

Abstract
The activity of cysteine cathepsin B increased markedly in lung homogenates and in bronchoalveolar lavage fluids (BALF) of the mouse model of bleomycin-induced lung fibrosis after 14 days of challenge. In contrast the level of the cysteine cathepsin inhibitor cystatin C was unaffected in BALF of wild-type and cathepsin B-deficient mice. Therefore, murine cystatin C is not a reliable marker of fibrosis during bleomycin-induced lung fibrosis. Current data are in sharp contrast to previous analysis carried on human BALF from patients with idiopathic pulmonary fibrosis, for which the level of cathepsin B remained unchanged while cystatin C was significantly increased.
Keywords: Bronchoalveolar lavage fluid, Bleomycin, Cathepsins, Cystatins, Fibrosis, Lung, Proteases
Findings
Cathepsin B, cystatin C and idiopathic pulmonary fibrosis: A quick focus
Idiopathic pulmonary fibrosis (IPF) is a chronic irreversible lung disease of unknown etiology characterized by an important deposition of extracellular matrix (ECM) components in the interstitial space and alveoli [1]. Most of in vivo studies relied on the murine model of bleomycin (BLM)-induced lung fibrosis [2]. Besides irradiation, fluorescein isothiocyanate (FITC) or silica model, the bleomycin model is the best characterized model with common characteristics of human IPF and is considered to be clinically relevant. Moreover, the experimental time frame is short and reproducible with the development of fibrosis occurring by day 14 (D14) as seen both biochemically and histologically [2]. However, fibrotic mechanisms for IPF and bleomycin-induced lung damage may be different, since experimental lung fibrosis is induced by a single dose of bleomycin applied via the intratracheal route and the role of inflammation is crucial, while human IPF is the result of repeated and diverse aggressions throughout the patient’s life and inflammatory episodes are of secondary importance. Moreover in the murine model, fibrosis is reversible as opposed to IPF that is a progressive and irreversible process leading to respiratory failure.
Cysteine cathepsins (11 members in humans) are proteases participating actively in ECM remodeling and in fibrotic disorders [3]. Cathepsin B (CatB) may contribute to lung myofibrogenesis by triggering the TGF-β1-driven Smad 2–3 pathway [4]. Conversely, pharmacological inhibition and genetic silencing of CatB diminished α-SMA expression, delayed fibroblast differentiation and led to an accumulation of intracellular pro-TGF-β1. In addition CatB drives activation of hepatic stellate cells, and participates in liver fibrogenesis. Recently, a conclusive trial (phase I) for the treatment of hepatic fibrosis (VBY-376, a CatB inhibitor from Virobay, Menlo Park, Ca, USA) supported the notion that the use of CatB inhibitors could be appropriate for therapy of lung fibrosis.
The proteolytic activity of cysteine cathepsins is regulated by their natural endogenous inhibitors belonging to the cystatin family [5, 6]. Among this family, cystatin C is the most potent inhibitor of cysteine cathepsins. Cystatin C is synthesized and secreted by an extensive variety of human cells with a widespread distribution in body fluids and tissues [7]. A significant increase of immunoreactive cystatin C was found in human bronchoalveolar lavage fluids (BALF) from IPF patients, raising the question of its use as a potential marker of IPF [8]. On the other hand, mouse and human cystatin C share 71 % of identity on amino acid level implying highly similar structural and functional properties of the homologues. Mouse cystatin C is expressed by all tissues with a relative content very similar to that of human tissues and has a widespread distribution in body fluids [9]. In terms of regulation cystatin C is rather seen as a housekeeping gene, because previous work could not establish its transcriptional regulation in response to various stimuli (e.g. bacterial and viral infections, stress, cytokines, growth factors) [10].
