

Abstract
Background
Overgrowth syndromes represent clinically and genetically heterogeneous conditions characterized by a wide spectrum of malformations, tall stature, intellectual disability and/or macrocephaly.
Results
In a cohort of four clinically characterized patients with overgrowth syndrome without known causative gene mutation, we performed an Illumina SNP-array analysis to identify the pathogenic copy number variations. We identified two rare copy number variations harboring overgrowth syndrome related genes. Patient 1 was Malan syndrome with a 1.4 Mb 19p13.2-13.13 microdeletion including NFIX, and Patient 2 was identified as Sotos syndrome with a 1.6 Mb 5q35.2 microdeletion encompassing NSD1.
Conclusions
We identified two patients associated with Manlan syndrome and Sotos syndrome respectively. We also discuss the use of the microarrays-based candidate gene strategy in Mendelian disease-gene identification.
Electronic supplementary material
The online version of this article (doi:10.1186/s13039-016-0282-4) contains supplementary material, which is available to authorized users.
Keywords: Deletion 19p13.2-13.13, Deletion 5q35.2, NSD1, SNP-array, Epigenetic diseases, Microarrays-based candidate gene strategy
Background
Overgrowth with multiple birth defects is useful phenotypic sign in diagnostic categorization of congenital diseases. Overgrowth syndromes (OGSs) are a group of heterogeneous disorders including Sotos syndrome (SOTOS1, OMIM#117550), Beckwith-Wiedemann syndrome (BWS; OMIM#130650), Simpson-Golabi-Behmel syndrome (SGBS1, OMIM#312870), Proteus syndrome (PS, OMIM#176920), Bannayan-Riley-Ruvalcaba syndrome (BRRS, OMIM#153480) and Weaver Syndrome (WVS, OMIM #277590) [1]. Recently the molecular causes of several OGSs have been revealed [2]. Despite these advances, the underlying genetic mechanisms for 20–40 % of overgrowth individuals remain unknown [2].
Malan syndrome, also named as Sotos-like syndrome or Sotos syndrome 2 (SOTOS2, OMIM#614753), is a recently introduced clinical condition characterized by tall stature, intellectual disability and/or macrocephaly [2–4]. This syndrome has been associated with the NFIX gene on chromosomal locus 19p13.2. NFIX encodes the nuclear factor I X (CCAAT-binding transcription factor) protein [5]. It has been suggested that either a nonsense mutation in the NFIX gene or microdeletion encompassing NFIX underlies this syndrome [2, 6].
As part of a larger study on the identification of pathogenic copy number variations in children with birth defects, four overgrowth children carrying multiple birth defects with undetermined genetic reasons were enrolled in this study. High-resolution SNP-array analysis (Illumina, San Diego, CA, USA) was performed to analyze the genomes of these patients. Here, we present two patients with overgrowth syndromes, Patient 1 was diagnosed as Malan syndrome with a 1.4 Mb deletion in chromosomal region 19p13.2-13.13, and Patient 2 was identified as Sotos syndrome with a 1.6 Mb 5q35.2 microdeletion.
Clinical report
Patient 1
This male patient was two-year and seven-month old at the time of genetic evaluation. He was the second child of a healthy unrelated couple. Family history of birth defects or genetic disorders was denied. He was born at 39 weeks of gestation by normal delivery. His birth weight was 4000 g (75-90th centile) with a birth length of 51 cm (50th centile) and a head circumference of 37 cm (75–90th centile). Due to respiratory failure, the patient was treated with intubation and artificial respiration and admitted to the neonatal intensive care unit (NICU). At six months, he attended to the Second Xiangya Hospital because of development delay. At this time, he weighed 8000 g (75–90th centile) with a length of 72 cm (50th centile) and a head circumference of 43 cm (75–90th centile). Mildly reduced volume of the white matter, and the thin corpus callosum were revealed by brain MRI. Electroencephalogram (EEG) showed no definite abnormality. At present, his length is 79 cm (50th centile), weight 12 kg (50th centile) and OFC 48.6 cm (75–90th centile), suggesting that his head is relatively large compared with his stature. He showed frontal bossing, high forehead, deep-set eyes, sparse eyebrows, a flat nasal bridge, low-set ears, high-arched palate, bilateral strabismus and pectus excavatum (Fig. 1). He could not sit up and walk by himself. Also he could not speak any meaningful words. His psychomotor development was evaluated by the GESELL Developmental Diagnosis Scale (GDDS) and the developmental quotient (DQ) was determined to be 19. Neurological examination revealed generalized hypotonia and reduced deep tendon reflexes. Peripheral nerve conduction velocities showed no abnormalities. Conventional G-banding analyses showed no abnormalities.
