Vizcaya, Spain.
dIstituto Zooprofilattico Sperimentale de Piemont, Torino (IZSTO) Italy (We than Dr. Pier Luigi Acutis for providing the Goat-Sc I15 isolate).
eLaboratório de Patologia; Instituto Nacional de Investigação Agrária e Veterinária (INIAV); Lisboa, Portugal.
Western blotting
Brain tissue was homogenized in 5% glucose in distilled water in grinding tubes (Bio-Rad) and adjusted to 10% (w/v) by using a TeSeETM Precess 48TM homogenizer (Bio-Rad) following the manufacturer’s instructions. To determine the presence of PrPres in transgenic mouse brains, 100 µL of 10% brain homogenate were analyzed by Western blotting as previously described [7] with some modifications. Briefly, digestion was done with 40 µg/mL of proteinase K in buffer 5% sarkosyl, 5% Triton X100, 1 M Urea and 16 mM Tris–HCl (pH 9.6) at 60 °C for 15 min. Samples were electrophoresed in 12% Criterion XT Bis–Tris Gel (BioRad). For immunoblotting, membranes were incubated with 12B2 PrP monoclonal antibody (epitope 93WGQGG97 of the sheep PrP sequence) at a final concentration of 1 μg/mL. Immunocomplexes were detected with horseradish peroxidase-conjugated anti-mouse IgG (Amersham Pharmacia Biotech) after incubating the membranes for 1 h, and blots were developed with chemiluminescent substrate ECL Select (GE Healthcare Amersham Biosciences). Images were captured using ChemiDoc XRS + System and then processed using Image Lab 5.2.1 Software.
Histopathological analysis
Formalin-fixed brains were trimmed, dehydrated, embedded in paraffin-wax, cut to 4 µm thickness, de-waxed and rehydrated by standard procedures. For IHC demonstration of PrPSc accumulation, tissue sections were subjected to antigen retrieval and quenching of hydrogen peroxide, as described previously [8] and incubated with polyclonal PrP antibody R486 (APHA, Weybridge, UK; 240SPPVILLISFLIFLI254 epitope of the sheep PrP sequence); the subsequent steps of the IHC procedure were performed by a commercial immunoperoxidase technique (Vector-Elite ABC kit, Vector Laboratories) as per manufacturer’s instructions and sections were finally counterstained with Mayer’s haematoxylin. The PrPSc types considered included the intraneuronal (ITNR), intraglial (ITGL, intra-microglial and intra-astrocytic combined), stellate (STEL), fine particulate (PART), coalescing (COAL), linear (LINR) and plaques (PLAQ, vascular and non-vascular combined) as described elsewhere [8]. Plaques were only considered as such when they showed a homogeneous core and a radiate periphery (mature plaques), while in the absence of these features the dense accumulations of PrPSc were termed “coalescing”, without prejudice to them actually corresponding to primitive plaques.
Results
Tg mouse lines expressing the goat wild type PrPC (Q222-Tg501) and the K222-PrPC variant (K222-Tg516) were intracerebrally challenged with 3 natural cases of atypical scrapie (Table 1) and their susceptibilities assessed and compared following previously described methods [5].
Both Q222-Tg501 and K222-Tg516 mice succumbed to the inoculation of all atypical scrapie isolates with 100% attack rates (Table 2). For each isolate, Q222-Tg501 and K222-Tg516 mice displayed similar survival times which were longer than 300 dpi in all cases (Table 2).
Table 2.
Transmission of atypical scrapie to Q 222 -Tg501 and K 222 -Tg516 mice
| Isolate | Mean survival time in days ± SD (n/n0) | |
|---|---|---|
| Q222-Tg501 | K222-Tg516 | |
| Sh-Sc M45 | 443 ± 70 (9/9) | 441 ± 131 (10/10) |
| Goat-Sc I15 | 552 ± 78 (6/6) | 552 ± 64 (6/6) |
| Goat-Sc 12-09106 | 331 ± 15 (7/7) | 309 ± 34 (6/6) |
n/n 0 attack rate expressed as the proportion of positive mice among the total inoculated mice
PrPres was detected in the brains of all the inoculated Q222-Tg501 and K222-Tg516 mice by Western blot. PrPres glycoprofiles were characterized by a ladder-like pattern and a low unglycosylated band of around 7 kDa similar to that observed in their respective original sheep or goat isolates (Figure 1).
