

Abstract
Objective
A novel algorithm to identify fetal microdeletion events in maternal plasma has been developed and used in clinical laboratory‐based noninvasive prenatal testing. We used this approach to identify the subchromosomal events 5pdel, 22q11del, 15qdel, 1p36del, 4pdel, 11qdel, and 8qdel in routine testing. We describe the clinical outcomes of those samples identified with these subchromosomal events.
Methods
Blood samples from high‐risk pregnant women submitted for noninvasive prenatal testing were analyzed using low coverage whole genome massively parallel sequencing. Sequencing data were analyzed using a novel algorithm to detect trisomies and microdeletions.
Results
In testing 175 393 samples, 55 subchromosomal deletions were reported. The overall positive predictive value for each subchromosomal aberration ranged from 60% to 100% for cases with diagnostic and clinical follow‐up information. The total false positive rate was 0.0017% for confirmed false positives results; false negative rate and sensitivity were not conclusively determined.
Conclusion
Noninvasive testing can be expanded into the detection of subchromosomal copy number variations, while maintaining overall high test specificity. In the current setting, our results demonstrate high positive predictive values for testing of rare subchromosomal deletions. © 2015 The Authors. Prenatal Diagnosis published by John Wiley & Sons Ltd.
Short abstract
What's already known about this topic?
Circulating cell free DNA is a powerful clinical tool that can detect whole chromosomal aneuploidies in a fetus as early as 10 weeks with high specificity and high sensitivity. Subchromosomal events in the fetus have been shown to be detected via ccfDNA via whole genome sequencing.
What does this study add?
This study shows that ccfDNA whole genome analysis for fetal subchromosomal aneuploidies including clinically significant microdeletions can be tested clinically with high specificity and high positive predictive value (PPV).
Introduction
In recent years, several studies have demonstrated the utility of fetal aneuploidy detection using whole‐genome sequencing of circulating cell‐free DNA (ccfDNA).1, 2, 3, 4, 5 Since the launch of a laboratory developed test in 2011, this technology has been rapidly included in routine clinical management of high‐risk pregnancies.6 More recently, this methodology has been expanded to include subchromosomal events.7, 8, 9
Historically, noninvasive maternal serum screening using biochemical analysis for markers of fetal chromosomal abnormalities has focused on the detection of trisomy 21, which, at a rate of 14.2 per 10 000 live births in the United States, is by far the most common chromosomal abnormality seen in humans.10 The same biochemical markers that are used to screen for trisomy 21 can also detect cases of trisomy 18, albeit with less sensitivity, and have not been as useful in screening for trisomy 13 or sex chromosome aneuploidies. The addition of ultrasound and nuchal translucency measurement to biochemical screening in the first trimester has improved performance and enabled earlier detection of trisomy 21 and trisomy 18 as well as detection of some fetuses with trisomy 13 and monosomy X.
Together, trisomies 13, 18, and 21, along with sex chromosome aneuploidies, make up 82% of karyotypic abnormalities observed in amniocentesis samples.11 While other types of chromosomal abnormalities and subchromosomal events are individually more rare than the common autosomal and sex chromosome aneuploidies, they are collectively common as demonstrated by Wapner et al. in a study of microarray analysis, which indicated that clinically relevant subchromosomal events could occur in as many as 1.6% of pregnancies.12
Noninvasive prenatal detection of these subchromosomal events is important for many reasons. Parents of a child with a microdeletion/microduplication syndrome may not receive a specific diagnosis of the condition for several years into development,13, 14, 15, 16, 17, 18, 19 resulting in the so‐called ‘diagnostic odyssey’. This time period can be financially draining and physically and emotionally distressing to the child and his or her family. Additionally, enrollment into early intervention programs, combined with improvements in medical techniques to treat certain physical conditions associated with these syndromes may lead to long‐term improvements in the health and quality of life of affected individuals.20
The focus of this study was to investigate the clinical outcomes of specific subchromosomal deletions detected by noninvasive ccfDNA testing. Between October 2013 and July 2014, subchromosomal analysis was performed on 123 096 commercial samples for the following microdeletions: 1p36, 5p‐ (Cri‐du‐chat syndrome), 15q‐ (Prader–Willi/Angelman syndrome), and 22q11.2 (DiGeorge syndrome/velocardiofacial syndrome). In August 2014, three additional microdeletions were added: 4p‐ (Wolf–Hirschhorn syndrome), 8q‐ (Langer–Giedion syndrome), and 11q‐ (Jacobsen syndrome). In the following time period between August 2014 and October 2014, an additional 52 297 samples were analyzed for all seven microdeletions. The following is a description of data obtained for the set of 55 cases that received a test report suggestive of the presence of a microdeletion in this cohort.
