

ABSTRACT
Guanidine compounds act as ion channel modulators. In the case of Cys-loop receptors, the guanidine compound amiloride antagonized the heteromeric GABA-A, glycine, and nicotinic acetylcholine receptors. However, amiloride exhibits characteristics consistent with a positive allosteric modulator for the human GABA-A (hGABA-A) ρ1 receptor. Site-directed mutagenesis revealed that the positive allosteric modulation was influenced by the GABA-A ρ1 second transmembrane domain 15′ position, a site implicated in ligand allosteric modulation of Cys-loop receptors. There are a variety of amiloride derivatives that provide opportunities to assess the significance of amiloride functional groups (e.g., the guanidine group, the pyrazine ring, etc.) in the modulation of the GABA-A ρ1 receptor activity. We utilized 3 amiloride derivatives (benzamil, phenamil, and 5-(N, N-Hexamethylene) amiloride) to assess the contribution of these groups toward the potentiation of the GABA-A ρ1 receptor. Benzamil and phenamil failed to potentiate on the wild type GABA-A ρ1 GABA-mediated current while HMA demonstrated efficacy only at the highest concentration studied. The hGABA-A ρ1 (I15'N) mutant receptor activity was potentiated by lower HMA concentrations compared to the wild type receptor. Our findings suggest that an exposed guanidine group on amiloride and amiloride derivatives is critical for modulating the GABA-A ρ1 receptor. The present study provides a conceptual framework for predicting which amiloride derivatives will demonstrate positive allosteric modulation of the GABA-A ρ1 receptor.
KEYWORDS: Cys-loop receptor, GABA-A receptor, guanidine compound, positive allosteric modulation
Introduction
Ion channel families have a complex pharmacology where it is common to see an agonist for one subtype be a partial agonist or antagonist in another. The GABA-A receptors are examples of this contrast for ligand activity. These receptors are members of the larger Cys-loop family of pentameric ion channels, along with the nicotinic acetylcholine receptors, serotonin type 3 receptors, and the glycine receptors making up the principal members of this family.1 The interaction of different GABA-A receptor ligands is dependent on the receptor's subunit composition. For example, benzodiazepine sensitivity is dependent on the presence of specific α subunits along with the γ subunit.2 GABA-A receptors with the α4 or α6 subunits are insensitive to classical 1,4-benzodiazepines, but remain sensitive to neurosteroids and ethanol.3 The α6 subunit is integral in the effects of the barbiturate pentobarbital, conveying both a higher affinity and efficacy for the direct activation of the GABA-A receptor.4 This observed complex pharmacology extends to other GABA-A receptor ligands, including guanidine compounds, like guanidinoacetic acid.5,6 Furthermore, the guanidine compound amiloride is an antagonist for GABA-A αβγ receptors with higher potency in α6βγ receptors.7
Of this large group of GABA-A receptors, the GABA-A ρ1 receptor consists of either 5 identical subunits or a combination of subunits. The GABA-A ρ1 receptor is found primarily in the retina, where the protein is involved in mediating inhibitory signals for sight.8 The GABA-A ρ1 receptor retains GABA as the full agonist and is sensitive to picrotoxin, the GABA-A receptor noncompetitive antagonist and convulsant.9 However, the homomeric GABA-A ρ1 receptors have a different pharmacology than the other GABA-A αβγ receptors. The GABA-A αβγ receptor antagonist bicuculine is not efficacious in the GABA-A ρ1 receptor.10 Furthermore, those ligands that have efficacy in the GABA-A αβγ receptor do not have similar pharmacology as the GABA-A ρ1 receptor. This differential pharmacology is of interest due to the accumulating evidence that the GABA-A ρ1 receptor subunit is found in non-retinal tissue and has a physiological role.11-13
In a recent report, we have shown that amiloride has different pharmacological properties on the GABA-A ρ1 receptor compared to the heteromeric GABA-A receptors.14 Unlike heteromeric GABA-A αβγ receptors, amiloride potentiated the GABA-mediated current in the hGABA-A ρ1 receptor, which is highly expressed in the retina.15 This potentiation of current was highest at concentrations above 100 μM and the introduction of the GABA-A ρ1 (I15'N) mutation abolished this potentiation. The GABA-A ρ1 (I15'N) mutation replaced the isoleucine for an asparagine. An asparagine at the second transmembrane domain 15' position is present in the GABA-A receptor β2, and β3 receptor subunits. This residue is crucial for conferring sensitivity to the barbiturates, such as pentobarbital.16 The differences in the second transmembrane domain 15′ positions may play a role in channel sensitivity to allosteric modulators. Thus, we proposed categorizing amiloride as a positive allosteric modulator for the GABA-A ρ1 receptor.
