

Summary
Recent studies demonstrate robust molecular cross talk and signaling between hydrogen sulfide (H2S) and nitric oxide (NO). Heart failure (HF) patients are deficient in both H2S and NO, two molecules that are critical for cardiovascular homeostasis. A phase I clinical trial of a novel H2S prodrug (SG1002) was designed to assess safety and changes in H2S and NO bioavailability in healthy and HF subjects. Healthy subjects (n = 7) and heart failure patients (n = 8) received oral SG1002 treatment in escalating dosages of 200, 400, and 800 mg twice daily for 7 days for each dose. Safety and tolerability were assessed by physical examination, vital signs, and ECG analysis. Plasma samples were collected during a 24‐h period each week for H2S and NO analysis. BNP and glutathione levels were analyzed as markers of cardiac health and redox status. Administration of SG1002 resulted in increased H2S levels in healthy subjects. We also observed increased H2S levels in HF subjects following 400 mg SG1002. Nitrite, a metabolite of NO, was increased in both healthy and HF patients receiving 400 mg and 800 mg SG1002. HF subjects treated with SG1002 displayed stable drug levels over the course of the trial. SG1002 was safe and well tolerated at all doses in both healthy and HF subjects. These data suggest that SG1002 increases blood H2S levels and circulating NO bioavailability. The finding that SG1002 attenuates increases in BNP in HF patients suggests that this novel agent warrants further study in a larger clinical study.
Keywords: Nitrite, Oxidative stress, Phase 1 clinical trial, Sulfide
Introduction
In the United States, HF has become the most common discharge diagnosis in patients over 65 years of age 1, 2. Treatments remain inadequate and heart transplant options are severely limited. Therefore, pharmacotherapies designed to coincide with standard means of care are needed to prevent cardiac remodeling and attenuate the extensive cardiac injury and dysfunction associated with late‐stage heart failure (HF). Our laboratory and others have shown that subjects with congestive heart failure (CHF) have a deficit in circulating hydrogen sulfide (H2S) levels, and that sulfide levels are inversely correlated with severity of CHF 3, 4, 5.
In mammals, hydrogen sulfide is enzymatically generated by cystathionine β‐synthase (CBS), cystathionine γ‐lyase (CSE) or 3‐mercaptopyruvate sulfur‐transferase (3‐MST) 6. H2S and its metabolites are found throughout the body, including the heart, liver, kidney, brain, nervous system, lung, airway tissues, gastrointestinal tract, reproductive organs, skeletal muscle, pancreas, synovial joints, connective tissue, cochlea, and adipose tissues 7. Dysfunction of CSE, resulting in impaired H2S generation, has been cited as a significant contributor to pathology in numerous disease states 8, 9, 10. Furthermore, H2S plays an important role in the normal physiology of the cardiovascular system with important actions on both the heart and the circulation 6, 11. In preclinical models, H2S therapy decreases disease severity by numerous mechanisms, including antioxidant activity, promoting angiogenesis, modulating mitochondrial function, reducing inflammation, upregulating antioxidant gene programs, inhibiting cell death, and attenuating fibrosis 6, 12, 13, 14.
A second endogenous gaseous messenger, nitric oxide (NO), is produced throughout the body and is critical for cardiovascular homeostasis 15, 16. Our laboratory and others have previously shown that NO is highly cytoprotective and that maintaining NO bioavailability protects against the development and progression of HF 3, 17, 18. Similarly to H2S, NO levels are diminished in HF patients compared with healthy controls 3. Recently, H2S has been shown to elevate circulating levels of NO by increasing the activity of endothelial nitric oxide synthase (eNOS) 9, 12. In addition, a genetic deficiency of CSE resulting in H2S deficit is associated with eNOS dysfunction and perturbed downstream NO‐dependent signaling, but is corrected with administration of an H2S donor 9.
In preclinical studies, the novel long‐acting H2S prodrug, SG1002, attenuates left ventricular (LV) remodeling and dysfunction in a pressure‐overload model of HF 12. Administration of SG1002 significantly increased both H2S and NO levels and decreased numerous indices of disease 12. Given the dramatic cardioprotective actions of SG1002 in preclinical studies, here we examined the safety of this pluripotent compound in healthy and CHF patients in a placebo‐controlled dose‐escalation phase I clinical trial. The purpose of this trial was to evaluate initial safety and maximum tolerated oral doses of SG1002. SG1002 significantly augments circulating H2S and plasma nitrite levels in healthy subjects and HF patients. SG1002 was also well tolerated and safe at all doses tested.
Methods
Inclusion/Exclusion Criteria for Healthy Subjects
Inclusion: Healthy male volunteers between 25 and 34 years of age with a body mass index (BMI) between 19 and 30 kg/m2, and no clinically significant findings in the medical history and physical examination; no clinically significant laboratory values and urinalysis; normal electrocardiogram (ECG), blood pressure and heart rate; willing to use contraception (single barrier methods); and willing and able to provide written consent. Exclusion: subjects could not meet any of the following exclusion criteria: have received blood products within 1 month prior to screening; have received any investigational research agent within 30 days or 5 half‐lives (whichever is longer) prior to the first dose of trial drug; have a history of thyroidectomy or thyroid disease that required medication within the past 12 months; have had serious angioedema episodes within the previous 3 years; have HIV, hepatitis B or C positive; have a history of or current clinically significant GI, hepatic, renal, cardiovascular, respiratory, endocrine, oncological, immunodeficiency, neurological, metabolic, hematological, or autoimmune disorder; have a history of or current tuberculosis, epilepsy, diabetes, or glaucoma; hypersensitivity to sulfur compounds, unable to provide repeated blood samples without undue trauma or distress; or anticipate surgery within the trial period.
Inclusion/Exclusion Criteria for Congestive Heart Failure Subjects
Inclusion: Aged between 40 and 71 years; have symptomatic HF, with New York Heart Association (NYHA) classification of II or III; ambulatory; left ventricular ejection fraction of less than 40%; normal hemoglobin screening; and CHF has been stable for the previous 3 months. Exclusion: subjects could not meet any of the following exclusion criteria: pregnant or breastfeeding; myocardial infarction, unstable angina, stroke, cerebrovascular accident, percutaneous coronary intervention, open heart surgery or transient ischemic attack within 3 months of screening; hypotension; poorly controlled hypertension; serious liver disease; life expectancy less than 6 months; evidence of drug or alcohol abuse; and HIV, or hepatitis B or C positive. SG1002‐treated HF subjects had the following comorbidities: type two diabetes (33.3%), dyslipidemia (50%), hypertension (33.3%), ischemic heart disease (16.7%), cardiomyopathy (66.7%), atrial fibrillation (16.7%), implantable defibrillator (100%), acute myocardial infarction (16.7%), obesity (50%). Placebo‐treated HF subjects had the following comorbidities: type two diabetes (50%), dyslipidemia (100%), hypertension (50%), nonischemic heart disease (50%), atrial fibrillation (50%), implantable defibrillator (100%), and acute myocardial infarction (50%).
