

Abstract
Aims
To evaluate the efficacy and tolerability of sitagliptin in subjects with impaired glucose tolerance (IGT).
Methods
In a double‐blind, parallel‐group study, 242 Japanese subjects with IGT, determined by a 75‐g oral glucose tolerance test (OGTT) at week −1, were randomized (1 : 1 : 1) to placebo (n = 83), sitagliptin 25 mg (n = 82) or 50 mg (n = 77) once daily for 8 weeks. Glycaemic variables were assessed using another OGTT at week 7 and meal tolerance tests (MTTs) at weeks 0 and 8. Primary and secondary endpoints were percent change from baseline in glucose total area under the curve 0–2 h (AUC0 –2 h) during the MTT and OGTT, respectively.
Results
Least squares mean percent change from baseline in glucose AUC0 –2 h during the MTT were −2.4, −9.5 and −11.5%, and during the OGTT were −3.7, −21.4 and −20.1% with placebo, sitagliptin 25 mg once daily, and 50 mg once daily, respectively (p < 0.001 for either sitagliptin dose vs placebo in both tests). Sitagliptin treatment enhanced early insulin response during the OGTT and decreased total insulin response, assessed as the total AUC0 –2 h during the MTT. Sitagliptin treatment also suppressed glucagon response during the MTT. The incidence of adverse events, including hypoglycaemia, was low and generally similar in all treatment groups.
Conclusions
Treatment with sitagliptin significantly reduced glucose excursions during both an MTT and an OGTT; this effect was associated with an increase in early insulin secretion after oral glucose loading as well as a blunted glucagon response during an MTT. Sitagliptin was generally well tolerated in subjects with IGT.
Keywords: dipeptidyl peptidase‐4 inhibitors, DPP‐4, incretins, MK‐0431
Introduction
Impaired glucose tolerance (IGT), defined as fasting plasma glucose (FPG) concentration <7.0 mmol/l (126 mg/dl) combined with elevated 2‐h plasma glucose concentration [≥7.8 mmol/l (140 mg/dl) and <11.1 mmol/l (200 mg/dl)] after a 75‐g glucose load in an oral glucose tolerance test (OGTT), is associated with an increased risk of type 2 diabetes (T2DM) 1 and macrovascular disease 2, 3, 4. Progression to T2DM is associated with a higher risk of macrovascular and microvascular complications, progressive deterioration of glycaemic control and, ultimately, requirement for multiple medications 5, 6, 7.
Prevention or delay in the onset of T2DM could have important public health implications. A first step towards prevention of the disease is the identification of patients with dysglycaemia, who are at risk of progressing to T2DM 8, followed by modification of diet and exercise. Complementary to this is assessment of the potential for therapies to slow or eliminate disease onset in patient populations not sufficiently responsive to lifestyle modification. A variety of oral antihyperglycaemic agents used in the management of T2DM have been evaluated in patients with IGT, with varying effect sizes with regard to slowing progression to diabetes 9. Metformin 10, rosiglitazone 11, pioglitazone 12, acarbose 13 and voglibose 14 have been shown to delay the onset of T2DM, although safety and tolerability issues (e.g. gastrointestinal side effects for metformin and α‐glucosidase inhibitors, oedema and increased risk of fracture for thiazolidinediones) may limit use in some patients. Current guidelines from the American Diabetes Association 15, the International Diabetes Federation 16 and the Japan Diabetes Society (JDS) 17 recommend diet and exercise as the primary treatment for individuals with IGT; however, these guidelines also recommend pharmacological intervention for those who have a higher risk of T2DM, or if lifestyle intervention has not achieved the desired weight loss and/or improved glucose tolerance goals.
Dipeptidyl peptidase‐4 (DPP‐4) inhibitors stabilize the circulating levels of the incretins glucagon‐like peptide‐1 and gastric inhibitory polypeptide, which improves insulin secretion and reduces glucagon secretion, thereby reducing hyperglycaemia 18, 19. These incretin effects are glucose‐dependent, thus minimizing the risk of hypoglycaemia. In light of the particular relevance of incretins to postprandial glucose homeostasis, the use of incretin‐based therapies is of interest in subjects with mild postprandial glycaemic excursions, but without overt T2DM. The DPP‐4 inhibitor vildagliptin has been shown to reduce postprandial glucose excursions in subjects with IGT during a meal tolerance test (MTT) 20. In a study in patients hospitalized for cardiovascular disease, sitagliptin was shown to reduce glucose excursions during an OGTT in patients newly diagnosed with IGT or T2DM 21. In the present study, we evaluated the effects of treatment with sitagliptin on glucose excursions and plasma insulin levels during an MTT or an OGTT, on circulating glucagon levels during the MTT, and on FPG and glycated haemoglobin (HbA1c) levels, in Japanese subjects with IGT.
Materials and Methods
This was a multicentre, randomized, placebo‐controlled, parallel‐group, double‐blind, dose–response study (Figure S1). After an 8‐week observation period, including diet/exercise therapy, and a single‐blind, 1‐week placebo run‐in period, subjects were randomized in a 1 : 1 : 1 ratio to once‐daily sitagliptin 25 mg, sitagliptin 50 mg or placebo, before breakfast for 8 weeks. The maximum sitagliptin dose was chosen based on the standard initial dose of sitagliptin approved in Japan for the treatment of T2DM.
Japanese patients, aged ≥20 years with suspected IGT were identified for screening by investigators. Individuals were eligible for the study if, at week −1, while on diet/exercise therapy, they had an FPG value <7.0 mmol/l (126 mg/dl), an OGTT 2‐h plasma glucose value ≥7.8 mmol/l (140 mg/dl) and <11.1 mmol/l (200 mg/dl), and an HbA1c value <6.5% [converted to the National Glycohemoglobin Standardization Program (NGSP) value from the JDS value used in screening, using the formula HbA1c (NGSP; %) = 1.02 × HbA1c (JDS; %) + 0.25%] 22. Written informed consent was obtained from all patients before they underwent any study procedure.
