

Abstract
Studies that investigated the genetic basis of source and sink related traits have been widely conducted. However, the vascular system that links source and sink received much less attention. When maize was domesticated from its wild ancestor, teosinte, the external morphology has changed dramatically; however, less is known for the internal anatomy changes. In this study, using a large maize‐teosinte experimental population, we performed a high‐resolution quantitative trait locus (QTL) mapping for the number of vascular bundle in the uppermost internode of maize stem. The results showed that vascular bundle number is dominated by a large number of small‐effect QTLs, in which a total of 16 QTLs that jointly accounts for 52.2% of phenotypic variation were detected, with no single QTL explaining more than 6% of variation. Different from QTLs for typical domestication traits, QTLs for vascular bundle number might not be under directional selection following domestication. Using Near Isogenic Lines (NILs) developed from heterogeneous inbred family (HIF), we further validated the effect of one QTL qVb9‐2 on chromosome 9 and fine mapped the QTL to a 1.8‐Mb physical region. This study provides important insights for the genetic architecture of vascular bundle number in maize stem and sets basis for cloning of qVb9‐2.
Keywords: Fine‐mapping, genetic architecture, maize, quantitative trait locus, vascular bundle number
Edited by: Sanwen Huang, The Institute of Vegetable and Flowers, CAAS, China
INTRODUCTION
The grain yield of crop is highly dependent on the source‐sink relationship in which grain or kernel is the photosynthetic sink while the leaves are the primary source. The vascular bundle is the transport system that links source and sink, and it determines how plants efficiently transport photosynthetic products, mineral nutrients and water (Housley and Peterson 1982). Significant correlations between vascular bundle system and grain yield have been observed in crops (Evans et al. 1970; Peterson et al. 1982; Nátrová 1991). The capacity of the vascular bundle system for efficiently transporting assimilates from the source to the sink has been shown as a limiting factor for yield potential realization (Housley and Peterson 1982). Understanding the genetic basis of vascular bundle system will provide useful information for the genetic improvement of yield potential.
Over the past decade, considerable progress has been made in understanding the developmental and physiological programs involved in the formation and function of the plant vascular bundle system (Lucas et al. 2013; Qiang et al. 2013; Sawchuk and Scarpella 2013). Many studies have been conducted to identify genes related to vascular bundle system in plants. In model species Arabidopsis, through characterizing mutants with defects in vascular system, a number of genes involved in vascular patterning have been identified, such as PHB, PHV, REC, and MP (Hardtke and Berleth 1998; Emery et al. 2003; Zhong and Ye 2004). In crops, wide natural variation in vascular bundle system has been observed. Using the strategy of QTL mapping, a number of QTLs for vascular bundle system have been mapped in tomato (Coaker et al. 2002), wheat (Sang et al. 2010), and rice (Sasahara et al. 1999; Zhang et al. 2002; Cui et al. 2003; Bai et al. 2012). Notably, several QTLs that affect the vascular bundle system in rice have been further cloned by positional cloning. For instance, genes such as APO1 and NAL1 have been shown to involve in enhanced translocation capacity of vascular bundles (Qi et al. 2008; Terao et al. 2010; Fujita et al. 2013). In contrast, the genetic studies for vascular bundle system in maize received much less attention.
Additionally, the differentiation in vascular bundle system is an important parameter that defines differentiation between species or subspecies. For example, in rice, indica varieties tend to have more large vascular bundles in peduncle than japonica varieties, which is highly related to the fact that indica varieties generally have more grains per panicle than japonica (Sasahara et al. 1982; Fukuyama and Takayama 1995; Zhang et al. 2002). Maize (Zea mays ssp. mays) was domesticated from its wild ancestor, Balsas teosinte (Zea mays ssp. parviglumis) approximately 9,000 years ago in southwestern Mexico (Matsuoka et al. 2002). During the domestication process, the plant architecture and inflorescence morphology have changed dramatically (Doebley 2004). These external radical morphological changes might cause synergetic changes in the internal anatomy, especially in vascular bundle system that interconnects all plant organs and provides mechanical support for the plants. Over the past few years, several genes controlling the critical morphological transformations during maize domestication have been cloned (Studer et al. 2011; Wills et al. 2013; Yang et al. 2013); however, the associated changes in vascular bundle system following domestication and the underlying genetic architecture have not been investigated yet. Characterizing the genetic changes in the vascular bundle system might shed new insights into the maize domestication process.
In this study, using a large population of 866 maize‐teosinte BC2S3 recombinant inbred lines (RILs) that has been previously genotyped at 19,838 SNP markers (Shannon 2012), we performed a high‐resolution QTL mapping for the vascular bundle number of the uppermost internode of maize stem and characterized the underlying genetic architecture. Through developing NILs from HIF that only segregates for the target QTL and employing a within‐family comparison strategy, we further validated the QTL effect of qVb9‐2 and narrowed down the QTL to a 1.8‐Mb physical region.
RESULTS
Phenotypic analysis
Wide phenotypic variation in the vascular bundle number of the uppermost internode of maize stem was observed in BC2S3 RILs population, with the minimum vascular bundle number of 69 and the maximum vascular bundle number of 172 (Figure 1). Because the BC2S3 RIL population was derived from a cross between a maize inbred W22 and a natural accession of teosinte (Zea mays ssp. parviglumis), the teosinte parent is now not available to score the vascular bundle number in stems. The maize parent W22 has 124 vascular bundles on average (Figure 1). The frequency distribution of vascular bundle number in BC2S3 RILs population exhibited a continuous variation (Figure 1), suggesting that vascular bundle number is a typical quantitative trait controlled by polygenes.
Figure 1.
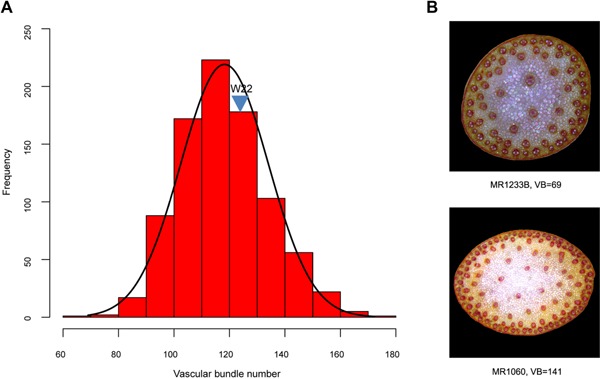
Phenotypic variation of vascular bundle number of maize stem in the maize‐teosinte BC2S3 recombinant inbred line (RIL) population (A) Phenotypic distribution of vascular bundle number in the uppermost internode of maize stem in BC2S3 RIL population. The x‐axis shows the vascular bundle number, and the y‐axis represents the number of RILs in each group. The vascular bundle number of W22 was indicated by triangle. (B) The number of vascular bundles in the uppermost internode of stem is shown for two lines. RILs MR1233B and MR1060 contain 69 and 141 vascular bundles, respectively.