Although this mutation abolished amiloride potentiation, it is unclear if the guanidine group or the pyrazine ring is necessary for this potentiation of amiloride on the hGABA-A ρ1 receptor. Understanding the contribution of the amiloride molecule groups has pharmacological implications. Amiloride's diuretic effect has contributed to antagonism of epithelial sodium channels, which resulted in the inhibition of sodium reabsorption.17 Additionally, amiloride inhibits acid–sensing ion channels (ASIC), which are expressed in the retina.18 In 2013, amiloride was proposed as a pharmacological intervention to relieve retinal degeneration in ischemic disorders, such as diabetic retinopathy, through the antagonism of retinal ASICs. It was concluded that amiloride (at 100 μM) reduced retinal degeneration. Inexplicably, amiloride at higher concentrations led to a worsening of retinal degeneration.19 This effect could be due to interaction with other retina-expressed receptors, such as the GABA-A ρ1 receptor. Substitutions on the amiloride molecule may alter the interaction between amiloride and the GABA-A ρ1 receptor.
Here, we investigated the importance of the molecular substitutions in amiloride for potentiation of the hGABA-A ρ1 receptor activity utilizing whole cell patch-clamp electrophysiology. The amiloride derivatives studied here are phenamil, benzamil, and 5-(N, N-Hexamethylene)amiloride (HMA). When co-applied with GABA to the hGABA-A ρ1 receptor, HMA potentiated GABA-mediated current at the highest concentration studied (1 mM) while the wild-type receptor was insensitive to phenamil and benzamil. Combined with previous findings, the data suggested that an exposed guanidine group in amiloride derivatives is critical for the positive allosteric modulation of the GABA-A ρ1 receptor. The hGABA-A ρ1 (I15'N) mutant receptor increased the receptor's sensitivity to HMA, suggesting that this residue affects channel activity rather than serve as an amiloride-binding site. Finally, our data point toward the importance of the guanidine group in amiloride and amiloride derivatives for influencing hGABA-A ρ1 receptor activity.
Results
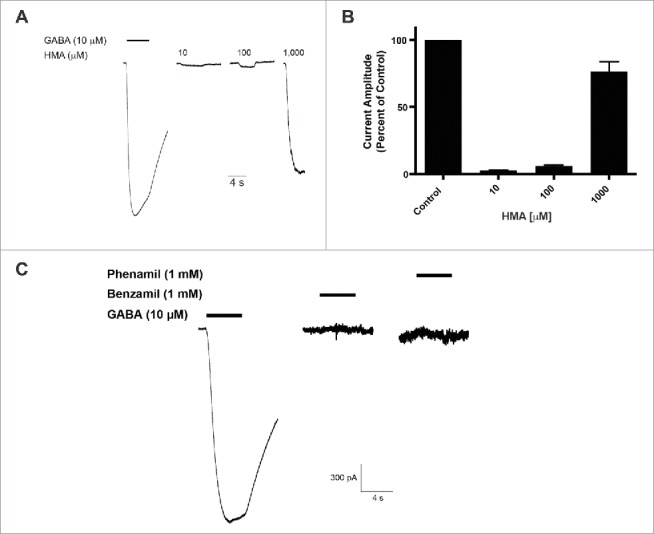
We compared the role of molecular substitutions on the amiloride molecule using whole cell patch-clamp electrophysiology and transient expression of hGABA-A ρ1 receptor. Increasing concentrations of HMA, ranging from 0.3 to 1,000 µM, were co-applied simultaneously 10 mM GABA (Fig. 1A). The 10 μM GABA concentration is an approximate GABA EC50 for the GABA-A ρ1 receptor that was reported previously.14 Upon co-application of the amiloride derivative and GABA, we observed significant potentiation of the GABA-mediated current at 1,000 μM HMA (Fig. 1B). In contrast to our previous study, amiloride potentiated GABA-mediated current at concentrations as low as 10 μM.14 Potentiation of the GABA-mediated current was not significant at lower concentrations of HMA (0.3 to 300 µM). In order to obtain a 1,000 μM concentration of HMA, we utilized DMSO as the solvent to get a 10 mM stock solution. However, concentrations higher than 1,000 μM HMA were not performed due to the diluted concentration of DMSO in the test solutions would be greater than 1%.
Figure 1.
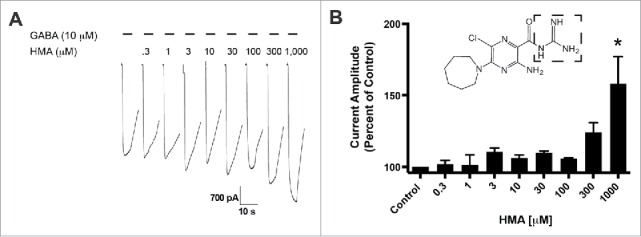
5-(N,N-Hexamethylene) amiloride (HMA) potentiates the hGABA-(A)ρ1 receptor. (A) Resulting representative wild type hGABA-A ρ1 receptor current generated by the co-application of GABA (10 µM) with increasing concentrations of HMA are shown. (B) Summary of potentiation of the hGABA-A ρ1 receptor in the presence of increasing HMA. Data are presented as the mean ± SEM of n ≥ 4 cells. Significance is indicated by: *, p ≤ 0.05 using a One-way ANOVA comparison with Bonferroni post-hoc test compared to the control. Dashed square highlights the guanidine group.