Subjects were excluded if they had overt diabetes, another disease causing secondary glucose intolerance, a history of treatment to prevent diabetes or treatment with any antihyperglycaemic agent or unstable cardiovascular disease, uncontrolled severe hypertension, active liver disease or chronic renal disease. Subjects with aspartate aminotransferase and/or alanine aminotransferase >twofold above the upper limit of normal, creatinine >0.13 mmol/l in men and >0.11 mmol/l in women, or haemoglobin <6.83 mmol/l in men and <6.21 mmol/l in women, were also excluded.
The study (Sitagliptin Protocol 105; ClinicalTrials.gov : NCT01405911) was conducted in accordance with principles of Good Clinical Practice and was approved by the appropriate institutional review boards.
Efficacy Assessments
At weeks −1 and 7, after a fast of ≥10 h (at week 7, 30 min after taking placebo or study drug), subjects underwent an OGTT, initiated by consumption of a 75‐g glucose solution. At weeks 0 and 8, after a fast of ≥10 h (at week 8, 30 min after taking placebo or study drug), subjects underwent an MTT, initiated by consumption of a 500 kcal meal (carbohydrate 60%, fat 25%, protein 15%) during a 15‐min period. For both the OGTT and the MTT, blood for measurement of glucose and insulin was obtained just before initiation of glucose or meal loading (t = 0) and at 30‐min intervals thereafter, up to 120 min. In the MTT, this blood was also used for the measurement of glucagon. FPG and HbA1c were measured at weeks 0 and 8.
The primary efficacy endpoint was percent change from baseline (week 0) in the week 8 MTT glucose total area under the curve 0–2 h (AUC0–2 h). The secondary efficacy endpoint was percent change from baseline (week −1) in the week 7 OGTT glucose total AUC0–2 h. Change from baseline in other glycaemic variables, including OGTT 2‐h plasma glucose, FPG and HbA1c were exploratory endpoints. In addition, change from baseline in insulin and glucagon during MTT and in insulinogenic index (= ΔInsulin0–30/ΔGlucose0–30) during OGTT, were assessed as exploratory endpoints. Change from baseline in 30‐min insulin during OGTT, and return to normoglycaemic status [defined as FPG <6.1 mmol/l (110 mg/dl) and OGTT 2‐h plasma glucose <7.8 mmol/l (140 mg/dl)] were assessed as post hoc analyses.
Safety Assessments
Safety measurements included adverse events (AEs), laboratory tests, body weight, vital signs (blood pressure and pulse rate) and ECG. Laboratory tests were performed at screening/week −8, at baseline/week 0, and at every 4‐week visit during the treatment period or at discontinuation. Body weight, blood pressure and pulse rate were measured and recorded at every visit (except for week 7/visit 5) or at discontinuation. ECG (12‐lead) was performed at week −8/visit 1, week 0/visit 3 and week 8/visit 6 or at discontinuation, at study sites, and recorded. AEs of symptomatic hypoglycaemia, as assessed by investigators, were prespecified as events of interest. Hypoglycaemic events were identified based on records kept by each patient in a log book, including patients' self‐monitoring blood glucose tests, performed twice weekly and in the event of any hypoglycaemic symptoms.
Laboratory Analyses
Laboratory tests for this study were performed by SRL, Inc. (Tokyo, Japan). Insulin was measured using a chemiluminescent enzyme immunoassay with Lumipulse Presto Insulin (Fuji Rebio, Tokyo, Japan) and glucagon was assayed using a radioimmunoassay KIT (Millipore, St. Charles, MO, USA; Cat. # GL‐32K). DPP‐4 activity was measured by Merck & Co., Inc., in Rahway, NJ, USA, using a high plasma concentration assay 23.
Statistical Analyses
For the primary and key secondary endpoint efficacy analyses, a constrained longitudinal data analysis method 24 was used. This repeated measures model included terms for treatment, time, and the interaction of time by treatment, with the restriction of a common baseline mean across treatment groups. The primary and secondary hypotheses were evaluated by a step‐down trend test, comparing doses of sitagliptin with placebo at week 7 or 8, depending on the variable. Multiplicity among linear contrast testing was controlled through the step‐down testing procedure. The primary population for all efficacy analyses comprised all subjects who received at least one dose of study therapy and had at least one efficacy measurement (baseline or post‐randomization).
All randomized patients who received at least one dose of study therapy were included in the safety analysis population. The AE of symptomatic hypoglycaemia was prespecified as an event of interest. The Miettinen and Nurminen method 25 was used to calculate the p values and 95% confidence intervals (CIs) for between‐treatment differences in the percentage of subjects with events of symptomatic hypoglycaemia. For other AEs that had ≥4 patients in a treatment group, for predefined limits of change in laboratory variables, and for the overall AE summary, the between‐group differences and associated 95% CIs were calculated. Point estimates by treatment group were calculated for AEs that had <4 patients in all treatment groups and for change or percent change from baseline in laboratory measurements, ECG, lipids and vital signs.
Approximately 234 subjects (78 subjects per group) were to be randomized, anticipating that 75 subjects per group would be available for analysis for the primary hypothesis at week 8. Using a standard deviation of 13%, this sample size would provide 90% (80%) power to detect a true difference of 7.0% (6.0%) in the mean percent change from baseline of glucose total AUC0–2 h in MTT between two treatment groups (two‐sided test, α = 0.05).
Results
Patient Disposition and Characteristics
A total of 560 subjects were screened and 242 were randomized for this study (Figure 1). An additional 19 subjects, including nine randomized subjects, were not included in this accounting of patients or in study analyses, because the site at which they were enrolled was identified as non‐compliant with some requirements of Good Clinical Practice and therefore data from this site were deemed unreliable. Subjects were most commonly excluded from randomization for not meeting eligibility criteria (89.3%) or because of withdrawal of consent (7.9%). The study was completed by 234 of the 242 subjects randomized (96.7%). Most study discontinuations resulted from withdrawal of consent (Figure 1). With the exception of a slight skewing towards subjects with combined impaired fasting glucose (IFG)/IGT in the group receiving 50 mg sitagliptin, baseline demographics and efficacy characteristics were generally well balanced among the treatment groups (Table 1).
Figure 1.