Figure 1.
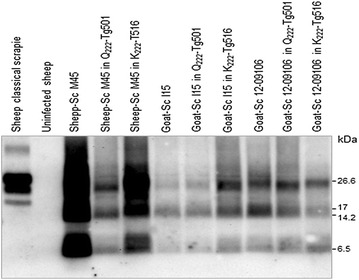
Brain PrP res from Q 222 -Tg501 and K 222 -Tg516 mice inoculated with an atypical scrapie isolate. Immunoblots of brain PrPres detected with 12B2 mAb of atypical scrapie isolates before and after transmission in either Q222-Tg501 or K222-Tg516 mice. Sheep classical scrapie and uninfected sheep brain were included on the right. Similar amounts of 10% brain homogenate were loaded in each lane for comparison. Both Q222-Tg501 and K222-Tg516 mice showed a similar PrPres ladder pattern characteristic of atypical scrapie. Molecular masses (kDa) are shown on the right.
All infected Q222-Tg501 and K222-Tg516 mice showed PrPSc in their brains with indistinguishable immunohistochemical phenotypes. PrPSc appeared as occasional, scattered coalescing or amorphous plaque-like deposits restricted to the molecular layer of the cerebellum while vacuolation was minimal in this area (Figure 2A, B). No PrPSc deposition or spongiform change were observed in any of the other brain areas examined, as exemplified by the hippocampus and thalamus in Figure 2C and D.
Figure 2.
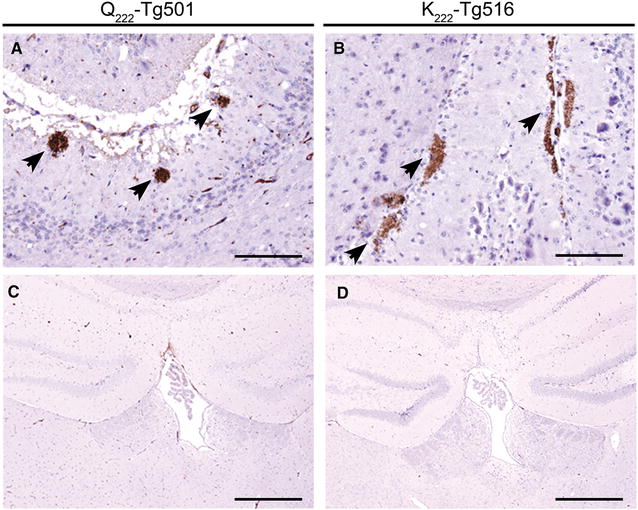
PrP Sc accumulation in the brain of Q 222 -Tg501 and K 222 -Tg516 mice infected with Sheep-Sc M45. Immunohistochemistry with R486 PrP antibody and haematoxylin counterstaining. PrPSc remained located to the molecular layer of the cerebellum as amorphous coalescing or plaque-like deposits (arrows) (A, B). Note the absence of PrPSc and vacuolation in the hippocampus and dorsal thalamus (C, D). Scale bars are 100 µm (A, B) and 500 µm (C, D).
Discussion
Intracerebral inoculations of prions in transgenic mice expressing different PRNP polymorphic variants are highly valuable to point to a specific amino acid substitution as the unique mutation responsible for the resistance/susceptibility of a species to a prion disease. Our results indicate that K222-PrPC variant replicates the atypical scrapie agent as efficiently as the wt-PrPC and thus, it might not be able to protect goats against the infection with this prion disease. However, this assumption must be confirmed by oral inoculation of Q/K222 goats since there is not always a correlation between intracerebral inoculations in transgenic mouse models and field infections in natural hosts [9].