Methods
This study analyzed extracted ccfDNA fragments from maternal plasma, which were then subject to whole‐genome sequencing and algorithmic analysis for chromosomal aneuploidies and subchromosomal under‐representation in specified regions. Both fetal and maternal fragments were sequenced and mapped to unique regions of the genome. The unique reads were assigned to a 50 kb bin, normalized across the genome, and counted. An under‐ or over‐representation of fragments in the 50 kb bin are indicative of a gain or loss in the genome. For autosomal trisomy analysis, this technique analyzes for over‐representation of DNA along the entire chromosome and is described in Jensen et al. 21 To determine the presence of subchromosomal deletions, this study used a statistical method to search for consistently under‐represented regions followed by a decision tree to differentiate whole‐chromosome events from deletions and is described at length in Zhao et al. 7 The criteria for the identification of positive subchromosomal deletion findings were z‐scoreCBS < −3.95 and an LORCBS >10. Samples meeting these criteria were flagged for additional review and, in some cases, were reanalyzed to confirm positive findings.
Microdeletion results were designated as ‘additional findings’, meaning the test only reports on the presence of an event but it does not provide a negative result when an event is not detected. Referring physicians were alerted if the microdeletion was suspected of being a maternal event based on the degree of under‐representation relative to the fetal fraction. All clinical outcomes where a microdeletion was reported were tracked by board‐certified genetic counselors staffed by the laboratory. Outcomes were designated as ‘confirmed’ if diagnostic testing on specimens obtained via amniocentesis, chorionic villus sampling, or whole blood was performed and the deletion was detected in the pregnant woman, fetus, or both. The most common diagnostic tests performed as follow‐up were chromosomal microarray analysis (CMA) or fluorescence in situ hybridization (FISH). Outcomes that were designated as ‘suspected’ were those in which phenotypic data via prenatal ultrasound or clinical examination after birth were consistent with the phenotypic presentation common to the deletion and the patient declined any further testing. A deletion detected by ccfDNA analysis was designated as falsely positive if diagnostic testing via CMA, karyotype, or FISH was negative for the deletion in either the fetus or the pregnant woman.
Results
Of the 175 393 specimens from high‐risk pregnancies that underwent microdeletion analysis from October 2013 through October 2014, 55 (0.03%) were found to have one of the tested microdeletions. Out of the 55 reported cases, diagnostic testing results and/or clinical phenotypes were available for 53 pregnancies, (96%); two patients (4%) were lost to follow‐up (Table 1). Diagnostic testing via karyotype, CMA, or FISH confirmed the presence of the deletion in the fetus, the pregnant woman, or both in 41/53 (77.4%) cases (Supplementary Table 1). Nine of the 53 cases (17.0%) did not have confirmatory testing by karyotype, CMA, or FISH but did demonstrate clinical features consistent with the deletion as determined by ultrasound or phenotype evaluation (Supplementary Table 2). False positive results were noted in three cases, one case of 1p36 deletion and two cases of 5p deletion (Supplementary Table 3).
Table 1.
