

Abstract
Purpose of review
Human and experimental research has identified cardioautonomic and respiratory dysfunction as a frequent accompaniment in human and animal model events of sudden unexpected death in epilepsy (SUDEP). This review aims to provide an overview of the scientific evidence behind the currently accepted risk factors and working hypotheses regarding SUDEP pathophysiology.
Recent findings
Epidemiological analysis of public health burden of SUDEP has shown that it rates second only to stroke in the years of potential life lost. Clinical and experimental studies uncovered the dynamic cardiorespiratory dysfunction interictally and imminently to SUDEP, and model systems have facilitated discoveries in SUDEP mechanistic understanding and application of pilot therapeutic interventions. Pilot molecular profiling of human SUDEP has uncovered complex genomic structure in the candidate gene network.
Summary
Extensive clinical and experimental work has established a rationale for the conceptual thinking about SUDEP mechanisms. The application of the global molecular profiling will be invaluable in unraveling the individually unique genomic complexities and interactions that underlie the physiological signature of each patient. At the same time, sophisticated model systems will be critical in the iterative translation of human genetics, physiology, pharmacological interventions, and in testing preventive interventions.
Keywords: epilepsy, gene, mechanisms, sudden unexpected death in epilepsy
INTRODUCTION
Sudden unexpected death in epilepsy (SUDEP) is a sudden unexpected witnessed or unwitnessed mortality in otherwise healthy patients with epilepsy with or without evidence of a seizure and excluding documented status epilepticus, in which postmortem examination does not reveal a cause of death [1]. If a patient is successfully resuscitated following a cardiopulmonary arrest, the event is classified as near-SUDEP [1]. SUDEP is the most frequent epilepsy-related cause of death, second only to stroke in the years of productive life lost [2■■]. Although SUDEP is most common among young adults with frequent, medically refractory seizures [3], it can affect children [4] and patients with seemingly well controlled epilepsy [5]. Therefore, our efforts for prevention critically depend upon understanding SUDEP physiology and the individually relevant profile of clinical and molecular risk factors. This review aims to provide an overview of the clinical and experimental evidence behind the currently accepted risk factors and working hypotheses regarding SUDEP pathophysiology.
SUDDEN UNEXPECTED DEATH IN EPILEPSY RISK FACTORS AND CANDIDATE MECHANISMS
Role of epilepsy and seizures
Lack of seizure remission has been the major risk factor identified in many epidemiological studies [3]. As few as one to two annual generalized tonic–clonic seizures triple the SUDEP risk, whereas mortality hazard in patients with yearly frequency of 50 or more generalized tonic–clonic seizures is increased more than 14 times [3]. Direct observation of rare SUDEP and near-SUDEP instances in patients in the epilepsy monitoring units showed that death would usually occur following a partial or generalized seizure and at night [6,7■■].
Role of the postictal generalized electroencephalographic suppression in sudden unexpected death in epilepsy
Postictal generalized electroencephalographic suppression (PGES) is a relatively frequent accompaniment of generalized tonic seizures [8]. In adults, it is often accompanied by elevated end-tidal CO2 and often profound and prolonged hypoxemia [9]. In children, it tends to be shorter [8] and associated with both peri-ictal tachycardia and hypoxemia [10■]. Freitas et al. [8] found that the PGES duration negatively correlated with the extent of depression in the vagally mediated heart rate variability (HRV), although this connection was not replicated by others [11]. PGES-related SUDEP risk attribution is currently unclear [12,13].
Role of peri-ictal cardiopulmonary complications in sudden unexpected death in epilepsy
Studies in healthy individuals showed that both hypoxemia and hypercapnia can affect cardiac repolarization and shift autonomic balance toward the enhanced sympathetic drive on the sinus node [14,15]. Seyal et al. [16] found a positive correlation between the extent of ictal oxygen desaturation and the degree of cardiac repolarization abnormalities in epilepsy patients and the SUDEP case review in the Mortality in Epilepsy Monitoring Unit Study (MORTEMUS) study [7■■] revealed complex and ultimately lethal cardiopulmonary dysfunction early following a seizure (Fig. 1). Review of cardiopulmonary functions in 101 seizures in 26 children uncovered that approximately 50% of seizures were associated with apnea, and oxygen desaturation was associated with ictal bradycardia [17].
FIGURE 1.
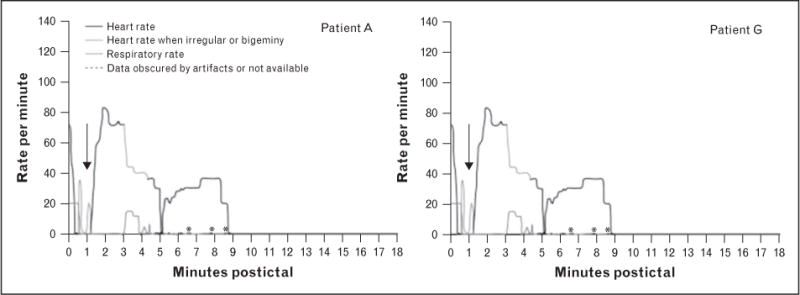
Example of two patients with SUDEP-onset captured during monitoring in an epilepsy monitoring unit. Of note, visual inspection of video recording together with respiration-induced artifacts on the electroencephalogram were used as surrogate markers of a respiratory effort. Black arrows indicate the early cardiorespiratory collapse observed in all SUDEP cases within the first 3 min postictally leading to immediate death in patient A, whereas in patient G there was a transient and partial restoration of the cardiorespiratory functions until the onset of terminal apnea followed by terminal asystole. SUDEP, sudden unexpected death in epilepsy. Reproduced with permission from [7■■].