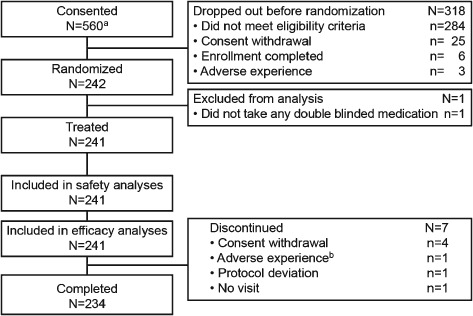
Patient disposition. q.d., once daily; ALT, alanine aminotransferase. aAn additional 19 subjects, including 9 who were randomized (3, 4, and 2 subjects in placebo, sitagliptin 25 and 50 mg q.d. groups, respectively), are not included in this disposition accounting and were not included in study analyses because the site at which they were enrolled was identified as noncompliant with some requirement of Good Clinical Practice and therefore data from this site were deemed to be unreliable. bDiscontinuation due to ALT increased at baseline (not on treatment).
Table 1.
Patient demographics and baseline characteristics
| Variable | Placebo | Sitagliptin 25 mg once daily | Sitagliptin 50 mg once daily | |
|---|---|---|---|---|
| N | 83 | 82 | 77 | |
| Age, years | 61.9 ± 10.6 | 63.1 ± 9.5 | 61.9 ± 9.3 | |
| Male/female, % | 57.8/42.2 | 53.7/46.3 | 58.4/41.6 | |
| Body mass index, kg/m2 | 25 ± 3 | 26 ± 3 | 25 ± 4 | |
| Isolated IGT/IFG plus IGT, % | 60.2/39.8 | 61.0/39.0 | 51.9/48.1 | |
| MTT | Glucose total AUC0−2 h, mmol · h/l | 15.67 ± 2.03 | 15.62 ± 2.10 | 15.83 ± 2.03 |
| OGTT | Glucose total AUC0−2 h, mmol · h/l | 19.77 ± 2.84 | 19.45 ± 2.30 | 20.09 ± 2.73 |
| 2‐h glucose, mmol/l | 9.21 ± 0.98 | 9.19 ± 0.82 | 9.34 ± 0.93 | |
| Fasting | Glucose, mmol/l | 5.87 ± 0.49 | 5.86 ± 0.51 | 5.92 ± 0.56 |
| Insulinogenic index, pmol/mmol | 61.28 ± 53.19 | 62.70 ± 50.68 | 56.51 ± 51.09 | |
| HbA1c, % (mmol/mol) | 5.98 ± 0.27 | 6.01 ± 0.25 | 6.02 ± 0.28 | |
| (41.79 ± 2.90) | (42.16 ± 2.68) | (42.28 ± 3.06) | ||
AUC0–2 h, area under the curve 0–2 h; IFG, impaired fasting glucose; IGT, impaired glucose tolerance; MTT, meal tolerance test; OGTT, 75‐g oral glucose tolerance test.
All values are mean ± standard deviation unless otherwise indicated; to convert insulinogenic index from SI unit to conventional units, divide by 109.
Efficacy
Glucose Excursion During Meal Tolerance Test
At week 8, a significant reduction in the primary endpoint, percent change from baseline (week 0) in glucose total AUC0–2 h during the MTT, was observed across active treatment groups compared with placebo (Table 2; p < 0.001). Compared with baseline, 8 weeks of daily treatment with 25 or 50 mg sitagliptin reduced the least squares (LS) mean MTT‐associated glucose excursions by ∼10% (Figure 2A; Table 2). When calculated as an increment of baseline Time 0 value, the MTT glucose excursion in both sitagliptin treatment groups was reduced by >35% (Table S1).
Table 2.
Change from baseline in efficacy endpoints
| Variable | Placebo (N = 82) | Sitagliptin, 25 mg once daily (N = 82) | Sitagliptin, 50 mg once daily (N = 77) |
|---|---|---|---|
| MTT | |||
| Glucose, total AUC0–2 h, mmol · h/l | |||
| Baseline | 15.67 ± 2.05 | 15.62 ± 2.10 | 15.83 ± 2.03 |
| Week 8 | 15.32 ± 2.25 | 14.07 ± 1.71 | 13.96 ± 1.57 |
| % change from baseline‡ | −2.42 (−4.48, −0.31) | −9.52 (−11.43, −7.75) | −11.49 (−13.40, −9.55) |
| Difference from placebo | — | −7.11* (−9.85, −4.36) | −9.08* (−11.82, −6.33) |
| Insulin, total AUC0–2 h, pmol · h/l | |||
| Baseline | 660.77 ± 418.53 | 657.18 ± 324.64 | 643.88 ± 370.78 |
| Week 8 | 660.59 ± 425.44 | 584.78 ± 336.79 | 585.88 ± 365.59 |
| Change from baseline‡ | 17.70 (−34.32, 69.73) | −73.44 (−125.5, −21.42) | −64.22 (−117.2, −11.20) |
| Difference from placebo | — | −91.14† (−163.9, −18.42) | −81.92† (−155.4, −8.48) |
| Glucagon, total AUC0–2 h, ng · h/l | |||
| Baseline | 162.11 ± 48.41 | 163.78 ± 42.48 | 161.89 ± 47.58 |
| Week 8 | 174.14 ± 49.38 | 162.25 ± 39.46 | 166.48 ± 46.95 |
| Change from baseline‡ | 11.82 (5.88, 17.75) | −0.59 (−6.53, 5.35) | 5.19. (−0.85, 11.24) |
| Difference from placebo | –– | −12.41† (−20.64, −4.17) | −6.63 (−14.94, 1.69) |
| OGTT | |||
| Glucose, total AUC0–2 h, mmol · h/l | |||
| Baseline | 19.76 ± 2.86 | 19.45 ± 2.30 | 20.09 ± 2.73 |
| Week 7 | 19.16 ± 3.54 | 15.42 ± 2.52 | 16.04 ± 2.52 |
| % change from baseline‡ | −3.68 (−6.66, −0.60) | −21.38 (−23.81, −18.87) | −20.09 (−22.61, −17.48) |
| Difference from placebo | –– | −17.70* (−21.55, −13.86) | −16.41* (−20.32, −12.50) |
| 2‐h glucose, mmol/l | |||
| Baseline | 9.20 ± 0.99 | 9.19 ± 0.82 | 9.34 ± 0.93 |
| Week 7 | 8.98 ± 2.27 | 7.29 ± 1.67 | 7.18 ± 1.54 |
| Change from baseline‡ | −0.23 (−0.62, 0.17) | −1.93 (−2.33, −1.54) | −2.13 (−2.53, −1.73) |
| Difference from placebo | — | −1.70* (−2.26, −1.15) | −1.90* (−2.46, −1.34) |
| 30 min insulin, pmol/l | |||
| Baseline | 281.85 ± 213.99 | 270.56 ± 187.94 | 273.95 ± 225.93 |
| Week 7 | 278.20 ± 264.38 | 340.62 ± 252.34 | 334.32 ± 260.81 |
| Change from baseline‡ | 0.79 (−37.47, 39.04) | 67.87 (29.62, 106.13) | 59.53 (20.53, 98.53) |
| Difference from placebo | — | 67.09† (13.07, 121.10) | 58.74† (4.19, 113.29) |
| Insulinogenic index, pmol/mmol | |||
| Baseline | 61.68 ± 53.39 | 62.70 ± 50.68 | 56.51 ± 51.09 |
| Week 8 | 61.18 ± 94.05 | 116.53 ± 130.64 | 99.80 ± 92.06 |
| Change from baseline‡ | −0.44 (−21.08, 20.19) | 52.70 (32.07, 73.34) | 43.02 (21.94, 64.09) |
| Difference from placebo | — | 53.14* (23.96, 82.33) | 43.46† (13.96, 72.95) |
AUC0–2 h, area under the curve 0–2 h; MTT, meal tolerance test; OGTT, oral glucose tolerance test.