QTLs for vascular bundle number
Using the multiple QTL model implemented in R/qtl (Broman et al. 2003), a total of 16 QTLs were detected for the vascular bundle number of the uppermost internodes of maize plants (Figure 2, Table 1). These QTLs were distributed across all 10 chromosomes and could jointly account for 52.2% of the total phenotypic variation. Significant additive effects were detected at all of the mapped QTLs, with additive effects ranging from 2.6 to 6.9 (Table 1). Only four QTLs, including qVb2‐1, qVb4‐1, qVb9‐1, and qVb9‐2, showed both significant additive and dominance effects (Table 1). The other 12 QTLs were due to additive effects only (Table 1). No single QTL accounted for a large proportion of phenotypic variation. The phenotypic variation explained by individual QTL ranged from 1.7% for qVb6‐1 to 5.6% for qVb9‐2, with an average of 3.3% (Table 1). The largest‐effect QTL qVb9‐2 is located at the region between 114.8 and 127.5 Mb on chromosome 9 (2‐LOD support interval), with the teosinte allele increasing 6.9 vascular bundles relative to the maize allele. The 2‐LOD support intervals for all QTLs range in genetic size from 3cM for qVb4‐1 to 22.6cM for qVb6‐1, with an average support interval size of 6.8cM (Table 1). In physical interval size, these QTLs range from 1.7 Mb for qVb4‐1 to 89.3 Mb for qVb6‐1. The teosinte alleles at 10 of 16 QTLs (62.5%) showed the effects of increasing vascular bundle number (Table 1).
Figure 2.
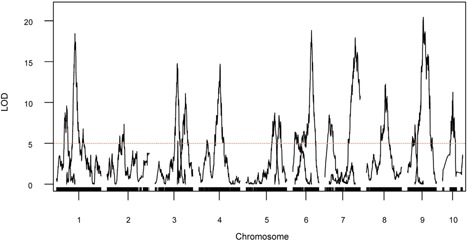
Genome‐wide logarithm of odds (LOD) plots for quantitative trait loci (QTLs) controlling vascular bundle number in BC2S3 recombinant inbred line (RIL) population Tick marks along the bottom axis represent genetic markers, curves represent LOD scores for QTL at each genomic position. The x‐axis shows the genetic position along the chromosomes in cM. The dotted horizontal line at LOD of 5 represents the threshold for significance as determined by 10,000 permutations of the data.
Table 1.
Summary of quantitative trait loci (QTLs) for vascular bundle number in the BC2S3 recombinant inbred line (RIL) population
| Chr | QTL | Left genetic position (cM) | Right genetic position (cM) | 2‐LOD support interval (cM) | 2‐LOD support interval (Mb) | LOD | Additive effect | P‐value of additive effect | Dominance effect | P‐value of dominance effect | PVE (%) |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | qVb1‐1 | 41.5 | 47.1 | 5.6 | 4.7 | 9.68 | 3.16 | 5.65E‐08 | 1.85 | 0.14 | 2.57 |
| 1 | qVb1‐2 | 77 | 82.1 | 5.1 | 5.5 | 18.65 | 5.54 | 4.22E‐18 | −1.06 | 0.40 | 5.08 |
| 2 | qVb2‐1 | 64.1 | 72.9 | 8.8 | 2 | 7.33 | 2.64 | 2.66E‐04 | 3.89 | 0.01 | 1.94 |
| 3 | qVb3‐1 | 93 | 97 | 4 | 2.2 | 14.67 | 5.76 | 6.14E‐14 | −1.14 | 0.41 | 3.95 |
| 3 | qVb3‐2 | 126.3 | 132.6 | 6.3 | 2.3 | 11.04 | −4.35 | 4.57E‐11 | 0.32 | 0.82 | 2.94 |
| 4 | qVb4‐1 | 88.5 | 91.5 | 3 | 1.7 | 14.85 | 4.35 | 8.23E‐11 | 2.91 | 0.04 | 4.00 |
| 5 | qVb5‐1 | 117.3 | 125.5 | 8.2 | 10.7 | 8.75 | 4.06 | 8.45E‐07 | 2.65 | 0.11 | 2.32 |
| 5 | qVb5‐2 | 136.5 | 143.5 | 7 | 1.9 | 8.45 | −3.8 | 1.11E‐07 | −1.57 | 0.30 | 2.24 |
| 6 | qVb6‐1 | 7.3 | 29.9 | 22.6 | 89.3 | 6.4 | 2.73 | 1.87E‐06 | 1.02 | 0.46 | 1.68 |
| 6 | qVb6‐2 | 77.8 | 81 | 3.2 | 3.9 | 18.9 | 5.44 | 4.02E‐18 | −0.49 | 0.73 | 5.15 |
| 7 | qVb7‐1 | 15.9 | 23.7 | 7.8 | 2.2 | 8.37 | −3.36 | 1.47E‐07 | −0.91 | 0.52 | 2.22 |
| 7 | qVb7‐2 | 127.1 | 132.6 | 5.5 | 2.1 | 17.89 | −4.62 | 1.13E‐14 | −1.77 | 0.18 | 4.86 |
| 8 | qVb8‐1 | 79.4 | 83 | 3.6 | 7.6 | 12.43 | −4.14 | 1.35E‐11 | −0.18 | 0.89 | 3.33 |
| 9 | qVb9‐1 | 30.5 | 35 | 4.5 | 3.6 | 7.2 | −3.61 | 2.51E‐08 | 3.96 | 4.00E‐03 | 1.90 |
| 9 | qVb9‐2 | 64.9 | 75 | 10.1 | 12.7 | 20.48 | 6.91 | 3.32E‐21 | −4.04 | 5.43E‐03 | 5.60 |
| 10 | qVb10‐1 | 42.2 | 45.3 | 3.1 | 10.6 | 11.21 | 6.74 | 7.65E‐11 | −1.96 | 0.29 | 2.99 |
The physical and genetic positions of the 2‐LOD support intervals for each QTL are shown. LOD scores, additive effect, P‐value of additive effect, dominance effect, P‐value of dominance effect and percent variance explained (PVE) were calculated for each QTL. Positive additive effect (+) indicates that the teosinte allele increased the number of vascular bundles. Negative additive effect (−) indicates that the maize allele increased the number of vascular bundles.