Clinical outcomes of noninvasive prenatal testing identified deletions
| Deletion; associated syndrome (n) | Total identified | Confirmed true positive | Suspected due to clinical findings | No additional information | Confirmed false positive | PPV lower–upper estimate (95% confidence) (%) |
|---|---|---|---|---|---|---|
| 22q11.2‐; DiGeorge (175 393) | 32 | 23 | 8 | 1 | 0 | 96.9–100 (82.0–100) |
| 1p36‐ (175 393) | 5 | 3 | 0 | 1 | 1 | 60.0–80.0 (17.0–98.9) |
| 15q‐; Prader–Willi/Angelman (175 393) | 9 | 8 | 1 | 0 | 0 | 100.0 (59.8–100.0) |
| 5p‐; Cri‐du‐chat (175 393) | 6 | 4 | 0 | 0 | 2 | 66.7 (24.1–94.0) |
| 4p‐; Wolf–Hirschhorn (52 297) | 1 | 1 | 0 | 0 | 0 | 100.0 (5.5–100.0) |
| 11q‐; Jacobsen (52 297) | 1 | 1 | 0 | 0 | 0 | 100.0 (5.5–100.0) |
| 8q‐; Langer–Giedion (52 297) | 1 | 0 | 1 | 0 | 0 | 100.0 (5.5 – 100.0) |
Confirmed cases were determined by diagnostic testing (chromosomal microarray analysis, karyotyping, or fluorescence in situ hybridization) of specimens obtained by an invasive procedure in fetal, maternal, or both. Suspected cases were those where diagnostic testing was declined and phenotypic data via clinical presentation in person or by ultrasound were consistent with the deletion. Positive predictive value lower estimate is calculated presuming all patients with no additional information are confirmed falsely positive. Positive predictive value (PPV) upper estimate is calculated presuming all patients with no additional information are confirmed true positives. The 95% confidence level is calculated according to the efficient‐score method and corrected for continuity.
Deletion of 22q11.2 was reported in 32 patient samples; for 20 of those patients, non‐invasive prenatal testing (NIPT) indicated a maternal component (Figure 1). Karyotype, CMA, or FISH confirmed the presence of the deletion in the fetus, the pregnant woman, or both in 23 (71.9%) cases. Eight additional pregnancies were suspected because of ultrasound findings consistent with the 22q11.2 deletion, including tetralogy of Fallot, or women with clinical features of 22q11.2 deletion syndrome but who declined further testing. One patient with a suspected maternal deletion was lost to follow‐up. Twelve samples with a positive result did not indicate a maternal deletion. In nine cases, the fetus was confirmed to have the deletion by CMA or FISH (Supplementary Table 1). Three other fetuses were strongly suspected to be affected based on the presence of complex heart defects and other ultrasound anomalies (Supplementary Table 2). Of the 20 maternally derived deletions, we would expect approximately ten affected fetuses based on autosomal dominant inheritance. We observed five affected fetuses of affected mothers confirmed by either CMA or FISH, and four additional fetuses were strongly suspected to have inherited the deletion from an affected mother based on ultrasound findings or phenotype. In all, nine of 21 (42.8%) affected or presumed affected fetuses inherited the mutation from the mother. Paternal samples were not tested, so it not clear whether the other 12 affected or presumed affected fetuses inherited the deletion from the father or whether those deletions were de novo. Previous studies have suggested that 8–28% of 22q11.2 deletions are inherited.22, 23, 24, 25
Figure 1.
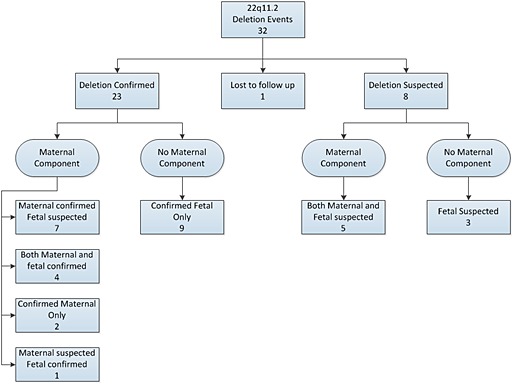
Breakdown of 22q11 deletions. Maternal component cases are indicated when a majority of circulating cell‐free DNA contains the deletion event
The 15q deletion encompassing the Prader–Willi and Angelman syndrome region was identified in nine patients. The deletion was confirmed by birth outcome, CMA, or FISH in four patients. The providers for four additional patients were notified of smaller <1.5 Mb deletions in 15q that did not fully encompass the Prader–Willi and Angelman syndrome critical region but that were clinically relevant as the distal region of this loci is noted to have an association with autism spectrum disorders in patients who have had CMA.26 All four of the smaller <1.5 Mb deletions were maternal and were confirmed by CMA. A suspected 15q deletion pregnancy was complicated by intrauterine growth restriction of the fetus and severe polyhydramnios, which are non‐specific but common findings in pregnancies with Prader–Willi syndrome.27
Five 1p36 deletions were detected in the 175 393 samples included in this cohort. Of these, three were confirmed in the fetus by CMA or FISH, one was lost to follow‐up, and one was a false positive result. One fetus lost to follow‐up was also positive by ccfDNA testing for trisomy 21. Trisomy 21 was confirmed by karyotype, and the pregnancy was terminated without additional analysis for the 1p36 deletion. The false positive result had single nucleotide polymorphism CMA, which showed loss of heterozygosity in the same 4.6 Mb region that appeared in NIPT to be deleted. This could indicate a post‐zygotic rescue event of the deleted region. This patient subsequently had premature rupture of the membranes at 23 weeks' gestation and later delivered a premature infant with features of Goldenhar syndrome.