Fig. 1.
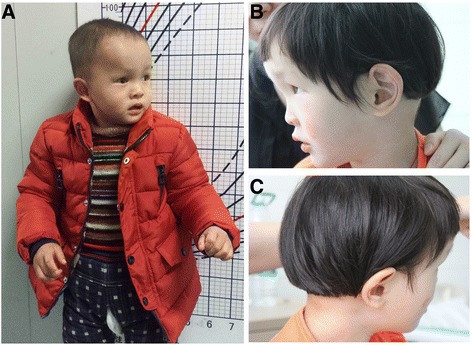
Clinical photographs of the probands. a: Patient 1 with Malan syndrome shows frontal bossing, high forehead, deep-set eyes, sparse eyebrows, a flat nasal bridge, low-set ears, high-arched palate, and bilateral strabismus. b, c: lateral view of the Patient 2 with Sotos syndrome. Frontal bossing, high forehead, deep-set eyes, heterochromia and congenital microphthalmia were observed
Patient 2
The patient was referred to the genetic service at the age of three years with intellectual disability, macrocephaly and congenital heart defect (patent ductus arteriosus, PDA). This girl was born to non-consanguineous, healthy parents. At her birth, her mother and father were both at 36 years old. She was born by cesarean at 39 weeks plus 4 days with a birth weight 3,780 g (70–90th centile); birth length of 52 cm (90th centile) and a head circumstance of 36.8 cm. Frontal bossing, high forehead, deep-set eyes and congenital heart disease were observed. Eye anomalies, heterochromia and congenital microphthalmia were also observed. Conventional G-banding analyses showed no abnormalities.
Patient 3
Patient 3 attended to our hospital due to congenital heart disease. Family history was absent. He was six-year old and had several distinct phenotypes, i.e. ASD (atrial septal defect), tall stature and behavior problems (self-hugging and frequent temper outbursts).
Patient 4
This patient was a five-year old boy. He was the second child of unrelated parents without obvious family history of inherited disease. Congenital heart defect (PDA), frontal bossing and high forehead, macrocephaly, big toe and also behavior problems (impulsiveness, frequent temper tantrums and outbursts) were observed.
Methods
The Review Board of the Second Xiangya Hospital of the Central South University approved this study. All subjects consented to this study.
Chromosome analysis was performed on peripheral blood of the patient and the parents by conventional G-banded techniques (550-band resolution). 5 ml peripheral blood was collected for each individual. All samples were subjected to lymphocyte culture according to standard cytogenetic protocol.
The genomic DNA was prepared from peripheral blood of the patient and the parents. Genomic DNA was prepared using a DNeasy Blood & Tissue Kit (Qiagen, Valencia, CA) on the QIAcube automated DNA extraction robot (Qiagen, Hiden, Germany).
Genomic DNA samples of the patient and parents were adjusted to a final concentration of 50 ng/μl. The HumanOmni1-Quad Chip (Illumina Inc., San Diego, USA) and the Illumina BeadScan genotyping system (Beadstation Scanner) were employed to obtain the signal intensities of SNP probes [7, 8]. Human Omni1-Quad Beadchip contains over 1.1 million loci across the human genome. The GenomeStudio V2011 software was used to analyze the genotypes (human genome build 37/Hg19 for analysis) and evaluate the experimental quality. The call rates of the samples are greater than 99.5 %.
