

Abstract
Bupropion is a widely used antidepressant, smoking cessation aid, and weight-loss therapy. It is administered as a racemic mixture, but the pharmacokinetics and activity of bupropion are stereoselective. The activity and side effects of bupropion are attributed to bupropion and its metabolites S,S- and R,R-OH-bupropion, threohydrobupropion, and erythrohydrobupropion. Yet the stereoselective metabolism in vitro and the enzymes contributing to the stereoselective disposition of bupropion have not been characterized. In humans, the fraction of bupropion metabolized (fm) to the CYP2B6 probe metabolite OH-bupropion is 5–16%, but ticlopidine increases bupropion exposure by 61%, suggesting a 40% CYP2B6 and/or CYP2C19 fm for bupropion. Yet, the CYP2C19 contribution to bupropion clearance has not been defined, and the enzymes contributing to overall bupropion metabolite formation have not been fully characterized. The aim of this study was to characterize the stereoselective metabolism of bupropion in vitro to explain the stereoselective pharmacokinetics and the effect of drug-drug interactions (DDIs) and CYP2C19 pharmacogenetics on bupropion exposure. The data predict that threohydrobupropion accounts for 50 and 82%, OH-bupropion for 34 and 12%, erythrohydrobupropion for 8 and 4%, and 4′-OH-bupropion for 8 and 2% of overall R- and S-bupropion clearance, respectively. The fm,CYP2B6 was predicted to be 21%, and the fm,CYP2C19, 6% for racemic bupropion. Importantly, ticlopidine was found to inhibit all metabolic pathways of bupropion in vitro, including threohydrobupropion, erythrohydrobupropion, and 4′OH-bupropion formation, explaining the in vivo DDI. The stereoselective pharmacokinetics of bupropion were quantitatively explained by the in vitro metabolic clearances and in vivo interconversion between bupropion stereoisomers.
Introduction
Bupropion is an antidepressant also used as a smoking cessation aid and weight loss therapy. Bupropion is cleared via oxidation by cytochrome P450s (P450s) to OH-bupropion and 4′-OH-bupropion (Faucette et al., 2000; Benowitz et al., 2013; Sager et al., 2016) and reduction by 11β-hydroxysteroid dehydrogenase-1 (11β-HSD-1) and aldoketoreductase(s) to threohydrobupropion and erythrohydrobupropion (Meyer et al., 2013; Skarydova et al., 2014; Connarn et al., 2015) (Fig. 1). All four metabolites undergo glucuronidation, and threo- and erythrohydrobupropion are also hydroxylated to threo-4′-OH- and erythro-4′-OH-hydrobupropion (Gufford et al., 2016; Sager et al., 2016). OH-bupropion, threohydrobupropion, and erythrohydrobupropion have plasma concentrations that exceed bupropion concentrations by 3- to 30-fold (Benowitz et al., 2013; Masters et al., 2016), and these metabolites are believed to contribute to bupropion’s pharmacological activity and toxicity (Bondarev et al., 2003; Damaj et al., 2004). However, higher bupropion and lower metabolite concentrations correlate with antidepressant response (Preskorn, 1983; Golden et al., 1988). Smoking cessation success also has a strong dose-response relationship (Hurt et al., 1997), but OH-bupropion concentrations, rather than bupropion, are predictive of smoking cessation outcome (Zhu et al., 2012). Bupropion side effects, such as dry mouth and insomnia, correlate with bupropion metabolite concentrations (Johnston et al., 2001), and based on mouse models, OH-bupropion is significantly more likely to cause seizures than threohydrobupropion and erythrohydrobupropion, with bupropion having the lowest potency to induce seizures (Silverstone et al., 2008). Based on these data, processes that affect bupropion metabolism resulting in altered metabolite and parent concentrations will directly impact the safety and efficacy of bupropion.
Fig. 1.

Metabolic pathways of bupropion stereoisomers. Bupropion is administered as a racemic mixture and undergoes stereoselective metabolism. In addition to the shown metabolic pathways, all metabolites undergo extensive glucuronidation. AKR, Aldo-keto reductase.
Bupropion is administered as a racemic mixture of S- and R-bupropion, which undergo chiral inversion (Coles and Kharasch, 2008). In humans, circulating concentrations of R-bupropion are 3- to 6-fold higher than S-bupropion, and R,R-OH-bupropion exposures are 15- to 65-fold higher than S,S-OH-bupropion (Kharasch et al., 2008; Masters et al., 2016). However, despite its lower exposure, S,S-OH-bupropion is believed to be mainly responsible for the nicotine cessation activity of bupropion (Damaj et al., 2004). S,S-OH-bupropion is more active than bupropion, whereas R,R-OH-bupropion is inactive toward inhibition of dopamine and norepinephrine uptake (Damaj et al., 2004). Both S,S- and R,R-OH-bupropion have similar activity as bupropion as acetylcholine receptors antagonists (Damaj et al., 2004). Taken together, these data suggest that stereoselective disposition has important implications to the activity and toxicity of bupropion. Yet, the overall stereoselective clearance pathways of bupropion have not been established.
The metabolism of bupropion to OH-bupropion is mainly mediated by CYP2B6 (Faucette et al., 2000; Hesse et al., 2000), and the intrinsic clearance of S-bupropion by CYP2B6 is 3-fold higher than that of R-bupropion (Coles and Kharasch, 2008). Similarly, the glucuronidation of bupropion metabolites is stereoselective (Gufford et al., 2016). However, these data on stereoselective metabolism cannot explain the in vivo ratios of bupropion stereoisomers and the variability in stereoisomer ratios observed between studies. It is likely that stereoselective formation of erythrohydrobupropion and threohydrobupropion as suggested by in vivo data (Masters et al., 2016), and possible stereoselective metabolism of bupropion by CYP2C19, which appears to contribute to bupropion clearance based on pharmacogenetic data (Zhu et al., 2014), contribute to the in vivo stereoselective disposition of bupropion. In addition, the chiral inversion of bupropion stereoisomers may confound some in vitro to in vivo extrapolations of direct stereoselective clearance values. Hence, thorough in vitro characterization of stereoselective metabolism of bupropion is necessary to establish the basis for the stereoselective disposition and activity of bupropion.
Bupropion is an accepted in vitro and in vivo probe of CYP2B6, but the possible confounding effects of stereochemistry and various clearance pathways of bupropion and its metabolites in assessing CYP2B6 induction and inhibition have been recently highlighted (Masters et al., 2016). Overall, the in vivo contributions of specific metabolic enzymes to bupropion clearance are not quantitatively understood. In vivo, clopidogrel and ticlopidine cause 36 and 61% increases in bupropion area under the plasma concentration-time curve (AUC), respectively (Turpeinen et al., 2005), which has been attributed to CYP2B6 inhibition. However, inhibition of CYP2B6-mediated formation of OH-bupropion cannot explain these drug-drug interactions (DDIs) due to the minor contribution of OH-bupropion to bupropion elimination (Sager et al., 2016). Similarly, the low fraction metabolized (fm) to 4′OH-bupropion, the likely CYP2C19-mediated metabolite of bupropion (Sager et al., 2016) cannot explain the 13% increase in bupropion AUC in CYP2C19 intermediate metabolizers (Zhu et al., 2014). Taken together, these data show that bupropion elimination pathways are inadequately characterized. The aim of this study was to determine the overall contribution of CYP2B6 and CYP2C19 to bupropion stereoisomer clearance, and to characterize the stereoselective metabolic pathways of bupropion that can explain the observed in vivo stereoselective disposition of bupropion.
Materials and Methods
Materials.
Bupropion, R-bupropion, S-bupropion, OH-bupropion, R,R-OH-bupropion, S,S-OH-bupropion, threohydrobupropion, erythrohydrobupropion, bupropion-d9, OH-bupropion-d6, and threohydrobupropion-d9 were purchased from Toronto Research Chemicals (Toronto, Canada). 4′-OH-bupropion, threo-4′-OH-hydrobupropion, and erythro-4′-OH-hydrobupropion were synthesized as previously described (Sager et al., 2016). Hydrochloric acid, formic acid, and sodium hydroxide were purchased from Sigma-Aldrich (St. Louis, MO). Optima grade acetonitrile, methanol, water, and pooled human liver microsomes (Gibco, mixed gender, n = 50) were purchased from Fisher Scientific (Waltham, MA). Pooled S9 fractions (n = 20, mixed gender) and recombinant P450 enzymes coexpressed with cytochrome P450 reductase and cytochrome b5 were purchased from BD Biosciences (San Jose, CA). CYP2B6 inhibitory monoclonal antibody (MAB-CYP2B6) was purchased from Corning (Corning, NY).
Liquid Chromatography–Tandem Mass Spectrometry Quantification Methods.