The HMA molecule has an exposed and unsubstituted guanidine group. To assess the influence of this guanidine group, we characterized the activity of amiloride derivatives with inaccessible guanidine groups for potentiation of GABA-induced current in the GABA-A ρ1 receptor. We applied 10 μM GABA with increasing concentrations of phenamil (Fig. 2A). Using a range of phenamil concentrations (1 to 1,000 μM), we failed to observe a significant effect on the measured peak current amplitudes of the GABA and phenamil recordings compared to the control (Fig. 2B). The co-application of GABA and benzamil failed to show significance effects compared to control GABA-mediated peak current amplitudes (Fig. 2C and 2D). Compared to HMA, the availability of the guanidine group may be critical in mediating enhancement of the GABA-mediate current.
Figure 2.
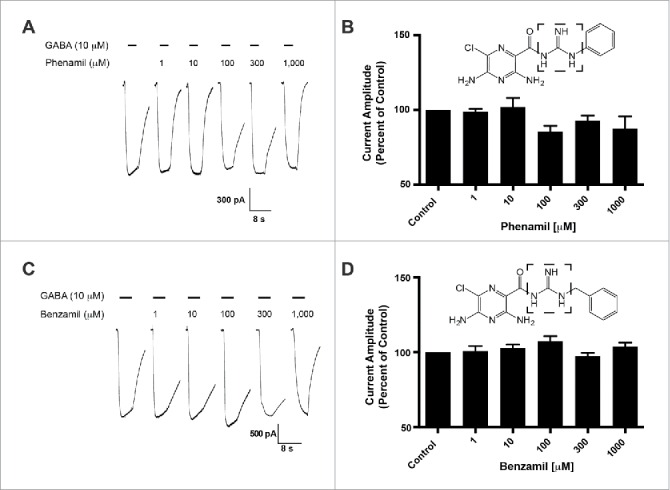
Phenamil and benzamil fails to elicit concentration-dependent effects on the hGABA-(A)ρ1 receptor. (A) Resulting representative traces generated by the co-application of GABA (10 µM) with increasing concentrations of phenamil in the presence of the wild type hGABA-A ρ1 receptor are shown. (B) Summary of phenamil activity on hGABA-A ρ1 receptor mediated peak current amplitude. Response to GABA and phenamil were normalized to the peak current amplitude of the control. (C) Resulting representative responses generated by the co-application of GABA (10 µM) with increasing concentrations of benzamil in the presence of the wild type hGABA-A ρ1 receptor are shown. (D) Summary of benzamil activity on hGABA-A ρ1 receptor mediated peak current amplitude. Responses to GABA and benzamil were normalized to the peak current amplitude of the GABA. Benzamil failed to elicit significant effects on the GABA-A ρ1 receptor. Data are presented as the mean ± SEM of n ≥ 4 cells. Dashed square highlights the guanidine group.
Previously, we reported that the GABA-A ρ1 (I15'N) mutant receptor was insensitive to amiloride.14 This site within the interface of 2 neighboring GABA-A ρ1 receptor subunits is an appealing candidate for an amiloride derivative site of action. To further probe this site, we assessed the mutant receptor for HMA, phenamil, and benzamil activity. Upon co-application of increasing concentrations of HMA with 3 μM GABA, the previously reported hGABA-A ρ1 (I15'N) receptor GABA EC50, we observed an increased peak current amplitude of the GABA-mediated current (Fig. 3A). HMA increased the peak current amplitude of GABA-mediated current at 30 (p < 0.05), 100 (p < 0.001), 300 (p < 0.001), and 1,000 μM (p < 0.001) concentrations. A concentration response profile for HMA was generated and the amiloride derivative had an EC50 of 16.55 ± 0.16 μM with a Hill coefficient of 1.37 ± 0.71 (Fig. 3B). In contrast, phenamil failed to elicit a change in peak current amplitude compared to the mutant receptor GABA EC50 control response (Fig. 4A). Furthermore, phenamil failed to change the peak current amplitude of the GABA-A ρ1 (I15'N) mutant receptor response (Fig. 4B). Additionally, benzamil caused an increase in the peak current amplitude at a single concentration in the mutant receptor compared to the GABA control response (Fig. 4C). The 100 μM benzamil concentration was the only concentration that resulted in a significant increased in peak current amplitude when compared to the control (Fig. 4D). Taken in context with the rest of the concentrations studied, there does not appear to be a concentration-dependent relationship for benzamil potentiation of GABA-mediated current in the hGABA-A ρ1 (I15'N) mutant receptor.
Figure 3.