Results
Conventional G-banding analyses showed no abnormalities in four children with overgrowth syndromes. To identify the patients' genetic lesions, we employed SNP-array system (HumanOmni1-Quad Chip) to analyze the possibility of pathogenic copy number variations (pCNVs). A de novo 1.4 Mb deletion ranging from 19p13.2 to 19p13.13 (chr19: 12,157,839-13,518,462/hg19) was identified in Patient 1 with unique clinical phenotypes i.e. macrocephaly, overgrowth and intellectual disability. This chromosome region is gene-rich and contains several OMIM genes NFIX, CACNA1A and CALR (Fig. 2). Consistent with previous reports that deletions of 19p13.2 are associated with overgrowth and other abnormalities, this patient got a clinically diagnosis as Malan syndrome (19p13.2 microdeletion syndrome). In addition, a de novo 1.6 Mb deletion on chromosomal locus 5q35.2 encompassing NSD1 (chr5:175,548,190-177,145,695/hg19) was revealed in Patient 2 with Sotos syndrome. SNP-array analysis showed no pathogenic deletions or duplications in Patient 3 and Patient 4, hence these two patients remain undiagnosed.
Fig. 2.
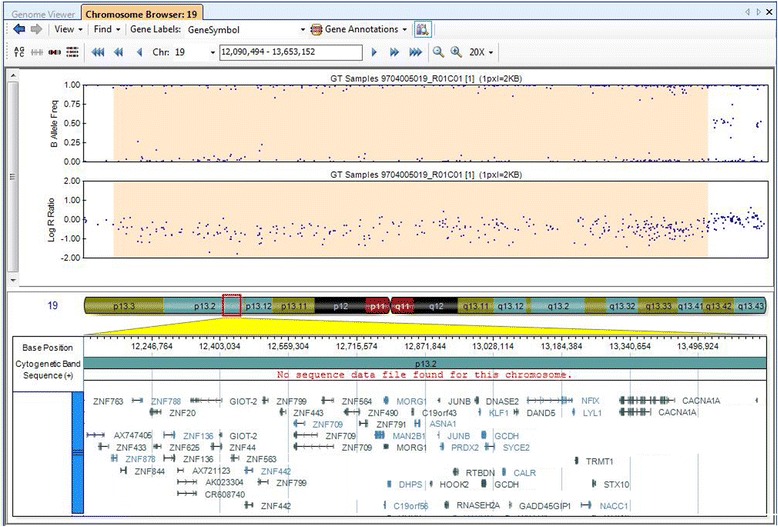
Human Omni1-Quad SNP-array result of the 19p13.2-p13.13 in Patient 1 with Malan syndrome. SNP-based array shows a de novo 1.4 Mb deletion (chr19: 12,157,839-13,518,462/hg19). Log R ratio and B allele frequencies are showed in upper panel; the lower panel shows genes in the deleted region
Discussion
Overgrowth syndromes are a group of conditions with heterogeneity and share several cardinal clinical features, i.e., overgrowth, intellectual disability and/or macrocephaly. The complexity of phenotypic features makes the differential diagnoses of overgrowth syndromes difficult. Recent advances in genetic studies have facilitated the molecular delineation of overgrowth syndromes. As a result, several overgrowth syndromes have been genetically elucidated in last decade. In this study, we report two patients with distinct overgrowth syndromes, Malan syndrome (19p13.2 deletion of NFIX) and Sotos Syndrome (5q35.2 deletion of NSD1).