Unless otherwise specified, an equal volume of acetonitrile (incubations) or 2 volumes of 1:3 methanol:acetonitrile (plasma and blood) containing 100 nM OH-bupropion-d6 and threohydrobupropion-d9 as internal standards was added to each sample. Samples were centrifuged for 15 minutes at 3,000g, and the supernatant was transferred to a 96-well plate for liquid chromatography–tandem mass spectrometry (LC-MS/MS) analysis. The concentrations of bupropion, OH-bupropion, threohydrobupropion, erythrohydrobupropion, 4′-OH-bupropion, erythro-4′-OH-hydrobupropion, threo-4′-OH-hydrobupropion, and ticlopidine were determined using an LC-MS/MS system consisting of an API 4500 triple quadrupole mass spectrometer (AB Sciex, Foster City, CA) coupled to an LC-20AD ultra-fast liquid chromatography system (Shimadzu Co., Kyoto, Japan). The turbo ion spray interface was operated in positive ion mode. For measurement of OH-bupropion formation in supersomes and for quantification of ticlopidine concentrations, analytes were separated using a ZORBAX XDB-C18 column (50 × 2.1 mm, 5 µM; Agilent, Santa Clara, CA) as previously described (Sager et al., 2016). In S9 fraction incubations, recombinant CYP2C19 incubations, and inhibition assays, OH-bupropion, threohydrobupropion, erythrohydrobupropion, erythro-4′-OH-hydrobupropion, threo-4′-OH-hydrobupropion, and 4′-OH-bupropion were separated using an Agilent ZORBAX XDB-C18 column (150 × 4.6 mm, 5 µM) with an isocratic elution at a flow rate of 0.8 ml/min with a mobile phase consisting of 35% methanol and 65% water with 0.4% formic acid, according to a previously described method (Connarn et al., 2015; Sager et al., 2016). Bupropion enantiomer concentrations in plasma were determined stereoselectively using an α-acid glycoprotein column (100 × 2 mm, 5 µm) with a guard cartridge (10 × 2 mm, 5µm; ChromTech, Apple Valley, MO). The mobile phase consisted of 20 mM ammonium formate (pH 5.7) (A) and methanol (B). A gradient elution at 0.22 ml/min starting at 10% B and increasing to 21.2% B by 25 minutes and 23.5% B by 34 minutes was used with re-equilibration to initial conditions by 45 minutes. Bupropion and its metabolites were detected using positive ion electrospray ionization using the following mass transitions (m/z): 240→184 (bupropion), 249→189 (bupropion-d9), 256→238 (OH-bupropion), 262→244 (OH-bupropion-d6), 242→168 (threo- and erythrohydrobupropion), 251→168 (threohydrobupropion-d9), 256→182 (4′-OH-bupropion), and 258→184 (threo- and erythro-4′-OH-bupropion). Ticlopidine was detected using the mass transition (m/z) 264→154. Data analysis was performed using Analyst software version 1.6.2 (AB Sciex).
General Incubation Conditions.
All incubations were performed as triplicates in 100 mM potassium phosphate buffer (pH 7.4), with a final incubation volume of 100 µl. With the exception of the IC50 shift experiments, all metabolite formation experiments were initiated with NADPH (1 mM) after a 5-minute preincubation at 37°C. For rCYP and inhibitor panels, all substrate concentrations were 1 μM. Activity assays were allowed to proceed for 7 minutes prior to termination of the reaction with an equal volume of acetonitrile containing 100 nM OH-bupropion-d6 and threohydrobupropion-d9. Reaction times were sufficiently short to ensure chiral inversion of S- and R-bupropion was <5%. The metabolite formation was linear in relation to incubation time and protein content, and no substrate depletion was observed in the incubations. Microsomal protein binding at all protein concentrations was determined to be negligible. All incubations including each concentration point in kinetic experiments were performed in triplicate together with control incubations without NADPH.
Recombinant Cytochrome P450 Incubations.
R-bupropion (1 or 100 µM) and S-bupropion (1 or 100 µM) were incubated in triplicate with 5 pmol recombinant CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, CYP2E1, and CYP3A4 in 100 mM potassium phosphate buffer (pH 7.4). For determination of OH-bupropion formation kinetics, R- and S-bupropion were first incubated with CYP2B6, CYP3A4, and CYP2C19 at seven concentrations between 1 and 500 µM to evaluate the saturability and approximate Km values for bupropion enantiomers with each enzyme. Based on the preliminary experiments, the estimated Km values were >100 µM for R- and S-bupropion with CYP2B6 and CYP3A4, and therefore, only intrinsic clearance (CLint) values were determined for R- and S-bupropion with CYP2B6 and CYP3A4. The CLint. values for OH-bupropion formation from R- and S-bupropion were determined at clinically relevant concentrations of bupropion ([S]<<Km) from the slope of the substrate concentration versus velocity plot, as described previously (Peng et al., 2011), after incubations of six to seven concentrations of R- or S-bupropion between 1 and 20 µM and 1 pmol CYP2B6 or 5 pmol CYP3A4. For determination of the CYP2C19 Km value for OH-bupropion and 4′OH-bupropion formation, R- and S-bupropion (eight concentrations between 0.5 and 48 µM) were incubated with 5 pmol CYP2C19. For determination of threo-4′-OH-hydrobupropion and erythro-4′-OH-hydrobupropion formation kinetics, threohydrobupropion (seven concentrations between 5 and 500 µM) and erythrohydrobupropion (seven concentrations between 5 and 250 µM) were incubated with 5 pmol CYP2C19, and the Michaelis-Menten kinetic values were determined. Intrinsic clearance values in recombinant enzyme systems were scaled to hepatic intrinsic clearances assuming 137 pmol CYP3A4/mg microsomal protein, 17 pmol CYP2C19/mg microsomal protein for CYP2C19 (Stringer et al., 2009), 14.2 pmol CYP2B6/mg microsomal protein (Totah et al., 2008), and 40 mg of microsomal protein/g liver (Brown et al., 2007). Assuming 70-kg body weight and 21.4 g liver/kg body weight, 1500 g was used as the liver weight (Ito and Houston, 2005). The fractional contribution of CYP2B6, CYP2C19, and CYP3A4 to the formation of R,R- and S,S-OH-bupropion was calculated by dividing the scaled hepatic intrinsic clearance for the enzyme of interest by the sum of the scaled hepatic intrinsic clearances for the enzymes forming the metabolite.
Inhibition of Bupropion Metabolite Formation by Specific P450 Inhibitors, a CYP2B6 Inhibitory Antibody, and Ticlopidine.
To further evaluate the contribution of specific P450 enzymes to the formation of OH-bupropion, 4′-OH-bupropion, threohydrobupropion, and erythrohydrobupropion, troleandomycin (CYP3A4, 50 μM), furafylline (CYP1A2, 10 μM), (+)-N-3-benzylnirvanol (CYP2C19, 2 μM), montelukast (CYP2C8, 1.5 μM), sulfaphenazole (CYP2C9, 30 μM), and quinidine (CYP2D6, 4 μM) were used as P450 specific inhibitors in human liver microsome (HLM) incubations as previously described (Peng et al., 2011). Time-dependent inhibitors troleandomycin and furafylline were preincubated for 15 minutes in the presence of NADPH prior to the addition of substrate. Inhibition studies with the CYP2B6 inhibitory antibody (MAB-2B6) were conducted by preincubating HLM (0.5 mg/ml) with antibody (2 μl per 100 μg of HLM) or Tris buffer (control) on ice for 20 minutes. Following the preincubation, 100 mM KPi buffer (pH 7.4) and substrate were added, and the samples were preincubated for 5 minutes at 37°C. All incubations with inhibitors were performed at 1 μM substrate concentration and as triplicates. The reactions were initiated by the addition of NADPH (1 mM) and quenched after 7 minutes for LC-MS/MS analysis.
To assess the potential for time-dependent inhibition of OH-bupropion, 4′-OH-bupropion, threohydrobupropion, and erythrohydrobupropion formation by ticlopidine, ticlopidine (seven concentrations between 0.1 and 100 µM) was incubated with 0.5 mg/ml HLM in 100 mM KPi buffer (100 µl final volume) at 37°C for 30 minutes in the presence or absence of 1 mM NADPH before bupropion (1 µM) or bupropion + NADPH was added. To control for ticlopidine depletion during the 30-minute preincubation, ticlopidine concentrations were measured prior to the bupropion metabolism assay, and the measured concentrations were used to determine the IC50 values. The magnitude of the IC50 shift was determined from the ratio of the IC50 value measured following preincubation without NADPH and the IC50 value measured with NADPH in the preincubation. A shift ≥1.5 was considered to indicate a potential for irreversible inhibition.
Determination of In Vitro Intrinsic Clearances for Bupropion Metabolites in S9 Fractions.