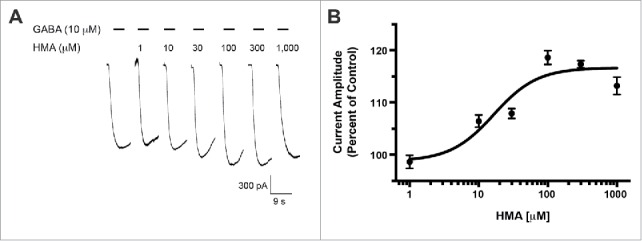
HMA potentiates the hGABA-(A)ρ1 I15'N mutant receptor. (A) Representative responses to increasing concentrations of HMA co-applied with GABA (3 µM) on the hGABA-A ρ1 (I15'N) mutant receptor are shown. (B) Concentration-response profile of HMA potentiation of GABA-mediated current is shown. The resulting HMA EC50 was 16.55 ± 0.16 mM with a Hill coefficient of 1.37 ± 0.71. Data are presented as the mean ± SEM of n ≥ 4 cells.
Figure 4.
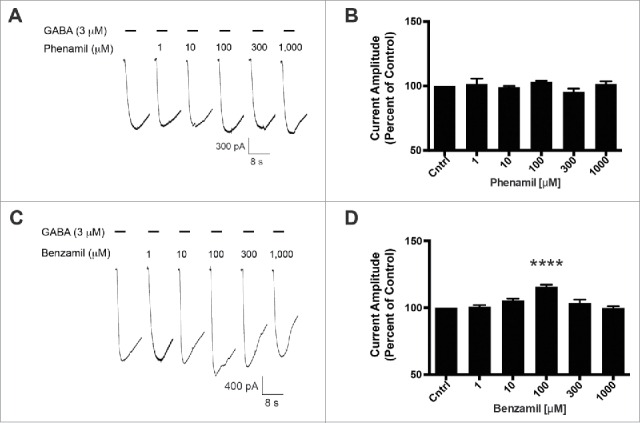
Phenamil and benzamil fail to elicit a concentration-dependent increase in GABA-induced current on the hGABA-A ρ1 (I15'N) mutant receptor. (A) Representative current traces of increasing concentrations of phenamil co-applied with 3 µM GABA on the hGABA-A ρ1 (I15'N) mutant receptor are shown. (B) Comparison of normalized response of co-application of control GABA and phenamil in the presence of the hGABA-A ρ1 I15'N mutant receptor is presented. (C) Representative current traces of increasing concentrations of benzamil co-applied with 3 µM GABA in the presence of the hGABA-A ρ1 I15'N mutant receptor are shown. (D) Comparison of normalized response of co-application of control GABA and increasing concentrations of benzamil. Data are presented as the mean ± SEM of n ≥ 4 cells. Significance was determined using One-way ANOVA and Bonferroni post-hoc analysis (****, p ≤ 0.0001 compared to the control response).
Finally, we screened each of the amiloride derivatives for direct activating activity in the GABA-A ρ1 receptor. An application of 1,000 µM HMA resulted in an observed inward current that was approximately 80% of control peak current amplitude (Figs 5A and 5B). Phenamil and benzamil at 1,000 μM failed to generate a response in cells expressing the hGABA-A ρ1 receptor (Fig 5C). HMA has been shown to modulate ion channels outside of the Cys-loop receptor family. The transfection expression system cells HEK293t may express some of these ion channels. Thus, we cannot rule out that the observed HMA mediated current is generated by a different channel other than the expressed GABA-A ρ1 receptor. Thus, the observed HMA response can be described as “GABA-A like” at this time.
Figure 5.
HMA generates a GABA-(A)like current in transfected HEK293T cells expressing hGABA-(A)ρ1 receptor. (A) Pooled responses of HMA mediating a GABA-A like current in transfected HEK293T cells are shown and scaled for comparison according to their respective controls. (B) A summary of the normalized responses of HMA-mediated GABA-A like current is shown. Data are the mean ± SEM of n ≥ 3 cells. (C) Benzamil and phenamil failed to elicit a response in hGABA-A ρ1 receptor transfected cells.
Discussion
We examined 3 amiloride derivatives to identify the chemical determinants of interaction with the hGABA-A ρ1 receptor. The amiloride derivatives phenamil and benzamil, which had inaccessible guanidine groups, failed to elicit a concentration-dependent potentiation in the GABA-mediated current. HMA, which has an accessible guanidine group, enhanced GABA-mediated current at the highest concentration tested. Our data suggest that an exposed guanidine group in amiloride derivatives is critical for interacting with the hGABA-A ρ1 receptor.
Five-(N, N-Hexamethylene) amiloride (HMA), benzamil, and phenamil have been synthesized utilizing amiloride as a backbone, but pharmacologically have different receptor interactions. These three amiloride derivatives target the Na+/ H+ exchanger,20 epithelial sodium channels, and acid-sensing ion channels. In the case of HMA, the structure contains a hexamethylene group substituted for 2 amino hydrogens, forming an azapane ring.21,22 In contrast, benzamil and phenamil have either a hydrophobic benzene ring or a hydrophobic phenyl group substituted for a hydrogen in the guanidine group, respectively. These amiloride derivatives provided an opportunity to assess the role of the exposed guanidine group for the modulation of the hGABA-A ρ1 receptor.