In 2010, Malan et al. combined chromosomal microarrays and candidate gene approaches and successfully discovered nonsense mutations or microdeletion of NFIX in patients with Sotos-like overgrowth syndrome [6]. Mutations in NFIX had been linked to a known clinical condition, Marshall-Smith syndrome (MSS, OMIM#602535)[9]. It is suggested that different types of NFIX variants underlie distinct clinical phenotypes [6]. Specifically, frameshift and splice mutations in NFIX resulted in Marshall-Smith syndrome (MSS) while NFIX deletions and nonsense mutations cause a Sotos-like overgrowth syndrome. This phenomenon that distinct mutations result in strikingly different phenotypes has also been observed in other syndromes i.e. PTEN mutations cause Cowden syndrome (CS) and Bannayan-Riley-Ruvalcaba syndrome (BRRS) and Proteus syndrome (PS)[10], while FBN1 underlies Marfan syndrome, Weill-Marchesani syndrome (WMS) and acromicric dysplasia.[11, 12]. Subsequently, several reports confirmed NFIX mutations in patients with Sotos syndrome-like features resembling the patients previously reported by Malan et al. [2, 3, 13, 14]. In 2015, Klaassens et al., reported six additional patients with point mutation or deletion of NFIX [2]. Literature review of patients with 19p13.2-19p13.13 deletions showed similar clinical features [15–20]. Finally this medical condition was collectively grouped as Malan syndrome (19p13.2 deletion syndrome).
Common and prominent features are shared by different types of overgrowth syndromes, which imply that these syndromes might share similar molecular mechanisms. Since the two overgrowth disease-related genes, NFIX and NSD1, are both transcription factor, we suspect that they might be involved in similar molecular processes. The transcription factor nuclear factor I X plays a pivotal role during the development of brain and skeleton [5]. The Nfix(-/-) mice have retarded growth and most would die after postnatal day 21 (P21) in addition to malformations in brain ventricles and the corpus callosum [5]. NSD1 encodes a protein containing a SET domain, four plant homeodomain (PHD) finger regions and a proline-rich region. The SET Domain of NSD1 protein has intrinsic histone methyltransferase activity [21]. Based on these observations, Sotos syndrome and Sotos-like overgrowth syndrome have been suggested to be epigenetic diseases caused by loss-of-function defects of epigenetic readers/writers that modify the H3K36 histone mark [22]. Consistent with this idea, mutations in the DNA methyltransferase gene DNMT3A, SETD2 and EZH2 have also been shown to cause overgrowth syndromes [22–24]. Most recent NFIX function studies focus on its role in the brain malformations, whether NFIX is involved in epigenetic regulation remains to be elucidated [25, 26].
The discovery of NFIX as a causal gene for Malan syndrome also provides a success example for the microarrays-based candidate gene strategy in the identification of Mendelian disease-genes. Various mapping approaches rely upon karyotyping, linkage analysis, homozygosity mapping, GWAS and the recently introduced chromosomal microarrays (copy number variation analysis) and WES/WGS (whole exome/genome sequencing) [27]. An overview of literatures suggests that 13 disease-causing genes have been successfully identified by microarrays-based approaches including array CGH and SNP-array (Table 1). The disease gene number is relatively small but shows great growing potential. More importantly, with the accumulated copy number variation database, microarrays-based candidate gene strategy would be more productive in combination with exome sequencing data (Next Generation Sequencing, NGS).
Table 1.