Heterogeneous Inbred Family (HIF) for qVb9‐2
To fine map the largest‐effect QTL qVb9‐2, a RIL (MR1480) that is heterozygous across the 2‐LOD support interval of qVb9‐2 was selected for developing NILs (Figure 3). In MR1480, in addition to the heterozygous introgression at qVb9‐2, there are another six homozygous or heterozygous introgressions, but those segments do not contain any mapped QTLs for vascular bundle number. Therefore, progeny from selfing MR1480 will be only segregating for qVb9‐2. This strategy of using Heterogeneous Inbred Family (HIF) (Tuinstra et al. 1997) has been widely used in QTL fine mapping.
Figure 3.
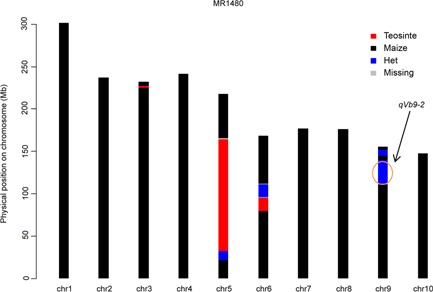
Graphical genotypes of a heterogeneous inbred family (HIF) (MR1480) used for constructing Near Isogenic Lines (NILs) for fine mapping of qVb9‐2 Black indicates regions homozygous for W22; red indicates regions homozygous for teosinte; blue indicates heterozygous regions; grey indicates unknown regions. The introgressions at target QTL region are indicated by arrows.
The phenotypic effects of qVb9‐2
Using the NIL pair (NIL_maize and NIL_teosinte, homozygous for maize and teosinte at qVb9‐2, respectively), the phenotypic effects of qVb9‐2 on a number of important agronomic traits were evaluated. As shown in Table 2, NIL_teosinte contain 15 more vascular bundle than NIL_maize (P = 8.5E‐11), consistent with the mapping results in BC2S3 population. Significant phenotypic difference was detected in total leaf number and tassel branch number between NIL_maize and NIL_teosinte. No significant difference was detected in leaf width, cob diameter, 100 kernel weight, 20 kernel length, 20 kernel width, and kernel row number between NIL_maize and NIL_teosinte.
Table 2.
The phenotypic effects of qVb9‐2
| Trait | NIL_maize | NIL_teosinte | P‐value |
|---|---|---|---|
| Vascular bundle number | 137 (8.68) | 152 (12.19) | 8.50E‐11 |
| Leaf width (cm) | 10.17 (0.56) | 10.2 (0.57) | 0.7695 |
| Total leaf number | 18.6 (0.66) | 19.35 (0.79) | 5.62E‐07 |
| Tassel branch number | 14.93 (3.01) | 18 (3.1) | 4.59E‐07 |
| Cob diameter (mm) | 18.07 (2.16) | 17.48 (2.28) | 0.199 |
| 100 Kernel weight (g) | 16.49 (2.18) | 16.31 (1.9) | 0.7724 |
| 20 Kernel length (cm) | 16.31 (0.78) | 16.31 (0.63) | 0.9986 |
| 20 Kernel width (cm) | 12.98 (0.62) | 12.94 (0.55) | 0.8253 |
| Kernel row number | 13.04 (1.37) | 13.22 (1.4) | 0.6573 |
NIL_maize and NIL_teosinte are the NIL homozygous for maize and teosinte at qVb9‐2, respectively. The values are shown in Mean (Sd) with n > 30 in maize_NIL and teosinte_NIL. P‐values are the significance of phenotypic difference between NIL_maize and NIL_teosinte (t‐tests).
Fine mapping of qVb9‐2
To further resolve the position of qVb9‐2, an F2 population containing 1,036 plants was created by self‐fertilizing the HIF family MR1480. Using markers M114.9 and M123.8 that flank the 2‐LOD support interval of qVb9‐2, a total of 80 recombinants were identified from the F2 population. To more precisely determine the recombination breakpoints, four additional markers between markers M114.9 and M123.8 were developed and used to genotype all recombinants. According to the marker genotypes at six indel markers, the 80 recombinants can be divided into 10 genotype groups. Due to the limited field space of Beijing trial in 2014, for each genotype group, only one recombinant family that contained more than 300 F3 seeds was selected and planted in Beijing for phenotypic testing.
Within each F3 family, homozygous recombinant (HR) and homozygous non‐recombinant (HNR) plants were identified with appropriate markers and evaluated for vascular bundle number in stem. Phenotypic difference in vascular bundle number was tested between HR and HNR pair within each family and the difference in significance was used to determine the genotype of gene in the parental recombinant (see Methods for details). Following this approach, a stepwise substitution mapping was conducted across 10 recombinant families to delimit the causal region of qVb9‐2.
As shown in Figure 4, genotype group IV, IX, and X carried recombination events between marker M114.9 and M116.2. Significant phenotypic difference in vascular bundle number was detected between HR and HNR pair within the family of group IV and X, whereas no significant phenotypic difference was detected within group IX. The phenotypic comparisons between HR and HNR pair within these three families consistently indicated that the causal variant of qVb9‐2 is located in a region downstream of marker M114.9. Group III and VIII carried recombinations between marker M116.2 and M118.5. Significant phenotypic difference was detected between HR and HNR pair within both groups, consistently suggesting that the causal region of qVb9‐2 is located in the downstream of marker M116.2. With a similar procedure, group V that carried a recombination between marker M120.9 and M123.8 placed qVb9‐2 to a region upstream of M123.8. Group I and VI that carried recombinations between marker M120.2 and M120.9 placed qVb9‐2 to a region upstream of M120.2. The most informative comparisons are from the group II and VII that carried recombination events between marker M118.5 and M120.9. Both groups delimited qVb9‐2 to a 1.8 Mb region between markers M118.5 and M120.2 (Figure 4).
Figure 4.
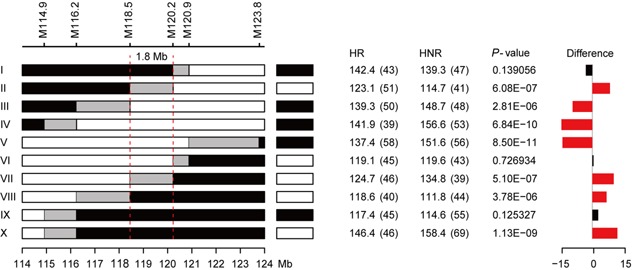
Fine mapping of qVb9‐2 The recombinants identified in F2 population were grouped into ten groups based on their genotypes. Graphical genotypes of identified homozygous recombinants (HR) at the target region within each recombinant group are shown on left. Black or white boxes next to the HR indicate the corresponding homozygous non‐recombinants (HNR) identified within each recombinant family. Black, white and gray boxes indicate homozygous for maize allele, homozygous for teosinte allele, and regions where recombination occurred, respectively. The relevant markers are displayed at the top. The graphs on the right are the phenotypic difference comparison between HR and HNR pair for each recombinant group. The values in the columns of HR and HNR are the phenotypic mean for homozygous recombinants and homozygous nonrecombinants. The values in parentheses are the sample size for HR and HNR. The P‐value column is the exact P‐value for the comparison between HR and HNR pair. Red bars indicate significant difference at P < 0.01 after Bonferroni correction. Black bars indicate no significant difference observed between HR and HNR pair.