Values are mean ± standard deviation, unless noted; to convert insulinogenic index in pmol/mmol to conventional unit, divide by 109.
p < 0.001 versus placebo;
p < 0.05 versus placebo;
LS mean (95% CI).
Figure 2.
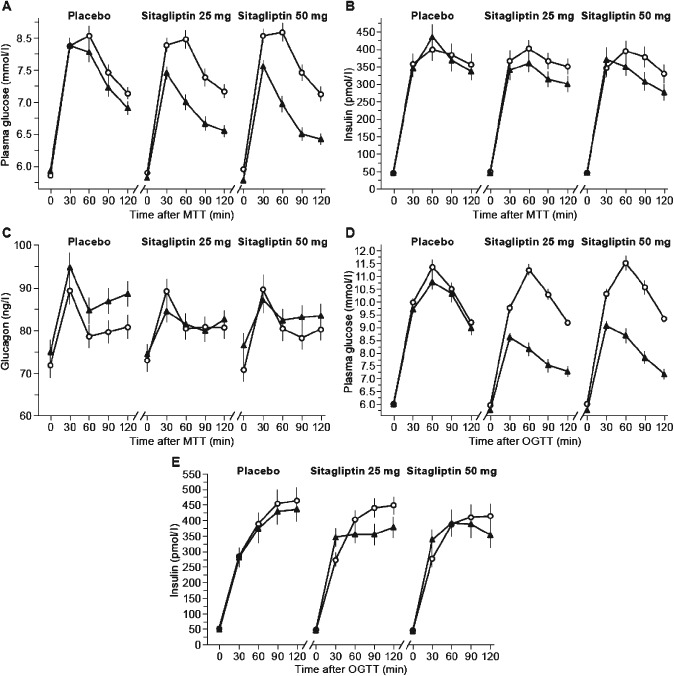
Treatment effects on plasma glucose (A), insulin (B) and glucagon (C) levels during meal tolerance test (MTT) at baseline ( ) and at week 8 of treatment (
) and at week 8 of treatment ( ) and on glucose (D) and insulin (E) during oral glucose tolerance test (OGTT) at baseline (
) and on glucose (D) and insulin (E) during oral glucose tolerance test (OGTT) at baseline ( ) and at week 7 of treatment (
) and at week 7 of treatment ( ). Data shown are mean ± standard error.
). Data shown are mean ± standard error.
Glucose Excursion During Oral Glucose Tolerance Test
Similarly, a significant reduction in the secondary endpoint, percent change from baseline (week −1) in glucose total AUC0–2 h during the OGTT at week 7, was observed across active treatment groups compared with placebo (Table 2; p < 0.001). Compared with baseline, 7 weeks of daily treatment with either sitagliptin dose reduced the LS mean OGTT glucose excursion by ∼20% (Figure 2D; Table 2). When calculated as an increment from the baseline Time 0 value, the OGTT glucose excursion in both sitagliptin treatment groups was reduced by ∼50% (Table S1).
Treatment with either dose of sitagliptin for 7 weeks reduced the LS mean plasma 2‐h OGTT glucose level by ∼2 mmol/l (Figure 2D; Table 2). Compared with placebo, treatment with either sitagliptin dose significantly reduced the LS mean 2‐h OGTT plasma glucose level (Table 2; p < 0.001).
Insulin and Glucagon Responses During Meal Tolerance Test
Compared with baseline, treatment with 25 or 50 mg sitagliptin for 8 weeks reduced LS mean MTT plasma insulin total AUC0–2 h by >60 pmol · h/l (Figure 2B and Table 2). Compared with placebo, treatment with 25 or 50 mg sitagliptin significantly reduced LS mean MTT insulin total AUC0–2 h (Figure 2B; Table 2). Consistent results were observed when calculated as an increment of baseline MTT Time 0 value (Table S1).
Sitagliptin treatment for 8 weeks reduced MTT plasma glucagon (Figure 2C, Table 2); the LS mean change from baseline plasma glucagon total AUC0–2 h associated with the 25 mg dose was significant compared with placebo (p = 0.003), while the LS mean change in plasma glucagon total AUC0–2 h associated with the 50 mg dose, compared with placebo, did not reach statistical significance. When calculated as an increment of baseline Time 0 value, MTT plasma glucagon decreased from baseline with both treatment doses (Table S1). Compared with placebo, there was a significant change from baseline in glucagon incremental AUC0–2 h associated with either sitagliptin dose (Table S1; p < 0.001).