The 5p deletion related to Cri‐du‐chat syndrome was detected in six pregnancies. The deletion was confirmed by CMA in four of the pregnancies. In two cases, the ccfDNA 5p microdeletion was not confirmed by CMA from amniocentesis.
Deletion analysis of 4p, 8q, and 11q was performed on 52 297 samples in this cohort. One case each of 4p, 11q, and 8q deletions was detected. The one case of 4p deletion, consistent with Wolf–Hirschhorn syndrome, was confirmed by CMA. CcfDNA testing detected a single 8q deletion that was suspected of being maternal in nature. This was confirmed by CMA to be maternal and was not inherited by the fetus. The deletion detected on 11q was small (1.89 Mb) and distal to the Jacobsen region but was part of an 11;15 translocation detected by karyotype and CMA of amniocytes.
Ultrasound finding was the most common indication for testing in those samples found to have a microdeletion, at 48.2% (Figure 2). This is higher than typically seen with autosomal trisomy positive samples and in the general high‐risk NIPT population, 19.2% and 11.9%, respectively. Advanced maternal age was the indication for testing in 32.1% of samples in which a deletion was detected, which is half as frequent as the high‐risk NIPT population with 66.8%. Abnormal serum biochemical screening as an indication for testing accounted for only 5.4% of samples with a microdeletion. Of the five microdeletion samples that had at least one indication of personal or family history on the test request form, all five were categorized as 22q11.2 deletions with a maternal contribution.
Figure 2.
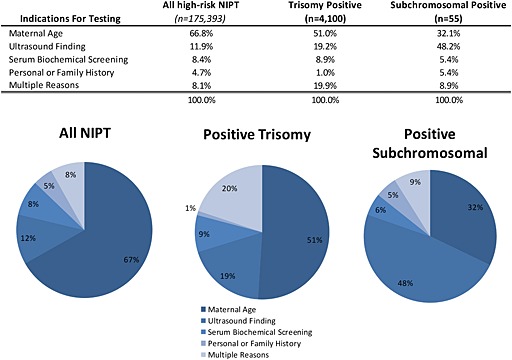
Indications for noninvasive prenatal testing (NIPT) testing by positive result. Multiple indications included patients in which more than one indication for testing was marked. Trisomy positive results include trisomies 21, 18, and 13. Subchromosomal positive results included all patients in which a deletion was detected and reported
Discussion
As expected, the most common microdeletion observed in this large cohort was 22q11.2 because of its estimated prevalence of 1 in 4000 births.22 Surprisingly, 20 of the 32 detected deletions had a maternal contribution. Previously reported reproductive fitness of individuals with 22q11.2 deletion syndrome has been estimated between 8% and 28%.22, 23, 24, 25 At this fitness and a prevalence of 1 out of 4000, we would expect anywhere from 3 to 12 women with a 22q11.2 deletion to be identified in a randomly selected population of 175 393 pregnant women. Instead, 20 pregnant women were identified by ccfDNA to carry the 22q11.2 deletion, which was either confirmed by diagnostic testing or strongly suspected based on clinical presentation, with one lost to follow‐up.