Increase of CatB activity in both lung homogenates and bronchoalveolar lavage fluids
BALF and lung homogenates from wild type (C57BL/6 strain) and CatB-deficient (CatB-/-) mice [11] were collected at D1 or D14 post-BLM treatment. To our knowledge consequences of the genetic deletion of CatB in bleomycin-induced fibrosis have not been reported or studied elsewhere. In this study, CatB-/- mice served primarily as control for assessment of the CatB-dependent peptidase activity. Samples were handled as described previously [8]. At D1, the overall cathepsin activity in lung homogenates remained unchanged for BLM- and saline-treated WT mice (Fig. 1a). Pre-incubation with CA-074, a selective CatB inhibitor [12], showed that CatB is the prevailing active cathepsin. Consistently, the peptidase activity decreased dramatically (~85 %) in lungs from saline- and BLM-treated CatB-/- mice. At D14, BLM instillation resulted in a 3-fold increase of the cathepsin activity in WT mice. Addition of CA-074 indicated that this activity is mostly CatB-dependent. Again cathepsin activity in BLM-treated CatB-/- mice was markedly decreased (~80 %), confirming that BLM administration to WT mice induces an overexpression of CatB at D14 (Fig. 1a). Results correlate with a recent article uncovering an increase of lung cathepsins after BLM administration [13]. In this elegant study, the use of an optical probe revealed a specific and maximal labeling (D14) of cathepsins at sites of fibrotic lesions correlated with the extent of disease burden [13].
Fig. 1.
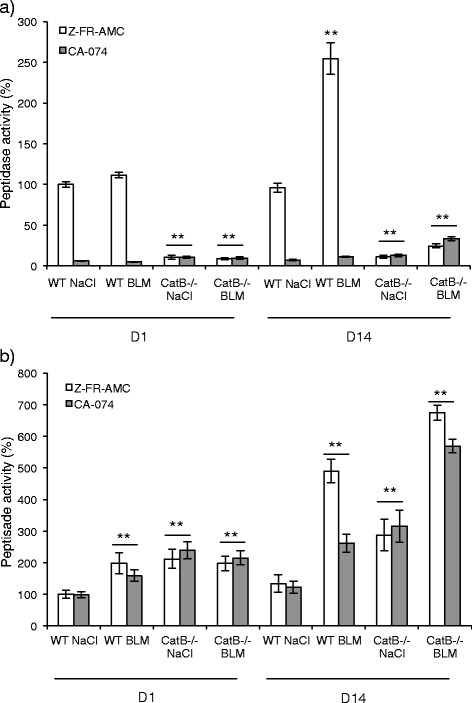
Peptidase activity of cysteine cathepsins after bleomycin administration to wild type and cathepsin B-deficient mice. Mice (C57BL/6 strain) were challenged with BLM (7.5 mg/kg for day 1; 3 mg/kg for day 14) or saline solution by intranasal instillation and sacrificed after 1 and 14 days [19]. Lung homogenates and BALF were prepared as described earlier by Gasse et al. ([19]) and Kasabova et al. ([8]). a Lung homogenates and (b) BALF were incubated in the activity buffer (0.1 M sodium acetate pH 5.5, 2 mM DTT, 2 mM EDTA, 0.01 % Brij35) for 5 min. at 30 °C. The overall cathepsin activity was measured toward Z-FR-AMC (50 μM, λexc = 350 nm and λem = 460 nm) at day 1 (D1) and day 14 (D14) (Gemini spectrofluorimeter, Molecular Devices). White bars: endopeptidase cathepsin activity. Grey bars: control experiments. Samples were preincubated in the presence of CA-074 (10 μM) for 30 min prior to measuring the peptidase activity. Data are expressed as mean values ± SEM. **p < 0.05 (Kruskall and Wallis non-parametric test) (WT NaCl, n = 5; WT BLM, n = 6; CatB-/- NaCl, n = 5; CatB-/- BLM, n = 8)
Cysteine cathepsin activity was next considered in BALF. At D1 post-BLM treatment, a ~2-fold increase of the activity was observed in BLM-treated WT mice compared to saline control. Pre-incubation with CA-074 demonstrated that active CatB, in contrast to the measurements in lung homogenates, is not the major cathepsin found in BALF at D1. The equivalent overall cathepsin activity in BALF from CatB-/- saline control and BLM-treated CatB-/- mice corroborated this statement (Fig. 1b). A 4-fold increase of cathepsin activity was assessed for WT mice at D14 post-BLM treatment and ~50 % of the peptidase activity related to CatB contrary to that observed at D1. Moreover, unlike lung homogenates, the cathepsin activity increased ~6-fold in CatB-/- mice after BLM challenge (Fig. 1b), supporting functional redundancy and/or the establishment of compensatory mechanisms between cysteine cathepsins [14, 15]. Present results differ from those observed in human BALF where no significant difference in cathepsin activity, including CatB, was observed between non-fibrotic and IPF patients [8]. A key point is that a similar amount of macrophages, which are the primary source of cysteine cathepsins in BALF, was found for both groups of patients [8]. Conversely in the murine BLM-induced lung fibrosis activated macrophages (with a M2/M2-like phenotype) are predominant in the immune infiltrate and BALF at D14, in association with the presence of markers of the Th2 profibrotic response [13]. Nevertheless recruited macrophages have intermediate proinflammatory and profibrotic phenotypes [16] and in overall the changes in the levels of key cytokines and chemokines upon bleomycin-induced fibrosis were consistent with those observed in human IPF [17].