Due to previous reports of minor cytosolic contributions to threo- and erythrohydrobupropion formation (Meyer et al., 2013; Skarydova et al., 2014; Connarn et al., 2015), all incubations to assess the relative contributions of metabolite formation were performed in human liver S9 fractions. To determine the intrinsic formation clearance of threohydrobupropion, erythrohydrobupropion, and OH-bupropion in human liver S9 fractions, R- and S-bupropion (seven concentrations between 0.4 and 16 µM) were added to KPi containing 1 mg/ml human liver S9 fractions, and the reactions were initiated by the addition of NADPH. All incubations were conducted as triplicates. For determination of formation clearances at substrate concentrations below 1 µM, which was needed for the measurement of 4′-OH-bupropion CLint, S9 fractions (5 mg/ml) were incubated with R- or S-bupropion at four concentrations between 0.2 and 1 µM. The intrinsic clearance values measured in S9 fractions were scaled to hepatic intrinsic clearance (CLint,H) using 96.1 mg of S9 protein/g liver (Watanabe et al., 2009). Assuming 70-kg body weight and 21.4 g liver/kg body weight, 1500 g was used as the liver weight (Ito and Houston, 2005), and total hepatic intrinsic clearance for R- and S-bupropion (CLint,R,H, CLint,S,H) was calculated as the sum of individual liver metabolite intrinsic clearance. The fraction of R- and S-bupropion metabolism (fm) attributed to each metabolic pathway was determined from the ratio of the formation clearance of a given metabolite(s) in S9 fractions to the sum of the intrinsic clearances measured for all of the metabolites formed from the respective substrate in S9 fractions.
The fm,CYP2B6 values for R-bupropion, S-bupropion, and racemic bupropion were calculated according to eqs. 1, 2, and 3, respectively, using the intrinsic clearance values scaled to the whole liver:
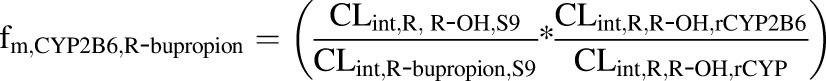 |
(1) |
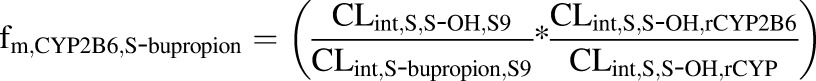 |
(2) |
| (3) |
where CLintR,R-OH,S9 and CLintS,S-OH,S9 are the CLint values determined for the formation of the OH-bupropion stereoisomers in S9 fractions after scaling to the whole liver, and CLint,R-bupropion,S9 and CLint,S-bupropion,S9 are the ∑CLint values for all of the detected metabolites for R-bupropion and S-bupropion in S9 fractions after scaling to the whole liver. CLint,R,R-OH,rCYP2B6 and CLint,S,S-OH,rCYP2B6 are the intrinsic clearances to OH-bupropion stereoisomers in rCYP2B6 after scaling to the whole liver. CLint,R,R-OH,rCYP and CLint,S,S-OH,rCYP are the sums of the intrinsic clearances for the formation of R,R-OH-bupropion and S,S-OH-bupropion by all rCYPs evaluated (CYP2B6, CYP3A4, and CYP2C19) after scaling to the whole liver. The fm,CYP2C19 was calculated similarly according to eqs. 4, 5, and 6:
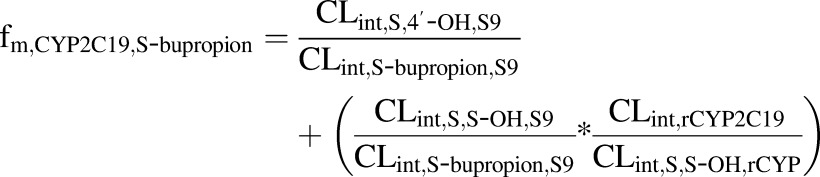 |
(4) |
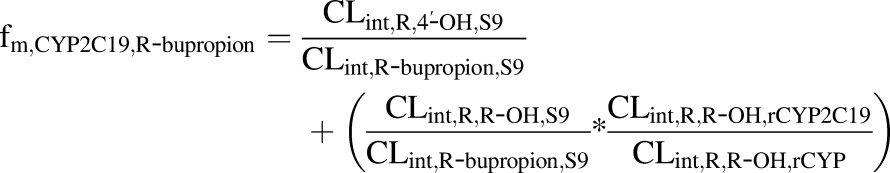 |
(5) |
| (6). |
Determination of Unbound Fractions, Bupropion Blood-to-Plasma Ratio, and Chiral Inversion Rates of R- and S-bupropion.
The protein binding of bupropion (1 µM) in plasma and in S9 fractions (5 mg/ml) and ticlopidine (4 µM) in HLM (0.5 mg/ml) was determined using ultracentrifugation, as described previously (Shirasaka et al., 2013). Blood-to-plasma ratio was measured in fresh blood. Blood was collected into heparinized tubes, and bupropion (0.5 µM) was added to the whole blood and allowed to equilibrate. Half of the blood was collected for measurement of bupropion concentration in whole blood, and half was centrifuged at 1,000g for 10 minutes to separate plasma from blood. Plasma was collected for analysis. Blood and plasma proteins were precipitated using 2 volumes of 1:3 methanol:acetonitrile containing 100 nM OH-bupropion-d6 and threohydrobupropion-d9 as internal standards, as described in the Liquid Chromatography–Tandem Mass Spectrometry Quantification Methods section. The blood-to-plasma concentration ratio was calculated as the ratio of concentration of bupropion measured in whole blood versus plasma. Due to bupropion isomerization (Coles and Kharasch, 2008), racemic bupropion (0.5 µM) was used to determine the plasma protein binding and blood-to-plasma ratio of the enantiomers of bupropion. The individual enantiomers were measured by LC-MS/MS. For determination of the isomerization rate of R- and S-bupropion in plasma, blank human plasma (100 µl) containing 500 nM R- or S-bupropion was incubated at 37°C. At 0, 15, 30, 45, and 60 minutes, plasma proteins were precipitated samples placed on ice until LC-MS/MS analysis. The rate of isomerization was determined using eq. 7, where S0 and St are the substrate concentrations at time zero and t, respectively, and k is the first-order rate constant for the isomerization:
| (7). |
Prediction of Hepatic Clearance and Simulation of the Steady-State Concentration Ratios of R- and S-bupropion.
The hepatic blood clearances of R- and S-bupropion (CLint,h,R, CLint,h,S) were predicted using the well stirred model according to eqs. 8 and 9, respectively:
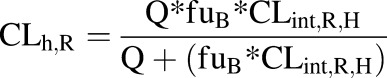 |
(8) |
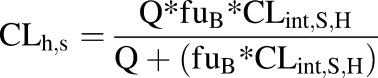 |
(9) |
where CLint,R,H and CLint,S,H are the R- and S-bupropion hepatic intrinsic clearances predicted from S9 fraction incubations. The fraction unbound in blood (fuB) was determined by multiplying the fraction unbound in plasma by the plasma:blood ratio. A hepatic blood flow (Q) of 90 l/h was used. The hepatic extraction ratio was calculated from the quotient of CLh and Q. The oral clearance of bupropion stereoisomers was calculated as the product of plasma unbound fraction (fu) and hepatic intrinsic clearance for the given enantiomer (CLint,R,H or CLint,S,H).
The effect of chiral inversion of bupropion enantiomers to steady-state R- and S-bupropion plasma concentrations was simulated using Simbiology (Mathworks, Natick, MA) according to eqs. 10 and 11, where CR is the concentration of R-bupropion and CS is the concentration of S-bupropion:
| (10) |
| (11) |
A constant rate input (Rin) of R- and S-bupropion (75 mg/day) was used with elimination as the hepatic clearance of R-bupropion (CLh,R) and S-bupropion (CLh,S). The R/S concentration ratio was simulated in the absence of isomerization and in the presence of variable in vivo isomerization rates (18 rates between 0.2 and 23 l/h).
Data Analysis.
For kinetic analyses, the formation velocity was plotted against the substrate concentration and fit via linear or nonlinear regression in GraphPad Prism (GraphPad Software, San Diego, CA). When saturable kinetics were observed over the substrate concentration range, the catalytic rate constant or the maximum metabolite formation velocity (kcat for recombinant enzymes) and Vmax for S9 fractions and the Michaelis-Menten affinity constant (Km) were determined using eq. 12:
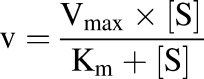 |
(12). |
The metabolite intrinsic formation clearance was calculated as the ratio of the Vmax (or kcat) and Km. In cases where product formation was linear over the concentration range, CLint was determined from the slope of the substrate concentration versus velocity plots using linear regression. One-way analysis of variance with post-hoc Tukey test was used to test for significance of changes in metabolite formation in the P450 inhibitor panels.
Results
Characterization of the Enzymes Involved in Bupropion Metabolism.