Study Procedures
Eight healthy subjects were randomized 1:3 (placebo:active groups). One subject was dropped from the study following the first dose due to recreational drug use. The seven remaining subjects were randomly assigned to receive either placebo (n = 2) or SG1002 (n = 5). The study protocol is depicted in Figure 1. Initially, subjects received 200 mg oral capsules SG1002 or placebo twice daily (BID) for 7 days (visit 1, days 0–6); then increased to 400 mg SG1002 or placebo BID for 7 days (visit 2, days 7–13); then increased to 800 mg SG1002 or placebo BID for 7 days (visit 3, days 14–21). In an identical dose‐escalation format, CHF patients (n = 8) received either placebo (n = 2) or SG1002 (n = 6). CHF patients received 200 mg oral capsules SG1002 or placebo BID for 7 days (visit 1, days 0–6); then increased to 400 mg SG1002 or placebo BID for 7 days (visit 2, days 7–13); then increased to 800 mg SG1002 or placebo BID for 7 days (visit 3, days 14–21).
Figure 1.
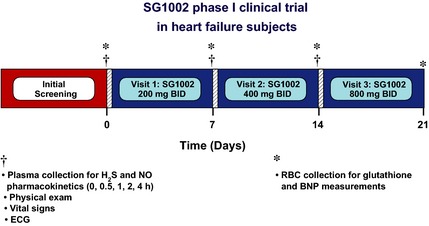
SG1002 phase I clinical trial in heart failure patients. Subjects received 200 mg oral capsules SG1002 or placebo twice daily (BID) for 7 days (visit 1, days 0–6); then increased to 400 mg SG1002 or placebo BID for 7 days (visit 2, days 7–13); then increased to 800 mg SG1002 or placebo BID for 7 days (visit 3, days 14–21). Blood was collected for pharmacokinetic analysis at 0, 0.5, 1, 2, and 4 h immediately following the administration of SG1002 or placebo at visits 1 and 2 and at 0, 0.5, 1, 2, 4, 6, 12, and 24 h during visit 3. Samples for the analysis of brain natriuretic peptide (BNP) and oxidative stress (glutathione) were taken on days 0, 7, 14, and 21 prior to the administration of SG1002. Parameters for safety, including physical examination, vital sign measurements, and electrocardiogram (ECG), were measured at baseline, 1, 2 and 3 weeks time points.
Safety Assessment
Parameters for safety of SG1002 included: physical examination, vital sign measurements, and electrocardiogram (ECG). Adverse event (AE) causality assessment was labeled: “definite” (>95% likely the trial drug caused the event), “highly likely” (75–95%), “probably” (50–74%), “possible” (25–49%), “unlikely” (<25%), and “unknown” if the case in not informative enough to assign the event to any of the above categories. AE severity classification was considered “mild” if there was an awareness of sign or symptom but easily tolerated and does not alter normal activity, “moderate” if the sign or symptom causes discomfort or interference with unusual activity, or “severe” if the sign or symptom causes significant impairment of function or incapacitation, or inability to perform normal activities.
Blood Sample Collection
Blood was collected for pharmacokinetic analysis at 0, 0.5, 1, 2, and 4 h immediately following the administration of SG1002 or placebo at visits 1 and 2 and at 0, 0.5, 1, 2, 4, 6, 12, and 24 h during visit 3. Samples for the analysis of brain natriuretic peptide (BNP) and oxidative stress (glutathione) were taken on days 0, 7, 14, and 21 prior to the administration of SG1002.
Measurement of Hydrogen Sulfide
Plasma free H2S levels were measured using gas chromatography chemiluminescence (7890 Series GC, G660XA Series Chemiluminescence Detector; Agilent, Santa Clara, CA, USA). Free H2S in plasma was liberated by incubating in 1M sodium citrate solution at 37°C for 10 min. The resultant headspace gases were analyzed using the GC system. For the measurement of H2S released from sulfane sulfur, 0.1 mL of sample homogenate and 0.1 mL of 15 mM DTT in 0.1 mM Tris/HCl, pH 9.0, were placed in a in a small glass vial, sealed, and incubated at 37°C for 50 min. After the incubation, 0.4 mL of 1 M sodium citrate buffer was injected through the rubber stopper and the mixture was incubated at 37°C for 10 min with shaking at 125 rpm on a rotary shaker to facilitate the release of H2S gas from the aqueous phase. After shaking, head‐space gas was injected into a gas chromatograph for quantification.
Measurement of Plasma Nitrite (NO2 −)
Plasma nitrite concentrations were quantified by an automated ion chromatography system (ENO30 Analyzer; Eicom San Diego, CA, USA). Nitrite was separated by a column (NO‐PAK with polystyrene polymer; Eicom). The mobile phase, delivered at a pump rate of 0.33 mL/min, was 10% methanol containing 0.15 mol/L NaCl‐NH4Cl and 0.5 g/L of tetrasodium EDTA. The Griess reagent, which was 1.25% HCl containing 5 g/L sulfanilamide with 0.25 g/L N‐naphthylethylenediamine, was delivered at a rate of 0.1 mL/min.
Glutathione Measurements
GSH and GSSG levels in patient red blood cells (RBCs) were determined using high‐performance liquid chromatography (HPLC). An equal volume of 10% TCA was added to the RBCs, and the samples were then vortexed and allowed to incubate overnight at 4°C. Following incubation, the samples were centrifuged at 10,000 rpm for 2 min at 4°C to pellet the precipitated proteins. The supernatant was removed and 80 mM iodoacetic acid (10% v/v) was added. The pH was adjusted to ~7.5–8 using 1 M potassium carbonate and allowed to incubate at room temperature for 1 h. Samples were then derivatized with 1‐fluoro‐2,4‐dinitrobenzene (DNFB), pH adjusted to 7 with 1M potassium carbonate and incubated at 4°C overnight in the dark. Samples were then centrifuged at 10,000 rpm at 4°C for 5 min and filtered through a 0.45‐micron syringe filter. Separation of GSH and GSSG derivatives was performed on a 250 mm × 4.6 mm Lichrosorb NH2 10 micron anion exchange column. GSH and GSSG were quantified by comparing standards derivatized in the same manner. RBC pellets were analyzed using Drabkin's assay to give final concentrations of GSH and GSSG in nmol/mg hemoglobin.