Insulin Response During Oral Glucose Tolerance Test
Compared with placebo, the change from baseline in the early (30‐min) LS mean insulin response to OGTT was increased significantly by 7 weeks of daily treatment with either dose of sitagliptin (Figure 2E; Table 2), while at later time points, plasma insulin levels were reduced after sitagliptin treatment (Figure 2E). Compared with placebo, the LS mean insulinogenic index was increased from baseline after 7 weeks of treatment with either dose of sitagliptin (Table 2; p < 0.05).
Glycated Haemoglobin and Fasting Plasma Glucose
Compared with placebo, daily treatment with either dose of sitagliptin for 8 weeks significantly reduced HbA1c from baseline [LS mean −0.17% (95% CI −0.21, −0.12) or −1.82 mmol/mol (−95% CI 2.33, −1.32) for 25 mg, and LS mean −0.18% (−0.23, −0.14) or −2.01 mmol/mol (95% CI −2.53, −1.50) for 50 mg; p < 0.001]. Compared with placebo, 50 mg sitagliptin significantly reduced FPG from baseline [−0.31 mmol/l (95% CI −0.46, −0.15); p < 0.001], while the reduction with 25 mg sitagliptin did not reach statistical significance [−0.10 mmol/l (95% CI −0.26, 0.05)]. The reduction from baseline in FPG associated with 50 mg sitagliptin treatment was greater than that for treatment with 25 mg sitagliptin by −0.20 mmol/l [(95% CI −0.36, −0.05); p = 0.011].
Overall Glycaemic Status
In a post hoc analysis, 19 of 82 patients (23.2%) who had IGT at baseline and then were treated for 7 weeks with placebo had become normoglycaemic, compared with 39 out 82 patients (47.6%) treated with sitagliptin 25 mg and with 41 of 77 patients (53.2%) treated with sitagliptin 50 mg.
DPP‐4 Activity
At week 8 of treatment, the trough geometric LS mean ± standard error percentage of plasma DPP‐4 activity inhibited was 0.4 ± 0.04, 81.5 ± 0.04 and 86.9 ± 0.05% for the placebo, sitagliptin 25 and 50 mg groups, respectively.
Safety
Over the 8‐week treatment period, no meaningful differences among treatment groups were observed in the incidence of AEs (Table 3). No serious AEs were reported. No deaths occurred during the study. Symptomatic hypoglycaemia, an AE of special interest, occurred only in the placebo group. AEs classified by system organ class were distributed similarly across all treatment groups. A small dose‐dependent increase was observed in the incidence of overall AEs; however, this increase was attributable to a variety of disparate AEs and not to a dose‐dependent increase in any specific AE or group of closely related AEs. The 95% CIs around the between‐group differences (vs placebo) in the incidences of overall AEs and of drug‐related AEs included zero. No serious AEs or AEs leading to discontinuation of study treatment occurred in this study. A patient in the sitagliptin 25 mg treatment group discontinued taking the study drug because of an AE of hepatic function abnormality, when exclusionary baseline alanine aminotransferase/aspartate aminotransferase values became available from the central laboratory.
Table 3.
Summary of adverse events
| Placebo | Sitagliptin 25 mg once daily | Sitagliptin 50 mg once daily | |
|---|---|---|---|
| AE summary | |||
| Subjects in population | N = 81 (%) | N = 82 (%) | N = 78* (%) |
| With ≥1 AE | 25 (30.9) | 28 (34.1) | 32 (41.0) |
| With no AE | 56 (69.1) | 54 (65.9) | 46 (59.0) |
| With drug‐related† AE | 10 (12.3) | 5 (6.1) | 7 (9.0) |
| With serious AE | 0 (0.0) | 0 (0.0) | 0 (0.0) |
| With serious drug‐related AE | 0 (0.0) | 0 (0.0) | 0 (0.0) |
| Who died | 0 (0.0) | 0 (0.0) | 0 (0.0) |
| Discontinued because of an AE | 0 (0.0) | 0 (0.0) | 0 (0.0) |
| Specific AEs | |||
| Hypoglycaemia | 7 (8.6) | 5 (6.1) | 4 (5.1) |
| Symptomatic hypoglycaemia | 2 (2.5) | 0 (0.0) | 0 (0.0) |
| AEs classified by system organ classes with incidence ≥4 in one or more treatment groups | |||
| Gastrointestinal disorders | 5 (6.2) | 4 (4.9) | 7 (9.0) |
| Infections and infestations | 8 (9.9) | 11 (13.4) | 12 (15.4) |
| Metabolism and nutrition disorders | 7 (8.6) | 5 (6.1) | 4 (5.1) |
| Respiratory, thoracic and mediastinal disorders | 3 (3.7) | 5 (6.1) | 1 (1.3) |
AE, adverse event.
One subject in the placebo group who took sitagliptin 50 mg once daily because of a prescription error was handled as a subject in the sitagliptin 50 mg once daily group in the safety analysis.
Determined by the investigator to be related to the drug.
In neither treatment group were predefined limits of change criteria met in ≥4 subjects for any specific laboratory analyte. No clinically relevant changes from baseline in the mean values for laboratory variables, body weight, vital signs and 12‐lead ECG variables were reported.
Discussion
The present report provides the first detailed analysis of the effects of sitagliptin, a selective DPP‐4 inhibitor 26, in a population of subjects with IGT. Sitagliptin has been shown to be an effective and well tolerated oral antihyperglycaemic agent when used as monotherapy or as add‐on therapy in patients with T2DM 27, but its effects in subjects with IGT have previously been evaluated only in a small cohort of patients newly diagnosed with IGT or those with T2DM identified after hospitalization for acute coronary syndrome 21.