To determine the enzymes contributing to R,R- and S,S-OH-bupropion formation, R- and S-bupropion were incubated with a panel of recombinant human P450 enzymes at 100 and 1 μM concentrations. At supraphysiological concentrations of R- and S-bupropion (100 µM), R,R-OH-bupropion and S,S-OH-bupropion formation was detected in CYP2B6, CYP2C19, CYP3A4, and CYP1A2 supersomes, but the relative formation rates of S,S-OH-bupropion and R,R-OH-bupropion in CYP3A4, CYP1A2, and CYP2C19 supersomes were only 2, 0.4, and 0.1% of those in CYP2B6 supersomes (Fig. 2). At clinically relevant concentrations of R- and S-bupropion (1 μM), formation of R,R-OH-bupropion and S,S-OH-bupropion was only observed with CYP2B6, CYP2C19, and CYP3A4 supersomes. At this concentration, CYP2B6 was still the most efficient enzyme forming R,R- and S,S-OH-bupropion, but the relative contributions of CYP2C19 and CYP3A4 were increased in comparison to those observed at 100 µM. At 1 μM substrate concentrations, product formation rates with CYP2C19 and CYP3A4 supersomes were 10 and 2% of the rates with CYP2B6 supersomes for S-bupropion and 7 and 2% for R-bupropion, respectively (Fig. 2).
Fig. 2.

Stereoselective metabolism of S- and R-bupropion to S,S- and R,R-OH-bupropion by recombinant P450 enzymes and human liver microsomes. (A and B) Formation of R,R- and S,S-OH-bupropion from 100 µM (A) or 1 µM (B) R- or S-bupropion with a P450 supersome panel. (C) Inhibition of R,R- and S,S-OH-bupropion formation from 1 µM R- or S-bupropion by P450 isoform-specific inhibitors and CYP2B6-specific inhibitory antibody (MAB-2B6). Control 1 is the control for the reversible inhibitors montelukast (2C8 MONT), sulfaphenazole (2C9 SULF), (+)-N-3-benzylnirvanol (2C19 N-BENZ), and quinidine (2D6 QUIN). Control 2 is the control for time-dependent inhibitors troleandomycin (3A4 TAO) and furafylline (1A2 FURA). *p < 0.05 in comparison with control, one-way analysis of variance. (D–F) Formation kinetics of R,R-OH-bupropion from R-bupropion by CYP2C19 (D), CYP3A4 (E), and CYP2B6 (F) supersomes. (G–I) Formation kinetics of S,S-OH-bupropion from S-bupropion by CYP2C19 (G), CYP3A4 (H), and CYP2B6 (I) supersomes. For CYP3A4 incubations with S-bupropion, the two lowest substrate concentrations did not result in sufficient product formation for quantification and these data points are not shown.
To further characterize the relative importance of CYP2B6 in R,R- and S,S-OH-bupropion formation, their formation kinetics were determined with recombinant P450s (Fig. 2; Table 1). As preliminary experiments using bupropion concentrations up to 500 µM suggested the Km values with CYP2B6 and CYP3A4 were between 100 and 500 µM (data not shown), only intrinsic clearance values were measured for these enzymes at clinically relevant concentrations. Significant stereoselectivity in OH-bupropion formation by recombinant P450s was observed. Formation of R,R- and S,S-OH-bupropion in CYP2B6 and CYP3A4 supersomes was linear over the clinically relevant substrate concentration range, confirming that the Km values for CYP2B6 and CYP3A4 greatly exceed 10 and 20 µM, respectively. The intrinsic formation clearance of S,S-OH-bupropion was 1.8-fold higher than that of R,R-OH-bupropion by CYP2B6 supersomes. In contrast, in CYP3A4 supersomes, R,R-OH-bupropion formation was 3.5-fold higher than that of S,S-OH-bupropion. Upon incubation of R- and S-bupropion with CYP2C19 supersomes, the formation of R,R- and S,S-OH-bupropion was saturable. Whereas both substrates had similar Km values (4.9 µM for R-bupropion and 4.0 µM for S-bupropion) with CYP2C19, the kcat and CLint values for S,S-OH-bupropion formation were 10-fold higher than those for R,R-OH-bupropion. When the supersome values were scaled to whole human liver based on P450 expression levels, CYP2B6 was predicted to contribute to 90 and 89% of R,R- and S,S-OH-bupropion formation, respectively (Table 1). CYP2C19 was predicted to contribute 3 and 9%, and CYP3A4 8 and 1% to R,R-OH-bupropion and S,S-OH-bupropion formation, respectively. To confirm the predicted contribution of individual P450 isoforms to R,R- and S,S-OH-bupropion formation, R- and S-bupropion were incubated with human liver microsomes in the presence of selective P450 inhibitors and an inhibitory CYP2B6 antibody (Fig. 2). The selective monoclonal antibody (MAB-2B6) decreased the formation of R,R- and S,S-OH-bupropion by 91 and 81%, respectively, confirming that CYP2B6 is the major enzyme involved in the formation of both OH-bupropion enantiomers (Fig. 2). No significant inhibition was observed by any of the chemical inhibitors, a finding in agreement with the predicted minor contributions of CYP2C19 and CYP3A4 to OH-bupropion formation. Interestingly, R,R- and S,S-OH-bupropion formation in the presence of N-benzylnirvanol in HLMs was 11 and 20% higher than the control, respectively (p < 0.05), and S,S-OH-bupropion formation in the presence of montelukast was 15% higher than control (p < 0.05), suggesting activation of some P450s by these inhibitors.
TABLE 1.
In vitro intrinsic clearance values for R-bupropion, S-bupropion, threohydrobupropion, and erythrohydrobupropion metabolism with recombinant P450s
Those enzymes found to form the metabolites of interest in a screen with a panel of P450 supersomes were evaluated. In vitro CLint values, scaled CLint,H values, and the fractional contributions of each P450 to the given metabolite formation were calculated as described in Materials and Methods.
| Substrate and Metabolite | Enzyme | CLint | CLint, H | Predicted fm,CYP,metabolite |
|---|---|---|---|---|
| μl/min/nmol P450 | l/h | |||
| R-bupropion | ||||
| R,R-OH-bupropion | CYP2B6 | 69.0 | 3.6 | 0.90 |
| CYP2C19 | 0.9 | 0.1 | 0.03 | |
| CYP3A4 | 0.5 | 0.3 | 0.08 | |
| 4′-OH-bupropion | CYP2C19 | 18.6 | 1.1 | 1 |
| S-bupropion | ||||
| S,S-OH-bupropion | CYP2B6 | 125.0 | 6.6 | 0.89 |
| CYP2C19 | 11.5 | 0.7 | 0.09 | |
| CYP3A4 | 0.1 | 0.1 | 0.01 | |
| 4′-OH-bupropion | CYP2C19 | 17.7 | 1.1 | 1 |
| Erythrohydrobupropion | ||||
| Erythro-4′-OH-hydrobupropion | CYP2C19 | 10.1 | 0.6 | 1 |
| Threohydrobupropion | ||||
| Threo-4′-OH-hydrobupropion | CYP2C19 | 42.3 | 2.6 | 1 |
To further evaluate the P450-mediated metabolism of R- and S-bupropion, the formation of 4′-OH-bupropion was evaluated using a supersome panel. The formation of 4′OH-bupropion from R- and S-bupropion was catalyzed exclusively by CYP2C19 (Fig. 3). Hence the formation kinetics of 4′-OH-bupropion from R- and S-bupropion were characterized only in CYP2C19 supersomes (Fig. 3; Table 1). Both R-bupropion and S-bupropion had similar affinity (Km = 5.1 and 4.3 µM, respectively) to CYP2C19, and the kcat for 4′-bupropion OH-formation was similar for both stereoisomers (94.8 pmol/min/nmol for R-bupropion and 75.5 pmol/min/nmol for S-bupropion). To confirm the importance of CYP2C19 in 4′-OH-bupropion formation, the effects of selective P450 inhibitors on 4′-OH-bupropion formation were determined (Fig. 3). Following incubation of R- or S-bupropion, N-benzylnirvanol (CYP2C19 inhibitor) inhibited 84 and 82% of the 4′-OH-bupropion formation from R- and S-bupropion, respectively. Minor inhibition (10%) of 4′-OH-bupropion formation was detected upon incubation of S-bupropion with MAB-CYP2B6, suggesting potential minor involvement of CYP2B6. 4′OH-bupropion was the main metabolite formed by CYP2C19 from both bupropion enantiomers. The CLint value by CYP2C19 for the formation of 4′OH-bupropion from R-bupropion was 20-fold greater than that of R,R-OH-bupropion formation by CYP2C19. For formation of 4′OH-bupropion from S-bupropion, the CLint value by CYP2C19 was 1.5-fold greater than that of S,S-OH-bupropion formation by CYP2C19. However, the overall hepatic formation clearance of R,R- and S,S-OH-bupropion was predicted to be 3- to 7-fold greater than the formation clearance of 4′OH-bupropion based on the supersome data and P450 expression levels in the liver. Threo- and erythrohydrobupropion formation was not observed in any incubations with P450 supersomes (data not shown).