Molecular mechanisms and genetics of respiratory dysfunction in sudden unexpected death in epilepsy
Studies of telemetry-monitored patients equipped with complete plethysmography have been invaluable in gaining insight into the mechanisms of the seizure-related respiratory distress [18,19]. Nashef et al. [18] found oxygen desaturations below 85% in 21% of all seizures and central apnea in 59% of patients and 42% of seizures. Bateman et al. [20] showed that ictal hypoxemia could last over 300 s, and the depth of desaturation correlated with seizure duration and spread to contralateral hemisphere. Moreover, oxygen desaturations below 85% were associated with the rise in the end-tidal CO2 that in some patients persisted despite an increase in respiratory rate [20], thus indicating a primary pulmonary dysfunction rather than hypoventilation [19]. Animal models have provided valuable insight into the molecular underpinnings of seizure-induced respiratory dysfunction with lethal outcome. Tendency for ictally induced respiratory arrest and death following a tonic extension phase of seizure was first noted in the Htr2c-deficient mice [21]. The important role of serotonin [5-hydroxytryptamine (5-HT)] in respiration, arousal, and epileptic seizures was subsequently elucidated through the genetically engineered Lmx1bf/f/p mice deficient in more than 99% of 5-HT neurons and affected by severe apnea, hypoventilation, diminished hypercapnic response, and compromised arousal from sleep [22]. The animals also exhibited lower threshold for maximal electroshock or pilocarpine-induced seizures and increased seizure-related mortality driven by terminal respiratory failure [23■]. Chemical inhibition of 5-HT synthesis in otherwise healthy mice reproduced the propensity for maximal electroshock induced seizures [23■]. Altered expression of the 5-HT2c, 5-HT3, 5-HT4, and 5-HT2B receptors was shown in the DBA/2 mouse prone to respiratory arrest and death triggered by audiogenic seizures [24]. The discoveries linking respiratory failure and SUDEP led to preliminary pharmacological explorations of the widely available serotonin reuptake inhibitors (SSRIs) with respect to the SUDEP risk [25,26]. The exogenous administration of fluoxetine ameliorated seizure severity and ictally induced respiratory arrest and death in the DBA model in a dose-dependent fashion [26], whereas it improved the inherent serotonin deficiency and low ventilatory sensitivity to hypercapnia in the Brown Norway rats [27]. The animal research was subsequently validated in a cohort of patients with epilepsy chronically exposed to the commonly used SSRIs [28] that exhibited a reduced severity of ictal desaturation less than 85% during a partial seizure, albeit the effect was not seen during the secondarily generalized seizures [28]. Re-evaluation of the DBA model showed that the SSRI effect may vary according to drug, dosing, medications kinetics, age, and sex [29,30■]. Although there is the translational evidence implicating the 5-hydroxytryptamine (5-HT) 2c receptor receptor in SUDEP, the role of the remaining 5-HT ligand-gated ion channels is largely unexplored. Large meta-analysis of patients affected by obstructive sleep apnea found that −1438G/A polymorphism in the HTR2A gene was a positive risk factor for obstructive sleep apnea in male patients [31], and a recent genetic analysis of a pediatric SUDEP case [32■■] suggested a contributory role of the inherited variants in the serotonergic receptor genes HTR3C and HTR3D that display altered expression in the DBA model [24]. Aside from the 5-HT pathways, the paired-like homeobox 2b (PHOX2B) gene has emerged as a candidate SUDEP molecule because of the previously described link to the central hypoventilation syndrome [33] and sudden infant death syndrome [34]. However, the initial screen of 68 Australian SUDEP cases indicates that detrimental mutations in this gene may not be a common SUDEP risk factor [35].
Molecular mechanisms and genetics of cardioautonomic dysfunction in sudden unexpected death in epilepsy
Cardioautonomic changes are observed in up to 42% of patients with refractory partial epilepsy monitored with a loop recorder [36]. Potentially serious dysrhythmias are seen in about 10% of patients and 6% of seizures [37]. Impaired cardiac recovery and repolarization abnormalities reflected in the depressed heart rate variability (HRV) and elevated values of the T-wave alternans were seen following the secondarily generalized seizures [38]. Meta-analysis of the HRV measure of epilepsy patients in 39 studies revealed consistent impairment of vagally driven activity with more profound HRV depression in individuals with refractory epilepsy [39]. Moreover, patients with refractory temporal lobe epilepsy (TLE) followed for an average of 6 years showed progressive depression of HRV measures over time in contrast to the well controlled TLE group [40]. Although the differences between patient groups did not reach statistical significance [40], this study was in line with the report of a SUDEP case with documented progressive deterioration of HRV variability and vagally mediated autonomic measure over several months preceding death [41]. Progressive autonomic changes were also noted in a recently published SUDEP case that displayed gradual rise in the parasympathetic activity over 7 months leading up to the terminal seizure. Additionally, ECG analysis revealed abnormally short QTc interval and clinically silent interictal episodes of supraventricular tachycardia [42■■].
Cardiac arrhythmia genes and sudden unexpected death in epilepsy
The first link between genetically predisposed cardiac arrhythmias and epilepsy was the discovery that the cardiac voltage-gated sodium channel SCN5A, underlying the long QT syndrome (LQTS) type 3 was expressed in the brain limbic regions [43]. Subsequent clinical case reports supported the concept of a combined neurocardiac phenotype triggered by a mutation in an ion channel dually expressed in the brain and in the heart [44,45]. The SCN5A nonsynonymous variant R523C with a predicted detrimental effect was found in a young woman affected by idiopathic epilepsy and peri-ictal cardiac palpitations [44] treated with lamotrigine and amitriptyline, medications with known effect on ion channels [46–49]. Her sudden death in the context of the SCN5A variant raised the possibility of an occult, pharmacologically triggered LQTS [44]. Another functionally detrimental de-novo SCN5A variant (R1623Q) was identified in a case of dual phenotype of neonatal seizures and long QT syndrome [45]. The initial mechanistic understanding of the complex phenotype of epilepsy, cardiac arrhythmias, and SUDEP came from transgenic mice carrying the human knock-in mutations in the most common LQT gene, the potassium channel KCNQ1 [50]. The gene showed regional expression in the murine hippocampus, thalamus, and the medullary dorsal motor nucleus of the vagus and nucleus ambiguus, the brainstem centers contributing preganglionic parasympathetic innervation via the vagal nerve to the heart. The animals displayed partial and generalized seizures together with a variety of cardiac arrhythmias, and more than two-thirds of the cardiac abnormalities occurred in association with epileptiform discharges [50]. The lockstep phenomenon of recurrent brief asystole triggered by interictal epileptiform discharges has also been observed in other genetic models [51] and clinically [52]. The link between the epilepsy phenotype and LQTS has been clinically validated [53,54]. A seizure phenotype was identified in 28% of cases with clinically evident and genetically confirmed LQTS caused by pathogenic variants in the KCNH2, KCNQ1, and SCN5A genes [54]. Molecular survey of these three genes in 68 SUDEP cases uncovered nonsynonymous variants of suspected functional significance in 10% of patients [55]. However, the molecular underpinnings of neurocardiac interactions encompass genes outside of the LQT syndrome channels [56■]. Catecholaminergic polymorphic ventricular tachycardia (CPVT) is a dysrhythmia of young individuals presenting with stress-induced syncopal events due to ventricular tachycardia at high risk for sudden death before the age of 30 [57]. The underlying defect is in the ryanodine receptor gene (RYR2) [58] expressed in cardiac myocytes and also in the Ammon’s horn and dentate gyrus of the mouse hippocampus [59]. A mutation leads to intracellular Ca2+ overload triggering early and delayed cardiac afterdepolarizations implicated as the pathological basis for the malignant bidirectional ventricular tachycardia [58]. The mouse model carrying the human mutation R2474S mirrored the human phenotype of exercise-induced ventricular arrhythmia, spontaneous convulsive seizures, and lethal arrhythmia triggered sudden death [60]. The combined phenotype of arrhythmias and seizures was observed in 12 of the 24 prospectively followed Dutch CPVT families affected by RYR2 mutations [57]. Moreover, molecular autopsy of an 8-year-old SUDEP patient with history of epilepsy and recurrent, exercise-induced syncope with normal resting electrocardiogram (ECG) revealed a missense mutation G4936A of the RYR2 channel in a screen of the principal LQT susceptibility genes [61]. The hyperpolarization-activated cyclic nucleotide-gated ion channels HCN1–4 are members of another dually expressed gene family. They are implicated in epilepsy and cardiac arrhythmias owing to their involvement in the generation of the cation (Na and K)-triggered Ih depolarizing current that facilitates action potential critical for spontaneous rhythmic activity in the neurons and pacemaking cardiomyocytes [62–66]. A mouse deficient in HCN2 displays the dual phenotype of absence epilepsy and sinus arrhythmia [67] albeit without an evidence of a reduced life span. Clinically confirmed dual epilepsy–arrhythmia phenotype has not yet been observed, although HCN channel mutational screen in 48 SUDEP cases uncovered nine coding variants, and in-silico prediction indicated functional significance for some [68]. As the biology of HCN channels in SUDEP remains to be defined, their involvement in clinically manifested arrhythmia or epilepsy warrants consideration of this gene family in the candidate SUDEP gene pool.