Mendelian causative genes identified by the microarrays-based candidate gene strategy
| Gene | Locus | Size (kb) | Syndrome/phenotype | Year | Reference |
|---|---|---|---|---|---|
| RAI1 | 17p11.2 | 4000 | Smith–Magenis syndrome | 2003 | Nat Genet.2003; 33:466-8. |
| CHD7 | 8q12 | 2300 | CHARGE syndrome | 2004 | Nat Genet. 2004; 36:955-7. |
| EHMT1 | 9q34 | 1200 | 9q34 deletion syndrome | 2005 | J Med Genet. 2005; 42:299-306. |
| MECP2 | Xq28 | 400–800 | Intellectual Disability | 2005 | Am J Hum Genet. 2005; 77:442-53. |
| MAPT | 17q21.31 | 600 | 17q21.31 microdeletion syndrome | 2006 | Nat Genet. 2006; 38:999-1001. |
| CHRNA7 | 15q13.3 | 1500 | 15q13.3 microdeletion syndrome | 2008 | Nat Genet. 2008; 40:322-8. |
| MBD5 | 2q23.1 | 200–4000 | 2q23.1 microdeletion syndrome | 2010 | Eur J Hum Genet. 2010; 18: 436-441. |
| NFIX | 19p13.2 | 100–200 | Sotos-like syndrome | 2010 | Am J Hum Genet. 2010; 87:189-98. |
| SHANK3 | 22q13.3 | 100–9000 | Phelan-McDermid Syndrome | 2011 | Nature. 2011; 472: 437-42. |
| KANSL1 | 17q21.31 | 68 | 17q21.31 microdeletion syndrome | 2012 | Nat Genet. 2012; 44:639-41. |
| ARIDIB | 6q25 | 2500 | Intellectual Disability | 2012 | Am J Hum Genet. 2012; 90:565-72 |
| KCTD13 | 16p11.2 | 600 | Intellectual Disability | 2012 | Nature. 2012; 485:363-7. |
| TGFB2 | 1q41 | 6500 | Thoracic aortic aneurysm | 2012 | Nat Genet. 2012; 44:922-7. |
| KDM6A | Xp11.3 | 45–816 | Kabuki syndrome | 2012 | Am J Hum Genet. 2012; 90:119-24. |
Both of our patients have eye problems. Patient 1 has strabismus which is typical findings in patients with Malan syndrome. Patinet 2 has congenital microphthalmia and heterochromia. The latter two symptoms are not commonly reported in Sotos syndrome. In addition, Patient 1 with Malan syndrome shows no heart defects, while Patient 2 has congenital heart disease that mostly associated with Sotos syndrome. Whether the cardiac defect of our Sotos syndrome patient is related to other deleted genes in this 5q35.2 deletion remains to be resolved.
Conclusion
In conclusion, we report two patients with overgrowth syndrome. Patient 1 is diagnosed as Malan syndrome with a 1.4 Mb 19p13.2-13.13 microdeletion including NFIX, while Patient 2 has Sotos syndrome with a 1.6 Mb 5q35.2 microdeletion encompassing NSD1. Our study contributes to the diagnosis and treatment of these diseases.
Acknowledgements
We thank the patients and their parents for participating in this study. We thank the State Key Laboratory of Medical Genetics of China for technical assistance.
Funding
This study was supported by the National Natural Science Foundation of China (81470445 to Tan ZP, 81370204 to Yang YF).
Availability of data and materials
The SNP-array data are included in the Additional file 1: Table S1, Additional file 2: Table S2, Additional file 3: Table S3 and Additional file 4: Table S4. More details are available on request.
Authors’ contributions
DHY and ZH completed the laboratory work, and co-wrote the manuscript with TZP; TZP supervised the laboratory work; HYQ and XL reviewed all laboratory results and patient data, YYF and WXY reviewed all the clinical data; WJ supervised the design of the SNP-array. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Ethics approval and consent to participate
The Review Board of the Second Xiangya Hospital of the Central South University approved this study.All patients consent to this study with signed statement. Consent was obtained to publish from the participant (or legal parent or guardian for children) to report individual patient data.