Fig. 3.

Stereoselective metabolism of S- and R-bupropion to 4′-OH-bupropion by recombinant P450 enzymes and human liver microsomes. (A) 4′-OH-bupropion formation from R- and S-bupropion (1 µM) in a panel of P450 supersomes. (B) Inhibition of 4′-OH-bupropion formation from R- and S-bupropion (1 µM) in HLM as percentage of control following incubation of R- or S-bupropion in the presence of selective P450 inhibitors or a CY2B6 inhibitory antibody (MAB-2B6). Control 1 is the control for the reversible inhibitors montelukast (2C8 MONT), sulfaphenazole (2C9 SULF), (+)-N-3-benzylnirvanol (2C19 N-BENZ), and quinidine (2D6 QUIN). Control 2 is the control for time-dependent inhibitors troleandomycin (3A4 TAO) and furafylline (1A2 FURA). *p < 0.05 in comparison to control, one-way analysis of variance. (C and D) Formation kinetics of 4′-OH-bupropion from R-bupropion (C) and S-bupropion (D) in CYP2C19 supersomes.
Similar to 4′OH-bupropion formation, it has been suggested that the formation of threo-4′-OH-hydrobupropion and erythro-4′-OH-hydrobupropion is catalyzed only by CYP2C19 (Sager et al., 2016). Incubation of erythrohydrobupropion and threohydrobupropion with recombinant P450 enzymes confirmed that threo- and erythro-4′OH-hydrobupropion were formed solely by CYP2C19 (Fig. 4), and the formation kinetics of erythro- and threo-4′OH-hydrobupropion were characterized in CYP2C19 supersomes. Michaelis-Menten kinetics were observed for both threo-4′-OH-hydrobupropion and erythro-4′-OH-hydrobupropion formation (Fig. 4). Based on the 4′OH-hydrobupropion formation kinetics, threohydrobupropion had nearly 3-fold higher affinity to CYP2C19 (Km = 13 µM) than erythrohydrobupropion (Km = 39 µM), but the kcat values for threo- and erythro-4′OH-hydrobupropion formation were similar (kcat = 552 and 399 pmol/min/pmol, respectively). The intrinsic clearance of threo-4′-OH-hydrobupropion formation was 4-fold higher than that of erythro-4′-OH-hydrobupropion (Fig. 4; Table 1). The major role of CYP2C19 in the formation of threo- and erythro-4′-OH-hydrobupropion was further confirmed by 87% inhibition of threo-4′-OH-hydrobupropion formation and 96% inhibition of erythro-4′-OH-hydrobupropion formation by N-benzylnirvanol in human liver microsomes (Fig. 4). Minor inhibition (15%) of erythro-4′-OH-hydrobupropion formation was detected upon incubation of erythrohydrobupropion with sulfaphenazole, suggesting potential minor involvement of CYP2C9 in erythro-4′OH-hydrobupropion formation. However, based on the 96% inhibition of metabolite formation by N-benzylnirvanol and the lack of metabolite formation in CYP2C9 supersomes, CYP2C19 is likely the only enzyme involved in erythro-4′-OH-hydrobupropion formation.
Fig. 4.

Metabolism of erythro- and threohydrobupropion by recombinant P450 enzymes and human liver microsomes. (A) Erythro- and threo-4′-OH-hydrobupropion formation from erythro- and threohydrobupropion in a panel of P450 supersomes. (B) Inhibition of erythro- and threo-4′-OH-hydrobupropion formation from erythro- and threohydrobupropion in HLM as percentage of control following incubation in the presence of selective P450 inhibitors or a CYP2B6 inhibitory antibody (MAB-2B6). Control 1 is the control for the reversible inhibitors montelukast (2C8 MONT), sulfaphenazole (2C9 SULF), (+)-N-3-benzylnirvanol (2C19 N-BENZ), and quinidine (2D6 QUIN). Control 2 is the control for time-dependent inhibitors troleandomycin (3A4 TAO) and furafylline (1A2 FURA). *p < 0.05 in comparison to control, one-way analysis of variance. (C and D) Formation kinetics of threo-4′-OH-hydrobupropion (C) and erythro-4′OH-hydrobupropion (D) in CYP2C19 supersomes.
Characterization of Bupropion Metabolism in Human Liver S9 Fractions.
To assess the relative importance of the metabolic pathways of bupropion in human liver, OH-bupropion, threohydrobupropion, erythrohydrobupropion, and 4′-OH-bupropion formation was measured in human liver S9 fractions from S- and R-bupropion (Fig. 5). Overall, marked stereoselectivity was observed in the formation of bupropion metabolites. In human liver S9 fractions, no saturation of metabolite formation was observed at the tested concentrations. At all tested concentrations, the formation of R,R-threohydrobupropion was the main metabolite observed from R-bupropion (50% of total metabolites), with R,R-OH-bupropion formation being nearly as efficient (34% of total metabolites). S,R-erythrohydrobupropion and 4′OH-bupropion formation (8 and 8% of total metabolites) were about 7 times less efficient than R,R-threohydrobupropion formation from R-bupropion (Fig. 5; Table 2). Similarly, S,S-threohydrobupropion was the predominant metabolite formed from S-bupropion in human liver S9 fractions (82% of total metabolic clearance), with nearly 10-fold lower formation of S,S-OH-bupropion (12% of total) and 4 and 2% of total metabolism accounted for by R,S-erythrohydrobupropion and 4′-OH-bupropion, respectively (Fig. 5; Table 2). The CLint,H for S-bupropion was greater than that of R-bupropion for all detected metabolites, resulting in a predicted 5-fold higher overall CLint,H for S-bupropion than for R-bupropion. S,S-OH-bupropion formation was nearly twice that of R,R-OH-bupropion, the CLint for S,S-threohydrobupropion was 8-fold higher than that of R,R-threohydrobupropion, and the CLint for R,S-erythrohydrobupropion was 2-fold higher than that of S,R-erythrohydrobupropion (Table 2). No difference was observed between the intrinsic formation clearance of 4′-OH-bupropion following incubation with R- and S-bupropion. Taking into account the total racemic bupropion CLint,H, threohydrobupropion is predicted to contribute to 66% of overall bupropion clearance, whereas the remaining clearance is attributed to OH-bupropion formation (23%), erythrohydrobupropion (6%), and 4′-OH-bupropion (5%) formation. Based on these values and the predicted fractional contribution of CYP2B6 to OH-bupropion formation (Table 1), the fm,CYP2B6 for R-bupropion, S-bupropion, and racemic bupropion was predicted to be 0.30, 0.10, and 0.21, respectively. Accounting for the contribution of CYP2C19 to the formation of OH-bupropion, as well as the fraction of bupropion metabolized to 4′-OH-bupropion, the fmCYP2C19 in bupropion clearance was predicted to be 0.09, 0.03, and 0.06 for R-bupropion, S-bupropion, and racemic bupropion, respectively.
Fig. 5.

Metabolism of R- and S-bupropion in human liver S9 fractions. (A) R-bupropion concentration-dependent formation of R,R-OH-bupropion, S,R-erythrohydrobupropion, R,R-threohydrobupropion, and 4′-OH-bupropion in human liver S9 fractions. (B) S-bupropion concentration-dependent formation of S,S-OH-bupropion, R,S-erythrohydrobupropion, S,S-threohydrobupropion, and 4′-OH-bupropion in human liver S9 fractions. For erythrohydrobupropion formation, two of the lowest substrate concentrations are not shown, as the metabolite formation was not quantifiable in those concentrations.
TABLE 2.
Kinetics of R- and S-bupropion metabolism in human liver S9 fractions
| Substrate and Metabolites |
CLint |
CLint,Ha |
fm,enantiomerb |
fmc |
|---|---|---|---|---|
| μl/min/mg S9 protein | l/h | |||
| R-bupropion | ||||
| R,R-OH-bupropion | 0.47 | 4.1 | 0.34 | 0.17 |
| R,R-threohydrobupropion | 0.69 | 6.0 | 0.50 | 0.25 |
| S,R-erythrohydrobupropion | 0.11 | 0.9 | 0.08 | 0.04 |
| 4′-OH-bupropion | 0.11 | 0.9 | 0.08 | 0.04 |
| Total R-bupropion CLint | 11.9 | |||
| S-bupropion | ||||
| S,S-OH-bupropion | 0.81 | 7.0 | 0.12 | 0.06 |
| S,S-threohydrobupropion | 5.61 | 48.5 | 0.82 | 0.41 |
| R,S-erythrohydrobupropion | 0.25 | 2.1 | 0.04 | 0.02 |
| 4′-OH-bupropion | 0.13 | 1.1 | 0.02 | 0.01 |
| Total S-bupropion CLint | 59.2 |
S9 fraction intrinsic clearance values were scaled to CLint,H using the scaling factor of 96.1 mg S9 protein/g liver (Watanabe et al., 2009). Assuming body weight of 70 kg and 21.4 g liver/kg body weight results in 1500 g liver weight (Ito and Houston, 2005). Protein binding in the incubations was negligible, and unbound fractions in the incubations were assumed to be 1.