Statistical Analysis
Data analysis was performed using Prism 6 software. Statistical significance was evaluated using a 1‐way ANOVA with a Bonferroni multiple comparison correction test. All statistical comparisons indicated are to baseline (pre‐treatment) values. A P‐value <0.05 was considered statistically significant. All data are expressed as mean ± SEM unless otherwise noted. SEM = standard error of the mean, SD = standard deviation.
Results
Demographics
All normal subjects were men, between the ages of 25 and 34, with an average age of 27.5 years and an average BMI of 23.6 (Table 1). One subject's (SG1002 treated group) participation in the study was terminated after visit 1 due to evidence of drug or alcohol abuse (Exclusion criteria #15). All other subjects completed the trial. HF subjects were Caucasian, between the ages of 40 and 71, with an average age of 57.7 years and an average BMI of 31.3. Six of the eight subjects were men, with both women randomized to the SG1002 treatment group (Table 1). Adherence was defined as taking at least 80% of the required study doses and was assessed at each visit. All healthy subjects and HF subjects adhered to investigational product usage throughout the study. Subject 05 (healthy subject) missed two capsules of placebo. Subject 09 (with HF) missed four capsules of placebo. Subject 11 (with CHF) missed three capsules of SG1002 (one 800 mg dose). All other subjects were 100% adherent to investigational product. Concomitant medications reported by healthy subjects included ascorbic acid, fish oil, ibuprofen, paracetamol and codeine. Concomitant medications reported by subjects with HF are listed in Table 2.
Table 1.
Participant demographics
| Demographic | Healthy subjects | Heart failure subjects | ||
|---|---|---|---|---|
| Placebo | SG1002 | Placebo | SG1002 | |
| n = 2 | n = 6 | n = 2 | n = 6 | |
| Age, years, mean (SD) | 27.0 (0.0) | 27.7 (3.4) | 56.0 (12.7) | 58.2 (14.2) |
| Sex, n (%) | ||||
| Male | 2 | 6 | 2 | 4 |
| Female | 0 | 0 | 0 | 2 |
| Race | ||||
| Caucasian | 2 | 5 | 2 | 6 |
| South Asian | 0 | 1 | ||
| Height, cm, mean (SD) | 179.0 (7.1) | 177.7 (3.8) | 171.5 (6.4) | 172.5 (7.3) |
| Weight, kg, mean (SD) | 73.0 (7.1) | 75.5 (5.7) | 99.0 (25.5) | 90.2 (22.2) |
| BMI, kg/m2, mean (SD) | 22.8 (0.4) | 23.9 (1.9) | 33.4 (6.2) | 30.6 (8.6) |
Table 2.
Concomitant medication use (Heart failure subjects)
| Subject # | Concomitant medication (preferred name) | Treatment |
|---|---|---|
| 9 | Panadeine co, fenofibrate, omeprazole, folic acid, metformin, clopidogrel bisulfate, mirtazapine, allopurinol, hydrochlorothiazide, potassium chloride, furosemide, spironolactone, carvedilol, methyldopa, warfarin, amlodipine besilate, amino acids with hydroxocobalamin, rosuvastatin, ramipril, hydralazine, ivabradine | Placebo |
| 10 | Acetylsalicylic acid, furosemide, simvastatin, gliclazide, isosorbide mononitrate, enalapril maleate, bisoprolol fumarate, warfarin, digoxin, potassium chloride, paracetamol, bumetanide, escitalopram oxalate | SG1002 |
| 11 | Warfarin, spironolactone, amiodarone, gemfibrozil, perindopril erbumine, carvedilol, tiotropium bromide, budesonide with formoterol fumarate | SG1002 |
| 12 | Salbutamol, spironolactone, pantoprazole, fluticasone propionate with salmeterol, desvenlafaxine, panadeine co, acetylsalicyclic acid, insulin aspart, fosinopril sodium, insulin glargine, furosemide, trimethoprim, rosuvastatin, colchicine, potassium chloride, allopurinol, vitamin D, | SG1002 |
| 13 | Carvedilol, spironolactone, levothyroxine sodium, vitamin B, perindopril, ivabradine hydrochloride | SG1002 |
| 14 | Glucosamine, acetylsalicylic acid, magnesium sulfate, fish oil, furosemide, ascorbic acid, metoprolol tartrate, spironolactone, rosuvastatin calcium, perindopril erbumine, ubidecarenone | SG1002 |
| 15 | Oxazepam, perindopril erbumine, spironolactone, furosemide, bisoprolol fumarate | SG1002 |
| 16 | Panadeine co, metformin, acetylsalicylic acid, esomeprazole sodium, bisoprolol, perindopril, rosuvastatin | Placebo |
Safety Data
There were a total of 10 AEs recorded during the study of healthy subjects (Table 3). All events resolved and no changes in study product dosing were required. There were a total of three AEs recorded during the study of subjects with HF, including two AEs that occurred prior to the first dose of investigational product. The pretreatment adverse events were moderate hyperkalemia (resolved) and mild upper respiratory tract infection (ongoing). The treatment emergent adverse event, mild bruising at the site of blood draw, occurred after the administration of the 200 mg dose and was considered unrelated. Mild gastrointestinal (GI) issues such as flatulence and nausea were observed in SG1002‐treated patients. However, the vast majority of preclinical studies show that H2S is GI protective. For example, H2S has been reported to enhance ulcer healing, reduce inflammation, and promotes colitis healing 19, 20, 21, 22. Additionally, all GI incidences were categorized as “mild” and not all were definitely caused by SG1002.
Table 3.