Epilepsy genes and sudden unexpected death in epilepsy
The potential number of novel candidate genes for SUDEP extends beyond those currently recognized in cardiac arrhythmias. The voltage-gated potassium channel KCNA1 is dually expressed in brain and in the vagus nerve, and Kcna1-null mice have seizures, cardiac arrhythmias, and vagal hyperexcitability, and they die prematurely [51]. In this model, seizures clearly exacerbated the cardiac abnormalities as evidenced by the five-fold increase in the atrioventricular conduction block rate. Pretreatment with atropine and β-blockers ameliorated the atrioventricular conduction blocks thus implicating the excessive parasympathetic tone in neurocardiac dysfunction [51]. This channel was also clinically validated in a SUDEP case affected by epileptic encephalopathy and suspected cardiac dysrhythmias carrying de-novo and novel KCNA1 intragenic duplication [32■■].
The Nav1.1 channel role in premature epilepsy mortality was first suspected in the context of a family affected by genetic epilepsy with febrile seizures plus syndrome, recurrent SUDEP, and a segregating novel, SCN1A (I1867T) variant [69]. Subsequently, Le Gal et al. [70] reported the first SUDEP case in a genetically confirmed Dravet syndrome. The sudden premature death in Dravet syndrome is currently estimated to affect about 4–12% of children with peak incidence before 4 years of age [71–73]. Moreover, analysis of Dravet syndrome cohort uncovered not only depressed HRV measures [74] but also increased P wave and QT interval dispersion [75]. The Scn1a-deficient models reproduced the complex human phenotype with the evidence of spontaneous seizures, autonomic instability, and seizure-driven vagal activation preceding sudden death [76,77■]. The administration of parasympatholytics reduced the incidence of ictal bradycardia and SUDEP [77■]. A knock-in mouse model of the human mutation SCN1A-R1407X [78■,79■■] offered insight into the neurocardiac effects of a patient-specific, disease causing, genetic variant. These mice displayed spontaneous seizures and a prolonged QT interval predisposing to a variety of cardiac dysrhythmias, including ventricular fibrillation and a model SUDEP [78■]. Interestingly, the cardiac arrhythmias often preceded the apparent convulsive seizures, thus indicating that SCN1A variants might predispose to sudden death through combined neurocardiac or sole cardiac mechanisms [78■]. Additionally, there is the experimental evidence that the risk of sudden death in Dravet syndrome may be influenced not only through a gene dosage but also via the affected neuronal cell type and regionally specific differences in Scn1a channel brain expression; the selective Nav1.1 deficiency in inhibitory GABAergic neurons resulted in a more severe epileptic phenotype and early and frequent sudden death as compared with mice with constitutive Scn1a defect [79■■]. Also, Scn1a deficiency in the forebrain excitatory neurons superimposed upon Nav1.1 defect in inhibitory GABAergic neurons mitigated the seizure phenotype and lessened the incidence of model SUDEP [79■■].
The discoveries linking KCNA1 and SCN1A to SUDEP brought attention to other epilepsy genes. The SCN1B gene encodes a voltage-gated sodium channel β subunit that is critical for proper gating and cell-surface expression of the voltage-gated sodium channel complex [80]. The phenotypic spectrum of SCN1B mutations includes genetic epilepsy with febrile seizures plus [81], TLE [82], and Dravet syndrome [83]. Although there has not been a report of human SUDEP linked to SCN1B, the Scn1b mouse model parallels some of the Scn1a animal model features with spontaneous seizures, prolonged QT and RR intervals on the ECG, and early mortality [84,85]. The increasing accessibility of whole-exome profiling facilitated the discovery of a functionally active de-novo variant in yet another epilepsy gene and a candidate SUDEP molecule, the SCN8A channel gene, in a child affected by epileptic encephalopathy and SUDEP [86]. Knock-in mouse model carrying the human missense variant p.Asn1768Asp showed early-onset seizures and premature mortality and the age of onset of seizures and death correlated with the mutant gene dosage [87■]. The value of translational animal models in pathophysiological analysis of SUDEP was recently validated in the report of the sentrin-specific protease 2 (SENP2)-deficient SUDEP mouse model. SENP2 molecule is a modifier of multiple potassium channels, including the Kv7 channel targeted by the channel opener, the antiepileptic drug, retigabine [88■■]. The SENP2-deficient animal develops epilepsy, vagally driven arrhythmias, and a model SUDEP [88■■] that were rectified by pretreatment with retigabine [88■■]. This work showed that the registry of candidate genetic risk factors in premature mortality will likely encompass an extensive network of interacting proteins, and carefully designed model systems will be invaluable in testing candidate therapeutic interventions.
Recent pilot work on adenosine connection to SUDEP deserves a mention. Adenosine exerts widespread modulatory effects systemically and in the nervous system via four types of guanine nucleotide binding (G) protein-coupled receptors A1, A2A, A2B, and A3 [89]. Seizure triggers a rise in the adenosine level, and the A1 receptors are important in the anticonvulsant and sleep induction effects [89]. However, activation of the adenosine receptors in the brainstem also triggers severe respiratory depression [90■■]. Preliminary work in a kainic acid seizure model indicated that pretreatment with caffeine, an adenosine receptor antagonist, improved survival in this otherwise lethal pharmacological model of status epilepticus [91]. Although there is insufficient data to draw conclusions regarding adenosine role in SUDEP, this molecular pathway ought to be considered in the candidate pathophysiological molecular network of premature mortality.
Oligogenic predisposition to human sudden unexpected death in epilepsy
There is growing evidence that complex genetic interactions influence the phenotypic expressions of cardiac arrhythmias [92■], epilepsy [93–95], and SUDEP [32■■]. Although SCN8A gain-of-function mutations are implicated in epileptic encephalopathies and SUDEP [86,96,97], loss-of-function variants were shown to ameliorate model seizure phenotype [98,99]. Similarly, crossing the Kcna1 model affected by early-onset severe spontaneous seizures and frequent sudden death with a Cacna1a absence seizure model results in a mild seizure phenotype and improved survival [100]. Combined deficiency of the gene Mapt encoding microtubule-binding protein τ and Kcna1 channel results in improvement in seizure frequency, severity, and survival [101■■]. Similar effect is seen in the Scn1a/Mapt digenic model [102]. A human example of genomic complexity in SUDEP was recently illustrated by a pediatric patient who was affected by Dravet syndrome, with severe seizures often accompanied by apnea, and suspected cardiac arrhythmias [32■■] (Fig. 2). The detailed genomic analysis uncovered complex combinations of single nucleotide polymorphisms and copy number variants in genes expressed in both neurocardiac and respiratory control pathways, including SCN1A, KCNA1, RYR3, and HTR2C [32■■].
FIGURE 2.