The fraction metabolized to each metabolite from the individual bupropion enantiomer (fm, enantiomer) calculated from CLint,H data.
The fraction of racemic bupropion metabolized to the specific metabolite (fm).
Inhibitory Effects of Ticlopidine on the Formation of Bupropion Metabolites.
Ticlopidine is a known mechanism-based inhibitor of CYP2C19 and CYP2B6 (Ha-Duong et al., 2001; Richter et al., 2004; Nishiya et al., 2009a,b) and causes a 61% increase in bupropion AUC in vivo (Turpeinen et al., 2005). As expected, ticlopidine was found to inhibit the formation of OH-bupropion (CYP2B6) and 4′-OH-bupropion (CYP2C19), with IC50 values of 0.53 and 0.87 µM, respectively (Fig. 6; Table 3). To assess the potential of ticlopidine to inhibit 11β-HSD-1, threo- and erythrohydrobupropion formation was measured in the presence and absence of ticlopidine. Interestingly, ticlopidine inhibited the formation of threohydrobupropion (IC50 = 10.20 µM) and erythrohydrobupropion (IC50 = 11.82 µM) in HLM when preincubated for 30 minutes in the absence of NADPH (Fig. 6; Table 3). Furthermore, 7.6-fold (OH-bupropion), 6.2-fold (4′-OH-bupropion), 2.4-fold (threohydrobupropion), and 2.3-fold (erythrohydrobupropion) shifts in the IC50 values for the specific metabolite formation were observed following a 30-minute preincubation of ticlopidine in the presence of NADPH, suggesting a potential for time-dependent inhibition of all bupropion metabolic pathways by ticlopidine. This inhibition of threo- and erythrohydrobupropion formation by ticlopidine is unlikely to be due to P450-mediated formation of erythro- and threohydrobupropion, as formation of these metabolites was not observed in any of the incubations of R- or S-bupropion with recombinant P450 enzymes in any of the described experiments (data not shown).
Fig. 6.

Inhibition of bupropion metabolism in human liver microsomes by ticlopidine. Ticlopidine IC50 values toward erythrohydrobupropion (A), threohydrobupropion (B), OH-bupropion (C), and 4′-OH-bupropion (D) formation were characterized following a 30-minute preincubation of HLMs with ticlopidine in the presence (filled circles) and absence (filled squares) of NADPH. Significant NADPH-dependent shifts were observed in the IC50 values for each metabolite formation.
TABLE 3.
IC50 values of ticlopidine towards bupropion metabolite formation in the presence and absence of a 30-minute preincubation with NADPH in HLM
IC50-values are shown with the 95% confidence intervals. OH-bupropion and 4′-OH-bupropion formation was used as a measure of CYP2B6 and CYP2C19 activity, respectively. Based on previous reports, the formation of threohydrobupropion and erythrohydrobupropion in HLM was attributed to 11β-HSD-1 (Meyer et al., 2013; Skarydova et al., 2014). Ticlopidine IC50 values were based on measured ticlopidine concentrations prior to the 7-minute bupropion incubation. Ticlopidine fu in 0.5 mg/ml HLM was 0.13, so the unbound IC50 values were 0.07 (OH-bupropion), 0.12 (4′-OH-bupropion), 1.3 (threohydrobupropion), and 1.5 µM (erythrohydrobupropion).
| Metabolite |
Primary Enzyme |
IC50 −NADPH |
IC50 +NADPH |
Fold Shift |
|---|---|---|---|---|
| µM | µM | |||
| OH-bupropion | CYP2B6 | 0.53 (0.40––0.70) | 0.07 (0.06–0.08) | 7.6 |
| 4′-OH-bupropion | CYP2C19 | 0.87 (0.25–3.04) | 0.14 (0.06–0.35) | 6.2 |
| Threohydrobupropion | 11β-HSD-1 | 11.82 (4.04–24.57) | 4.91 (2.41–10.02) | 2.4 |
| Erythrohydrobupropion | 11β-HSD-1 | 10.20 (4.41–23.59) | 4.39 (2.10–9.29) | 2.3 |
Prediction of the Stereoselective Disposition of Bupropion in vivo.
The unbound hepatic intrinsic clearances of R- and S-bupropion were predicted using the intrinsic clearances obtained in S9 fractions (CLint,H, Table 2), the measured plasma unbound fractions (fu), and the measured blood-to-plasma ratio of 0.42. The plasma fu values were 0.5 and 0.6 for R- and S-bupropion, respectively, and the resulting hepatic unbound intrinsic clearances (fu*CLint) were 6.0 l/h for R-bupropion and 35.5 l/h for S-bupropion, corresponding to hepatic blood unbound intrinsic clearances of 14.2 l/h for R-bupropion and 84.5 l/h for S-bupropion. Based on these values and assuming no chiral inversion of bupropion and equal absorption and gut extraction (Fa and Fg = 1) of the stereoisomers, the R/S AUC ratio after oral administration of bupropion was predicted to be 5.9. The overall hepatic blood clearance of bupropion was predicted using the well stirred model. The predicted hepatic clearance values for S- and R-bupropion were 43.5 and 12.2 l/h, respectively, suggesting a hepatic extraction ratio of 0.48 for S-bupropion and 0.14 for R-bupropion.
To evaluate whether chiral inversion of bupropion may confound the predicted R/S exposure ratios due to elimination of the lower-clearance R-enantiomers via chiral inversion to S-bupropion, the R/S ratio was simulated at different chiral inversion rates. Experimentally, upon incubation of R- or S-bupropion in human plasma, the rate of chiral inversion was 0.54 hour−1. Scaling this value to blood volume (5 l) resulted in an estimated in vivo isomerization CL of 2.7 l/h. If this chiral inversion was scaled to total body water (42 l), an estimated in vivo isomerization CL of 22.7 l/h was obtained. Based on this, the R/S ratio was simulated using systemic hepatic clearances of S- and R-bupropion and isomerization rates between 0 and 23 l/h. With increasing isomerization rates, the R/S steady-state concentration ratio decreased from 3.56 (at no isomerization) to 2 (at isomerization rate of 10 l/h) and was approximately 1 at an isomerization rate of 23 l/h. This suggests that, in vivo, the chiral inversion occurs predominantly in plasma, and that chiral inversion has a negligible role in the clearance of bupropion. Based on the simulation, the isomerization also had no effect on the predicted fraction of bupropion metabolized to each metabolite. The predicted fm values were 0.21 (OH-bupropion), 0.68 (threohydrobupropion), 0.06 (erythrohydrobupropion), and 0.04 (4′-OH-bupropion) from the model.
Discussion
Bupropion is a clinically important antidepressant and smoking cessation aid. Similarly, it is a valuable CYP2B6 probe in in vitro DDI risk assessment and in clinical DDI studies. However, both efficacy and safety of bupropion and the validity of bupropion as a CYP2B6 probe are dependent on stereoselective metabolism and disposition (Damaj et al., 2004; Kharasch et al., 2008; Masters et al., 2016). Therefore, thorough characterization of the stereoselective metabolism of bupropion in vitro is critically needed.
The results of this study show that S-bupropion has significantly higher clearance than R-bupropion, leading to a higher hepatic extraction ratio of S-bupropion than R-bupropion. The higher hepatic clearance of S-bupropion is predominantly due to the nearly 5-fold higher formation clearance of threohydrobupropion from S-bupropion than from R-bupropion. The in vivo R/S-bupropion AUC-ratio is likely driven by the combination of differences in the first-pass hepatic metabolism of R- and S-bupropion and the stereoselective systemic clearance. Recently, an R/S-bupropion AUC ratio of 6 was reported following administration of 100 mg of immediate-release racemic bupropion (Masters et al., 2016). This value is in excellent agreement with the predicted R/S ratio of 5.9 obtained in this study based on the predicted R/S-bupropion oral clearance ratio. This suggests that differences in hepatic extraction alone are sufficient to explain the stereoselective disposition of bupropion, although reduction of bupropion to threo- and erythrohydrobupropion in extrahepatic sites has been shown (Connarn et al., 2015). Interestingly, a lower mean R/S-bupropion AUC ratio of 2.97 was reported following a 150-mg single dose of bupropion (Kharasch et al., 2008). This R/S ratio is close to the ratio that would be predicted using just systemic hepatic clearances of R- and S-bupropion (3.56) but less than what is predicted when first-pass hepatic extraction is considered. It is possible that the observed R/S ratios in the clinical studies are due to the chiral inversion of bupropion, as the simulations conducted in this study show that higher chiral inversion clearance in vivo will result in a lower R/S-bupropion ratio. It is also possible that CYP2B6 and CYP2C19 genotypic variability between the studies contributes to the different R/S ratios observed, as all of 4′OH-bupropion and a fraction of OH-bupropion are formed by CYP2C19, and CYP2B6 contributes to different fractions of R- and S-bupropion clearance.