In Japanese subjects with IGT, sitagliptin treatment for 7–8 weeks resulted in reductions in post‐challenge glucose excursions during both an MTT and an OGTT. The observation that treatment with sitagliptin increased the early insulin response to the glucose load during the OGTT, and reduced circulating glucagon levels during the MTT, may help explain the effects of sitagliptin in the clinical context of IGT. Both compromised early insulin release and lack of glucagon suppression in response to glucose challenge have been identified as key aspects of IGT. These defects contribute to the hyperglycaemia seen in patients with IGT, and to the progression of worsening hyperglycaemia, leading to the onset of T2DM 28. In the present study, improvement in glycaemic control was accompanied by improvement in both of these defects. The mean insulin level measured 30 min after the glucose challenge during the OGTT in subjects treated with either 25 or 50 mg sitagliptin was significantly higher than that of subjects treated with placebo. Consistent with this finding, in sitagliptin‐treated subjects plasma glucose levels were reduced compared with placebo at the earliest measured time points after the glucose challenge during the OGTT. The improved early insulin response to glucose may be responsible for the reduction in magnitude of the late insulin response in both the OGTT and the MTT, the reduction in plasma insulin AUC0–2h during the MTT, and the improvement in the insulinogenic index calculated from data gathered during the OGTT, all associated with sitagliptin treatment. Glucose‐lowering, and hence reduction in total plasma insulin, may also result from the effect of sitagliptin treatment on plasma glucagon levels. During the 2‐h MTT, incremental plasma glucagon levels were reduced compared with placebo treatment, consistent with the effects of sitagliptin on the stabilization of active glucagon‐like peptide‐1 29.
Compared with placebo, treatment with 50 mg sitagliptin for 8 weeks yielded a small, but statistically significant, reduction in FPG, while treatment with 25 mg sitagliptin treatment did not reduce FPG. Both active treatments yielded small, though statistically significant, reductions in HbA1c. In the cases of both FPG and HbA1c, the modest reductions in these glycaemic measures are consistent with the modest degree of hyperglycaemia present in the study population at baseline. Consistent with the improvements observed in individual efficacy endpoints, a greater percentage of patients could be considered to have normoglycaemia after 7 weeks of treatment with either dose of sitagliptin compared with placebo.
No consistent dose‐dependent effects of sitagliptin on the measured glycaemic variables were seen; however, this study was not powered to distinguish between small differences in response to treatment that might be expected to occur with 25 versus 50 mg sitagliptin in a mildly hyperglycaemic patient population. It is possible that observation of significant dose‐dependent improvements in FPG or in glycaemic markers during an MTT or an OGTT will require a study in a larger patient population.
All active treatments were well tolerated, and no increases in the incidence of hypoglycaemia or gastrointestinal AEs were observed. This observation is consistent with previous evaluations of the safety and tolerability of sitagliptin 30.
Previous evaluation of another DPP‐4 inhibitor, vildagliptin, in patients with IGT found that after 12 weeks of treatment, there was a reduction from baseline in prandial glucose excursions and in HbA1c 20; however, unlike the experience with 50 mg sitagliptin reported in the present study, 12 weeks of treatment with vildagliptin did not reduce FPG 20. In studies of patients with IFG, 8 weeks of treatment with sitagliptin 100 mg daily did not alter either fasting or postprandial glucose 31, while 6 weeks of treatment with vildagliptin 100 mg daily significantly decreased the incremental area under the glucose curve during an MTT but did not change FPG 32. While there are observed differences in these studies, cross‐study comparison should not be made because of differences in study design, subject population and other factors.
The effects of sitagliptin in subjects with IGT are consistent with its effect in subjects with T2DM, in whom it improves not only glycaemic control but also β‐cell function 18, 19, 33. Other antihyperglycaemic agents effective in patients with T2DM have been reported to significantly reduce progression of subjects with IGT to T2DM 9. The results of these studies cannot be compared with the current exploratory evaluation, as all had duration of 0.9–3.2 years, compared with the present 8‐week study. In Japan, voglibose has been approved for suppression of onset of T2DM 17, and the American Diabetes Association consensus statement highlights metformin as an option in selected individuals with IGT 15. Given the results of the present study, and considering the safety and tolerability profile of sitagliptin 30, it is possible that sitagliptin treatment would provide a safe and effective reduction in progression to T2DM in patients with IGT; however, evaluating the effect on IGT in a broader population and over a more extended period of time will be required to evaluate this hypothesis.
Conflict of Interest