Intracerebral transmission of atypical scrapie in both Q222-Tg501 and K222-Tg516 mouse lines resulted in a prion disease characterized by noticeable long survival times (>300 dpi) and poor pathology in brain. These long survival times are consistent with the old age at which sheep and goats normally develop atypical scrapie either naturally (over 5 years of age) [4] or after intracerebral transmission (around 2.5 years of age) [10] and suggests that atypical scrapie is a slow infectious agent in its natural PrPC species context. In line with this view, mice overexpressing 8- to 10-fold the sheep PrPC in their brains succumbed to the intracerebral inoculation of multiple French atypical scrapie isolates only after more than 400 dpi [11]. However, prolonged survival times are not exclusive of atypical scrapie agents since particular classical scrapie isolates can produce survival times longer than 500 dpi even after serial intracerebral passages in homozygous Q222-Tg501 mice (unpublished data).
The histological and immunohistochemical examinations of the brains from all the atypical scrapie infected Q222-Tg501 and K222-Tg516 mice consistently revealed a mild accumulation of PrPSc restricted to the cerebellum with little or no vacuolation also limited to this brain area. These outcomes clearly contrast with the widespread PrPSc and intense vacuolation observed in classical scrapie infected Q222-Tg501 mice or in BSE infected K222-Tg516 mice [5]; instead they correlate well with the particularly prominent PrPSc deposition in the cerebellum of sheep and goats with natural atypical scrapie [12]. The PrPres glycoprofiles of all the atypical scrapie isolates were also maintained in both Q222-Tg501 and K222-Tg516 mouse lines which, together with the survival times and the histological outcomes, further demonstrates the validity of our animal models to characterize prion strains of sheep and goats.
Susceptibility to prions and disease phenotypes are likely modulated by conformational properties of prion strains and the PrPC and PrPSc amino acid sequence [13]. Some host PrPC amino acid sequences may have greater “plasticity” or ability to misfold—as proposed for the unique susceptibility of bank voles to a wide variety of prion diseases [14] and to “in vitro” conversion experiments [15]—while others may show the opposite situation, locking PrPC and preventing its conversion by different prion strains. Nonetheless, the plasticity of the host PrPC would not explain why certain amino acid substitutions inhibit the replication of some prion strains but not others. A clear example is the goat K222 variant which provides high resistance against classical scrapie and bovine spongiform encephalopathy (BSE) from cattle—being protective even in heterozygous mice and goats [5, 6, 9]—but it is as efficient as the goat wt PrPC in replicating sheep and goat-BSE [5] and atypical scrapie when intracerebrally inoculated. The resistance of the K222 variant to classical scrapie was associated to the additional positive charge at codon 222 provided by the lysine amino acid, which could interfere with the PrPC:PrPSc interaction, abolishing or lowering the conversion rates of PrPC [16]. Similarly, the perturbations in the PrPC surface charge distribution and structural rearrangements mainly localized at the β2–α2 loop region (residues 169–179 in goat PrPC numbering) are likely to underlie the resistance of the human E/K219 variant against CJD [17].
More recently, the existence of critical initial PrPC–PrPSc interaction sites during the templating of PrPC by different prions was proposed [18]. The involvement of specific PrPC areas in the strain-dependent replication of prions could account for the variable role of the K222-PrPC variant in the susceptibility to scrapie. While for classical scrapie polymorphic variants along the PrPC have been associated to modulate the susceptibility to the disease, for atypical scrapie only few have been reported—A/V136, F/L141, R/H154 and Q/R171 [4, 19]— that, interestingly, are all located within β1–α1 and β2–α2 loops. This area could be the critical segment for atypical scrapie interaction with the host PrPC and therefore any amino acid exchange outside this segment would not affect the capacity of replication of the atypical scrapie, as observed in our transmission study in goat wt and K222-PrPC mice. In fact this area seems to be accessible in the abnormally folded aggregate of the atypical scrapie agent as suggested by the observation that the C-terminal part of atypical scrapie PrPSc can be specifically trimmed by PK [20]. In conclusion, although more information is still needed to decipher the mechanisms of prion conversion, our study points to the involvement of specific PrPC areas in the strain-dependent replication of prions. Thus, strategies to fight prion diseases based on genotype breeding programs should be prion strain targeted.