The finding that the formation of S,S-OH-bupropion is more efficient than the formation of R,R-OH-bupropion by CYP2B6 is in agreement with previous work (Coles and Kharasch, 2008). The predicted hepatic clearances based on the recombinant P450 enzyme data were also in excellent agreement with the predicted hepatic clearances obtained from the S9 fractions for OH-bupropion and 4′OH-bupropion stereoisomers. Importantly, based on the liver S9 fraction data, the predicted overall fraction of R-bupropion metabolized to OH-bupropion was 2.8-fold greater (34%) than that observed from S-bupropion (12%). As CYP2B6 contributes similarly to the R,R-OH- and S,S-OH-bupropion formation (90% of OH-bupropion formation is by CYP2B6), these data suggest that R,R-OH-bupropion formation clearance or R,R-OH-bupropion/R-bupropion metabolic ratio may be a more sensitive and better probe of CYP2B6 activity than S,S-OH-bupropion/S-bupropion formation clearance/metabolic ratio. Previously, S,S-OH-bupropion formation clearance was suggested as a possibly more sensitive probe of CYP2B6 activity than R,R-OH-bupropion or racemic OH-bupropion (Kharasch et al., 2008), a notion not supported by the in vitro data. However, different clearances of OH-bupropion stereoisomers may affect validity of in vivo probes. In vivo R,R-OH-bupropion to S,S-OH-bupropion ratios of 44–65 have been reported (Kharasch et al., 2008; Masters et al., 2016). If R,R-OH- and S,S-OH-bupropion had similar in vivo clearances, the in vitro data presented here would predict an R,R-OH- to S,S-OH-bupropion ratio of 3.5 based on an in vivo R/S-bupropion AUC ratio of 6 and a ratio of the formation clearances of 0.58. Therefore, these data suggest that S,S-OH-bupropion clearance is much higher than that of R,R-OH-bupropion, pointing to a necessity to better understand the differences in the clearance of OH-bupropion enantiomers.
The predicted 2.8-fold greater fraction of R-bupropion metabolized to OH-bupropion than S-bupropion is in agreement with the observed 3.1-fold greater recovery of R,R-OH-bupropion in urine when compared with S,S-OH-bupropion (Masters et al., 2016). However, the overall fraction of bupropion dose recovered in urine as OH-bupropion, 5% over the dosing interval at steady state (Sager et al., 2016) or 5.4% following a single dose (Masters et al., 2016), is much less than the 23% of the total clearance predicted here. This is likely due to incomplete absorption of bupropion in humans, as only 39% of the total dose is recovered in urine as bupropion and its metabolites, and 15% of the known metabolites recovered in urine were OH-bupropion or its conjugates (Sager et al., 2016). It is also possible that additional subsequent metabolites of OH-bupropion exist that have not been detected in the in vivo studies, or that part of the OH-bupropion and its conjugates are cleared via biliary secretion. However, the relatively minor role of OH-bupropion formation in overall bupropion clearance is in agreement with the observed reduced OH-bupropion formation without alteration in bupropion concentrations in carriers of lower-activity CYP2B6 alleles CYP2B6*6 and *18 (Benowitz et al., 2013).
Overall, the relative contributions of individual bupropion metabolites to bupropion clearance based on urinary recovery data (Sager et al., 2016) are well predicted from the in vitro data. Threohydrobupropion formation is predicted to account for 68% of the total bupropion metabolism, and 4′-OH-formation is predicted to account for 4%. In patients taking bupropion, 67% of the drug-related material was recovered in urine as threohydrobupropion and its metabolites and 2% as 4′-OH-bupropion and its conjugates (Sager et al., 2016). Following a single dose of bupropion to healthy volunteers, a smaller fraction of the bupropion-related species, approximately 40%, was recovered in urine as threohydrobupropion (Masters et al., 2016). This lower percentage is most likely due to the lack of accounting for the threo-4′OH-hydrobupropion metabolite, as 4′-hydroxylation was shown to account for 20% of threohydrobupropion clearance (Sager et al., 2016). 4′-Hydroxylation is also a major elimination pathway for erythrohydrobupropion, accounting for 70% of erythrohydrobupropion clearance (Sager et al., 2016). Our findings show that CYP2C19 catalyzes the formation of threo- and erythro-4′-OH-hydrobupropion, a finding consistent with the observation that threo- and erythrohydrobupropion exposure is increased by 30–40% in individuals carrying at least one copy of CYP2C19*2 (Zhu et al., 2014). However, our data predict a smaller fraction of erythrohydrobupropion formation of total bupropion metabolism (6%) than what is observed in vivo (15%) (Sager et al., 2016). This discrepancy is likely due to extrahepatic formation of erythrohydrobupropion that is not accounted for in the in vitro system.
Bupropion’s fm,CYP2B6 has been proposed to be 26–40% based on changes in bupropion exposure following clopidogrel or ticlopidine administration (Turpeinen et al., 2005). Furthermore, CYP2C19 has been proposed to be a quantitatively important elimination pathway for bupropion based on a 13% increase in bupropion AUC in CYP2C19 intermediate metabolizers compared with extensive metabolizers (Zhu et al., 2014) and in vitro metabolism of bupropion by CYP2C19 (Chen et al., 2010; Sager et al., 2016). It is well established that ticlopidine and clopidogrel are time-dependent inhibitors of CYP2B6 and CYP2C19 (Ha-Duong et al., 2001; Richter et al., 2004; Nishiya et al., 2009a,b). However, CYP2C19-mediated formation of 4′-OH-bupropion is only predicted to contribute to 5% of bupropion clearance, and was observed to account for 2% of all bupropion metabolites recovered in urine in vivo (Sager et al., 2016). Thus, inhibition of CYP2B6 and CYP2C19 by clopidogrel and ticlopidine cannot explain the magnitude of the in vivo DDIs. This study shows that ticlopidine also inhibits threo- and erythrohydrobupropion formation likely by a time-dependent mechanism as shown by the significant IC50 shifts observed. This inhibition is likely due to inhibition of 11β-HSD-1 by ticlopidine, as bupropion reduction to threo- and erythrohydrobupropion is mainly mediated by 11β-HSD-1 (Meyer et al., 2013; Connarn et al., 2015). Based on a 19% contribution of CYP2B6 and 5% contribution of CYP2C19 to bupropion elimination, and assuming complete inhibition of these pathways by ticlopidine, a 22% inhibition of threo- and erythrohydrobupropion formation by ticlopidine in vivo will explain the 40% inhibition of bupropion clearance observed in vivo. Further studies are needed to evaluate the inhibition of 11β-HSD-1 by ticlopidine and clopidrogrel in vivo and in vitro.
In conclusion, this study demonstrates the overall stereoselective metabolism of bupropion in vitro, explaining the observed stereoselectivity in bupropion disposition in vivo. The in vitro intrinsic clearance data show that CYP2B6 and CYP2C19 are minor elimination pathways of bupropion, and that time-dependent inhibition of 11β-HSD-1 by ticlopidine likely explains why the fm,CYP2B6 has been overestimated based on the DDI data. Furthermore, this study shows that CYP2C19 contributes to threohydrobupropion and erythrohydrobupropion elimination possibly affecting the steady-state concentrations of threo- and erythrohydrobupropion, and that in vivo chiral inversion is a minor factor in altering bupropion R/S ratios and is unlikely to confound measures of CYP2B6 activity in vivo.
Abbreviations
- AUC
area under the plasma concentrations-time curve
- DDI
drug-drug interaction
- fm
fraction metabolized
- HLM
human liver microsomes
- 11β-HSD-1
11β-hydroxysteroid dehydrogenase-1
- KPi
potassium phosphate
- LC-MS/MS
liquid chromatography–tandem mass spectrometry
- MAB
monoclonal antibody
- P450
cytochrome P450
- rCYP
recombinant cytochrome P450
Authorship Contributions
Participated in research design: Sager, Isoherranen.
Conducted experiments: Sager, Price.
Performed data analysis: Sager, Price, Isoherranen.
Wrote or contributed to the writing of the manuscript: Sager, Isoherranen.
Footnotes
This work was supported in part by the National Institutes of Health [Grants P01 DA032507 (N.I.) and T32 GM007750 (J.E.S.)].