There are several explanations for the higher‐than‐anticipated rates of maternal 22q11.2 deletion in this cohort. Small deletions are more common in these samples, which likely represents women with less severe clinical features. Whole‐genome sequencing of ccfDNA more easily identifies maternal deletions than fetal deletions. Because roughly 90% of the DNA fragments are estimated to be maternal in origin, those undiagnosed women with more mild phenotypes will increase the maternal prevalence rate. Additionally, we cannot exclude the possibility that reproductive fitness has increased for women with 22q11.2 deletion with improvements in healthcare that can more effectively treat congenital heart disease and also with improvements in early intervention programs for persons with intellectual disabilities or learning difficulties.
Although ccfDNA screening is designed to ascertain risk to a fetus, as illustrated previously, maternal deletions are more readily detected than fetal, an observation acknowledged recently in a statement from the International Society of Prenatal Diagnostics.28 In all, 25 of the 55 identified microdeletions had a maternal contribution based on the criteria that a majority of ccfDNA contained the event. Identification of these maternal events is clinically relevant for multiple reasons. Women with a microdeletion have a 50% chance of passing on the deletion to the fetus, which is itself an indication for invasive diagnostic testing and a significant risk factor for future pregnancies. Additionally, diagnosis of a microdeletion in the pregnant woman could be pertinent to the mother's own medical care. Pregnant women with a 22q11.2 deletion can be at increased risk for health complications during pregnancy. Knowing that these patients have the 22q11.2 deletion enables additional monitoring of the woman during her pregnancy by a maternal fetal medicine specialist as well as other appropriate specialists to improve her chances of a healthy pregnancy for herself and delivery for her fetus.
One primary concern with adding subchromosomal conditions to a noninvasive prenatal test has been the potential that it could result in an increase in the test's overall false positive rate, leading to additional diagnostic testing.29 Three of the 55 identified deletions were confirmed to be false positive results, and two patients were lost to follow‐up. Assuming all cases lost to follow‐up were false positive, the false positive rate of this cohort is 0.0029% with a positive predictive value of 90.9% (confidence interval (CI): 79.3–96.6%). Although this marks a slight increase in invasive testing when compared with NIPT without these additional findings, the clinical population tested in this study was all high‐risk and, in the absence of NIPT, would have been recommended to undergo invasive testing. It is important to note that these calculations were performed including all confirmed events.
The classification criteria for subchromosomal deletion screening favored a higher specificity with a lower sensitivity to reduce false positives. Clinical outcomes were largely unavailable for the 175 393 pregnancies in which no deletion was detected; therefore, sensitivity cannot accurately be determined. Although this precludes a formal calculation of sensitivity, one can approximate sensitivities either based on the clinical feedback of reported false negatives, assuming complete reporting, or from the birth prevalence, assuming the theoretical expected value as the true positive value. For clinical feedback, there were three confirmed false negative patients for the eight subchromosomal events. All false negative patients were fetal 22q11.2 deletion patients with an average size of 2.2 Mb. Sensitivity estimates based on this feedback are >99.99% for all events except 22q11.2, which is estimated at 91.2% (CI: 75.2–97.7%). Estimating sensitivity for 22q11.2 based on a 1 in 4000 prevalence results in 44 expected cases with 32 identified in this cohort, yielding a sensitivity of 70.5% (CI: 54.6–82.8%). However, because 20 of those 32 findings were maternal and not all were confirmed in the fetus, an estimated sensitivity range would be 50–70% for an average 22q11 finding of 2.1 Mb. For the more rare conditions in which prevalence is less well known, estimating sensitivity in this manner is of limited value.
One factor that impacts the detection of subchromosomal events in whole‐genome sequencing of ccfDNA is the size of the deletion. The larger the event is in the fetus, the easier it is to detect against a background of normal maternal DNA. The sensitivity of detecting microdeletions with this method is estimated based on deletion size. For deletions that are <7 Mb, the sensitivity of the method at this level of sequencing coverage is estimated to be 60–85%, with increasing sensitivity as fetal fraction increases. For larger deletions, such as those that might be detected on a G‐banded karyotype, >7 Mb, the sensitivity is estimated to be >85% at the level of coverage used in this testing.7
Nearly half of all samples in which a deletion was detected were referred for testing due to abnormal ultrasound findings, double the frequency of those in which an autosomal aneuploidy was detected. This indicates that adding microdeletion screening, especially in cases with abnormal ultrasound findings, can aid clinicians in detection of clinically relevant findings that will guide pregnancy management and care of the neonate after birth. However, while a positive finding may be helpful in directing management, a negative one does not exclude the possibility of a pathogenic chromosomal rearrangement, and invasive testing may still be required to confirm chromosomal normality.