Candidate genes in the 1.8 Mb target region
Based on the gene annotation of B73 reference genome (Schnable et al. 2009), there are 32 genes located in the 1.8 Mb target region (Table 3). Among these genes, five of them are transcription factors. Of them, AC197901.3_FG003, GRMZM2G161255, GRMZM2G345155, and AC201740.3_FG003 belong to C2H2 zinc‐finger family protein. GRMZM2G098179 belongs to MYB family. Another gene, GRMZM2G424075 encodes sterol methyltransferase 2 (SMT2).
Table 3.
Candidate genes within 1.8‐Mb physical regions
| Gene id | Chromosome | Gene start (bp) | Gene end (bp) | Gene annotation in Arabidopsis or rice |
|---|---|---|---|---|
| GRMZM2G113873 | 9 | 118,450,767 | 118,456,524 | Pyridoxal phosphate (PLP)‐dependent transferases superfamily protein |
| AC197901.3_FG003 | 9 | 118,759,325 | 118,759,666 | Zinc finger protein 2 |
| GRMZM2G161255 | 9 | 118,825,230 | 118,825,766 | Zinc finger protein 2 |
| GRMZM2G345155 | 9 | 118,900,667 | 118,901,349 | C2H2 and C2HC zinc fingers superfamily protein |
| AC201740.3_FG003 | 9 | 118,908,308 | 118,908,703 | C2H2 and C2HC zinc fingers superfamily protein |
| GRMZM2G424075 | 9 | 118,929,862 | 118,930,502 | Sterol methyltransferase 2 |
| GRMZM2G126120 | 9 | 118,937,736 | 118,942,951 | RNA‐binding KH domain‐containing protein |
| GRMZM2G126216 | 9 | 118,946,617 | 118,948,213 | Protein of unknown function (DUF594) |
| AC194940.3_FG001 | 9 | 119,056,647 | 119,056,907 | Unknown function |
| GRMZM2G028921 | 9 | 119,131,712 | 119,134,566 | Polypyrimidine tract‐binding protein 1 |
| GRMZM2G500076 | 9 | 119,145,197 | 119,145,531 | Unknown function |
| AC190743.3_FG012 | 9 | 119,202,475 | 119,202,907 | Polypyrimidine tract‐binding protein 1 |
| GRMZM2G097987 | 9 | 119,288,007 | 119,290,748 | GDA1/CD39 nucleoside phosphatase family protein |
| GRMZM2G098079 | 9 | 119,300,411 | 119,307,758 | Transferases, transferring acyl groups |
| GRMZM2G098179 | 9 | 119,310,947 | 119,312,449 | Myb domain protein 96 |
| GRMZM2G417255 | 9 | 119,425,016 | 119,426,143 | Unknown function |
| GRMZM2G117680 | 9 | 119,429,148 | 119,432,667 | S‐adenosyl‐L‐methionine‐dependent methyltransferases superfamily protein |
| GRMZM2G045518 | 9 | 119,443,263 | 119,444,157 | S‐adenosyl‐L‐methionine‐dependent methyltransferases superfamily protein |
| GRMZM2G052654 | 9 | 119,473,033 | 119,477,758 | Mucin‐related |
| GRMZM5G832108 | 9 | 119,492,893 | 119,495,217 | Ribosomal protein S4 |
| GRMZM5G854500 | 9 | 119,591,895 | 119,593,809 | 3‐ketoacyl‐CoA synthase 12 |
| GRMZM2G451861 | 9 | 119,658,310 | 119,660,247 | Unknown function |
| GRMZM2G151476 | 9 | 119,658,410 | 119,660,324 | 3‐ketoacyl‐CoA synthase 12 |
| GRMZM2G151387 | 9 | 119,710,078 | 119,715,669 | SAP domain‐containing protein |
| GRMZM2G093092 | 9 | 119,779,040 | 119,780,565 | O‐methyltransferase family protein |
| GRMZM2G106172 | 9 | 119,838,646 | 119,840,122 | O‐methyltransferase family protein |
| GRMZM2G013934 | 9 | 119,893,667 | 119,899,665 | DNAJ heat shock family protein |
| GRMZM2G035517 | 9 | 119,945,144 | 119,948,254 | Serine carboxypeptidase‐like 40 |
| GRMZM2G035531 | 9 | 119,969,236 | 119,970,106 | VQ motif‐containing protein |
| GRMZM2G128491 | 9 | 120,079,591 | 120,082,284 | Unknown function |
| GRMZM2G093096 | 9 | 120,156,896 | 120,162,159 | Dentin sialophosphoprotein‐related |
| GRMZM2G000209 | 9 | 120,228,025 | 120,230,405 | Nodulin MtN21/EamA‐like transporter family protein |
DISCUSSION
In this study, a previously well‐characterized maize‐teosinte BC2S3 RIL population (Shannon 2012) was used to dissect the genetic architecture of vascular bundle number of the uppermost internode of maize stem. This BC2S3 population consisted of 866 RILs and has been genotyped at 19,838 SNP markers (Shannon 2012). Multiple generations of recombination, large population size and high‐density molecular markers allowed for QTL mapping with enhanced statistical power and mapping resolution. The mapping results showed that the average 2‐LOD QTL support interval for vascular bundle number is only 6.8cM in genetic size and 10.2 Mb in physical size. This mapping resolution is much higher than that of most previous QTL mapping studies that generally used a population size of 100–200 lines and hundreds of markers for mapping (Salvi and Tuberosa 2005; Price 2006). Relatively short initial mapping intervals provide promising targets for further experimental validation and saves time for further fine mapping. In addition to the increased mapping resolution, this large BC2S3 RIL population enabled more accurate inference on the genetic architecture of trait of interest. It is well known that small population is underpowered in detecting small‐effect QTLs and tend to overestimate QTL effects (known as Beavis effect; Beavis 1998). Limited molecular marker density further limits to separate closely linked QTLs. In small populations genotyped by limited number of markers, the lack of power in identifying small‐effect QTLs and the biased effect estimation cause biased inference on the genetic architecture of the trait of interest. It has been shown by simulation and empirical data that the large maize‐teosinte BC2S3 RIL population is powerful in dissecting the genetic architecture of complex traits (Shannon 2012).