Abbreviations
- ASD
Atrial septal defect
- CACNA1A
Calcium voltage-gated channel subunit alpha1 A
- CALR
Calreticulin
- CGH
Comparative genomic hybridization
- CHD
Congenital heart disease
- DNMT3A
DNA methyltransferase 3 alpha
- DQ
Developmental quotient
- EEG
Electroencephalogram
- EZH2
Enhancer of zeste 2 polycomb repressive complex 2 subunit
- FBN1
Fibrillin 1
- GWAS
Genome-wide association study
- NFIX
Nuclear factor I X
- NSD1
Nuclear receptor binding SET domain protein 1
- OFC
Occipital frontal circumference
- OGS
Overgrowth syndromes
- OMIM
Online Mendelian Inheritance in Man
- pCNVs
Pathogenic copy number variations
- PDA
Patent ductus arteriosus
- PHD
Plant homeodomain
- PTEN
Phosphatase and tensin homolog
- SETD2
SET domain containing 2
- SNP
Single nucleotide polymorphism
- WES/WGS
Whole exome/genome sequencing
Additional files
SNP-array data of Patient 1. (XLSX 13 kb)
SNP-array data of Patient 2. (XLSX 26 kb)
SNP-array data of Patient 3. (XLSX 30 kb)
SNP-array data of Patient 4. (XLSX 29 kb)
Contributor Information
Hai-Yun Dong, Email: donghaiyun2005@126.com.
Hui Zeng, Email: 382770404@qq.com.
Yi-Qiao Hu, Email: huyiqiao@sklmg.edu.cn.
Li Xie, Email: csuxieli@gmail.com.
Jian Wang, Email: wangjian.19860820@163.com.
Xiu-Ying Wang, Email: songxiu2007@yahoo.cn.
Yi-Feng Yang, Email: yyf627@163.com.
Zhi-Ping Tan, Phone: +86 731 8529 2178, Email: tanzhiping@gmail.com.
References
- 1.Baujat G, Rio M, Rossignol S, Sanlaville D, Lyonnet S, Le Merrer M, Munnich A, Gicquel C, Colleaux L, Cormier-Daire V. Clinical and molecular overlap in overgrowth syndromes. Am J Med Genet C Semin Med Genet. 2005;137C(1):4–11. doi: 10.1002/ajmg.c.30060. [DOI] [PubMed] [Google Scholar]
- 2.Klaassens M, Morrogh D, Rosser EM, Jaffer F, Vreeburg M, Bok LA, Segboer T, van Belzen M, Quinlivan RM, Kumar A, et al. Malan syndrome: Sotos-like overgrowth with de novo NFIX sequence variants and deletions in six new patients and a review of the literature. Eur J Hum Genet. 2015;23(5):610–615. doi: 10.1038/ejhg.2014.162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gurrieri F, Cavaliere ML, Wischmeijer A, Mammi C, Neri G, Pisanti MA, Rodella G, Lagana C, Priolo M. NFIX mutations affecting the DNA-binding domain cause a peculiar overgrowth syndrome (Malan syndrome): a new patients series. Eur J Med Genet. 2015;58(9):488–491. doi: 10.1016/j.ejmg.2015.06.009. [DOI] [PubMed] [Google Scholar]
- 4.Shimojima K, Okamoto N, Tamasaki A, Sangu N, Shimada S, Yamamoto T. An association of 19p13.2 microdeletions with Malan syndrome and Chiari malformation. Am J Med Genet A. 2015;167A(4):724–730. doi: 10.1002/ajmg.a.36959. [DOI] [PubMed] [Google Scholar]
- 5.Driller K, Pagenstecher A, Uhl M, Omran H, Berlis A, Grunder A, Sippel AE. Nuclear factor I X deficiency causes brain malformation and severe skeletal defects. Mol Cell Biol. 2007;27(10):3855–3867. doi: 10.1128/MCB.02293-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Malan V, Rajan D, Thomas S, Shaw AC, Louis Dit Picard H, Layet V, Till M, van Haeringen A, Mortier G, Nampoothiri S, et al. Distinct effects of allelic NFIX mutations on nonsense-mediated mRNA decay engender either a Sotos-like or a Marshall-Smith syndrome. Am J Hum Genet. 2010;87(2):189–198. doi: 10.1016/j.ajhg.2010.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tang M, Yang YF, Xie L, Chen JL, Zhang WZ, Wang J, Zhao TL, Yang JF, Tan ZP. Duplication of 10q22.3-q23.3 encompassing BMPR1A and NGR3 associated with congenital heart disease, microcephaly, and mild intellectual disability. Am J Med Genet A. 2015;167(12):3174–3179. doi: 10.1002/ajmg.a.37347. [DOI] [PubMed] [Google Scholar]