Treatment emergent adverse events (Healthy subjects)
| Adverse events | Placebo | SG1002 | Dose | Severitya | Causalityb |
|---|---|---|---|---|---|
| n = 2 | n = 6 | (bid) | |||
| Total AEs, n | 4 | 6 | |||
| Subjects with ≥1 AE, n (%) | 2 (100) | 3 (50.0) | |||
| Gastrointestinal disorders, n (%) | 3 (50.0) | ||||
| Diarrhea | 1 (16.7) | 200 mg | Mild | Possible | |
| Flatulence | 1 (16.7) | 800 mg | Mild | Probable | |
| Nausea | 1 (16.7) | 800 mg | Mild | Definite | |
| Vomiting | 1 (16.7) | 800 mg | Mild | Definite | |
| Infections and infestations, n (%) | 2 (100) | ||||
| URTI | 2 (100) | 800 mg pbo | Mild | Unlikely | |
| 200 mg pbo | Moderate | Unlikely | |||
| Nervous system disorders, n (%) | 1 (50.0) | 1 (16.7) | |||
| Headache | 1 (50.0) | 400 mg pbo | Moderate | Possible | |
| Lethargy | 1 (16.7) | 200 mg | Mild | Unlikely | |
| Syncope | 1 (16.7) | 200 mg | Mild | Unlikely | |
| Psychiatric disorders, n (%) | 1 (50.0) | ||||
| Libido increased | 1 (50.0) | 400 mg pbo | Mild | Possible |
Pbo: placebo.
bid: twice daily.
URTI: upper respiratory tract infection.
Severity as assessed by the investigator.
Causality is the investigator assessment of the likelihood that the investigational product caused the adverse event.
There were no notable changes in clinical chemistry test results in healthy subjects or subjects with HF (Table 4) nor were there notable changes in vital signs, including blood pressure or pulse (Table 5). The ECG results for healthy subjects were all reported as normal, and for subjects with HF, there were no new clinically significant abnormalities detected.
Table 4.
Liver and renal chemistry data in healthy and heart failure subjects following 200 mg (Visit 1), 400 mg (Visit 2), and 800 mg (Visit 3) SG1002
| ALT (U/L) | AST (U/L) | Creatinine (umol/L) | Urea (mmol/L) | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| BL | Visit 1 | Visit 2 | Visit 3 | BL | Visit 1 | Visit 2 | Visit 3 | BL | Visit 1 | Visit 2 | Visit 3 | BL | Visit 1 | Visit 2 | Visit 3 | |
| Healthy | ||||||||||||||||
| SG1002, Mean (SEM) | 22.8 (5.1) | 20.2 (6.1) | 18.6 (3.9) | 17.8 (3.2) | 20.2 (3.0) | 17.6 (3.3) | 20.4 (2.9) | 17.2 (2.1) | 78.2 (3.7) | 73.4 (3.6) | 74.6 (3.6) | 78.2 (4.3) | 4.56 (0.7) | 4.3 (0.4) | 4.92 (0.7) | 5.24 (0.6) |
| Placebo, Mean (SEM) | 17 (2.0) | 15.5 (0.5) | 21 (4.0) | 18 (3.0) | 22 (4.0) | 19.5 (1.5) | 34 (8.0) | 27.5 (4.5) | 77.5 (9.5) | 73 (11.0) | 84 (11.0) | 78.5 (11.5) | 3.75 (0.8) | 4.6 (0.2) | 5.75 (0.5) | 4.7 (1.0) |
| CHF | ||||||||||||||||
| SG1002, Mean (SEM) | 40.3 (21.9) | 35 (17.4) | 29.2 (10.8) | 37.7 (20.5) | 31 (10.1) | 30 (11.7) | 28.5 (9.2) | 35.8 (16.3) | 94.5 (19.7) | 88.2 (14.4) | 86.7 (15.8) | 88.3 (11.5) | 7.9 (1.6) | 7.6 (1.2) | 8.5 (1.6) | 8.4 (1.3) |
| Placebo, Mean (SEM) | 37.5 (11.5) | 25 (5.0) | 21 (1.0) | 29.5 (8.5) | 26.5 (9.5) | 22 (3.0) | 19.5 (0.5) | 25 (5.0) | 78 (2.0) | 72.5 (2.5) | 69.5 (0.5) | 83.5 (5.5) | 6.65 (0.2) | 6.15 (0.9) | 4.6 (0.1) | 5.45 (0.3) |
Table 5.
Heart rate and blood pressure data from healthy and HF subjects
| Day 0 | Day 7 | Day 14 | Day 21 | |
|---|---|---|---|---|
| Pulse rate (beats per minute) | ||||
| Placebo (Healthy) | 44.0 (1.4) | 56.5 (10.6) | 57.0 (5.7) | 54.0 (2.8) |
| SG1002 (Healthy) | 63.2 (12.3) | 62.8 (8.7) | 62.2 (9.0) | 59.7 (11.7) |
| Placebo (HF) | 75.0 (22.6) | 75.0 (5.7) | 76.5 (2.1) | 80.0 (26.9) |
| SG1002 (HF) | 75.2 (8.6) | 68.0 (5.9) | 72.0 5.9) | 73.2 (8.4) |
| Systolic blood pressure (mmHg) | ||||
| Placebo (Healthy) | 116.5 (0.7) | 125.5 (4.9) | 119.0 (0.0) | 119.0 (5.7) |
| SG1002 (Healthy) | 123.0 (3.8) | 116.8 (4.8) | 121.2 (3.5) | 122.3 (12.9) |
| Placebo (HF) | 116.5 (0.7) | 118.5 (0.7) | 116.5 (1.4) | 130.0 (0.0) |
| SG1002 (HF) | 114.0 (8.8) | 111.7 (16.4) | 108.5 (5.3) | 112.2 (8.8) |
| Diastolic blood pressure (mmHg) | ||||
| Placebo (Healthy) | 63.0 (11.3) | 79.5 (0.7) | 77.5 (2.1) | 72.5 (2.1) |
| SG1002 (Healthy) | 75.8 (5.9) | 69.8 (5.9) | 78.2 (6.3) | 75.5 (7.7) |
| Placebo (HF) | 76.0 (12.8) | 78.5 (0.7) | 81.5 (8.5) | 90.5 (9.2) |
| SG1002 (HF) | 68.0 (10.5) | 73.3 (16.7) | 67.5 (7.4) | 70.3 (6.3) |
SG1002 Elevated H2S Levels in Healthy and Heart Failure Subjects
Compared to baseline (pretreatment values), peak free‐H2S levels from each patient (between 0.5 and 4 h following SG1002 treatment) were increased at the 400 mg (P < 0.01) and 800 mg (P < 0.01) doses in healthy subjects (Figure 2A). In HF subjects, a significant increase in free‐H2S was observed following the 400 mg dose (P < 0.05), compared to baseline values (Figure 2B). No significant elevations in peak sulfane sulfur levels in healthy or HF subjects at any dose (Figure 2C,D). Pharmacokinetic curves following SG1002 treatment display a sustained elevation in sulfide levels in healthy patients (Figures 3 and 4). Placebo H2S and sulfane sulfur pharmacokinetics are depicted in Table S1.