Using the BC2S3 population, Shannon (2012) conducted a comprehensive genetic dissection for 16 important domestication related traits. The results showed that the genetic architecture varied across traits, and the genetic architecture can be classified into three classes. The first class of genetic architecture showed relatively simple inheritance, in which a single QTL can explain the majority of variation. Traits such as disarticulation and prolificacy belong to this class. A second class of genetic architecture is polygenic, involving a large number of small‐effect QTLs with no single large effect QTL detected. Traits such as ear length and kernel weight belong to this class. A final class is mixed including both a single large‐effect QTL plus a large number of QTLs with moderate to small effect. Traits including ear diameter and days to pollen belong to this type. In this study, using the same population, a total of 16 QTLs were identified for the number of vascular bundle in the uppermost internode of maize stem. Although these QTLs can jointly explain 52.2% of total phenotypic variation, individual QTL can only explain 3.3% of variation on average. No single QTL can account for a large proportion of phenotypic variation. The largest effect QTL qVb9‐2 can only explain 5.6% of phenotypic variation. These results suggested that vascular bundle number is under the control of the second class of genetic architecture described above. Namely, vascular bundle number is dominated by a large number of small‐effect QTLs. It has been shown that, for typical domestication traits, QTLs for them tend to be under directional selection during maize domestication (Shannon 2012). For example, all 21 QTLs for kernel weight acted in the same direction with the teosinte allele consistently conditioning lighter kernels (Shannon 2012). However, for vascular bundle number examined in this study, we detected a mix of positive and negative acting effects, with 62.5% of QTLs showing effects of increasing vascular bundle number and 37.5% decreasing vascular bundle number. This result might suggest that, different from typical domestication traits, the QTLs for vascular bundle number might not be under strong directional selection following maize domestication. This locus level analysis provided genetic evidence that the internal anatomy such as vascular bundle number was not a direct target of selection during domestication. The observed changes in vascular bundle number are more likely the indirect results from the strong direct selection on other traits such as plant architecture, inflorescence morphology and yield traits. More physiological and genetic studies need to be conducted in the future to clarify these relationships and the underlying genetic basis.
To identify causal genes underlying the variation of vascular bundle number, the largest‐effect QTL qVb9‐2, located on the long arm of chromosome 9, was selected for further characterization and fine mapping. A strategy of using heterogeneous inbred family (HIF) (Tuinstra et al. 1997) was used to develop NILs for fine mapping qVb9‐2 in this study. The residual heterozygosity preserved in BC2S3 population provided a unique advantage for finding appropriate HIF for QTL fine mapping. A RIL (MR1480) that is heterozygous across qVb9‐2 was selected for developing NILs for narrowing down the causal region. The use of within‐family comparison in fine mapping has been shown being powerful in controlling both family and environment effects (Hung et al. 2012). Within each recombinant‐derived F3 family, the homozygous recombinants and homozygous non‐recombinants are randomly distributed in the field plots of F3 family, which could efficiently minimize the environmental effects. Because the homozygous non‐recombinants shared the same genetic background as homozygous recombinants, homozygous non‐recombinants thus provide an ideal internal control for testing the phenotypic effects of homozygous recombinants.
Through substitution mapping, qVb9‐2 was finally narrowed down to a 1.8 Mb physical region. There are 32 annotated genes in the 1.8 Mb causal region, including four C2H2 zinc‐finger transcription factors, one MYB transcription factor and a gene encoding sterol methyltransferase 2 (SMT2). C2H2 zinc‐finger and MYB family proteins have been shown to play important roles in plant growth and development (Ciftci‐Yilmaz and Mittler 2008; Dubos et al. 2010). The most interesting candidate gene is GRMZM2G424075 which encodes sterol methyltransferase 2 (SMT2) that catalyze the reaction that distinguishes the synthesis of structural sterols from signaling brassinosteroid derivatives (Carland et al. 2002; Carland et al. 2010). In Arabidopsis, COTYLEDON VASCULAR PATTERNING1 (CVP1) encodes SMT2 and the deficiency of SMT2 in the CVP1 mutant results in aberrant cotyledon vascular patterning (Carland et al. 2002; Carland et al. 2010). Further fine‐mapping by identifying more recombinants in the 1.8 Mb region would help to finally determine the candidate gene controlling the vascular bundle number.
MATERIALS AND METHODS
Plant materials
A large population containing 866 maize‐teosinte BC2S3 RILs, derived from a cross between a typical temperate maize inbred line, W22 and a typical accession of Zea mays ssp. parviglumis, CIMMYT accession 8759, was obtained from Maize Coop Stock Center. The detailed information on this BC2S3 RILs population can be found in Shannon (2012). In brief, W22 was used as the recurrent parent for the two generations of backcrossing. After the second generation of backcrossing, three generations of self‐pollination were conducted. After BC2S3 generation, sib‐mating was conducted to preserve residual heterozygosity and increase seeds. The 866 BC2S3 RILs were genotyped using the genotyping‐by‐sequencing (GBS) technology (Elshire et al. 2011). A total of 51,770 genetic markers were obtained. Marker positions in the genome were determined based on the sequence tags in the B73 reference genome (AGPv2; http://www.maizesequence.org/index.html). Of these 51,770 markers, 32,392 were redundant or non‐informative. At last, a set of 19,838 informative markers remained, with each chromosome containing 2871, 2191, 2052, 2304, 2439, 1621, 1997, 2095, 1653, and 615 markers on chromosome 1–10, respectively. The genetic distances between markers were estimated using R/qtl (Broman et al. 2003). After several steps of imputation (Shannon 2012), the average missing genotype rate for GBS markers is 1.4% in the population. Large sample size and high‐density markers enable this population to be a powerful tool for traits dissection at high resolution, as evidenced by the recent studies on various domestication traits and identifying underlying genes of using this population (Hung et al. 2012; Lin et al. 2012; Wills et al. 2013; Lang et al. 2014).