The amiloride guanidine group is found with a positive charge at physiological pH. Here, the positive charge in HMA may facilitate interaction with the hGABA-A ρ1 receptor. Benzamil and phenamil are less protonated at physiological pH due to the substitution of hydrophobic groups for the guanidine hydrogen. The loss of the positively charged guanidine group may prevent interaction between phenamil and benzamil with the hGABA-A ρ1 receptor, resulting in no definitive effect. Furthermore, HMA showed characteristics for positive allosteric modulation on the hGABA-A ρ1 receptor (Fig. 1). Although we observed a potentiation of the GABA-mediated current, the potentiation was significant only at the higher concentration tested. The HMA azapine ring may account for the apparent reduction in potency compared to amiloride studied previously.14 The increase in size and retention of the exposed guanidine group in HMA provided additional information regarding the scale of a potential amiloride/amiloride-derivative binding site within the hGABA-A ρ1 receptor.
Previously, we observed that amiloride failed to potentiate the GABA-mediated current in the hGABA-A ρ1 (I15'N) mutant receptor.14 This second transmembrane domain site was an attractive target as this protein region has been implicated in the actions of known GABA-A αβγ receptor allosteric modulators.23 We sought to investigate if this site was necessary for HMA potentiation. Upon co-application of GABA with increasing concentrations of HMA, the hGABA-A ρ1 (I15'N) mutant receptor was sensitive to HMA within a 30-1000 μM concentration range (Fig. 3A and 3B). While there was potentiation of current, the observed current was less than that observed for HMA potentiation of the wild type hGABA-A ρ1 receptor (Fig. 1). This reduction in potentiation and shift toward insensitivity suggested that this residue, and the protein domain, plays a role in mediating HMA positive allosteric modulation. Benzamil and phenamil were inactive at the hGABA-A ρ1 (I15'N) mutant receptor. The positive allosteric modulatory activity of HMA in the mutant receptor was unexpected, as the same mutation abolished amiloride activity in a previous study.14 We speculate that the isoleucine to asparagine mutation facilitated HMA interaction, possibly through accommodating the HMA azapine ring within the site. Additionally, the I15'N mutation may alter the activity of the GABA-A ρ1 receptor and modify the amiloride/amiloride-derivative site, where the site is a distinct site separate from the channel's second transmembrane domain 15' site.
In addition to the HMA positive allosteric modulation, we observed a GABA-A like current following application of the amiloride derivative in cells expressing the GABA-A ρ1 receptor (Fig. 5). Previously, we reported that amiloride directly activated the hGABA-A ρ1 receptor.14 Further characterization is needed to provide support that HMA mediated this current by activating the GABA-A ρ1 receptor. If this current was generated through activation of the GABA-A ρ1 receptor, this increase in efficacy could be due to the substitution of an amidogen with hexamethylene on the 5-position of the amiloride pyrazine ring (Fig. 6). One possible explanation could be that amiloride occupies a portion of the proposed amiloride-binding site, which may accommodate larger amiloride-like molecules such as HMA. This accommodation results in direct activation of the receptor with higher efficacy (Fig. 5). Without additional evidence and the location of the amiloride and amiloride derivative binding-site remaining unknown, this explanation remains speculative.
Figure 6.
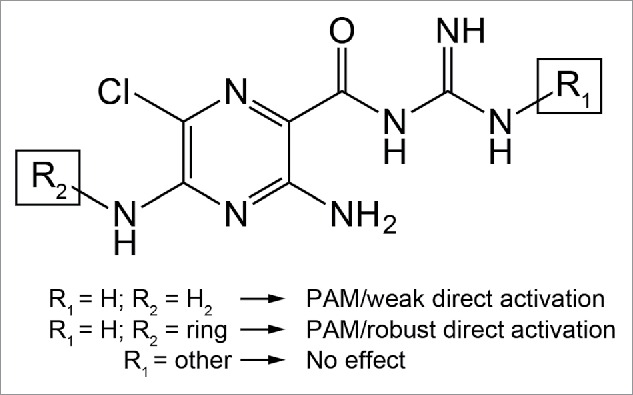
Proposed effects of amiloride derivation on the hGABA-(A)ρ1 receptor. When the guanidine group is accessible and the pyrazine ring remains unchanged, as in the case of amiloride, there is positive allosteric modulatory activity and weak direct activity on the hGABA-A ρ1 receptor. The HMA structure is produced from derivation of the pyrazine ring along with accessibility of the guanidine group. If there is a substitution on the guanidine group, however, the resulting derivative fails to elicit positive allosteric modulatory activity from the receptor, as in the case of benzamil and phenamil.