Figure 2.
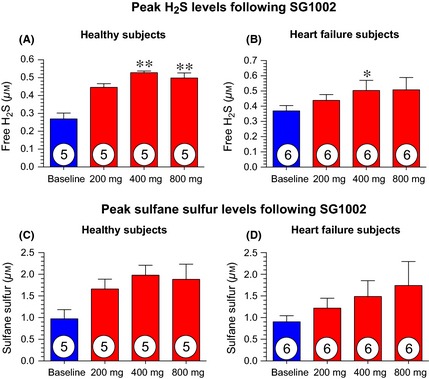
Peak sulfide levels for healthy and heart failure subjects following 1st dose of 200, 400 and 800 mg SG1002 (individual maximum concentrations reached 0.5–4 h post administration). (A) Peak free H2S levels following 200, 400 and 800 mg BID in healthy subjects. (B) Peak free H2S levels following 200, 400 and 800 mg BID in heart failure subjects. (C) Peak sulfane sulfur levels following 200, 400 and 800 mg BID in healthy subjects. (D) Peak sulfane sulfur levels following 200, 400 and 800 mg BID in heart failure subjects. *P < 0.05, **P < 0.01 compared to baseline (pretreatment values) using a 1‐way ANOVA with a Bonferroni multiple comparison correction test. Values are expressed as mean ± SEM.
Figure 3.
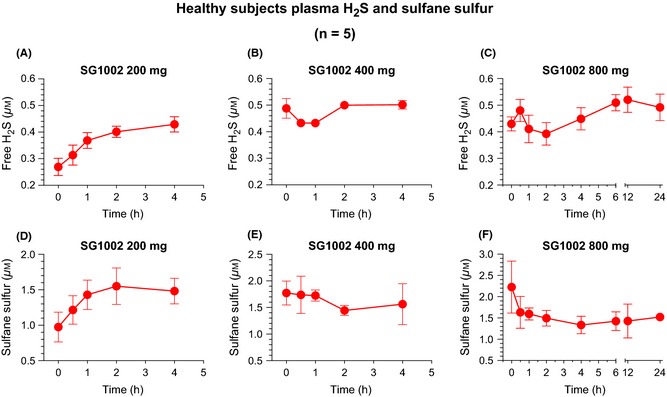
Pharmacokinetic analysis of free H2S and sulfane sulfur in healthy subjects. (A) Free H2S levels following 200 mg SG1002 BID, (B) 400 mg BID, and (C) 800 mg BID. (D) Free sulfane sulfur levels following 200 mg SG1002 BID, (E) 400 mg BID, and (F) 800 mg BID. Values are expressed as mean ± SEM. n = 5.
Figure 4.
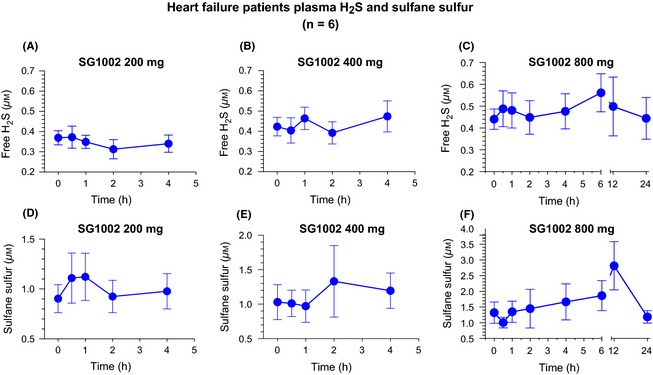
Pharmacokinetic analysis of free H2S and sulfane sulfur in heart failure subjects. (A) Free H2S levels following 200 mg SG1002 BID, (B) 400 mg BID, and (C) 800 mg BID. (D) Free sulfane sulfur levels following 200 mg SG1002 BID, (E) 400 mg BID, and (F) 800 mg BID. Values are expressed as mean ± SEM. n = 6.
SG1002 Promotes Nitric Oxide Bioavailability in Healthy and Heart Failure Subjects
Nitrite is an active storage form of NO and is an indicator of NO reserves and production. The prodrug, SG1002, promoted a sustained elevation in nitrite levels in healthy subjects compared to baseline values (Figure 5A–C). In these subjects, peak nitrite levels were significantly increased following the 400 mg (2.0‐fold, P < 0.05) and 800 mg (2.4‐fold, P < 0.001) doses compared to pretreatment values (Figure 6A). Similarly, peak nitrite levels in HF subjects following SG1002 therapy were elevated following 400 mg (P < 0.05) and 800 mg (P < 0.05) doses compared to baseline levels. Placebo nitrite pharmacokinetics are depicted in Table S1.
Figure 5.
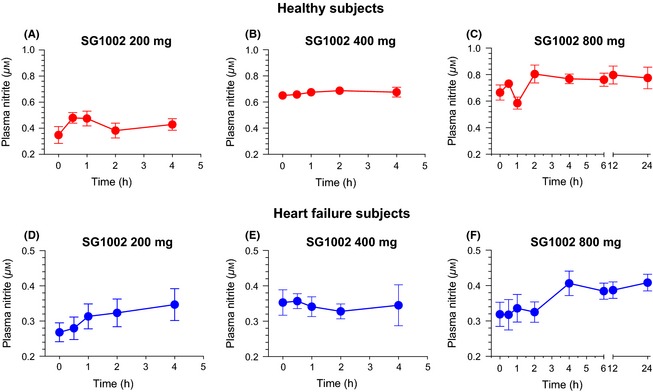
Pharmacokinetic analysis of nitrite in healthy and heart failure subjects. (A) Nitrite levels following 200 mg SG1002 BID, (B) 400 mg BID, and (C) 800 mg BID in healthy subjects (n = 5). (D) Plasma nitrite levels following 200 mg SG1002 BID, (E) 400 mg BID, and (F) 800 mg BID in HF subjects (n = 6). Values are expressed as mean ± SEM.