Field trials and phenotyping
The maize‐teosinte BC2S3 RILs were grown in Shangzhuang Experimental Station of China Agricultural University, Beijing (39.9°N, 116.4°E), China, in the summer of 2013. Due to the limited field space, the 866 BC2S3 RILs were planted in a single replication using an augmented incomplete randomized block design that has been widely used in phenotyping for large populations (Buckler et al. 2009; Kump et al. 2011; Tian et al. 2011). All RILs were divided into 43 incomplete blocks with each block containing 21 RILs. In each incomplete block, two inbreds W22 and Mo17 were randomly inserted as control. Each RIL was planted in three‐row plot, with 15 plants per row, 25 cm between plants within each row and 50 cm between rows. Through selfing a HIF family MR1480 that is heterozygous at QTL qVb9‐2 region (Figure 3), an F2 population containing 1,036 plants was planted to identify recombinants for fine mapping qVb9‐2 at Sanya (18°N, 109°E), Hainan Province, China, in the winter of 2013. NILs that are homozygous for W22 (NIL_maize) and homozygous for teosinte (NIL_teosinte) across the qVb9‐2 region, respectively, were identified using molecular markers flanking the QTL and were further used for evaluating the phenotypic effects of qVb9‐2 on a number of agronomic traits. F3 families derived from F2 recombinants were grown at the same site in Beijing in the summer of 2014. Within each recombinant‐derived F3 family, the homozygous recombinant (HR) and homozygous nonrecombinant (HNR) individuals were identified using markers flanking qVb9‐2. The planting manner of plots for fine mapping materials was the same as that of the BC2S3 population described above.
For BC2S3 population, the uppermost internodes of three plants for each RIL were collected and stored at 4 °C in preparation for scoring the number of vascular bundle. For each recombinant‐derived F3 family, the uppermost internodes of all homozygous recombinant and homozygous nonrecombinant individuals were harvested for scoring vascular bundle number. For each harvested internode, cross‐section slices at the middle part of the internodes were manually made. The slices were then fixed with 5% (g/ml) m‐trihydroxybenzene and stained with concentrated hydrochloric acid. At last, the stained slices were imaged by Leica DFC450 (Germany) microscope and the number of vascular bundle was counted manually. The mean value of vascular bundle number of three plants was calculated for each RIL and used for subsequent data analysis.
Phenotypic data analysis
Following the previous described method that minimizes the effects of environment variation (Buckler et al. 2009; Kump et al. 2011; Tian et al. 2011), a linear mixed model was fitted including the effects of genotypes, incomplete blocks, field ranges and field rows. To further account for the potential field spatial variation, in the mixed model, spatial correlations among residuals were modeled with first‐order autoregressive (AR1 × AR1) residual structures (Gilmour et al. 1997). Likelihood ratio tests were used to determine which effects to retain in the final model. Best linear unbiased predictor (BLUP) for each line was predicted with ASREML software (Gilmour et al. 2009) and then used as the phenotype in the subsequent QTL mapping.
DNA extraction and molecular marker analysis
DNA was extracted from fresh leaves according to the CTAB method (Murray and Thompson 1980) with minor modifications. To fine map qVb9‐2, new markers were developed based on the B73 reference genome v2.0 assembly (B73 RefGen_v2; http://www.maizegdb.org/). Primers were designed using software Primer 3.0 (http://bioinfo.ut.ee/primer3‐0.4.0/) and were then amplified from NIL_maize and NIL_teosinte. A total volume of 10 µl reaction mixture was used for PCR amplification, which is composed of 1 µl template DNA, 0.5 µl of each primer, 3 µl of ddH2O, and 5 µl of 2× Taq PCR StarMix (GenStar). Amplification was performed on program for the initial denaturing step with 95 °C for 5 min, followed by 35 cycles for 45 s at 95 °C, 45 s at 57–60 °C, 45 s at 72°C, with a final extension at 72 °C for 10 min. PCR products were directly sequenced in both directions. Sequence alignments were performed with BioEdit (version 7.0.9.0) and manually edited if necessary. Sequences that showed large insertions or deletions between NILs were further developed into indel markers. The primer sequences of indel markers used in fine mapping can be found in Table 4. The genotypes of recombinants and recombinant‐derived F3 families at indel markers were electrophoretically determined on a 3% agarose gel.
Table 4.
The primer sequence of six markers used for qVb9‐2 fine mapping
| Marker | Type | Chromosome | Physical position (Mb) | Forward primer | Reverse primer |
|---|---|---|---|---|---|
| M114.9 | indel | 9 | 114.92 | GGTTGTTCGTGATGATCCAG | GTGCAACAGCTGGCGACTA |
| M116.2 | indel | 9 | 116.24 | GAACGCCCACCAGGAATCT | TCCCGAGAAAGGGTGACTT |
| M118.5 | indel | 9 | 118.45 | CACGCAATACCGAGATGAGA | GCAGCAGAGGCGCTTATCTA |
| M120.2 | indel | 9 | 120.23 | ACTGCCTCTTGGTGGCTGTC | GATCGATTAACATAGACAGTGAAGTCC |
| M120.9 | indel | 9 | 120.88 | GCCATTGCCAACAACAGTAA | GGAGAGTTGCAACGGCGTA |
| M123.8 | indel | 9 | 123.78 | AACCGCCTAAGACCAAGAGG | GTTTGTCAAGGCCTACTCCG |
QTL mapping
Quantitative trait locus mapping was performed using a modified version of R/qtl, which takes into account the BC2S3 pedigree of the RILs (Shannon 2012). A previously described multiple QTL mapping procedure (Shannon 2012) was used to identify QTL for vascular bundle number. This mapping procedure has been shown to be a powerful method to identify small‐effect QTLs and separate linked QTLs in various phenotypic data (Hung et al. 2012; Lin et al. 2012; Shannon 2012; Wills et al. 2013). In detail, the simple interval mapping using Haley‐Knott regression was firstly conducted using the scanone command in R/qtl (Broman et al. 2003). An experiment‐wise significance threshold at the P = 0.05 level was determined using 10,000 permutations. The model from scanone scanning, consisting of a list of QTL positions, was then used as a starting point for the subsequent multiple QTL fitting. Each model was confirmed using a drop‐one ANOVA, such that only QTL with a LOD score above the threshold and an ANOVA P‐value less than 0.05 were retained. The refineqtl command was then used to further refine the positions of each QTL by fitting all QTLs in the model in which the likelihood ratio test was used to measure the improvement of the model in each iteration. Finally, additional QTLs that can improve the model were searched using the addqtl tool. If new QTL was added, the ANOVA and refineqtl procedure was repeated to evaluate the fit of new model. The entire process was repeated until no more significant QTLs could be added. The total phenotypic variation explained by all QTLs was calculated from a full model that fitted all QTL terms in the model using fitqtl function in R/qtl (Broman et al. 2003). The percentage of phenotypic variation explained by each QTL was estimated by a drop‐one‐ANOVA analysis implemented in fitqtl function in R/qtl (Broman et al. 2003). Confidence interval for each QTL was defined using a 2‐LOD support interval.