Looking to the future, it is evident that noninvasive screening for subchromosomal events can yield clinically relevant information with reasonably low false positive rates. While this study focused on a subset of a few known recurrent microdeletions, pathogenic subchromosomal events may occur across the genome, as such screening for these events should be a consideration for future noninvasive testing. Wider availability of subchromosomal screening should aid clinicians in recommending confirmatory diagnostic testing for a broader range of clinically significant genomic events, with potential implications for both the fetus and the pregnant woman.
What's Already Known About this Topic?
Circulating cell free DNA is a powerful clinical tool that can detect whole chromosomal aneuploidies in a fetus as early as 10 weeks with high specificity and high sensitivity. Subchromosomal events in the fetus have been shown to be detected via ccfDNA via whole genome sequencing.
What Does this Study Add?
This study shows that ccfDNA whole genome analysis for fetal subchromosomal aneuploidies including clinically significant microdeletions can be tested clinically with high specificity and high positive predictive value (PPV).
Supporting information
Supporting info item
Helgeson, J. , Wardrop, J. , Boomer, T. , Almasri, E. , Paxton, W. B. , Saldivar, J. S. , Dharajiya, N. , Monroe, T. J. , Farkas, D. H. , Grosu, D. S. , and McCullough, R. M. (2015) Clinical outcome of subchromosomal events detected by whole‐genome noninvasive prenatal testing. Prenat Diagn, 35: 999–1004. doi: 10.1002/pd.4640.
Funding sources: None
Conflicts of interest: None declared
References
- 1. Chiu RW, Chan KC, Gao Y, et al Noninvasive prenatal diagnosis of fetal chromosomal aneuploidy by massively parallel genomic sequencing of DNA in maternal plasma. Proc Natl Acad Sci U S A 2008;105:20458–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ehrich M, Deciu C, Zwiefelhofer T, et al. Noninvasive detection of fetal trisomy 21 by sequencing of DNA in maternal blood: A study in a clinical setting. Am J Obstet Gynecol 2011;204:205 e1–11. [DOI] [PubMed] [Google Scholar]
- 3. Fan HC, Blumenfeld YJ, Chitkara U, et al Noninvasive diagnosis of fetal aneuploidy by shotgun sequencing DNA from maternal blood. Proc Natl Acad Sci U S A 2008;105:16266–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Palomaki GE, Deciu C, Kloza EM, et al. DNA sequencing of maternal plasma reliably identifies trisomy 18 and trisomy 13 as well as Down syndrome: An international collaborative study. Genet Med 2012;14:296–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Palomaki GE, Kloza EM, Lambert‐Messerlian GM, et al DNA sequencing of maternal plasma to detect Down syndrome: An international clinical validation study. Genet Med 2011;13:913–20. [DOI] [PubMed] [Google Scholar]
- 6. McCullough RM, Almasri EA, Guan X, et al. Non‐invasive prenatal chromosomal aneuploidy testing ‐ clinical experience: 100,000 clinical samples. PLoS One 2014;1–9(10):e109173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Zhao C, Tynan J, Ehrich M, et al. Detection of fetal subchromosomal abnormalities by sequencing circulating cell‐free DNA from maternal plasma. Clin Chem 2015;61:608–16. [DOI] [PubMed] [Google Scholar]
- 8. Bayindir B, Dehaspe L, Brison N, et al. Noninvasive prenatal testing using a novel analysis pipeline to screen for all autosomal fetal aneuploidies improves pregnancy management. Eur J Hum Genet 2015. DOI:10.1038/ejhg.2014.282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Snyder MW, Simmons LE, Kitzman JO, et al. Copy-Number Variation and False Positive Prenatal Aneuploidy Screening Results. N Engl J Med 2015;372:1639–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Mai CT, Kucik JE, Isenburg J, et al. Selected birth defects data from population‐based birth defects surveillance programs in the United States, 2006 to 2010: Featuring trisomy conditions. Birth Defects Res A Clin Mol Teratol 2013;97:709–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ferguson‐Smith MA, Yates JR. Maternal age specific rates for chromosome aberrations and factors influencing them: Report of a collaborative European study on 52,965 amniocenteses. Prenat Diagn 1984;4:5–54. [DOI] [PubMed] [Google Scholar]