Figure 6.
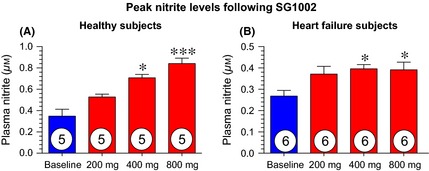
Peak nitrite levels for healthy and heart failure subjects following 1st dose of 200, 400, and 800 mg SG1002 (individual maximum concentrations reached 0.5–4 h post administration). (A) Peak nitrite levels following 200, 400, and 800 mg BID in healthy subjects (n = 5). (B) Peak nitrite levels following 200, 400, and 800 mg BID in heart failure subjects (n = 6). *P < 0.05, ***P < 0.001 compared to baseline (pretreatment values) using a 1‐way ANOVA with a Bonferroni multiple comparison correction test. Values are expressed as mean ± SEM.
SG1002 Maintains BNP Levels in Heart Failure Patients
Fold change in the stress responsive polypeptide, BNP, was calculated with paired correction to individual baseline levels. Circulating BNP concentrations were measure at baseline, day 7 (completion of 200 mg BID), day 14 (400 mg BID), and day 21 (800 mg BID). SG1002‐treated patients displayed steady BNP levels at all doses (Figure 7A).
Figure 7.
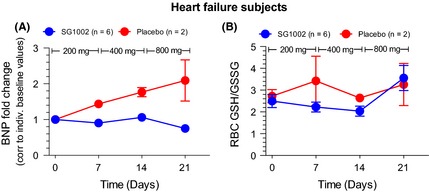
Markers of cardiac health following SG1002 treatment in heart failure subjects. (A) Fold change in the stress responsive polypeptide, BNP. Circulating BNP concentrations were measure at baseline, day 7 (completion of 200 mg BID), day 14 (400 mg BID), and day 21 (800 mg BID). (B) Relative reduced (GSH) and oxidized (GSSG) glutathione levels in RBCs at baseline, after 7 days of 200 mg BID (day 7), after 7 days of 400 mg BID (day 14), and after 7 days of 800 mg BID (day 21). Values are expressed as mean ± SEM.
SG1002 does not Alter Red Blood Cell Glutathione Content
We measured the reduced (GSH) and oxidized (GSSG) glutathione levels in RBCs at baseline, after 7 days of 200 mg BID (day 7), after 7 days of 400 mg BID (day 14), and after 7 days of 800 mg BID (day 21). At these time points and dosages, we failed to observe any significant change in GSH, GSSH, or the GSH:GSSG ratio in either group (Figure 7B).
Discussion
Recent preclinical studies have shown that H2S attenuates adverse cardiac remodeling and dysfunction in the setting of HF 12, 13. These studies build upon previous findings that revealed the protective action of H2S against ischemic injury (myocardial 23, hindlimb 24, hepatic 9, and stroke 25), gastrointestinal damage 26, atherosclerosis 27, and metabolic syndrome 28. The principal protective actions of H2S in these pathological states are as follows: potent antioxidant actions 29, pro‐angiogenesic properties 30, and preservation of mitochondrial function 23, 25. More recently, it has been reported that the administration of H2S restores redox balance and NO bioavailability via direct actions on eNOS to recouple this enzyme under pathological conditions 9. This restoration of NO levels results in cytoprotective signal transduction resulting in tissue preservation 9.
Novel, long‐acting, and controllable H2S‐based therapeutics (i.e., H2S donors, prodrugs, and H2S enzyme activators) may represent valuable candidates for drug development. Currently, H2S donors are very limited in scope for clinical development because of poor pharmacokinetics with a very short half‐life and uncontrolled release 6. For example, commercially available inorganic salts, Na2S and NaHS, rapidly increase H2S concentration, but the increase in free‐H2S is very short lived (seconds) and these compounds have a very small therapeutic dose window leading to potential toxicity 31. Use of these compounds for treating chronic disorders, such as HF, is not clinically feasible to supplement H2S levels in a sustained manner. Given the toxicity of supraphysiological H2S levels, it is critical that novel H2S agents with favorable pharmacokinetic profiles are developed for clinical application.
SG1002 is an α‐sulfur oral formulation H2S prodrug that has been examined in in vivo, preclinical studies of HF 12. This study revealed cardioprotective actions including reductions in myocardial fibrosis and increased myocardial vascularity as well as significant increases in NO bioavailability. Results from preclinical studies demonstrating the effects of SG1002 on LV ejection fraction following transverse aortic constriction prompted us to test the safety and ability of SG1002 to provide sustained, nontoxic elevations in circulating H2S in man.
Overall, the present study demonstrated that SG1002 was well tolerated and safe in a small cohort of healthy volunteers and HF patients. There were no safety concerns at all doses in the healthy and HF subjects. The peak H2S levels did not exceed 0.7 μM at any time point analyzed in the healthy subjects and reached a maximum of 1.1 μM in one of the HF subjects. Even these levels remain within physiological range and remain below cytotoxic concentrations 32. Free H2S plasma levels following SG1002 were elevated in the healthy subjects (Figure 2) to a greater extent than increases observed in heart failure patients. It is likely that H2S derived from SG1002 was more rapidly degraded in HF patients as a result of increased oxidative stress associated with heart failure. Preclinical studies indicate that these levels would provide cardioprotection 12. Due to the safety of the drug at the high dose, we believe that the 800 mg dose would be most appropriate in a phase II study. Similarly, nitrite levels rose to safe circulating levels. Although NO is a potent vasodilator (and H2S also exerts vasodilatory actions), the increases in NO and H2S bioavailability seen in both healthy and HF subjects did not accompany a drop in blood pressure. These findings are in accordance with other studies that show that long‐acting (i.e., extended release) NO donors (sodium nitrite) do not alter blood pressure when NO levels are kept within physiological levels 33. Based on preclinical studies 3, an increase in nitrite levels observed at the 800 mg dose would likely have physiological benefits in heart failure subjects.