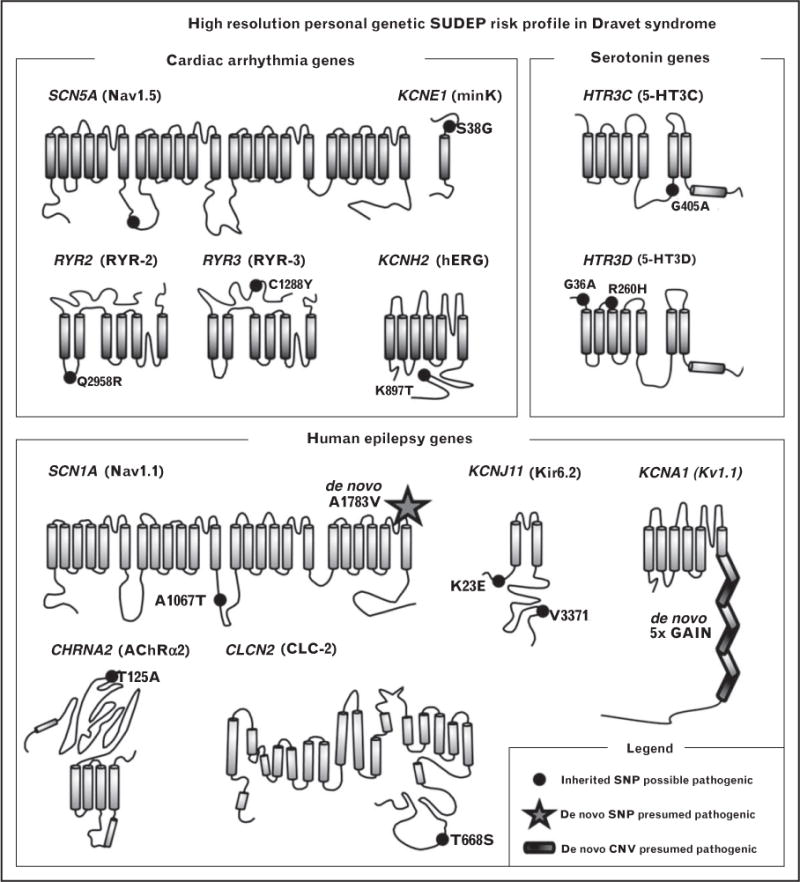
Complex genomic profiles in a SUDEP case affected by Dravet syndrome. The proband harbors an intricate combination of de-novo and inherited variants of variable pathogenic potential in multiple genes of the candidate SUDEP biological pathways. CNV, copy number variant; SNP, single nucleotide polymorphism; SUDEP, sudden unexpected death in epilepsy. Reproduced with permission from [32■■].
CONCLUSION
Our current mechanistic understanding of SUDEP was recently expertly summarized [90■■]. The lethal trigger might in some individuals be a life threatening cardiac arrhythmia initiated ictally or via a lockstep interictal epileptiform activity. Yet, in others, it may be ictally induced prolonged hypoxemia and hypercapnia resulting in acidosis that aids in seizure termination but at the same time predisposes to bradycardias or asystole in vulnerable individuals [19,90■■]. The extensive clinical and experimental work has been critical in formulating our current conceptual thinking about SUDEP mechanisms. It has also become evident that the application of currently available diagnostic modalities, such as ECG and respirometry, affords important insight into an individual cardiorespiratory physiology as it may relate to mortality risk assessment. The new cloud computing platforms now permit integration and analysis of large and highly complex physiological data [103], and are certain to facilitate extraction of physiological SUDEP biomarkers. As indicated by the published data, global molecular profiling in patients with epilepsy will be invaluable in unraveling the individually unique genomic complexities and interactions that underlie the physiological signature of each patient, thus improving the precision of SUDEP risk assessment in the individual. At the same time, sophisticated model systems will be an indispensable tool in the iterative translation of human genetics and physiology as well as in testing pharmacological interventions and preventive interventions. Many essential components for clinically relevant SUDEP research have recently come together as the Center for SUDEP Research (http://csr.case.edu/), a National Institute for Neurological Disorders and Stroke (NINDS) funded Center Without Walls for Collaborative Research in the Epilepsies. This unique collaborative network of researchers from the United States and Europe is exceptionally poised for making important discoveries related to SUDEP etiology and individually relevant and quantifiable risk factors, and for prompt clinical application of the findings.
KEY POINTS.
Cardioautonomic and respiratory dysfunctions have been firmly established as the currently known principal mechanisms leading up to SUDEP.
Clinical research has shown that comprehensive cardiopulmonary monitoring of patients with epilepsy is critical in understanding candidate pathophysiology of human SUDEP, and it will be important in identifying patients who are likely at increased SUDEP risk.
Translational animal models have been indispensable in unraveling molecular mechanisms and pathophysiology of the cardioautonomic and respiratory dysfunctions leading to lethal outcome.
Molecular profiling of human SUDEP cases and the analysis of genetic models have led to the identification of candidate SUDEP genes of which most are ion channels active along the neurocardiac, neuroautonomic, and neurorespiratory pathways. However, animal models deficient in MAPT and SENP2 genes have shown that candidate SUDEP molecules will likely involve much larger genic networks.
Advanced technological platforms have facilitated pilot comprehensive profiling of human SUDEP and aided in uncovering genomic complexities that parallel discoveries in epilepsy by several large-scale efforts.
Acknowledgments
None.
Financial support and sponsorship
This work was supported by the Department of Neurology, Baylor College of Medicine, Houston, Texas as well as by the research support provided by the National Institute of Neurological Disorders and Stroke grants NS067013, 1P20NS076916, NS090406, NS090362, and NS067013S; CURE; Fiorito Foundation; the Emma Bursick Memorial Fund.
Footnotes
Conflicts of interest
There are no conflicts of interest.