The ability for guanidine compounds such as HMA, along with the previously studied amiloride,14 suggest that there may be other compounds that may prove to be efficacious at the GABA-A ρ1 receptor. Our work has shown that guanidine compounds that have a planar geometry (amiloride and HMA) could be candidates for screening against the GABA-A ρ1 receptor and the rest of the GABA-A receptor family. Other guanidine compounds have shown to have efficacy in the GABA-A ρ1 receptors, such as guanidino acids.6 However, the guanidino acids appear to compete with GABA at the GABA-A ρ1 agonist-binding site. Older compound libraries that the pharmaceutical industry has generated may contain GABA-A ρ1 receptor modulators. Furthermore, ligands that modulate other ion channels distinct from the Cys-loop receptor family may be modulators for the GABA-A receptor family. For example, the acid-sensing ion channel is sensitive to guanidine compounds, and amiloride is an antagonist for these sodium channels. In 2009, amiloride-derivatives that included modifications on the pyrazine ring were reported to inhibit acid-sensing ion channel subtype 3 (ASIC3).24 The authors described over 50 amiloride derivatives that have an unmodified, exposed guanidine group. Other investigators published a report that identified an allosteric modulatory site within ASIC3 (the nonproton ligand site). In that ASIC3 modulatory site study, 255 guanidine compounds were screened for ASIC3 activity.25 We suspect that many of these compounds have GABA-A receptor efficacy, specifically efficacy at the GABA-A ρ1 receptor. Future studies should focus on screening these compounds for activity in the GABA-A receptor family, with particular interest in the GABA-A ρ1 receptor for positive allosteric modulation. Adding to the current library of ligands to probe for the functional presence of the GABA-A ρ1 receptor will be important for understanding the role of this channel in tissue outside of the retina and within the central nervous system.
Based on the described observations, an exposed guanidine group is critical for the amiloride-derivative positive allosteric modulation of the hGABA-A ρ1 receptor. Furthermore, we speculate that enhanced direct activation by amiloride derivatives may depend on the substitution found on the 5-position of the pyrazine ring. These results reveal a new line of inquiry for the identification and development of hGABA-A ρ1 receptor positive allosteric modulators.
Materials and methods
Cell culture and protein expression
Human embryonic kidney cells (HEK293T) were transfected with pcDNA3.1 vector with either the wild-type hGABA-A ρ1 or hGABA-A ρ1 (I15'N) receptor as described in a previous report.14 The HEK293T cell line was maintained in T25 tissue culture flasks at 37 °C in a 5% CO2 water-jacketed incubator. After reaching 80% confluency, HEK293T cells were subcultured onto glass coverslips 2-4 hours before transfection. Cells were ransfected with pNEGFP-EU (2 μg), hGABA-A ρ1 (2 μg) or hGABA-A ρ1 (I15'N) cDNA (2 µg) as described previously.14 Experiments were performed 18-24 hours following transfection.
Electrophysiology
Whole cell patch-clamp electrophysiology was conducted on an inverted fluorescence microscope with an Axopatch 200B amplifier. Cells were patched with borosilicate glass patch electrodes (resistance of 8-12 MΩ). Amiloride-derivatives were purchased from Sigma-Aldrich. These compounds were phenamil methanesulfonate salt (catalog number P203), benzamil hydrochloride hydrate (catalog number B2417), 5-(N, N-Hexamethylene)amiloride (HMA) (catalog number A9561), and γ-aminobutyric acid (GABA) (catalog number A2129). Stock solutions were made using water (for GABA) or DMSO (for amiloride derivatives) as the solvent. Stock solutions were stored at −20 °C while test solutions were prepared fresh daily from these stock solutions. The extracellular solution consisted of 137 mM NaCl, 5.4 mM KCl, 1.8 mM CaCl2, 1 mM MgCl2, and 5 mM HEPES with pH adjusted to pH 7.4. The internal solution consisted of 120 mM CsCl, 20 mM tetraehtylammonium chloride, 1 mM CaCl2, 2 mM MgCl2, 11 mM EGTA, and 10 mM HEPES with a pH of 7.2. These solutions were described previously.14 Test solution pH was checked and if deviated from pH 7.4, was re-established following dilution of the test compounds. An array of capillary tubes was used to expose patched cells to the test solutions. Cells were voltage-clamped at a −70 mV holding potential. Solution flow was controlled electronically using computer driven PTFE valves via a ValveLink8.2 controller (AutoMate Scientific).
Cells were bathed continuously in external solution. Upon successful establishment of the whole-cell patch-clamp configuration, the test solution was applied for 5 seconds, followed by a 90-second washout period to ensure complete recovery. The approximate GABA EC50for the wild type hGABA-A ρ1 and I15'N mutant receptors are 10 and 3 μM were used as controls, as determined and reported previously.14 Recordings were terminated if consecutive control peak current amplitudes varied by more that 10%. Concentration-response profiles were fit to a dose-response function Graphpad Prism 6.