Recent preclinical studies have shown that H2S increases the activity of eNOS and NO bioavailability 9. Under homeostatic conditions, eNOS is a coupled homodimer that can be readily phosphorylated to either activate (ser1177) or inactivate (thr495) the enzyme 34. Under pathological states, eNOS can uncouple to its monomeric form or undergo alterations in phosphorylation status of the enzyme, resulting in reduced NO levels 35. Diminished levels of H2S due to genetic deficiency of CSE result in eNOS uncoupling and an inactive phosphorylation status 9. In the present study, the H2S prodrug substantially augmented NO bioavailability that might be attributed to an increase in eNOS activity or a general decrease in systemic oxidative stress. Clinically, the direct effect of a therapeutic agent eNOS to increase NO bioavailability may be more advantageous than the transient burst of NO often associated with NO donors.
Increased oxidative stress results in cardiac contractile dysfunction and is one of the contributing factors in the transition from compensatory cardiac hypertrophy to decompensated HF 36. H2S is known to reduce oxidative stress by direct inactivation of oxidant species and via upregulation of endogenous antioxidant defenses 29, 37. NO similarly promotes antioxidant enzyme defenses 38. Finally, SG1002 may attenuate oxidative stress via a third mechanism by recoupling of eNOS. Uncoupled eNOS (as seen in HF) not only results in decreased NO generation, but leads to excessive peroxynitrite concentrations 39. Peroxynitrite can interact with lipids, DNA, and proteins. These reactions elicit many downstream actions that can result in extensive oxidative injury, resulting in cellular necrosis and apoptosis 40. We failed to observe a significant change in oxidative stress, as indicated by the concentration of the antioxidant glutathione in red blood cells. However, because this trial was primarily designed to examine safety, the sample size was not sufficient to rule out the antioxidant capacity of SG1002. A larger, phase II trial is required to examine whether SG1002 use in man will align with preclinical studies that more fully elucidate its capability of reducing free radical damage.
In summary, this trial revealed for the first time the safety of SG1002 in both healthy and HF patients. Furthermore, this novel H2S prodrug enhanced free H2S and H2S metabolites (sulfane sulfur) in humans. SG1002 significantly elevated NO bioavailability, as measured by plasma nitrite levels. The results from this trial are promising and indicate that the use of SG1002 to treat HF patients is worthy of further exploration. Currently, a larger, placebo‐controlled phase II study in HF patients is being designed to examine the ability of SG1002 to elevate H2S, reduce oxidative stress, and ultimately improve cardiac function.
Funding Sources
These studies were supported by Sulfagenix Australia Pty Ltd.
Disclosures
TG is a cofounder of Sulfagenix Australia Pty Ltd. and is president and CEO of Sulfagenix and is paid by Sulfagenix. TG has significant stock in Sulfagenix. GG Sr. and GG Jr. are both founders of Sulfagenix and have significant stock in Sulfagenix. GG Sr. is Chief Technical Officer of Sulfagenix and is paid by Sulfagenix. GG Jr. is on the Board of Directors of Sulfagenix. Sulfagenix is currently developing H2S‐based therapeutics for cardiovascular disease states.
Conflict of Interest
The authors declare no conflict of interest.
Supporting information
Table S1. Plasma H2S, sulfane sulfur, and nitrite pharmacokinetic data in placebo treated healthy and heart failure subjects.
Clinical Trial Registration: https://clinicaltrials.gov/ct2/show/NCT01989208. ID: NCT01989208
References
- 1. Foo RS, Mani K, Kitsis RN. Death begets failure in the heart. J Clin Invest 2005;115:565–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Cohen‐Solal A, Beauvais F, Logeart D. Heart failure and diabetes mellitus: Epidemiology and management of an alarming association. J Card Fail 2008;14:615–625. [DOI] [PubMed] [Google Scholar]
- 3. Bhushan S, Kondo K, Polhemus DJ, et al. Nitrite therapy improves left ventricular function during heart failure via restoration of nitric oxide‐mediated cytoprotective signaling. Circ Res 2014;114:1281–1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kovacic D, Glavnik N, Marinsek M, et al. Total plasma sulfide in congestive heart failure. J Card Fail 2012;18:541–548. [DOI] [PubMed] [Google Scholar]
- 5. Polhemus DJ, Calvert JW, Butler J, Lefer DJ. The cardioprotective actions of hydrogen sulfide in acute myocardial infarction and heart failure. Scientifica 2014;2014:768607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Polhemus DJ, Lefer DJ. Emergence of hydrogen sulfide as an endogenous gaseous signaling molecule in cardiovascular disease. Circ Res 2014;114:730–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Predmore BL, Lefer DJ, Gojon G. Hydrogen sulfide in biochemistry and medicine. Antioxid Redox Signal 2012;17:119–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Wang K, Ahmad S, Cai M, et al. Dysregulation of hydrogen sulfide producing enzyme cystathionine gamma‐lyase contributes to maternal hypertension and placental abnormalities in preeclampsia. Circulation 2013;127:2514–2522. [DOI] [PubMed] [Google Scholar]
- 9. King AL, Polhemus DJ, Bhushan S, et al. Hydrogen sulfide cytoprotective signaling is endothelial nitric oxide synthase‐nitric oxide dependent. Proc Natl Acad Sci USA 2014;111:3182–3187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Gallagher PM, Meleady R, Shields DC, et al. Homocysteine and risk of premature coronary heart disease. Evidence for a common gene mutation. Circulation 1996;94:2154–2158. [DOI] [PubMed] [Google Scholar]