REFERENCES AND RECOMMENDED READING
Papers of particular interest, published within the annual period of review, have been highlighted as:
■ of special interest
■■ of outstanding interest
- 1.Nashef L, So EL, Ryvlin P, Tomson T. Unifying the definitions of sudden unexpected death in epilepsy. Epilepsia. 2012;53:227–233. doi: 10.1111/j.1528-1167.2011.03358.x. [DOI] [PubMed] [Google Scholar]
- 2■■.Thurman DJ, Hesdorffer DC, French JA. Sudden unexpected death in epilepsy: assessing the public health burden. Epilepsia. 2014;55:1479–1485. doi: 10.1111/epi.12666. Meta-analysis of SUDEP public health burden of SUDEP and societal cost of epilepsy-related premature mortality. [DOI] [PubMed] [Google Scholar]
- 3.Hesdorffer DC, Tomson T, Benn E, et al. Combined analysis of risk factors for SUDEP. Epilepsia. 2011;52:1150–1159. doi: 10.1111/j.1528-1167.2010.02952.x. [DOI] [PubMed] [Google Scholar]
- 4.Sillanpaa M, Shinnar S. Long-term mortality in childhood-onset epilepsy. N Engl J Med. 2010;363:2522–2529. doi: 10.1056/NEJMoa0911610. [DOI] [PubMed] [Google Scholar]
- 5.LanganY, Nashef L, Sander JW. Sudden unexpected death in epilepsy: a series of witnessed deaths. J Neurol Neurosurg Psychiatry. 2000;68:211–213. doi: 10.1136/jnnp.68.2.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bateman LM, Spitz M, Seyal M. Ictal hypoventilation contributes to cardiac arrhythmia and SUDEP: report on two deaths in video-EEG-monitored patients. Epilepsia. 2010;51:916–920. doi: 10.1111/j.1528-1167.2009.02513.x. [DOI] [PubMed] [Google Scholar]
- 7■■.Ryvlin P, Nashef L, Lhatoo SD, et al. Incidence and mechanisms of cardiorespiratory arrests in epilepsy monitoring units (MORTEMUS): a retrospective study. Lancet Neurol. 2013;12:966–977. doi: 10.1016/S1474-4422(13)70214-X. Collaborative multicenter retrospective review of physiological correlates of SUDEP and near-SUDEP cases captured in the monitoring units. [DOI] [PubMed] [Google Scholar]
- 8.Freitas J, Kaur G, Fernandez GB, et al. Age-specific periictal electroclinical features of generalized tonic–clonic seizures and potential risk of sudden unexpected death in epilepsy (SUDEP) Epilepsy Behav. 2013;29:289–294. doi: 10.1016/j.yebeh.2013.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Seyal M, Hardin KA, Bateman LM. Postictal generalized EEG suppression is linked to seizure-associated respiratory dysfunction but not postictal apnea. Epilepsia. 2012;53:825–831. doi: 10.1111/j.1528-1167.2012.03443.x. [DOI] [PubMed] [Google Scholar]
- 10■.Moseley BD, So E, Wirrell EC, et al. Characteristics of postictal generalized EEG suppression in children. Epilepsy Res. 2013;106:123–127. doi: 10.1016/j.eplepsyres.2013.05.007. Retrospective review of physiological correlates of epileptic seizures in pediatric population. [DOI] [PubMed] [Google Scholar]
- 11.Lamberts RJ, Laranjo S, Kalitzin SN, et al. Postictal generalized EEG suppression is not associated with periictal cardiac autonomic instability in people with convulsive seizures. Epilepsia. 2013;54:523–529. doi: 10.1111/epi.12021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lhatoo SD, Faulkner HJ, Dembny K, et al. An electroclinical case–control study of sudden unexpected death in epilepsy. Ann Neurol. 2010;68:787–796. doi: 10.1002/ana.22101. [DOI] [PubMed] [Google Scholar]
- 13.Surges R, Strzelczyk A, Scott CA, et al. Postictal generalized electroencephalographic suppression is associated with generalized seizures. Epilepsy Behav. 2011;21:271–274. doi: 10.1016/j.yebeh.2011.04.008. [DOI] [PubMed] [Google Scholar]
- 14.Kiely DG, Cargill RI, Grove A, et al. Abnormal myocardial repolarisation in response to hypoxaemia and fenoterol. Thorax. 1995;50:1062–1066. doi: 10.1136/thx.50.10.1062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Roche F, Reynaud C, Pichot V, et al. Effect of acute hypoxia on QT rate dependence and corrected QT interval in healthy subjects. Am J Cardiol. 2003;91:916–919. doi: 10.1016/s0002-9149(03)00040-7. [DOI] [PubMed] [Google Scholar]
- 16.Seyal M, Pascual F, Lee CY, et al. Seizure-related cardiac repolarization abnormalities are associated with ictal hypoxemia. Epilepsia. 2011;52:2105–2111. doi: 10.1111/j.1528-1167.2011.03262.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Singh K, Katz ES, Zarowski M, et al. Cardiopulmonary complications during pediatric seizures: a prelude to understanding SUDEP. Epilepsia. 2013;54:1083–1091. doi: 10.1111/epi.12153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nashef L, Walker F, Allen P, et al. Apnoea and bradycardia during epileptic seizures: relation to sudden death in epilepsy. J Neurol Neurosurg Psychiatry. 1996;60:297–300. doi: 10.1136/jnnp.60.3.297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Seyal M, Bateman LM, Albertson TE, et al. Respiratory changes with seizures in localization-related epilepsy: analysis of periictal hypercapnia and airflow patterns. Epilepsia. 2010;51:1359–1364. doi: 10.1111/j.1528-1167.2009.02518.x. [DOI] [PubMed] [Google Scholar]
- 20.Bateman LM, Li CS, Seyal M. Ictal hypoxemia in localization-related epilepsy: analysis of incidence, severity and risk factors. Brain. 2008;131:3239–3245. doi: 10.1093/brain/awn277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tecott LH, Sun LM, Akana SF, et al. Eating disorder and epilepsy in mice lacking 5-HT2c serotonin receptors. Nature. 1995;374:542–546. doi: 10.1038/374542a0. [DOI] [PubMed] [Google Scholar]
- 22.Hodges MR, Wehner M, Aungst J, et al. Transgenic mice lacking serotonin neurons have severe apnea and high mortality during development. J Neurosci. 2009;29:10341–10349. doi: 10.1523/JNEUROSCI.1963-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23■.Buchanan GF, Murray NM, Hajek MA, Richerson GB. Serotonin neurons have anticonvulsant effects and reduce seizure-induced mortality. J Physiol. 2014;592:4395–4410. doi: 10.1113/jphysiol.2014.277574. Mechanistic insight into the role of serotonergic neurons in model seizure-related mortality. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Uteshev VV, Tupal S, Mhaskar Y, Faingold CL. Abnormal serotonin receptor expression in DBA/2 mice associated with susceptibility to sudden death due to respiratory arrest. Epilepsy Res. 2010;88:183–188. doi: 10.1016/j.eplepsyres.2009.11.004. [DOI] [PubMed] [Google Scholar]
- 25.Faingold CL, Tupal S, Randall M. Prevention of seizure-induced sudden death in a chronic SUDEP model by semichronic administration of a selective serotonin reuptake inhibitor. Epilepsy Behav. 2011;22:186–190. doi: 10.1016/j.yebeh.2011.06.015. [DOI] [PubMed] [Google Scholar]
- 26.Tupal S, Faingold CL. Evidence supporting a role of serotonin in modulation of sudden death induced by seizures in DBA/2 mice. Epilepsia. 2006;47:21–26. doi: 10.1111/j.1528-1167.2006.00365.x. [DOI] [PubMed] [Google Scholar]