Data analysis
Peak current amplitude in each whole cell patch-clamp electrophysiological experiment was measured and normalized to the peak current amplitude elicited by the receptor's respective control (the GABA EC50). Data is presented as the mean ± standard error of the mean (SEM) of individual cells. Statistical significance was determined using a One-way ANOVA and Bonferroni post-hoc test in comparison to the control (Graphpad Prism 6).
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
We would like to thank Dr. Nathalie Sumien for her assistance with statistical analysis.
Funding
This work was supported by grants from the Welch Foundation BK-1736, the UNTHSC Rainwater Grant, and the American Heart Association 12BGIA8820001 (to EBG). This work was supported by a UNTHSC Intramural Student Bridge Funding Grant and a NIH training Grant (NIA No. T32AG020494) (to HDS).
References
- [1].Olsen RW, Sieghart W. International Union of Pharmacology. LXX. Subtypes of gamma-aminobutyric acid(A) receptors: classification on the basis of subunit composition, pharmacology, and function. Update. Pharmacol Rev 2008; 60:243-60; PMID:18790874; http://dx.doi.org/ 10.1124/pr.108.00505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Milic M, Divljakovic J, Rallapalli S, van Linn ML, Timic T, Cook JM, Savić MM. The role of alpha1 and alpha5 subunit-containing GABAA receptors in motor impairment induced by benzodiazepines in rats. Behav Pharmacol 2012; 23:191-7; PMID:22327019; http://dx.doi.org/ 10.1097/FBP.0b013e3283512c85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Derry JM, Dunn SM, Davies M. Identification of a residue in the gamma-aminobutyric acid type A receptor α subunit that differentially affects diazepam-sensitive and -insensitive benzodiazepine site binding. J Neurochem 2004; 88:1431-8; PMID:15009644; http://dx.doi.org/ 10.1046/j.1471-4159.2003.02264.x [DOI] [PubMed] [Google Scholar]
- [4].Thompson SA, Whiting PJ, Wafford KA. Barbiturate interactions at the human GABAA receptor: dependence on receptor subunit combination. Br J Pharmacol 1996; 117:521-7; PMID:8821543; http://dx.doi.org/ 10.1111/j.1476-5381.1996.tb15221.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Neu A, Neuhoff H, Trube G, Fehr S, Ullrich K, Roeper J, Isbrandt D. Activation of GABA(A) receptors by guanidinoacetate: a novel pathophysiological mechanism. Neurobiol Dis 2002; 11:298-307; PMID:12505422; http://dx.doi.org/ 10.1006/nbdi.2002.0547 [DOI] [PubMed] [Google Scholar]
- [6].Chebib M, Gavande N, Wong KY, Park A, Premoli I, Mewett KN, Allan RD, Duke RK, Johnston GA, Hanrahan JR. Guanidino acids act as rho1 GABA(C) receptor antagonists. Neurochem Res 2009; 34:1704-11; PMID:19387831; http://dx.doi.org/ 10.1007/s11064-009-9968-x [DOI] [PubMed] [Google Scholar]
- [7].Fisher JL. Amiloride inhibition of gamma-aminobutyric acid(A) receptors depends upon the α subunit subtype. Mol Pharmacol 2002; 61:1322-8; PMID:12021393; http://dx.doi.org/ 10.1124/mol.61.6.1322 [DOI] [PubMed] [Google Scholar]
- [8].Bormann J, Feigenspan A. GABAC receptors. Trends Neurosci 1995; 18:515-9; PMID:8638289; http://dx.doi.org/ 10.1016/0166-2236(95)98370-E [DOI] [PubMed] [Google Scholar]
- [9].Greka A, Koolen JA, Lipton SA, Zhang D. Cloning and characterization of mouse GABA(C) receptor subunits. Neuroreport 1998; 9:229-32; PMID:9507960; http://dx.doi.org/ 10.1097/00001756-199801260-00010 [DOI] [PubMed] [Google Scholar]
- [10].Zhang J, Xue F, Chang Y. Structural determinants for antagonist pharmacology that distinguish the rho1 GABAC receptor from GABAA receptors. Mol Pharmacol 2008; 74:941-51; PMID:18599601; http://dx.doi.org/ 10.1124/mol.108.048710 [DOI] [PubMed] [Google Scholar]
- [11].Denter DG, Heck N, Riedemann T, White R, Kilb W, Luhmann HJ. GABAC receptors are functionally expressed in the intermediate zone and regulate radial migration in the embryonic mouse neocortex. Neuroscience 2010; 167:124-34; PMID:20123002; http://dx.doi.org/ 10.1016/j.neuroscience.2010.01.049 [DOI] [PubMed] [Google Scholar]