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
PAC, JCE, OA and JMT designed the experiments, conducted the analyses, and helped to draft the manuscript. LG performed the histological analysis. LO, LG and RJ contributed to the analysis of the data. All authors read and approved the final manuscript.
Acknowledgements
We thank the staff of the BSL-3 animal facility and the Biosafety Office at the CISA-INIA (Valdeolmos-Madrid) for their excellent animal care and work.
Funding
This work was supported by grants from the Spanish Ministerio de Ciencia e Innovación (AGL2009-11553-C02-02 and AGL2012-37988-C04-04) and by European Union projects (FOOD-CT-2006-36353, and 219235 ERA-NET EMIDA).
Contributor Information
Patricia Aguilar-Calvo, Email: p2aguilar@ucsd.edu.
Juan-Carlos Espinosa, Email: espinosa.juan@inia.es.
Olivier Andréoletti, Email: o.andreoletti@envt.fr.
Lorenzo González, Email: lontxi20@gmail.com.
Leonor Orge, Email: leonor.orge@iniav.pt.
Ramón Juste, Email: rjuste@neiker.eus.
Juan-María Torres, Email: jmtorres@inia.es.
References
- 1.Comoy EE, Mikol J, Luccantoni-Freire S, Correia E, Lescoutra-Etchegaray N, Durand V, Dehen C, Andreoletti O, Casalone C, Richt JA, Greenlee JJ, Baron T, Benestad SL, Brown P, Deslys JP. Transmission of scrapie prions to primate after an extended silent incubation period. Sci Rep. 2015;5:11573. doi: 10.1038/srep11573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cassard H, Torres JM, Lacroux C, Douet JY, Benestad SL, Lantier F, Lugan S, Lantier I, Costes P, Aron N, Reine F, Herzog L, Espinosa JC, Beringue V, Andréoletti O. Evidence for zoonotic potential of ovine scrapie prions. Nat Commun. 2014;5:5821. doi: 10.1038/ncomms6821. [DOI] [PubMed] [Google Scholar]
- 3.Fediaevsky A, Gasqui P, Calavas D, Ducrot C. Discrepant epidemiological patterns between classical and atypical scrapie in sheep flocks under French TSE control measures. Vet J. 2010;185:338–340. doi: 10.1016/j.tvjl.2009.06.019. [DOI] [PubMed] [Google Scholar]
- 4.Benestad SL, Arsac JN, Goldmann W, Noremark M. Atypical/Nor98 scrapie: properties of the agent, genetics, and epidemiology. Vet Res. 2008;39:19. doi: 10.1051/vetres:2007056. [DOI] [PubMed] [Google Scholar]
- 5.Aguilar-Calvo P, Espinosa JC, Pintado B, Gutierrez-Adan A, Alamillo E, Miranda A, Prieto I, Bossers A, Andreoletti O, Torres JM. Role of the goat K222-PrPC polymorphic variant in prion infection resistance. J Virol. 2014;88:2670–2676. doi: 10.1128/JVI.02074-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lacroux C, Perrin-Chauvineau C, Corbiere F, Aron N, Aguilar-Calvo P, Torres JM, Costes P, Bremaud I, Lugan S, Schelcher F, Barillet F, Andréoletti O. Genetic resistance to scrapie infection in experimentally challenged goats. J Virol. 2014;88:2406–2413. doi: 10.1128/JVI.02872-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Padilla D, Béringue V, Espinosa JC, Andreoletti O, Jaumain E, Reine F, Herzog L, Gutierrez-Adan A, Pintado B, Laude H, Torres JM. Sheep and goat BSE propagate more efficiently than cattle BSE in human PrP transgenic mice. PLoS Pathog. 2011;7:e1001319. doi: 10.1371/journal.ppat.1001319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gonzalez L, Martin S, Houston FE, Hunter N, Reid HW, Bellworthy SJ, Jeffrey M. Phenotype of disease-associated PrP accumulation in the brain of bovine spongiform encephalopathy experimentally infected sheep. J Gen Virol. 2005;86:827–838. doi: 10.1099/vir.0.80299-0. [DOI] [PubMed] [Google Scholar]