References
- Benowitz NL, Zhu AZX, Tyndale RF, Dempsey D, Jacob P., 3rd (2013) Influence of CYP2B6 genetic variants on plasma and urine concentrations of bupropion and metabolites at steady state. Pharmacogenet Genomics 23:135–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bondarev ML, Bondareva TS, Young R, Glennon RA. (2003) Behavioral and biochemical investigations of bupropion metabolites. Eur J Pharmacol 474:85–93. [DOI] [PubMed] [Google Scholar]
- Brown HS, Griffin M, Houston JB. (2007) Evaluation of cryopreserved human hepatocytes as an alternative in vitro system to microsomes for the prediction of metabolic clearance. Drug Metab Dispos 35:293–301. [DOI] [PubMed] [Google Scholar]
- Chen Y, Liu HF, Liu L, Nguyen K, Jones EB, Fretland AJ. (2010) The in vitro metabolism of bupropion revisited: concentration dependent involvement of cytochrome P450 2C19. Xenobiotica 40:536–546. [DOI] [PubMed] [Google Scholar]
- Coles R, Kharasch ED. (2008) Stereoselective metabolism of bupropion by cytochrome P4502B6 (CYP2B6) and human liver microsomes. Pharm Res 25:1405–1411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connarn JN, Zhang X, Babiskin A, Sun D. (2015) Metabolism of bupropion by carbonyl reductases in liver and intestine. Drug Metab Dispos 43:1019–1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Damaj MI, Carroll FI, Eaton JB, Navarro HA, Blough BE, Mirza S, Lukas RJ, Martin BR. (2004) Enantioselective effects of hydroxy metabolites of bupropion on behavior and on function of monoamine transporters and nicotinic receptors. Mol Pharmacol 66:675–682. [DOI] [PubMed] [Google Scholar]
- Faucette SR, Hawke RL, Lecluyse EL, Shord SS, Yan B, Laethem RM, Lindley CM. (2000) Validation of bupropion hydroxylation as a selective marker of human cytochrome P450 2B6 catalytic activity. Drug Metab Dispos 28:1222–1230. [PubMed] [Google Scholar]
- Golden RN, De Vane CL, Laizure SC, Rudorfer MV, Sherer MA, Potter WZ. (1988) Bupropion in depression. II. The role of metabolites in clinical outcome. Arch Gen Psychiatry 45:145–149. [DOI] [PubMed] [Google Scholar]
- Gufford BT, Lu JBL, Metzger IF, Jones DR, Desta Z. (2016) Stereoselective Glucuronidation of Bupropion Metabolites In Vitro and In Vivo. Drug Metab Dispos 44:544–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ha-Duong NT, Dijols S, Macherey AC, Goldstein JA, Dansette PM, Mansuy D. (2001) Ticlopidine as a selective mechanism-based inhibitor of human cytochrome P450 2C19. Biochemistry 40:12112–12122. [DOI] [PubMed] [Google Scholar]
- Hesse LM, Venkatakrishnan K, Court MH, von Moltke LL, Duan SX, Shader RI, Greenblatt DJ. (2000) CYP2B6 mediates the in vitro hydroxylation of bupropion: potential drug interactions with other antidepressants. Drug Metab Dispos 28:1176–1183. [PubMed] [Google Scholar]
- Hurt RD, Sachs DPL, Glover ED, Offord KP, Johnston JA, Dale LC, Khayrallah MA, Schroeder DR, Glover PN, Sullivan CR, et al. (1997) A comparison of sustained-release bupropion and placebo for smoking cessation. N Engl J Med 337:1195–1202. [DOI] [PubMed] [Google Scholar]
- Ito K, Houston JB. (2005) Prediction of human drug clearance from in vitro and preclinical data using physiologically based and empirical approaches. Pharm Res 22:103–112. [DOI] [PubMed] [Google Scholar]
- Johnston JA, Fiedler-Kelly J, Glover ED, Sachs DP, Grasela TH, DeVeaugh-Geiss J. (2001) Relationship between drug exposure and the efficacy and safety of bupropion sustained release for smoking cessation. Nicotine Tob Res 3:131–140. [DOI] [PubMed] [Google Scholar]
- Kharasch ED, Mitchell D, Coles R. (2008) Stereoselective bupropion hydroxylation as an in vivo phenotypic probe for cytochrome P4502B6 (CYP2B6) activity. J Clin Pharmacol 48:464–474. [DOI] [PubMed] [Google Scholar]
- Masters AR, Gufford BT, Lu JB, Metzger IF, Jones DR, Desta Z. (2016) Chiral Plasma Pharmacokinetics and Urinary Excretion of Bupropion and Metabolites in Healthy Volunteers. J Pharmacol Exp Ther 358:230–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer A, Vuorinen A, Zielinska AE, Strajhar P, Lavery GG, Schuster D, Odermatt A. (2013) Formation of threohydrobupropion from bupropion is dependent on 11β-hydroxysteroid dehydrogenase 1. Drug Metab Dispos 41:1671–1678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishiya Y, Hagihara K, Ito T, Tajima M, Miura S, Kurihara A, Farid NA, Ikeda T. (2009a) Mechanism-based inhibition of human cytochrome P450 2B6 by ticlopidine, clopidogrel, and the thiolactone metabolite of prasugrel. Drug Metab Dispos 37:589–593. [DOI] [PubMed] [Google Scholar]
- Nishiya Y, Hagihara K, Kurihara A, Okudaira N, Farid NA, Okazaki O, Ikeda T. (2009b) Comparison of mechanism-based inhibition of human cytochrome P450 2C19 by ticlopidine, clopidogrel, and prasugrel. Xenobiotica 39:836–843. [DOI] [PubMed] [Google Scholar]
- Peng C-C, Templeton I, Thummel KE, Davis C, Kunze KL, Isoherranen N. (2011) Evaluation of 6β-hydroxycortisol, 6β-hydroxycortisone, and a combination of the two as endogenous probes for inhibition of CYP3A4 in vivo. Clin Pharmacol Ther 89:888–895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Preskorn SH. (1983) Antidepressant response and plasma concentrations of bupropion. J Clin Psychiatry 44:137–139. [PubMed] [Google Scholar]
- Richter T, Mürdter TE, Heinkele G, Pleiss J, Tatzel S, Schwab M, Eichelbaum M, Zanger UM. (2004) Potent mechanism-based inhibition of human CYP2B6 by clopidogrel and ticlopidine. J Pharmacol Exp Ther 308:189–197. [DOI] [PubMed] [Google Scholar]
- Sager JE, Choiniere JR, Chang J, Stephenson-Famy A, Nelson WL, Isoherranen N. (2016) Identification and structural characterization of three new metabolites of bupropion in humans. ACS Med Chem Lett 7:791–796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirasaka Y, Sager JE, Lutz JD, Davis C, Isoherranen N. (2013) Inhibition of CYP2C19 and CYP3A4 by omeprazole metabolites and their contribution to drug-drug interactions. Drug Metab Dispos 41:1414–1424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silverstone PH, Williams R, McMahon L, Fleming R, Fogarty S. (2008) Convulsive liability of bupropion hydrochloride metabolites in Swiss albino mice. Ann Gen Psychiatry 7:19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skarydova L, Tomanova R, Havlikova L, Stambergova H, Solich P, Wsol V. (2014) Deeper insight into the reducing biotransformation of bupropion in the human liver. Drug Metab Pharmacokinet 29:177–184. [DOI] [PubMed] [Google Scholar]
- Stringer RA, Strain-Damerell C, Nicklin P, Houston JB. (2009) Evaluation of recombinant cytochrome P450 enzymes as an in vitro system for metabolic clearance predictions. Drug Metab Dispos 37:1025–1034. [DOI] [PubMed] [Google Scholar]
- Totah RA, Sheffels P, Roberts T, Whittington D, Thummel K, Kharasch ED. (2008) Role of CYP2B6 in stereoselective human methadone metabolism. Anesthesiology 108:363–374. [DOI] [PubMed] [Google Scholar]
- Turpeinen M, Tolonen A, Uusitalo J, Jalonen J, Pelkonen O, Laine K. (2005) Effect of clopidogrel and ticlopidine on cytochrome P450 2B6 activity as measured by bupropion hydroxylation. Clin Pharmacol Ther 77:553–559. [DOI] [PubMed] [Google Scholar]
- Watanabe T, Kusuhara H, Maeda K, Shitara Y, Sugiyama Y. (2009) Physiologically based pharmacokinetic modeling to predict transporter-mediated clearance and distribution of pravastatin in humans. J Pharmacol Exp Ther 328:652–662. [DOI] [PubMed] [Google Scholar]
- Zhu AZX, Cox LS, Nollen N, Faseru B, Okuyemi KS, Ahluwalia JS, Benowitz NL, Tyndale RF. (2012) CYP2B6 and bupropion’s smoking-cessation pharmacology: the role of hydroxybupropion. Clin Pharmacol Ther 92:771–777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu AZX, Zhou Q, Cox LS, Ahluwalia JS, Benowitz NL, Tyndale RF. (2014) Gene variants in CYP2C19 are associated with altered in vivo bupropion pharmacokinetics but not bupropion-assisted smoking cessation outcomes. Drug Metab Dispos 42:1971–1977. [DOI] [PMC free article] [PubMed] [Google Scholar]