K. K. has acted as advisory panel member for AstraZeneca K.K., Chugai Pharmaceutical Co., Ltd, Novo Nordisk Pharma Ltd, Sanofi K.K. and Takeda Pharmaceutical Co., Ltd., as a consultant for Astellas Pharma, Inc., MSD K.K., Sanwa Kagaku Kenkyusho Co. and Taisho Pharmaceutical Co, Ltd., and as a speaker for Dainippon Sumitomo Pharma Co., Ltd, Kowa Pharmaceutical Co., Ltd, MSD K.K., Novo Nordisk Pharma Ltd and Takeda Pharmaceutical Co., Ltd., and has obtained research support from AstraZeneca K.K., Chugai Pharmaceutical Co., Ltd, Daiichi Sankyo Co., Ltd, MSD K.K., Novo Nordisk Pharma Ltd, Sanofi K.K. and Takeda Pharmaceutical Co., Ltd. T. K. has acted as advisory panel member for AstraZeneca K.K., Chugai Pharmaceutical Co., Ltd, Mitsubishi Tanabe Pharma Corp., Sanofi K.K. and Takeda Pharmaceutical Co., Ltd, as a consultant for MSD K.K. and Ono Pharmaceutical Co., Ltd, as a speaker for Astellas Pharma, Inc., AstraZeneca K.K., Daiichi Sankyo Co., Ltd, Dainippon Sumitomo Pharma Co., Ltd, Eli Lilly Japan K.K., Kowa Pharmaceutical Co., Ltd, Kyowa Hakko Kirin Co., Mitsubishi Tanabe Pharma Corp., MSD K.K., Nippon Boehringer Ingelheim Co., Ltd, Novartis Pharma K.K., Ono Pharmaceutical Co., Ltd, Sanofi K.K., Sanwa Kagaku Kenkyusho Co., Taisho Pharmaceutical Co., Ltd and Takeda Pharmaceutical Co., Ltd, has obtained research support from Daiichi Sankyo Co., Ltd, Dainippon Sumitomo Pharma Co., Ltd, MSD K.K., Novartis Pharma K.K., Novo Nordisk Pharma Ltd, Ono Pharmaceutical Co., Ltd, Sanofi K.K. and Takeda Pharmaceutical Co., Ltd, and has been involved in research units endowed by MSD K.K., Nippon Boehringer Ingelheim Co., Ltd, Novo Nordisk Pharma Ltd, Takeda Pharmaceutical Co., Ltd, and Terumo Corp. Y. T. has acted as advisory panel member for Chugai Pharmaceutical Co., Ltd, Eli Lilly Japan K.K., Novo Nordisk Pharma Ltd and Sanofi K.K., as a consultant for Astellas Pharma, Inc., AstraZeneca K.K., Kowa Pharmaceutical Co., Ltd, MSD K.K., Shionogi & Co., Ltd and Takeda Pharmaceutical Co., Ltd, as a speaker for AstraZeneca K.K., Bayer Yakuhin, Ltd, Daiichi Sankyo Co., Ltd, Eli Lilly Japan K.K., Kowa Pharmaceutical Co., Ltd, Mitsubishi Tanabe Pharma Corp., MSD K.K., Nippon Boehringer Ingelheim Co., Ltd, Novartis Pharma K.K., Novo Nordisk Pharma Ltd, Ono Pharmaceutical Co., Ltd, Sanofi K.K., Sanwa Kagaku Kenkyusho Co., Ltd, Shionogi & Co., Ltd, Takeda Pharmaceutical Co., Ltd, and has obtained research support from AstraZeneca K.K., Astellas Pharma, Inc., Bayer Yakuhin, Ltd, Daiichi Sankyo Co., Ltd, Dainippon Sumitomo Pharma Co., Ltd, Eli Lilly Japan K.K., Mitsubishi Tanabe Pharma Corp., MSD K.K., Nippon Boehringer Ingelheim Co., Ltd, Novartis Pharma K.K., Novo Nordisk Pharma Ltd, Ono Pharmaceutical Co., Ltd, Pfizer Japan, Inc., Sanofi K.K., Sanwa Kagaku Kenkyusho Co., Ltd, Shionogi & Co., Ltd, and Takeda Pharmaceutical Co., Ltd. T. O, A. S., K. O., J. C. A. and B. J. G. are current or former employees of MSD K.K. or Merck & Co., Inc., Whitehouse Station, NJ, and may own stock/stock options in the company. No other potential conflicts of interest relevant to this article are reported.
K. K., T. K. and Y. T. contributed to finalization of the study protocol with provision of substantive suggestions for the study design, interpreted the results, provided substantive suggestions for the initial draft of the manuscript, critically reviewed subsequent iterations of the manuscript and reviewed and approved the final version of the manuscript. T. O. conceived, designed and planned the study, interpreted the results, wrote sections of the initial draft, provided substantive suggestions for revision, critically reviewed subsequent iterations of the manuscript, and reviewed and approved the final version of the manuscript. A. S., J. C. A. and B. J. G. conceived, designed and planned the study, interpreted the results, provided substantive suggestions for revision, critically reviewed subsequent iterations of the manuscript and reviewed and approved the final version of the manuscript. K. O. performed and supervised analyses, interpreted the results, provided substantive suggestions for revision, critically reviewed subsequent iterations of the manuscript, provided statistical expertise and reviewed and approved the final version of the manuscript. T. O. is the guarantor of this work and, as such, had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Parts of this study were presented in abstract form at the International Diabetes Federation Western Pacific Region meeting in November 2012.
Supporting information
Figure S1. Study design.
Table S1. Incremental change from baseline in efficacy endpoints.
Appendix S1. Study investigators.
Acknowledgements
The authors wish to thank the study investigators (Appendix S1) for their contributions to the execution of this study, and Edward A. O'Neill, Jennifer Rotonda, and Kristen Lewis (employees of Merck & Co., Inc., Whitehouse Station, NJ, USA) for editorial assistance and manuscript preparation. This study was sponsored by MSD KK, a subsidiary of Merck & Co., Inc., Whitehouse Station, NJ, the manufacturer of sitagliptin.
The copyright line for this article was changed on September 17, 2015 after original online publication.
References
- 1. Report of the expert committee on the diagnosis and classification of diabetes mellitus. Diabetes Care 1997; 20: 1183–1197. [DOI] [PubMed] [Google Scholar]
- 2. Glucose tolerance and cardiovascular mortality: comparison of fasting and 2‐hour diagnostic criteria. Arch Intern Med 2001; 161: 397–405. [DOI] [PubMed] [Google Scholar]
- 3. Nakagami T; DECODA Study Group . Hyperglycaemia and mortality from all causes and from cardiovascular disease in five populations of Asian origin. Diabetologia 2004; 47:385–394. [DOI] [PubMed] [Google Scholar]
- 4. Tominaga M, Eguchi H, Manaka H, Igarashi K, Kato T, Sekikawa A. Impaired glucose tolerance is a risk factor for cardiovascular disease, but not impaired fasting glucose. The Funagata diabetes study. Diabetes Care 1999; 22: 920–924. [DOI] [PubMed] [Google Scholar]
- 5. Kahn SE, Haffner SM, Heise MA et al. Glycemic durability of rosiglitazone, metformin, or glyburide monotherapy. N Engl J Med 2006; 355: 2427–2443. [DOI] [PubMed] [Google Scholar]
- 6. Stratton IM, Adler AI, Neil HA et al. Association of glycaemia with macrovascular and microvascular complications of type 2 diabetes (UKPDS 35): prospective observational study. Br Med J 2000; 321: 405–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Turner RC, Cull CA, Frighi V, Holman RR. Glycemic control with diet, sulfonylurea, metformin, or insulin in patients with type 2 diabetes mellitus: progressive requirement for multiple therapies (UKPDS 49). UK Prospective Diabetes Study (UKPDS) Group. J Am Med Assoc 1999; 281: 2005–2012. [DOI] [PubMed] [Google Scholar]