- 8.Xie L, Chen JL, Zhang WZ, Wang SZ, Zhao TL, Huang C, Wang J, Yang JF, Yang YF, Tan ZP. Rare de novo copy number variants in patients with congenital pulmonary atresia. PLoS One. 2014;9(5) doi: 10.1371/journal.pone.0096471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Adam MP, Hennekam RC, Keppen LD, Bull MJ, Clericuzio CL, Burke LW, Ormond KE, Hoyme EH. Marshall-Smith syndrome: natural history and evidence of an osteochondrodysplasia with connective tissue abnormalities. Am J Med Genet A. 2005;137(2):117–124. doi: 10.1002/ajmg.a.30580. [DOI] [PubMed] [Google Scholar]
- 10.Martinez F, Marin-Reina P, Sanchis-Calvo A, Perez-Aytes A, Oltra S, Rosello M, Mayo S, Monfort S, Pantoja J, Orellana C. Novel mutations of NFIX gene causing Marshall-Smith syndrome or Sotos-like syndrome: one gene, two phenotypes. Pediatr Res. 2015;78(5):533–539. doi: 10.1038/pr.2015.135. [DOI] [PubMed] [Google Scholar]
- 11.Cecchi A, Ogawa N, Martinez HR, Carlson A, Fan Y, Penny DJ, Guo DC, Eisenberg S, Safi H, Estrera A, et al. Missense mutations in FBN1 exons 41 and 42 cause Weill-Marchesani syndrome with thoracic aortic disease and Marfan syndrome. Am J Med Genet A. 2013;161A(9):2305–2310. doi: 10.1002/ajmg.a.36044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Eng C. PTEN: one gene, many syndromes. Hum Mutat. 2003;22(3):183–198. doi: 10.1002/humu.10257. [DOI] [PubMed] [Google Scholar]
- 13.Yoneda Y, Saitsu H, Touyama M, Makita Y, Miyamoto A, Hamada K, Kurotaki N, Tomita H, Nishiyama K, Tsurusaki Y, et al. Missense mutations in the DNA-binding/dimerization domain of NFIX cause Sotos-like features. J Hum Genet. 2012;57(3):207–211. doi: 10.1038/jhg.2012.7. [DOI] [PubMed] [Google Scholar]
- 14.Priolo M, Grosso E, Mammi C, Labate C, Naretto VG, Vacalebre C, Caridi P, Lagana C. A peculiar mutation in the DNA-binding/dimerization domain of NFIX causes Sotos-like overgrowth syndrome: a new case. Gene. 2012;511(1):103–105. doi: 10.1016/j.gene.2012.08.040. [DOI] [PubMed] [Google Scholar]
- 15.Lysy PA, Ravoet M, Wustefeld S, Bernard P, Nassogne MC, Wyns E, Sibille C. A new case of syndromic craniosynostosis with cryptic 19p13.2-p13.13 deletion. Am J Med Genet A. 2009;149A(11):2564–2568. doi: 10.1002/ajmg.a.33056. [DOI] [PubMed] [Google Scholar]
- 16.Jensen DR, Martin DM, Gebarski S, Sahoo T, Brundage EK, Chinault AC, Otto EA, Chaki M, Hildebrandt F, Cheung SW, et al. A novel chromosome 19p13.12 deletion in a child with multiple congenital anomalies. Am J Med Genet A. 2009;149A(3):396–402. doi: 10.1002/ajmg.a.32691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Marangi G, Orteschi D, Vigevano F, Felie J, Walsh CA, Manzini MC, Neri G. Expanding the spectrum of rearrangements involving chromosome 19: a mild phenotype associated with a 19p13.12-p13.13 deletion. Am J Med Genet A. 2012;158A(4):888–893. doi: 10.1002/ajmg.a.35254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dolan M, Mendelsohn NJ, Pierpont ME, Schimmenti LA, Berry SA, Hirsch B. A novel microdeletion/microduplication syndrome of 19p13.13. Genet Med. 2010;12(8):503–511. doi: 10.1097/GIM.0b013e3181e59291. [DOI] [PubMed] [Google Scholar]