The concentration of cystatin C is not affected by BLM challenge
We reported earlier a significant increase of immunoreactive cystatin C, in human BALF from IPF patients raising the question of its potential use as a new biomarker [8]. The increase of cystatin C level was significant for each of three IPF severity grades (stages I, II, II). Cystatin C has long been validated besides creatinine as a serum biomarker of glomerular filtration rate, but we did not measure a statistically confident variation in alveolar concentration of cystatin C between patients with a low (<60 ml/min) or a high (>60 ml/min) glomerular clearance (p > 0.1) and no co-morbidity in conjunction with cystatin C level was observed [8]. Here, cystatin C concentration remained unchanged both in lung extracts (Fig. 2a) and BALF (Fig. 2b) for BLM-treated and control mice at D1 and D14. The same level of BALF cystatin C was also measured for saline- and BLM-treated CatB-/- mice. Conversely to that observed in human BALF from IPF patients, cystatin C level is unaffected at D14 following BLM challenge, supporting clearly that murine cystatin C cannot be embraced as a marker in BLM-induced lung fibrosis.
Fig. 2.
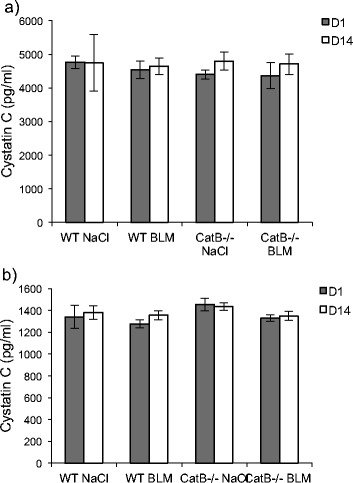
Cystatin C immunoassay. Dosage of immunoreactive cystatin C was performed after BLM (or saline solution) administration to wild-type and CatB-deficient mice (see legend to Fig. 1). The concentration of cystatin C was determined using the mouse/rat ELISA Quantikine kit according to the manufacturer’s instructions (R&D Systems Inc, Minneapolis, MN, USA). a Lung homogenates and (b) BALF. Data are expressed as mean values ± SEM (WT NaCl, n = 5; WT BLM, n = 6; CatB-/- NaCl, n = 5; CatB-/- BLM, n = 8)
Of interest bronchopulmonary dysplasia (BPD) is characterized by impaired alveolar development and widespread bronchial disease, and consecutive fibrotic changes are observed. Results in accordance with the present study were reported using a non-human primate model of BPD: mRNA and protein levels of CatB were significantly increased in the lung tissue of baboons with BPD. In contrast, both mRNA and protein levels of cystatin C remained unchanged in lung tissue lysates and BALF [18]. Although the rodent model of BLM-induced fibrosis is of mandatory concern to decipher proteolytic mechanisms of fibrogenesis, the present data confirm and point out that transposition of the results to human IPF should be done with caution.