- 8. Gerstein HC, Santaguida P, Raina P et al. Annual incidence and relative risk of diabetes in people with various categories of dysglycemia: a systematic overview and meta‐analysis of prospective studies. Diabetes Res Clin Pract 2007; 78: 305–312. [DOI] [PubMed] [Google Scholar]
- 9. Rosenstock J. Reflecting on type 2 diabetes prevention: more questions than answers!. Diabetes Obes Metab 2007; 9(Suppl. 1): 3–11. [DOI] [PubMed] [Google Scholar]
- 10. Knowler WC, Barrett‐Connor E, Fowler SE et al. Reduction in the incidence of type 2 diabetes with lifestyle intervention or metformin. N Engl J Med 2002; 346: 393–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Gerstein HC, Yusuf S, Bosch J et al. Effect of rosiglitazone on the frequency of diabetes in patients with impaired glucose tolerance or impaired fasting glucose: a randomised controlled trial. Lancet 2006; 368: 1096–1105. [DOI] [PubMed] [Google Scholar]
- 12. DeFronzo RA, Tripathy D, Schwenke DC et al. Pioglitazone for diabetes prevention in impaired glucose tolerance. N Engl J Med 2011; 364: 1104–1115. [DOI] [PubMed] [Google Scholar]
- 13. Chiasson JL, Josse RG, Gomis R, Hanefeld M, Karasik A, Laakso M. Acarbose for prevention of type 2 diabetes mellitus: the STOP‐NIDDM randomised trial. Lancet 2002; 359: 2072–2077. [DOI] [PubMed] [Google Scholar]
- 14. Kawamori R, Tajima N, Iwamoto Y, Kashiwagi A, Shimamoto K, Kaku K. Voglibose for prevention of type 2 diabetes mellitus: a randomised, double‐blind trial in Japanese individuals with impaired glucose tolerance. Lancet 2009; 373: 1607–1614. [DOI] [PubMed] [Google Scholar]
- 15. Standards of medical care in diabetes‐2015. Diabetes Care 2015; 38 (Suppl. 1):S1–S94. [DOI] [PubMed] [Google Scholar]
- 16. Alberti KG, Zimmet P, Shaw J. International Diabetes Federation: a consensus on type 2 diabetes prevention. Diabet Med 2007; 24: 451–463. [DOI] [PubMed] [Google Scholar]
- 17. Evidence‐Based Practice Guideline for the Treatment of Diabetes in Japan 2013. Tokyo: Nankodo Co., Ltd, 2013. [Google Scholar]
- 18. Xu L, Man CD, Charbonnel B et al. Effect of sitagliptin, a dipeptidyl peptidase‐4 inhibitor, on beta‐cell function in patients with type 2 diabetes: a model‐based approach. Diabetes Obes Metab 2008; 10: 1212–1220. [DOI] [PubMed] [Google Scholar]
- 19. Brazg R, Xu L, Dalla MC, Cobelli C, Thomas K, Stein PP. Effect of adding sitagliptin, a dipeptidyl peptidase‐4 inhibitor, to metformin on 24‐h glycaemic control and beta‐cell function in patients with type 2 diabetes. Diabetes Obes Metab 2007; 9: 186–193. [DOI] [PubMed] [Google Scholar]
- 20. Rosenstock J, Foley JE, Rendell M et al. Effects of the dipeptidyl peptidase‐IV inhibitor vildagliptin on incretin hormones, islet function, and postprandial glycemia in subjects with impaired glucose tolerance. Diabetes Care 2008; 31: 30–35. [DOI] [PubMed] [Google Scholar]
- 21. Hage C, Brismar K, Efendic S, Lundman P, Ryden L, Mellbin L. Sitagliptin improves beta‐cell function in patients with acute coronary syndromes and newly diagnosed glucose abnormalities‐the BEGAMI study. J Intern Med 2013; 273: 410–421. [DOI] [PubMed] [Google Scholar]
- 22. Kashiwagi A, Kasuga M, Araki E et al. International clinical harmonization of glycated hemoglobin in Japan: from Japan Diabetes Society to National Glycohemoglobin Standardization Program values. J Diabetes Invest 2012; 3: 39–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Tatosian DA, Guo Y, Schaeffer AK et al. Dipeptidyl peptidase‐4 inhibition in patients with type 2 diabetes treated with saxagliptin, sitagliptin, or vildagliptin. Diabetes Ther 2013; 4: 431–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Liang KY, Zeger SL. Longitudinal data analysis of continuous and discrete responses for pre‐post designs. Indian J Stat 2000; 62: 134–148. [Google Scholar]
- 25. Miettinen O, Nurminen M. Comparative analysis of two rates. Stat Med 1985; 4: 213–226. [DOI] [PubMed] [Google Scholar]
- 26. Thornberry NA, Weber AE. Discovery of JANUVIA (Sitagliptin), a selective dipeptidyl peptidase IV inhibitor for the treatment of type 2 diabetes. Curr Top Med Chem 2007; 7: 557–568. [DOI] [PubMed] [Google Scholar]
- 27. U.S. prescribing information for JANUVIA® (sitagliptin) tablets. February 2014. Available from URL: https://www.merckconnect.com/januvia/overview.html. Accessed 22 October 2014.
- 28. Mitrakou A, Kelley D, Mokan M et al. Role of reduced suppression of glucose production and diminished early insulin release in impaired glucose tolerance. N Engl J Med 1992; 326: 22–29. [DOI] [PubMed] [Google Scholar]
- 29. Herman GA, Bergman A, Stevens C et al. Effect of single oral doses of sitagliptin, a dipeptidyl peptidase‐4 inhibitor, on incretin and plasma glucose levels after an oral glucose tolerance test in patients with type 2 diabetes. J Clin Endocrinol Metab 2006; 91: 4612–4619. [DOI] [PubMed] [Google Scholar]
- 30. Engel SS, Round E, Golm GT, Kaufman KD, Goldstein BJ. Safety and tolerability of sitagliptin in type 2 diabetes: pooled analysis of 25 clinical studies. Diabetes Ther 2013; 4: 119–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Bock G, Dalla MC, Micheletto F et al. The effect of DPP‐4 inhibition with sitagliptin on incretin secretion and on fasting and postprandial glucose turnover in subjects with impaired fasting glucose. Clin Endocrinol (Oxf) 2010; 73: 189–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Utzschneider KM, Tong J, Montgomery B et al. The dipeptidyl peptidase‐4 inhibitor vildagliptin improves beta‐cell function and insulin sensitivity in subjects with impaired fasting glucose. Diabetes Care 2008; 31: 108–113. [DOI] [PubMed] [Google Scholar]
- 33. Ahren B. Use of DPP‐4 inhibitors in type 2 diabetes: focus on sitagliptin. Diabetes Metab Syndr Obes 2010; 3: 31–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Study design.
Table S1. Incremental change from baseline in efficacy endpoints.
Appendix S1. Study investigators.