- 9.Aguilar-Calvo P, Fast C, Tauscher K, Espinosa JC, Groschup MH, Nadeem M, Goldmann W, Langeveld J, Bossers A, Andreoletti O, Torres JM. Effect of Q211 and K222 PRNP polymorphic variants in the susceptibility of goats to oral infection with goat bovine spongiform encephalopathy. J Infect Dis. 2015;212:664–672. doi: 10.1093/infdis/jiv112. [DOI] [PubMed] [Google Scholar]
- 10.Simmons MM, Moore SJ, Lockey R, Chaplin MJ, Konold T, Vickery C, Spiropoulos J. Phenotype shift from atypical scrapie to CH1641 following experimental transmission in sheep. PLoS ONE. 2015;10:e0117063. doi: 10.1371/journal.pone.0117063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Arsac JN, Betemps D, Morignat E, Feraudet C, Bencsik A, Aubert D, Grassi J, Baron T. Transmissibility of atypical scrapie in ovine transgenic mice: major effects of host prion protein expression and donor prion genotype. PLoS ONE. 2009;4:e7300. doi: 10.1371/journal.pone.0007300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Moore SJ, Simmons M, Chaplin M, Spiropoulos J. Neuroanatomical distribution of abnormal prion protein in naturally occurring atypical scrapie cases in Great Britain. Acta Neuropathol. 2008;116:547–559. doi: 10.1007/s00401-008-0433-8. [DOI] [PubMed] [Google Scholar]
- 13.Torres JM, Espinosa JC, Aguilar-Calvo P, Herva ME, Relano-Gines A, Villa-Diaz A, Morales M, Parra B, Alamillo E, Brun A, Brun A, Castilla J, Molina S, Hawkins SA, Andreoletti O. Elements modulating the prion species barrier and its passage consequences. PLoS ONE. 2014;9:e89722. doi: 10.1371/journal.pone.0089722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Watts JC, Giles K, Patel S, Oehler A, Dearmond SJ, Prusiner SB. Evidence that bank vole PrP is a universal acceptor for prions. PLoS Pathog. 2014;10:e1003990. doi: 10.1371/journal.ppat.1003990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Orru CD, Groveman BR, Raymond LD, Hughson AG, Nonno R, Zou W, Ghetti B, Gambetti P, Caughey B. Bank vole prion protein as an apparently universal substrate for RT-QuIC-based detection and discrimination of prion strains. PLoS Pathog. 2015;11:e1004983. doi: 10.1371/journal.ppat.1004983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Eiden M, Soto EO, Mettenleiter TC, Groschup MH. Effects of polymorphisms in ovine and caprine prion protein alleles on cell-free conversion. Vet Res. 2011;42:30. doi: 10.1186/1297-9716-42-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Biljan I, Giachin G, Ilc G, Zhukov I, Plavec J, Legname G. Structural basis for the protective effect of the human prion protein carrying the dominant-negative E219 K polymorphism. Biochem J. 2012;446:243–251. doi: 10.1042/BJ20111940. [DOI] [PubMed] [Google Scholar]
- 18.Kurt TD, Jiang L, Fernandez-Borges N, Bett C, Liu J, Yang T, Spraker TR, Castilla J, Eisenberg D, Kong Q, Sigurdson CJ. Human prion protein sequence elements impede cross-species chronic wasting disease transmission. J Clin Invest. 2015;125:2548. doi: 10.1172/JCI82647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Moum T, Olsaker I, Hopp P, Moldal T, Valheim M, Benestad SL. Polymorphisms at codons 141 and 154 in the ovine prion protein gene are associated with scrapie Nor98 cases. J Gen Virol. 2005;86:231–235. doi: 10.1099/vir.0.80437-0. [DOI] [PubMed] [Google Scholar]
- 20.Pirisinu L, Nonno R, Esposito E, Benestad SL, Gambetti P, Agrimi U, Zou WQ. Small ruminant nor98 prions share biochemical features with human gerstmann-straussler-scheinker disease and variably protease-sensitive prionopathy. PLoS ONE. 2013;8:e66405. doi: 10.1371/journal.pone.0066405. [DOI] [PMC free article] [PubMed] [Google Scholar]