- 27.Hodges MR, Echert AE, Puissant MM, Mouradian GC., Jr Fluoxetine augments ventilatory CO2 sensitivity in Brown Norway but not Sprague Dawley rats. Respir Physiol Neurobiol. 2013;186:221–228. doi: 10.1016/j.resp.2013.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bateman LM, Li CS, Lin TC, Seyal M. Serotonin reuptake inhibitors are associated with reduced severity of ictal hypoxemia in medically refractory partial epilepsy. Epilepsia. 2010;51:2211–2214. doi: 10.1111/j.1528-1167.2010.02594.x. [DOI] [PubMed] [Google Scholar]
- 29.Faingold CL, Randall M. Effects of age, sex, and sertraline administration on seizure-induced respiratory arrest in the DBA/1 mouse model of sudden unexpected death in epilepsy (SUDEP) Epilepsy Behav. 2013;28:78–82. doi: 10.1016/j.yebeh.2013.04.003. [DOI] [PubMed] [Google Scholar]
- 30■.Faingold CL, Kommajosyula SP, Long X, et al. Serotonin and sudden death: differential effects of serotonergic drugs on seizure-induced respiratory arrest in DBA/1 mice. Epilepsy Behav. 2014;37:198–203. doi: 10.1016/j.yebeh.2014.06.028. Animal translational study evaluating the modifying effects of the selective SSRIs on seizure-related model mortality. [DOI] [PubMed] [Google Scholar]
- 31.Wu Y, Liu HB, Ding M, et al. Association between the −1438G/A and T102C polymorphisms of 5-HT2A receptor gene and obstructive sleep apnea: a meta-analysis. Mol Biol Rep. 2013;40:6223–6231. doi: 10.1007/s11033-013-2734-9. [DOI] [PubMed] [Google Scholar]
- 32■■.Klassen TL, Bomben VC, Patel A, et al. High-resolution molecular genomic autopsy reveals complex sudden unexpected death in epilepsy risk profile. Epilepsia. 2014;55:e6–e12. doi: 10.1111/epi.12489. The first gene-targeted comprehensive genomic survey in a pediatric SUDEP case. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Weese-Mayer DE, Berry-Kravis EM, Zhou L, et al. Idiopathic congenital central hypoventilation syndrome: analysis of genes pertinent to early autonomic nervous system embryologic development and identification of mutations in PHOX2b. Am J Med Genet A. 2003;123A:267–278. doi: 10.1002/ajmg.a.20527. [DOI] [PubMed] [Google Scholar]
- 34.Liebrechts-Akkerman G, Liu F, Lao O, et al. PHOX2B polyalanine repeat length is associated with sudden infant death syndrome and unclassified sudden infant death in the Dutch population. Int J Legal Med. 2014;128:621–629. doi: 10.1007/s00414-013-0962-0. [DOI] [PubMed] [Google Scholar]
- 35.Bagnall RD, Crompton DE, Cutmore C, et al. Genetic analysis of PHOX2B in sudden unexpected death in epilepsy cases. Neurology. 2014;83:1018–1021. doi: 10.1212/WNL.0000000000000781. [DOI] [PubMed] [Google Scholar]
- 36.Nei M, Sperling MR, Mintzer S, Ho RT. Long-term cardiac rhythm and repolarization abnormalities in refractory focal and generalized epilepsy. Epilepsia. 2012;53:e137–e140. doi: 10.1111/j.1528-1167.2012.03561.x. [DOI] [PubMed] [Google Scholar]
- 37.Opherk C, Coromilas J, Hirsch LJ. Heart rate and EKG changes in 102 seizures: analysis of influencing factors. Epilepsy Res. 2002;52:117–127. doi: 10.1016/s0920-1211(02)00215-2. [DOI] [PubMed] [Google Scholar]
- 38.Strzelczyk A, Adjei P, Scott CA, et al. Postictal increase in T-wave alternans after generalized tonic–clonic seizures. Epilepsia. 2011;52:2112–2117. doi: 10.1111/j.1528-1167.2011.03266.x. [DOI] [PubMed] [Google Scholar]
- 39.Lotufo PA, Valiengo L, Bensenor IM, Brunoni AR. A systematic review and meta-analysis of heart rate variability in epilepsy and antiepileptic drugs. Epilepsia. 2012;53:272–282. doi: 10.1111/j.1528-1167.2011.03361.x. [DOI] [PubMed] [Google Scholar]
- 40.Suorsa E, Korpelainen JT, Ansakorpi H, et al. Heart rate dynamics in temporal lobe epilepsy: a long-term follow-up study. Epilepsy Res. 2011;93:80–83. doi: 10.1016/j.eplepsyres.2010.10.005. [DOI] [PubMed] [Google Scholar]
- 41.Rauscher G, DeGiorgio AC, Miller PR, DeGiorgio CM. Sudden unexpected death in epilepsy associated with progressive deterioration in heart rate variability. Epilepsy Behav. 2011;21:103–105. doi: 10.1016/j.yebeh.2011.03.002. [DOI] [PubMed] [Google Scholar]
- 42■■.Jeppesen J, Fuglsang-Frederiksen A, Brugada R, et al. Heart rate variability analysis indicates preictal parasympathetic overdrive preceding seizure-induced cardiac dysrhythmias leading to sudden unexpected death in a patient with epilepsy. Epilepsia. 2014;55:e67–71. doi: 10.1111/epi.12614. Study provides insight into the potential mechanisms by which long-standing epilepsy and repetitive seizures can trigger progressive deterioration of autonomic cardiac function over time. [DOI] [PubMed] [Google Scholar]
- 43.Hartmann HA, Colom LV, Sutherland ML, Noebels JL. Selective localization of cardiac SCN5A sodium channels in limbic regions of rat brain. Nat Neurosc. 1999;2:593–595. doi: 10.1038/10147. [DOI] [PubMed] [Google Scholar]
- 44.Aurlien D, Leren TP, Tauboll E, Gjerstad L. New SCN5A mutation in a SUDEP victim with idiopathic epilepsy. Seizure. 2009;18:158–160. doi: 10.1016/j.seizure.2008.07.008. [DOI] [PubMed] [Google Scholar]
- 45.Heron SE, Hernandez M, Edwards C, et al. Neonatal seizures and long QT syndrome: a cardiocerebral channelopathy? Epilepsia. 2010;51:293–296. doi: 10.1111/j.1528-1167.2009.02317.x. [DOI] [PubMed] [Google Scholar]
- 46.Marshall JB, Forker AD. Cardiovascular effects of tricyclic antidepressant drugs: therapeutic usage, overdose, and management of complications. Am Heart J. 1982;103:401–414. doi: 10.1016/0002-8703(82)90281-2. [DOI] [PubMed] [Google Scholar]
- 47.Danielsson BR, Lansdell K, Patmore L, Tomson T. Effects of the antiepileptic drugs lamotrigine, topiramate and gabapentin on hERG potassium currents. Epilepsy Res. 2005;63:17–25. doi: 10.1016/j.eplepsyres.2004.10.002. [DOI] [PubMed] [Google Scholar]
- 48.Moore PW, Donovan JW, Burkhart KK, Haggerty D. A case series of patients with lamotrigine toxicity at one center from 2003 to 2012. Clin Toxicol. 2013;51:545–549. doi: 10.3109/15563650.2013.818685. [DOI] [PubMed] [Google Scholar]
- 49.Dibue M, Kamp MA, Neumaier F, et al. Cardiac phenomena during kainic-acid induced epilepsy and lamotrigine antiepileptic therapy. Epilepsy Res. 2014;108:666–674. doi: 10.1016/j.eplepsyres.2014.02.010. [DOI] [PubMed] [Google Scholar]
- 50.Goldman AM, Glasscock E, Yoo J, et al. Arrhythmia in heart and brain: KCNQ1 mutations link epilepsy and sudden unexplained death. Sci Transl Med. 2009;1:2r. doi: 10.1126/scitranslmed.3000289. a6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Glasscock E, Yoo JW, Chen TT, et al. Kv1.1 potassium channel deficiency reveals brain-driven cardiac dysfunction as a candidate mechanism for sudden unexplained death in epilepsy. J Neurosci. 2010;30:5167–5175. doi: 10.1523/JNEUROSCI.5591-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Nei M, Zangaladze AT, Sharan A, Ho RT. Interictal epileptiform discharges and asystole. Epilepsy Res. 2011;93:204–207. doi: 10.1016/j.eplepsyres.2010.12.001. [DOI] [PubMed] [Google Scholar]