- 19.Natiq A, Elalaoui SC, Miesch S, Bonnet C, Jonveaux P, Amzazi S, Sefiani A. A new case of de novo 19p13.2p13.12 deletion in a girl with overgrowth and severe developmental delay. Mol Cytogenet. 2014;7:40. doi: 10.1186/1755-8166-7-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jorge R, Silva C, Agueda S, Doria S, Leao M. Intellectual disability and overgrowth-A new case of 19p13.13 microdeletion syndrome with digital abnormalities. Am J Med Genet A. 2015;167(11):2839–2843. doi: 10.1002/ajmg.a.37280. [DOI] [PubMed] [Google Scholar]
- 21.Qiao Q, Li Y, Chen Z, Wang M, Reinberg D, Xu RM. The structure of NSD1 reveals an autoregulatory mechanism underlying histone H3K36 methylation. J Biol Chem. 2011;286(10):8361–8368. doi: 10.1074/jbc.M110.204115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Luscan A, Laurendeau I, Malan V, Francannet C, Odent S, Giuliano F, Lacombe D, Touraine R, Vidaud M, Pasmant E, et al. Mutations in SETD2 cause a novel overgrowth condition. J Med Genet. 2014;51(8):512–517. doi: 10.1136/jmedgenet-2014-102402. [DOI] [PubMed] [Google Scholar]
- 23.Tatton-Brown K, Seal S, Ruark E, Harmer J, Ramsay E, Del Vecchio DS, Zachariou A, Hanks S, O'Brien E, Aksglaede L, et al. Mutations in the DNA methyltransferase gene DNMT3A cause an overgrowth syndrome with intellectual disability. Nat Genet. 2014;46(4):385–388. doi: 10.1038/ng.2917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gibson WT, Hood RL, Zhan SH, Bulman DE, Fejes AP, Moore R, Mungall AJ, Eydoux P, Babul-Hirji R, An J, et al. Mutations in EZH2 cause Weaver syndrome. Am J Hum Genet. 2012;90(1):110–118. doi: 10.1016/j.ajhg.2011.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Heng YH, Zhou B, Harris L, Harvey T, Smith A, Horne E, Martynoga B, Andersen J, Achimastou A, Cato K, et al. NFIX Regulates Proliferation and Migration Within the Murine SVZ Neurogenic Niche. Cereb Cortex. 2015;25(10):3758–3778. doi: 10.1093/cercor/bhu253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Vidovic D, Harris L, Harvey TJ, Evelyn Heng YH, Smith AG, Osinski J, Hughes J, Thomas P, Gronostajski RM, Bailey TL, et al. Expansion of the lateral ventricles and ependymal deficits underlie the hydrocephalus evident in mice lacking the transcription factor NFIX. Brain Res. 2015;1616:71–87. doi: 10.1016/j.brainres.2015.04.057. [DOI] [PubMed] [Google Scholar]
- 27.Gilissen C, Hoischen A, Brunner HG, Veltman JA. Disease gene identification strategies for exome sequencing. Eur J Hum Genet. 2012;20(5):490–497. doi: 10.1038/ejhg.2011.258. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The SNP-array data are included in the Additional file 1: Table S1, Additional file 2: Table S2, Additional file 3: Table S3 and Additional file 4: Table S4. More details are available on request.