- 11. Kolluru GK, Shen X, Bir SC, Kevil CG. Hydrogen sulfide chemical biology: Pathophysiological roles and detection. Nitric Oxide 2013;35:5–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kondo K, Bhushan S, King AL, et al. H(2)s protects against pressure overload‐induced heart failure via upregulation of endothelial nitric oxide synthase. Circulation 2013;127:1116–1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Polhemus DJ, Kondo K, Bhushan S, et al. Hydrogen sulfide attenuates cardiac dysfunction after heart failure via induction of angiogenesis. Circ Heart Fail 2013;6:1077–1086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kimura Y, Kimura H. Hydrogen sulfide protects neurons from oxidative stress. FASEB J 2004;18:1165–1167. [DOI] [PubMed] [Google Scholar]
- 15. Wink DA, Mitchell JB. Chemical biology of nitric oxide: Insights into regulatory, cytotoxic, and cytoprotective mechanisms of nitric oxide. Free Radic Biol Med 1998;25:434–456. [DOI] [PubMed] [Google Scholar]
- 16. Schulz R, Rassaf T, Massion PB, Kelm M, Balligand JL. Recent advances in the understanding of the role of nitric oxide in cardiovascular homeostasis. Pharmacol Ther 2005;108:225–256. [DOI] [PubMed] [Google Scholar]
- 17. Cai H, Harrison DG. Endothelial dysfunction in cardiovascular diseases: The role of oxidant stress. Circ Res 2000;87:840–844. [DOI] [PubMed] [Google Scholar]
- 18. Elrod JW, Duranski MR, Langston W, et al. Enos gene therapy exacerbates hepatic ischemia‐reperfusion injury in diabetes: A role for enos uncoupling. Circ Res 2006;99:78–85. [DOI] [PubMed] [Google Scholar]
- 19. Motta JP, Flannigan KL, Agbor TA, et al. Hydrogen sulfide protects from colitis and restores intestinal microbiota biofilm and mucus production. Inflamm Bowel Dis 2015;21:1006–1017. [DOI] [PubMed] [Google Scholar]
- 20. Wallace JL, Dicay M, McKnight W, Martin GR. Hydrogen sulfide enhances ulcer healing in rats. FASEB J 2007;21:4070–4076. [DOI] [PubMed] [Google Scholar]
- 21. Wallace JL, Blackler RW, Chan MV, et al. Anti‐inflammatory and cytoprotective actions of hydrogen sulfide: Translation to therapeutics. Antioxid Redox Signal 2015;22:398–410. [DOI] [PubMed] [Google Scholar]
- 22. Wallace JL, Vong L, McKnight W, Dicay M, Martin GR. Endogenous and exogenous hydrogen sulfide promotes resolution of colitis in rats. Gastroenterology 2009;137:569–578, 578 e561 [DOI] [PubMed] [Google Scholar]
- 23. Elrod JW, Calvert JW, Morrison J, et al. Hydrogen sulfide attenuates myocardial ischemia‐reperfusion injury by preservation of mitochondrial function. Proc Natl Acad Sci USA 2007;104:15560–15565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wang MJ, Cai WJ, Li N, Ding YJ, Chen Y, Zhu YC. The hydrogen sulfide donor nahs promotes angiogenesis in a rat model of hind limb ischemia. Antioxid Redox Signal 2010;12:1065–1077. [DOI] [PubMed] [Google Scholar]
- 25. Kimura Y, Goto Y, Kimura H. Hydrogen sulfide increases glutathione production and suppresses oxidative stress in mitochondria. Antioxid Redox Signal 2010;12:1–13. [DOI] [PubMed] [Google Scholar]
- 26. Fiorucci S, Distrutti E, Cirino G, Wallace JL. The emerging roles of hydrogen sulfide in the gastrointestinal tract and liver. Gastroenterology 2006;131:259–271. [DOI] [PubMed] [Google Scholar]
- 27. Wang Y, Zhao X, Jin H, et al. Role of hydrogen sulfide in the development of atherosclerotic lesions in apolipoprotein e knockout mice. Arterioscler Thromb Vasc Biol 2009;29:173–179. [DOI] [PubMed] [Google Scholar]
- 28. Kaneko Y, Kimura Y, Kimura H, Niki I. L‐cysteine inhibits insulin release from the pancreatic beta‐cell: Possible involvement of metabolic production of hydrogen sulfide, a novel gasotransmitter. Diabetes 2006;55:1391–1397. [DOI] [PubMed] [Google Scholar]
- 29. Jha S, Calvert JW, Duranski MR, Ramachandran A, Lefer DJ. Hydrogen sulfide attenuates hepatic ischemia‐reperfusion injury: Role of antioxidant and antiapoptotic signaling. Am J Physiol Heart Circ Physiol 2008;295:H801–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Papapetropoulos A, Pyriochou A, Altaany Z, et al. Hydrogen sulfide is an endogenous stimulator of angiogenesis. Proc Natl Acad Sci USA 2009;106:21972–21977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Caliendo G, Cirino G, Santagada V, Wallace JL. Synthesis and biological effects of hydrogen sulfide (h2s): Development of h2s‐releasing drugs as pharmaceuticals. J Med Chem 2010;53:6275–6286. [DOI] [PubMed] [Google Scholar]
- 32. Reiffenstein RJ, Hulbert WC, Roth SH. Toxicology of hydrogen sulfide. Annu Rev Pharmacol Toxicol 1992;32:109–134. [DOI] [PubMed] [Google Scholar]
- 33. Greenway FL, Predmore BL, Flanagan DR, et al. Single‐dose pharmacokinetics of different oral sodium nitrite formulations in diabetes patients. Diabetes Technol Ther 2012;14:552–560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Mount PF, Kemp BE, Power DA. Regulation of endothelial and myocardial no synthesis by multi‐site enos phosphorylation. J Mol Cell Cardiol 2007;42:271–279. [DOI] [PubMed] [Google Scholar]
- 35. Chalupsky K, Cai H. Endothelial dihydrofolate reductase: Critical for nitric oxide bioavailability and role in angiotensin ii uncoupling of endothelial nitric oxide synthase. Proc Natl Acad Sci USA 2005;102:9056–9061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Dhalla AK, Hill MF, Singal PK. Role of oxidative stress in transition of hypertrophy to heart failure. J Am Coll Cardiol 1996;28:506–514. [DOI] [PubMed] [Google Scholar]
- 37. Calvert JW, Jha S, Gundewar S, et al. Hydrogen sulfide mediates cardioprotection through nrf2 signaling. Circ Res 2009;105:365–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kanner J, Harel S, Granit R. Nitric oxide as an antioxidant. Arch Biochem Biophys 1991;289:130–136. [DOI] [PubMed] [Google Scholar]
- 39. Kuzkaya N, Weissmann N, Harrison DG, Dikalov S. Interactions of peroxynitrite, tetrahydrobiopterin, ascorbic acid, and thiols: Implications for uncoupling endothelial nitric‐oxide synthase. J Biol Chem 2003;278:22546–22554. [DOI] [PubMed] [Google Scholar]
- 40. Estevez AG, Radi R, Barbeito L, Shin JT, Thompson JA, Beckman JS. Peroxynitrite‐induced cytotoxicity in pc12 cells: Evidence for an apoptotic mechanism differentially modulated by neurotrophic factors. J Neurochem 1995;65:1543–1550. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Plasma H2S, sulfane sulfur, and nitrite pharmacokinetic data in placebo treated healthy and heart failure subjects.