Acknowledgements
Not applicable.
Funding
This work was supported by la Région Centre-Val de Loire (FibroCat project; number: 201000049823). We acknowledge the Institut National de la Santé et de la Recherche Médicale (INSERM) for institutional fundings. MK held a doctoral fellowship from MENRT (Ministère de l’Education Nationale, de la Recherche et de la Technologie, France). Funding agencies had no role in the design of the study, in the analysis and interpretation of data, and in writing the manuscript.
Availability of data and material
All data generated or analysed during this study are included in this published article.
Authors’ contributions
MK, IC and GL planned and designed research; MK, BV and AG performed experiments; FL and SMA contributed analytic tools; TR provided genetically engineered mice; MK, FL, IC and GL analyzed data; AG, FL, TR, SMA and IC revised the paper; MK and GL wrote the paper; All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Not applicable.
Ethics approval and consent to participate
All animal experiments were examined and approved under the number D-45-234-6 by the Ethic committee for animal experimentation of CNRS and University of Orléans and complied with the French Government’s ethical and animal experiment regulations.
Abbreviations
- AMC
7-Amino-4-methylcoumarin
- BALF
Bronchoalveolar lavage fluid, BLM, Bleomycin
- CA-074
N-(L-3-trans-propylcarbamoyloxirane-2-carbonyl)-L-isoleucyl-L-proline
- Cat
Cathepsin
- ECM
Extracellular matrix
- IPF
Idiopathic pulmonary fibrosis
- WT
Wild type
- Z
Benzyloxycarbonyl
Contributor Information
Mariana Kasabova, Email: mariana.kasabova@ulg.ac.be.
Bérengère Villeret, Email: berengere.villeret@inserm.fr.
Aurélie Gombault, Email: agombaul@cnrs-orleans.fr.
Fabien Lecaille, Email: fabien.lecaille@univ-tours.fr.
Thomas Reinheckel, Email: thomas.reinheckel@uniklinik-freiburg.de.
Sylvain Marchand-Adam, Email: sylvain.marchand-adam@univ-tours.fr.
Isabelle Couillin, Email: couillin@cnrs-orleans.fr.
Gilles Lalmanach, Phone: (+33) 247366151, Email: gilles.lalmanach@univ-tours.fr.
References
- 1.Spagnolo P, Tzouvelekis A, Maher TM. Personalized medicine in idiopathic pulmonary fibrosis: facts and promises. Curr Opin Pulm Med. 2015;21:470–8. doi: 10.1097/MCP.0000000000000187. [DOI] [PubMed] [Google Scholar]
- 2.Moore BB, Hogaboam CM. Murine models of pulmonary fibrosis. Am J Physiol Lung Cell Mol Physiol. 2008;294:152–60. doi: 10.1152/ajplung.00313.2007. [DOI] [PubMed] [Google Scholar]
- 3.Lalmanach G, Saidi A, Marchand-Adam S, Lecaille F, Kasabova M. Cysteine cathepsins and cystatins: from ancillary tasks to prominent status in lung diseases. Biol Chem. 2015;396:111–30. doi: 10.1515/hsz-2014-0210. [DOI] [PubMed] [Google Scholar]
- 4.Kasabova M, Joulin-Giet A, Lecaille F, Gilmore BF, Marchand-Adam S, Saidi A, et al. Regulation of TGF-β1-Driven Differentiation of Human Lung Fibroblasts: Emerging Roles of Cathepsin B and Cystatin C. J Biol Chem. 2014;289:16239–51. doi: 10.1074/jbc.M113.542407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Abrahamson M, Alvarez-Fernandez M, Nathanson CM. Cystatins. Biochem Soc Symp. 2003;70:179–99. doi: 10.1042/bss0700179. [DOI] [PubMed] [Google Scholar]
- 6.Turk V, Stoka V, Turk D. Cystatins: biochemical and structural properties, and medical relevance. Front Biosci. 2008;13:5406–20. doi: 10.2741/3089. [DOI] [PubMed] [Google Scholar]