- 53.Johnson JN, Hofman N, Haglund CM, et al. Identification of a possible pathogenic link between congenital long QT syndrome and epilepsy. Neurology. 2009;72:224–231. doi: 10.1212/01.wnl.0000335760.02995.ca. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Anderson JH, Bos JM, Cascino GD, Ackerman MJ. Prevalence and spectrum of electroencephalogram-identified epileptiform activity among patients with long QT syndrome. Heart Rhythm. 2014;11:53–57. doi: 10.1016/j.hrthm.2013.10.010. [DOI] [PubMed] [Google Scholar]
- 55.Tu E, Bagnall RD, Duflou J, Semsarian C. Postmortem review and genetic analysis of sudden unexpected death in epilepsy (SUDEP) cases. Brain Pathol. 2011;21:201–208. doi: 10.1111/j.1750-3639.2010.00438.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56■.Wilde AA, Behr ER. Genetic testing for inherited cardiac disease. Nat Rev Cardiol. 2013;10:571–583. doi: 10.1038/nrcardio.2013.108. Comprehensive review of genetics of inherited cardiac arrhythmias in the context of clinical relevance. [DOI] [PubMed] [Google Scholar]
- 57.Postma AV, Denjoy I, Kamblock J, et al. Catecholaminergic polymorphic ventricular tachycardia: RYR2 mutations, bradycardia, and follow up of the patients. J Med Genet. 2005;42:863–870. doi: 10.1136/jmg.2004.028993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Liu N, Priori SG. Disruption of calcium homeostasis and arrhythmogenesis induced by mutations in the cardiac ryanodine receptor and calsequestrin. Cardiovasc Res. 2008;77:293–301. doi: 10.1093/cvr/cvm004. [DOI] [PubMed] [Google Scholar]
- 59.Mori F, Fukaya M, Abe H, et al. Developmental changes in expression of the three ryanodine receptor mRNAs in the mouse brain. Neurosc Lett. 2000;285:57–60. doi: 10.1016/s0304-3940(00)01046-6. [DOI] [PubMed] [Google Scholar]
- 60.Lehnart SE, Mongillo M, Bellinger A, et al. Leaky Ca2+ release channel/ryanodine receptor 2 causes seizures and sudden cardiac death in mice. J Clin Invest. 2008;118:2230–2245. doi: 10.1172/JCI35346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Johnson JN, Tester DJ, Bass NE, Ackerman MJ. Cardiac channel molecular autopsy for sudden unexpected death in epilepsy. J Child Neurol. 2010;25:916–921. doi: 10.1177/0883073809343722. [DOI] [PubMed] [Google Scholar]
- 62.Monteggia LM, Eisch AJ, Tang MD, et al. Cloning and localization of the hyperpolarization-activated cyclic nucleotide-gated channel family in rat brain. Brain Res Mol Brain Res. 2000;81:129–139. doi: 10.1016/s0169-328x(00)00155-8. [DOI] [PubMed] [Google Scholar]
- 63.Marionneau C, Couette B, Liu J, et al. Specific pattern of ionic channel gene expression associated with pacemaker activity in the mouse heart. J Physiol. 2005;562:223–234. doi: 10.1113/jphysiol.2004.074047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Meuth SG, Kanyshkova T, Meuth P, et al. Membrane resting potential of thalamocortical relay neurons is shaped by the interaction among TASK3 and HCN2 channels. J Neurophysiol. 2006;96:1517–1529. doi: 10.1152/jn.01212.2005. [DOI] [PubMed] [Google Scholar]
- 65.Benarroch EE. HCN channels: function and clinical implications. Neurology. 2013;80:304–310. doi: 10.1212/WNL.0b013e31827dec42. [DOI] [PubMed] [Google Scholar]
- 66.Stieber J, Wieland K, Stockl G, et al. Bradycardic and proarrhythmic properties of sinus node inhibitors. Mol Pharmacol. 2006;69:1328–1337. doi: 10.1124/mol.105.020701. [DOI] [PubMed] [Google Scholar]
- 67.Ludwig A, Budde T, Stieber J, et al. Absence epilepsy and sinus dysrhythmia in mice lacking the pacemaker channel HCN2. EMBO J. 2003;22:216–224. doi: 10.1093/emboj/cdg032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Tu E, Waterhouse L, Duflou J, et al. Genetic analysis of hyperpolarization-activated cyclic nucleotide-gated cation channels in sudden unexpected death in epilepsy cases. Brain Pathol. 2011;21:692–698. doi: 10.1111/j.1750-3639.2011.00500.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hindocha N, Nashef L, Elmslie F, et al. Two cases of sudden unexpected death in epilepsy in a GEFS+ family with an SCN1A mutation. Epilepsia. 2008;49:360–365. doi: 10.1111/j.1528-1167.2007.01439_2.x. [DOI] [PubMed] [Google Scholar]
- 70.Le Gal F, Korff CM, Monso-Hinard C, et al. A case of SUDEP in a patient with Dravet syndrome with SCN1A mutation. Epilepsia. 2010;51:1915–1918. doi: 10.1111/j.1528-1167.2010.02691.x. [DOI] [PubMed] [Google Scholar]
- 71.Genton P, Velizarova R, Dravet C. Dravet syndrome: the long-term outcome. Epilepsia. 2011;52(Suppl 2):44–49. doi: 10.1111/j.1528-1167.2011.03001.x. [DOI] [PubMed] [Google Scholar]
- 72.Sakauchi M, Oguni H, Kato I, et al. Retrospective multiinstitutional study of the prevalence of early death in Dravet syndrome. Epilepsia. 2011;52:1144–1149. doi: 10.1111/j.1528-1167.2011.03053.x. [DOI] [PubMed] [Google Scholar]
- 73.Skluzacek JV, Watts KP, Parsy O, et al. Dravet syndrome and parent associations: the IDEA League experience with comorbid conditions, mortality, management, adaptation, and grief. Epilepsia. 2011;52(Suppl 2):95–101. doi: 10.1111/j.1528-1167.2011.03012.x. [DOI] [PubMed] [Google Scholar]
- 74.Delogu AB, Spinelli A, Battaglia D, et al. Electrical and autonomic cardiac function in patients with Dravet syndrome. Epilepsia. 2011;52(Suppl 2):55–58. doi: 10.1111/j.1528-1167.2011.03003.x. [DOI] [PubMed] [Google Scholar]
- 75.Ergul Y, Ekici B, Tatli B, et al. QT and P wave dispersion and heart rate variability in patients with Dravet syndrome. Acta Neurol Belg. 2013;113:161–166. doi: 10.1007/s13760-012-0140-z. [DOI] [PubMed] [Google Scholar]
- 76.Cheah CS, Yu FH, Westenbroek RE, et al. Specific deletion of NaV1.1 sodium channels in inhibitory interneurons causes seizures and premature death in a mouse model of Dravet syndrome. Proc Natl Acad Sci U S A. 2012;109:14646–14651. doi: 10.1073/pnas.1211591109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77■.Kalume F, Westenbroek RE, Cheah CS, et al. Sudden unexpected death in a mouse model of Dravet syndrome. J Clin Invest. 2013;123:1798–1808. doi: 10.1172/JCI66220. Mechanistic insight into the SUDEP mechanisms in a knock-out mouse model of Dravet syndrome. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78■.Auerbach DS, Jones J, Clawson BC, et al. Altered cardiac electrophysiology and SUDEP in a model of Dravet syndrome. PloS One. 2013;8:e77843. doi: 10.1371/journal.pone.0077843. Detailed analysis of cardiac dysfunction related SUDEP mechanisms a knock-in mouse model of Dravet syndrome. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79■■.Ogiwara I, Iwasato T, Miyamoto H, et al. Nav1.1 haploinsufficiency in excitatory neurons ameliorates seizure-associated sudden death in a mouse model of Dravet syndrome. Hum Mol Genet. 2013;22:4784–4804. doi: 10.1093/hmg/ddt331. Experimental study demonstrating modulatory effect of regionally specific expression of mutant Scn1a channel on seizure severity and SUDEP risk. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Brackenbury WJ, Yuan Y, O’Malley HA, et al. Abnormal neuronal patterning occurs during early postnatal brain development of Scn1b-null mice and precedes hyperexcitability. Proc Natl Acad Sci U S A. 2013;110:1089–1094. doi: 10.1073/pnas.1208767110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Wallace RH, Wang DW, Singh R, et al. Febrile seizures and generalized epilepsy associated with a mutation in the Na+-channel beta1 subunit gene SCN1B. Nat Genet. 1998;19:366–370. doi: 10.1038/1252. [DOI] [PubMed] [Google Scholar]