- 12. Wapner RJ, Martin CL, Levy B, et al Chromosomal microarray versus karyotyping for prenatal diagnosis. N Engl J Med 2012;367:2175–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Oskarsdottir S. Presenting phenotype in 100 children with the 22q11 deletion syndrome. Eur J Pediatr 2005;3:146–53. [DOI] [PubMed] [Google Scholar]
- 14. Mainardi PC. The natural history of Cri du Chat syndrome. A report from the Italian Register. Eur J Human Genet 2006;49:363–83. [DOI] [PubMed] [Google Scholar]
- 15. Torrado M. Clinical‐etiologic correlation in children with Prader–Willi syndrome (PWS): An interdisciplinary study. Am J Med Genet A 2007; 143:460–8. [DOI] [PubMed] [Google Scholar]
- 16. Mertz LG. Angleman syndrome In Denmark. Birth incidence, genetic findings, and age at diagnosis. Am J Med Genet 2013;9:2197–203. [DOI] [PubMed] [Google Scholar]
- 17. Battaglia A. Further delineation of deletion 1p36 syndrome in 60 patients: A recognizable phenotype and common cause of developmental delay and mental retardation. Pediatrics 2008;121: 404–10. [DOI] [PubMed] [Google Scholar]
- 18. Blanco‐Lago R. Wolf–Hirschhorn syndrome. A series of 27 patients: Their epidemiological and clinical characteristics. The current situation of the patients and the opinions of their caregivers regarding the diagnostic process. Rev Neurol 2013;57:49–56. [PubMed] [Google Scholar]
- 19. Slavotinek A, Shaffer LG, Shapira SK. Monosomy 1p36. Amer J Med Genet 1999;36:657–63. [PMC free article] [PubMed] [Google Scholar]
- 20. Cheung ENM, George SR, Andrade DM, et al. Neonatal hypocalcemia, neonatal seizures, and intellectual disability in 22q11.2 deletion syndrome. Genet Med 2014;16:40–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Jensen TJ, Zwiefelhofer T, Tim RC, et al. High‐throughput massively parallel sequencing for fetal aneuploidy detection from maternal plasma. PLoS One 2013;1–8(3):e57381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Wilson DI, Cross IE, Wren C, et al. Minimum prevalence of chromosome 22q11 deletions. Am J Hum Genet 1994;55:A169. [Google Scholar]
- 23. Digilio MC, Marino B, Giannotti A, Dallapiccola B. Familial deletions of chromosome 22q11. Am J Med Genet 1997;73:95–6. [PubMed] [Google Scholar]
- 24. Leana‐Cox J, Pangkanon S, Eanet KR, et al. Familial DiGeorge/velocardiofacial syndrome with deletions of chromosome area 22q11.2: Report of five families with a review of the literature. Am J Med Genet 1996;65:309–16. [DOI] [PubMed] [Google Scholar]
- 25. Thompson PW, Davies SJ. Frequency of inherited deletions of 22q11. J Med Genet 1998;35:789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kim SJ, Miller JL, Kuipers PJ, et al Unique and atypical deletions in Prader–Willi syndrome reveal distinct phenotypes. Eur J Hum Genet 2012 Mar;20(3):283–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Gross N, Rabinowitz R, Gross‐Tsur V, et al. Prader–Willi syndrome can be diagnosed prenatally. Amer J Med Genet 2014;167:80–5. [DOI] [PubMed] [Google Scholar]
- 28. Benn P, Borrell A, Chiu R, et al Position statement from the Chromosome Abnormality Screening Committee on behalf of the Board of the International Society for Prenatal Diagnosis. Prenat Diagn 2015;35(8):724–34, DOI:10.1002/pd.4608. [DOI] [PubMed] [Google Scholar]
- 29. Dondorp W, Wert G, Bombard Y, et al Non‐invasive prenatal testing for aneuploidy and beyond: Challenges of responsible innovation in prenatal screening. Eur J Hum Genet 2015. DOI:10.1038/ejhg.2015.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting info item