- [12].Rosas-Arellano A, Parodi J, Machuca-Parra AI, Sanchez-Gutierrez A, Inestrosa NC, Miledi R, Martínez-Torres A. The GABA(A)rho receptors in hippocampal spontaneous activity and their distribution in hippocampus, amygdala and visual cortex. Neurosci Lett 2011; 500:20-5; PMID:21683123; http://dx.doi.org/ 10.1016/j.neulet.2011.05.235 [DOI] [PubMed] [Google Scholar]
- [13].Blednov YA, Benavidez JM, Black M, Leiter CR, Osterndorff-Kahanek E, Johnson D, Borghese CM, Hanrahan JR, Johnston GA, Chebib M, et al.. GABAA receptors containing rho1 subunits contribute to in vivo effects of ethanol in mice. PloS One 2014; 9:e85525; PMID:24454882; http://dx.doi.org/ 10.1371/journal.pone.0085525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Snell HD, Gonzales EB. Amiloride and GMQ Allosteric Modulation of the GABA-A rho1 Receptor: Influences of the Intersubunit Site. J Pharmacol Exp Ther 2015; 353:551-9; PMID:25829529; http://dx.doi.org/ 10.1124/jpet.115.222802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Cutting GR, Lu L, O'Hara BF, Kasch LM, Montrose-Rafizadeh C, Donovan DM, Shimada S, Antonarakis SE, Guggino WB, Uhl GR, et al.. Cloning of the gamma-aminobutyric acid (GABA) rho 1 cDNA: a GABA receptor subunit highly expressed in the retina. Proc Natl Acad Sci U S A 1991; 88:2673-7; PMID:1849271; http://dx.doi.org/ 10.1073/pnas.88.7.2673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Pistis M, Belelli D, McGurk K, Peters JA, Lambert JJ. Complementary regulation of anaesthetic activation of human (alpha6beta3gamma2L) and Drosophila (RDL) GABA receptors by a single amino acid residue. J Physiol 1999; 515(Pt 1):3-18; PMID:9925873; http://dx.doi.org/ 10.1111/j.1469-7793.1999.003ad.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Loffing J, Kaissling B. Sodium and calcium transport pathways along the mammalian distal nephron: from rabbit to human. Am J Physiol Renal Physiol 2003; 284:F628-43; PMID:12620920; http://dx.doi.org/ 10.1152/ajprenal.00217.2002 [DOI] [PubMed] [Google Scholar]
- [18].Chu XP, Xiong ZG. Physiological and pathological functions of acid-sensing ion channels in the central nervous system. Curr Drug Targets 2012; 13:263-71; PMID:22204324; http://dx.doi.org/ 10.2174/138945012799201685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Miyake T, Nishiwaki A, Yasukawa T, Ugawa S, Shimada S, Ogura Y. Possible implications of acid-sensing ion channels in ischemia-induced retinal injury in rats. Jpn J Ophthalmol 2013; 57:120-5; PMID:23152156; http://dx.doi.org/ 10.1007/s10384-012-0213-9 [DOI] [PubMed] [Google Scholar]
- [20].Masereel B, Pochet L, Laeckmann D. An overview of inhibitors of Na(+)/H(+) exchanger. Eur J Med Chem 2003; 38:547-54; PMID:12832126; http://dx.doi.org/ 10.1016/S0223-5234(03)00100-4 [DOI] [PubMed] [Google Scholar]
- [21].Giansanti V, Santamaria G, Torriglia A, Aredia F, Scovassi AI, Bottiroli G, Croce AC. Fluorescence properties of the Na(+)/H(+)exchanger inhibitor HMA (5-(N,N-hexamethylene)amiloride) are modulated by intracellular pH. Eur J Histochem 2012; 56:e3; PMID:22472891; http://dx.doi.org/ 10.4081/ejh.2012.e3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Darboux I, Lingueglia E, Champigny G, Coscoy S, Barbry P, Lazdunski M. dGNaC1, a gonad-specific amiloride-sensitive Na+ channel. J Biol Chem 1998; 273:9424-9; PMID:9545267; http://dx.doi.org/ 10.1074/jbc.273.16.9424 [DOI] [PubMed] [Google Scholar]
- [23].Saunders PA, Ho IK. Barbiturates and the GABAA receptor complex. Prog Drug Res 1990; 34:261-86; PMID:2173020 [DOI] [PubMed] [Google Scholar]
- [24].Kuduk SD, Di Marco CN, Chang RK, Dipardo RM, Cook SP, Cato MJ, Jovanovska A, Urban MO, Leitl M, Spencer RH, et al.. Amiloride derived inhibitors of acid-sensing ion channel-3 (ASIC3). Bioorg Med Chem Lett 2009; 19:2514-8; PMID:19339181; http://dx.doi.org/ 10.1016/j.bmcl.2009.03.029 [DOI] [PubMed] [Google Scholar]
- [25].Yu Y, Chen Z, Li WG, Cao H, Feng EG, Yu F, Liu H, Jiang H, Xu TL. A nonproton ligand sensor in the acid-sensing ion channel. Neuron 2010; 68:61-72; PMID:20920791; http://dx.doi.org/ 10.1016/j.neuron.2010.09.001 [DOI] [PubMed] [Google Scholar]