- 7.Wallin H, Bjarnadottir M, Vogel LK, Wassélius J, Ekström U, Abrahamson M. Cystatins-Extra- and intracellular cysteine protease inhibitors: High-level secretion and uptake of cystatin C in human neuroblastoma cells. Biochimie. 2010;92:1625–34. doi: 10.1016/j.biochi.2010.08.011. [DOI] [PubMed] [Google Scholar]
- 8.Kasabova M, Joulin-Giet A, Lecaille F, Saidi A, Marchand-Adam A, Lalmanach G. Human cystatin C: A new biomarker of idiopathic pulmonary fibrosis? Proteomics Clin Appl. 2014;8:447–53. doi: 10.1002/prca.201300047. [DOI] [PubMed] [Google Scholar]
- 9.Hakansson K, Huh C, Grubb A, Karlsson S, Abrahamson M. Mouse and rat cystatin C: Escherichia coli production, characterization and tissue distribution. Comp Biochem Physiol B Biochem Mol Biol. 1996;114:303–11. doi: 10.1016/0305-0491(96)00025-9. [DOI] [PubMed] [Google Scholar]
- 10.Xu Y, Ding Y, Li X, Wu X. Cystatin C is a disease-associated protein subject to multiple regulation. Immunol Cell Biol. 2015;93:442–51. doi: 10.1038/icb.2014.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Halangk W, Lerch MM, Brandt-Nedelev B, Roth W, Ruthenbuerger M, Reinheckel T, et al. Role of cathepsin B in intracellular trypsinogen activation and the onset of acute pancreatitis. J Clin Invest. 2000;106:773–81. doi: 10.1172/JCI9411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rawlings ND, Barrett AJ, Finn R. Twenty years of the MEROPS database of proteolytic enzymes, their substrates and inhibitors. Nucleic Acids Res. 2016;44:D343–50. doi: 10.1093/nar/gkv1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Withana NP, Ma X, McGuire HM, Verdoes M, van der Linden WA, Ofori LO, et al. Non-invasive Imaging of Idiopathic Pulmonary Fibrosis Using Cathepsin Protease Probes. Sci Rep. 2016;6:19755. doi: 10.1038/srep19755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nägler DK, Ménard R. Family C1 cysteine proteases: biological diversity or redundancy? Biol Chem. 2003;384:837–43. doi: 10.1515/BC.2003.094. [DOI] [PubMed] [Google Scholar]
- 15.Akkari L, Gocheva V, Quick ML, Kester JC, Spencer AK, Garfall AL, et al. Combined deletion of cathepsin protease family members reveals compensatory mechanisms in cancer. Genes Dev. 2016;30:222–32. doi: 10.1101/gad.270439.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.François A, Gombault A, Villeret B, Alsaleh G, Fanny M, Gasse P, et al. B cell activating factor is central to bleomycin- and IL-17-mediated experimental pulmonary fibrosis. J Autoimmun. 2015;56:1–11. doi: 10.1016/j.jaut.2014.08.003. [DOI] [PubMed] [Google Scholar]
- 17.Bergeron A, Soler P, Kambouchner M, Loiseau P, Milleron B, Valeyre D, et al. Cytokine profiles in idiopathic pulmonary fibrosis suggest an important role for TGF-beta and IL-10. Eur Respir J. 2003;22:69–76. doi: 10.1183/09031936.03.00014703. [DOI] [PubMed] [Google Scholar]
- 18.Altiok O, Yasumatsu R, Bingol-Karakoc G, Riese RJ, Stahlman MT, Dwyer W, et al. Imbalance between cysteine proteases and inhibitors in a baboon model of bronchopulmonary dysplasia. Am J Respir Crit Care Med. 2006;173:318–26. doi: 10.1164/rccm.200503-425OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gasse P, Riteau N, Charron S, Girre S, Fick L, Pétrilli V, et al. Uric acid is a danger signal activating NALP3 inflammasome in lung injury inflammation and fibrosis. Am J Respir Crit Care Med. 2009;179:903–913. doi: 10.1164/rccm.200808-1274OC. [DOI] [PubMed] [Google Scholar]