QTL fine mapping
A total of 1,036 F2 plants from selfing MR1480 that is heterozygous at QTL qVb9‐2 region were planted for identifying recombinants using markers flanking qVb9‐2. A within‐family comparison strategy that has been successfully applied in QTL fine mapping (Hung et al. 2012) was used for fine mapping qVb9‐2. Briefly, in recombinant‐derived F3 family, homozygous recombinant (HR) and homozygous non‐recombinant (HNR) plants were identified using markers flanking the QTL. Phenotypic difference was tested between HR and HNR pair within each family. Any significant phenotypic difference between HR and HNR pair suggests that the parental F2 recombinant is heterozygous for the target QTL (segregating in recombinant‐derived F3 family), otherwise is homozygous for either parent (not segregating in F3 family). T‐tests with Bonferroni corrections for multiple testing were used to test the significance of the vascular bundle number difference between HR and HNR pair (P < 0.05). The substitution mapping procedure widely used in fine mapping (Paterson et al. 1990) was used to delimit the causal QTL region.
ACKNOWLEDGEMENTS
This research was supported by the National Hi‐Tech Research and Development Program of China (2012AA10A307), National Natural Science Foundation of China (31322042), the Recruitment Program of Global Experts and the Fundamental Research Funds for the Central Universities.
Huang C, Chen Q, Xu G, Xu D, Tian J, Tian F ( 2015) Identification and fine mapping of quantitative trait loci for the number of vascular bundle in maize stem. J Integr Plant Biol 58: 81–90
Available online on Apr. 3, 2015 at www.wileyonlinelibrary.com/journal/jipb
REFERENCES
- Bai X, Wu B, Xing Y ( 2012) Yield‐related QTLs and their applications in rice genetic improvement. J Integr Plant Biol 54: 300–311 [DOI] [PubMed] [Google Scholar]
- Beavis WD (1998) QTL analyses: Power, precision, and accuracy In: Paterson A, ed. Molecular Dissection of Complex Traits. CRC Press, New York: pp. 145–162 [Google Scholar]
- Broman KW, Wu H, Sen Ś, Churchill GA ( 2003) R/qtl: QTL mapping in experimental crosses. Bioinformatics 19: 889–890 [DOI] [PubMed] [Google Scholar]
- Buckler ES, Holland JB, Bradbury PJ, Acharya CB, Brown PJ, Browne C, Ersoz E, Flint‐Garcia S, Garcia A, Glaubitz JC ( 2009) The genetic architecture of maize flowering time. Science 325: 714–718 [DOI] [PubMed] [Google Scholar]
- Carland F, Fujioka S, Nelson T ( 2010) The sterol methyltransferases SMT1, SMT2, and SMT3 influence Arabidopsis development through nonbrassinosteroid products. Plant Physiol 153: 741–756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carland FM, Fujioka S, Takatsuto S, Yoshida S, Nelson T ( 2002) The identification of CVP1 reveals a role for sterols in vascular patterning. Plant Cell 14: 2045–2058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciftci‐Yilmaz S, Mittler R ( 2008) The zinc finger network of plants. Cell Mol Life Sci 65: 1150–1160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coaker GL, Meulia T, Kabelka EA, Jones AK, Francis DM ( 2002) A QTL controlling stem morphology and vascular development in Lycopersicon esculentum × Lycopersicon hirsutum (Solanaceae) crosses is located on chromosome 2. Am J Bot 89: 1859–1866 [DOI] [PubMed] [Google Scholar]
- Cui K, Peng S, Xing Y, Yu S, Xu C, Zhang Q ( 2003) Molecular dissection of the genetic relationships of source, sink and transport tissue with yield traits in rice. Theor Appl Genet 106: 649–658 [DOI] [PubMed] [Google Scholar]
- Doebley J ( 2004) The genetics of maize evolution. Annu Rev Genet 38: 37–59 [DOI] [PubMed] [Google Scholar]
- Dubos C, Stracke R, Grotewold E, Weisshaar B, Martin C, Lepiniec L ( 2010) MYB transcription factors in Arabidopsis . Trends Plant Sci 15: 573–581 [DOI] [PubMed] [Google Scholar]
- Elshire RJ, Glaubitz JC, Sun Q, Poland JA, Kawamoto K, Buckler ES, Mitchell SE ( 2011) A robust, simple genotyping‐by‐sequencing (GBS) approach for high diversity species. PLoS ONE 6: e19379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emery JF, Floyd SK, Alvarez J, Eshed Y, Hawker NP, Izhaki A, Baum SF, Bowman JL ( 2003) Radial patterning of Arabidopsis shoots by class III HD‐ZIP and KANADI genes. Curr Biol 13: 1768–1774 [DOI] [PubMed] [Google Scholar]
- Evans L, Dunstone R, Rawson H, Williams R ( 1970) The phloem of the wheat stem in relation to requirements for assimilate by the ear. Aust J Biol Sci 23: 743–752 [Google Scholar]
- Fujita D, Trijatmiko KR, Tagle AG, Sapasap MV, Koide Y, Sasaki K, Tsakirpaloglou N, Gannaban RB, Nishimura T, Yanagihara S ( 2013) NAL1 allele from a rice landrace greatly increases yield in modern indica cultivars. Proc Natl Acad Sci USA 110: 20431–20436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukuyama T, Takayama T ( 1995) Variations of the vascular bundle system in Asian rice cultivars. Euphytica 86: 227–231 [Google Scholar]
- Gilmour AR, Cullis BR, Verbyla AP ( 1997) Accounting for natural and extraneous variation in the analysis of field experiments. J Agric Biol Env Stat 2: 269–293 [Google Scholar]
- Gilmour AR, Gogel B, Cullis B, Thompson R ( 2009) ASReml User Guide Release 3.0. VSN Int Ltd, Hemel Hempstead, UK: [Google Scholar]
- Hardtke CS, Berleth T ( 1998) The Arabidopsis gene MONOPTEROS encodes a transcription factor mediating embryo axis formation and vascular development. EMBO J 17: 1405–1411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Housley TL, Peterson DM ( 1982) Oat stem vascular size in relation to kernel number and weight. I. Controlled environment. Crop Sci 22: 259–263 [Google Scholar]
- Hung H‐Y., Shannon LM, Tian F, Bradbury PJ, Chen C, Flint‐Garcia SA, McMullen MD, Ware D, Buckler ES, Doebley JF ( 2012) ZmCCT and the genetic basis of day‐length adaptation underlying the postdomestication spread of maize. Proc Natl Acad Sci USA 109: E1913–E1921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kump KL, Bradbury PJ, Wisser RJ, Buckler ES, Belcher AR, Oropeza‐Rosas MA, Zwonitzer JC, Kresovich S, McMullen MD, Ware D ( 2011) Genome‐wide association study of quantitative resistance to southern leaf blight in the maize nested association mapping population. Nat Genet 43: 163–168 [DOI] [PubMed] [Google Scholar]