- 82.Scheffer IE, Harkin LA, Grinton BE, et al. Temporal lobe epilepsy and GEFS+ phenotypes associated with SCN1B mutations. Brain. 2007;130:100–109. doi: 10.1093/brain/awl272. [DOI] [PubMed] [Google Scholar]
- 83.Ogiwara I, Nakayama T, Yamagata T, et al. A homozygous mutation of voltage-gated sodium channel beta(I) gene SCN1B in a patient with Dravet syndrome. Epilepsia. 2012;53:e200–e203. doi: 10.1111/epi.12040. [DOI] [PubMed] [Google Scholar]
- 84.Chen C, Westenbroek RE, Xu X, et al. Mice lacking sodium channel beta1 subunits display defects in neuronal excitability, sodium channel expression, and nodal architecture. J Neurosci. 2004;24:4030–4042. doi: 10.1523/JNEUROSCI.4139-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Lopez-Santiago LF, Meadows LS, Ernst SJ, et al. Sodium channel Scn1b null mice exhibit prolonged QT and RR intervals. J Mol Cell Cardiol. 2007;43:636–647. doi: 10.1016/j.yjmcc.2007.07.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Veeramah KR, O’Brien JE, Meisler MH, et al. De novo pathogenic SCN8A mutation identified by whole-genome sequencing of a family quartet affected by infantile epileptic encephalopathy and SUDEP. Am J Hum Genet. 2012;90:502–510. doi: 10.1016/j.ajhg.2012.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87■.Wagnon JL, Korn MJ, Parent R, et al. Convulsive seizures and SUDEP in a mouse model of SCN8A epileptic encephalopathy. Hum Mol Genet. 2015;24:506–515. doi: 10.1093/hmg/ddu470. Experimental study demonstrating differential effect of gain vs. loss of function mutation of a single gene on epilepsy expressivity and SUDEP risk. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88■■.Qi Y, Wang J, Bomben VC, et al. Hyper-SUMOylation of the Kv7 potassium channel diminishes the M-current leading to seizures and sudden death. Neuron. 2014;83:1159–1171. doi: 10.1016/j.neuron.2014.07.042. Study of SENP2 gene uncovered novel category of channel modulating molecules as candidate for SUDEP genetic risk factors. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Benarroch EE. Adenosine and its receptors: multiple modulatory functions and potential therapeutic targets for neurologic disease. Neurology. 2008;70:231–236. doi: 10.1212/01.wnl.0000297939.18236.ec. [DOI] [PubMed] [Google Scholar]
- 90■■.Massey CA, Sowers LP, Dlouhy BJ, Richerson GB. Mechanisms of sudden unexpected death in epilepsy: the pathway to prevention. Nat Rev Neurol. 2014;10:271–282. doi: 10.1038/nrneurol.2014.64. Comprehensive and in-depth review of current concepts and molecular mechanisms of SUDEP. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Shen HY, Li T, Boison D. A novel mouse model for sudden unexpected death in epilepsy (SUDEP): role of impaired adenosine clearance. Epilepsia. 2010;51:465–468. doi: 10.1111/j.1528-1167.2009.02248.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92■.Mullally J, Goldenberg I, Moss AJ, et al. Risk of life-threatening cardiac events among patients with long QT syndrome and multiple mutations. Heart Rhythm. 2013;10:378–382. doi: 10.1016/j.hrthm.2012.11.006. Study demonstrates the oligogenic interactions that influence the character and severity of inherited cardiac arrhythmias. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.EuroEPINOMICS-RES Consortium; Epilepsy Phenome/Genome Project; Epi4K Consortium. De novo mutations in synaptic transmission genes including DNM1 cause epileptic encephalopathies. Am J Hum Genet. 2014;95:360–370. doi: 10.1016/j.ajhg.2014.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Epi4K Consortium; Epilepsy Phenome/Genome Project. Allen AS, Berkovic SF, Cossette P, et al. De novo mutations in epileptic encephalopathies. Nature. 2013;501:217–221. doi: 10.1038/nature12439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Klassen T, Davis C, Goldman A, et al. Exome sequencing of ion channel genes reveals complex profiles confounding personal risk assessment in epilepsy. Cell. 2011;145:1036–1048. doi: 10.1016/j.cell.2011.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Carvill GL, Heavin SB, Yendle SC, et al. Targeted resequencing in epileptic encephalopathies identifies de novo mutations in CHD2 and SYNGAP1. Nat Genet. 2013;45:825–830. doi: 10.1038/ng.2646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Veeramah KR, Johnstone L, Karafet TM, et al. Exome sequencing reveals new causal mutations in children with epileptic encephalopathies. Epilepsia. 2013;54:1270–1281. doi: 10.1111/epi.12201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Hawkins NA, Martin MS, Frankel WN, et al. Neuronal voltage-gated ion channels are genetic modifiers of generalized epilepsy with febrile seizures plus. Neurobiol Dis. 2011;41:655–660. doi: 10.1016/j.nbd.2010.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.O’Brien JE, Meisler MH. Sodium channel SCN8A (Nav1.6): properties and de novo mutations in epileptic encephalopathy and intellectual disability. Front Genet. 2013;4:213. doi: 10.3389/fgene.2013.00213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Glasscock E, Qian J, Yoo JW, Noebels JL. Masking epilepsy by combining two epilepsy genes. Nat Neurosc. 2007;10:1554–1558. doi: 10.1038/nn1999. [DOI] [PubMed] [Google Scholar]
- 101■■.Holth JK, Bomben VC, Reed JG, et al. Tau loss attenuates neuronal network hyperexcitability in mouse and Drosophila genetic models of epilepsy. J Neurosci. 2013;33:1651–1659. doi: 10.1523/JNEUROSCI.3191-12.2013. Digenic knock-out mouse model demonstrating lesser mortality risk paradoxically to higher gene loss burden. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Gheyara AL, Ponnusamy R, Djukic B, et al. Tau reduction prevents disease in a mouse model of Dravet syndrome. Ann Neurol. 2014;76:443–456. doi: 10.1002/ana.24230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Sahoo SS, Jayapandian C, Garg G, et al. Heart beats in the cloud: distributed analysis of electrophysiological ’Big Data’ using cloud computing for epilepsy clinical research. J Am Med Inform Assoc. 2014;21:263–271. doi: 10.1136/amiajnl-2013-002156. [DOI] [PMC free article] [PubMed] [Google Scholar]