- Lang Z, Wills DM, Lemmon ZH, Shannon LM, Bukowski R, Wu Y, Messing J, Doebley JF ( 2014) Defining the role of prolamin‐box binding factor1 gene during maize domestication. J Hered 105: 576–582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Z, Li X, Shannon LM, Yeh C‐T., Wang ML, Bai G, Peng Z, Li J, Trick HN, Clemente TE ( 2012) Parallel domestication of the Shattering1 genes in cereals. Nat Genet 44: 720–724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucas WJ, Groover A, Lichtenberger R, Furuta K, Yadav SR, Helariutta Y, He XQ, Fukuda H, Kang J, Lucas WJ, Groover A, Lichtenberger R, Furuta K, Yadav SR, Helariutta Y, He XQ, Fukuda H, Kang J, Brady SM, Patrick JW, Sperry J, Yoshida A, Ĺopez‐Milĺan AF, Grusak MA, Kachroo P ( 2013) The plant vascular system: Evolution, development and functions. J Integr Plant Biol 55: 294–388 [DOI] [PubMed] [Google Scholar]
- Matsuoka Y, Vigouroux Y, Goodman MM, Sanchez J, Buckler E, Doebley J ( 2002) A single domestication for maize shown by multilocus microsatellite genotyping. Proc Natl Acad Sci USA 99: 6080–6084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray M, Thompson WF ( 1980) Rapid isolation of high molecular weight plant DNA. Nucleic Acids Res 8: 4321–4326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nátrová Z ( 1991) Anatomical characteristics of the uppermost internode of winter wheat genoypes differing in stem length. Biol Plant 33: 491–494 [Google Scholar]
- Paterson AH, DeVerna JW, Lanini B, Tanksley SD ( 1990) Fine mapping of quantitative trait loci using selected overlapping recombinant chromosomes, in an interspecies cross of tomato. Genetics 124: 735–742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson DM, Housley TL, Luk TM ( 1982) Oat stem vascular size in relation to kernel number and weight. II. Field environment. Crop Sci 22: 274–278 [Google Scholar]
- Price AH ( 2006) Believe it or not, QTLs are accurate! Trends Plant Sci 11: 213–216 [DOI] [PubMed] [Google Scholar]
- Qi J, Qian Q, Bu Q, Li S, Chen Q, Sun J, Liang W, Zhou Y, Chu C, Li X ( 2008) Mutation of the rice Narrow leaf1 gene, which encodes a novel protein, affects vein patterning and polar auxin transport. Plant Physiol 147: 1947–1959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiang Y, Wu J, Han H, Wang G ( 2013) CLE peptides in vascular development. J Integr Plant Biol 55: 389–394 [DOI] [PubMed] [Google Scholar]
- Salvi S, Tuberosa R ( 2005) To clone or not to clone plant QTLs: Present and future challenges. Trends Plant Sci 10: 297–304 [DOI] [PubMed] [Google Scholar]
- Sang Y, Deng Z, Zhao L, Zhang K, Tian J, Ye B ( 2010) QTLs for the vascular bundle system of the uppermost internode using a doubled haploid population of two elite Chinese wheat cultivars. Plant Breed 129: 605–610 [Google Scholar]
- Sasahara H, Fukuta Y, Fukuyama T ( 1999) Mapping of QTLs for vascular bundle system and spike morphology in rice, Oryza sativa L. Breed Sci 49: 75–82 [Google Scholar]
- Sasahara T, Takahashi M, Kambayashi M ( 1982) Studies on structure and function of the rice ear, 3: Final ear weight, and increasing rate of ear weight and decreasing rate of straw weight at the maximum increasing period of ear weight. Jap J Crop Sci 51: 18–25 [Google Scholar]
- Sawchuk MG, Scarpella E ( 2013) Polarity, continuity, and alignment in plant vascular strands. J Integr Plant Biol 55: 824–834 [DOI] [PubMed] [Google Scholar]
- Schnable PS, Ware D, Fulton RS, Stein JC, Wei F, Pasternak S, Liang C, Zhang J, Fulton L, Graves TA ( 2009) The B73 maize genome: Complexity, diversity, and dynamics. Science 326: 1112–1115 [DOI] [PubMed] [Google Scholar]
- Shannon LM ( 2012) The Genetic Architecture of Maize Domestication and Range Expansion. PhD Dissertation. The University of Wisconsin‐Madison
- Studer A, Zhao Q, Ross‐Ibarra J, Doebley J ( 2011) A transposon insertion was the causative mutation in the maize domestication gene tb1 . Nat Genet 43: 1160–1163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terao T, Nagata K, Morino K, Hirose T ( 2010) A gene controlling the number of primary rachis branches also controls the vascular bundle formation and hence is responsible to increase the harvest index and grain yield in rice. Theor Appl Genet 120: 875–893 [DOI] [PubMed] [Google Scholar]
- Tian F, Bradbury PJ, Brown PJ, Hung H, Sun Q, Flint‐Garcia S, Rocheford TR, McMullen MD, Holland JB, Buckler ES ( 2011) Genome‐wide association study of leaf architecture in the maize nested association mapping population. Nat Genet 43: 159–162 [DOI] [PubMed] [Google Scholar]
- Tuinstra M, Ejeta G, Goldsbrough P ( 1997) Heterogeneous inbred family (HIF) analysis: A method for developing near‐isogenic lines that differ at quantitative trait loci. Theor Appl Genet 95: 1005–1011 [Google Scholar]
- Wills DM, Whipple CJ, Takuno S, Kursel LE, Shannon LM, Ross‐Ibarra J, Doebley JF ( 2013) From many, one: Genetic control of prolificacy during maize domestication. PLoS Genet 9: e1003604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Q, Li Z, Li W, Ku L, Wang C, Ye J, Li K, Yang N, Li Y, Zhong T ( 2013) CACTA‐like transposable element in ZmCCT attenuated photoperiod sensitivity and accelerated the postdomestication spread of maize. Proc Natl Acad Sci USA 110: 16969–16974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang ZH, Li P, Wang LX, Tan CJ, Hu ZL, Zhu YG, Zhu LH ( 2002) Identification of quantitative trait loci (QTLs) for the characters of vascular bundles in peduncle related to indica‐japonica differentiation in rice (Oryza sativa L.). Euphytica 128: 279–284 [PubMed] [Google Scholar]
- Zhong R, Ye ZH ( 2004) Amphivasal vascular bundle 1, a gain‐of‐function mutation of the IFL1/REV gene, is associated with alterations in the polarity of leaves, stems and carpels. Plant Cell Physiol 45: 369–385 [DOI] [PubMed] [Google Scholar]