

Abstract
Background. The pharmacokinetics and pharmacodynamics of lumefantrine, a component of the most widely used treatment for malaria, artemether-lumefantrine, has not been adequately characterized in young children.
Methods. Capillary whole-blood lumefantrine concentration and treatment outcomes were determined in 105 Ugandan children, ages 6 months to 2 years, who were treated for 249 episodes of Plasmodium falciparum malaria with artemether-lumefantrine.
Results. Population pharmacokinetics for lumefantrine used a 2-compartment open model with first-order absorption. Age had a significant positive correlation with bioavailability in a model that included allometric scaling. Children not receiving trimethoprim-sulfamethoxazole with capillary whole blood concentrations <200 ng/mL had a 3-fold higher hazard of 28-day recurrent parasitemia, compared with those with concentrations >200 ng/mL (P = .0007). However, for children receiving trimethoprim-sulfamethoxazole, the risk of recurrent parasitemia did not differ significantly on the basis of this threshold. Day 3 concentrations were a stronger predictor of 28-day recurrence than day 7 concentrations.
Conclusions. We demonstrate that age, in addition to weight, is a determinant of lumefantrine exposure, and in the absence of trimethoprim-sulfamethoxazole, lumefantrine exposure is a determinant of recurrent parasitemia. Exposure levels in children aged 6 months to 2 years was generally lower than levels published for older children and adults. Further refinement of artemether-lumefantrine dosing to improve exposure in infants and very young children may be warranted.
Keywords: Malaria, population pharmacokinetics, lumefantrine, artemisinin combination therapy, antimalarial, nonlinear mixed effects modeling, pharmacodynamics, trimethoprim-sulfamethoxazole
Despite major strides in control of malaria, an estimated 214 million cases occurred in 2015, mainly in sub-Saharan African children [1]. Artemisinin-based combination therapies (ACTs) are the mainstay of therapy, with artemether-lumefantrine (AL) used as first- or second-line therapy in >40 countries in Africa [1]. AL combines artemether, a fast-acting, highly efficacious, short–half-life drug, with lumefantrine, a long–half-life (approximately 4.5 days) partner drug that eliminates residual parasites and reduces the risk of developing resistance [2].
In Asia, growing resistance to artemisinins and partner drugs, particularly piperaquine, has been reported [3–5]. Improper dosing of ACTs is an important determinant of inadequate efficacy and contributes to the development of resistance [6]. Among the groups at highest risk of underdosing are young children [7–10]. For AL, a recent meta-analysis of individual day 7 lumefantrine concentrations suggested that exposure was lowest in children <3 years of age, particularly those who were underweight (low weight for age) [8]. In addition, a day 7 lumefantrine concentration of <200 ng/mL was most predictive of the risk of recurrent malaria [8].
The overwhelming majority of studies of young children have involved drug sampling only on day 7. A very small number of children in this age range have undergone population pharmacokinetic (PK) analysis, and even fewer underwent PK combined with pharmacodynamic (PD) analysis [8, 11–13]. The purpose of this study was to characterize the population PKs and PDs of lumefantrine in a cohort of Ugandan children 6 months to 2 years of age. This investigation included the evaluation of clinical and demographic covariate effects on lumefantrine exposure, including the use of trimethoprim-sulfamethoxazole (TMP-SMZ) prophylaxis for those infected with or exposed to human immunodeficiency virus (HIV) [14, 15], as well as the relationship between concentration and clinical outcome.
METHODS
Study Area and Patient Enrollment
The study was conducted in a high-transmission area of Uganda [16]. PK/PD study participants were part of a larger randomized clinical trial comparing the antimalarial efficacy of AL versus that of dihydroartemisinin-piperaquine in young children (Supplementary Materials) [17–19]. Daily TMP-SMZ prophylaxis was given to all HIV-infected participants. HIV-exposed participants received TMP-SMZ until completion of breast-feeding, and if they remained HIV-uninfected, they were randomly assigned to continue TMP-SMZ therapy until 2 years of age or to discontinue prophylaxis [18]. HIV-infected participants were provided triple-drug antiretroviral therapy (ART) with nevirapine, lamivudine, and either stavudine or zidovudine, according to national guidelines. The PK/PD subtudy population consisted of children randomly assigned to receive AL for each episode of malaria occurring during longitudinal follow-up. Supplementary Figure 1 depicts the breakdown of HIV status, ART use, and TMP-SMZ use among substudy participants.
Diagnosis and Treatment of Malaria
Uncomplicated P. falciparum malaria was diagnosed in patients with both a positive thick blood smear (regardless of parasite density) and either a documented fever (tympanic temperature, >38.0°C) or a 24-hour history of fever. Clinical evaluation was made on days 0 (the day of diagnosis), 1, 2, 3, 7, 14, 21, and 28. Primary outcome was the 28-day risk of recurrent parasitemia. Sixty-three-day risk of recurrent malaria (parasitemia with a documented fever or a 24-hour history of fever) and polymerase chain reaction genotyping to distinguish new from recrudescent infections were assessed as secondary outcomes [20]. Children were eligible for repeat PK sampling with each malaria episode during follow-up.
Treatment Regimen
AL (Coartem, Novartis Pharmaceuticals) was administered as a fixed dose according to body weight at the time of diagnosis. All children received the same dose—20 mg of artemether and 120 mg of lumefantrine (1 tablet) twice daily over 3 days—because all body weights were <14 kg. All morning clinic doses were administered in the clinic as crushed tablets dispersed in approximately 5 mL of water, followed by administration of 150 mL of reconstituted cow's milk containing approximately 5 g of fat or breast-feeding, to ensure optimal and consistent absorption of lumefantrine [21]. Evening doses were provided to parents with milk powder. Full doses were readministered if vomiting occurred within 30 minutes.
Sample Collection and Analysis
A total of 100 µL of capillary whole-blood was collected into heparinized microtubes on day 0 (before the first dose), day 2 (before the fifth dose), day 3 (12 hours after the last dose), day 7 (108 hours after the last dose), and day 14 (276 hours after the last dose); Supplementary Figure 1. Blood was expelled onto Whatman 31ET Chr Chromatography paper pretreated with 0.75 M tartaric acid per validated methods [22] and later shipped and analyzed in the laboratory of one of the authors (Y. B.) with a lower limit of quantification (LLOQ) of 132 ng/mL and a limit of detection (LOD) of 52 ng/mL [22].
Pharmacokinetic Analysis
Population PK analysis with nonlinear mixed-effects modeling was conducted using Monolix software, version 4.3 (Lixoft, Orsay, France; Supplement) [23]. The structural model was defined as a 2-compartment open model with first-order absorption and linear elimination. Owing to limited data in the absorption phase, the rate constant of absorption (ka) was fixed to the previously reported value of 0.45 h−1 [24]. Disposition parameters of the model were capillary whole-blood clearance/bioavailability (CL/F), central volume of distribution/F (V1/F), intercompartmental clearance/F (Q/F), and peripheral volume of distribution/F (V2/F). A 1-compartment model was also tested. Data did not support testing of more complex structural models (eg, a 3-compartment model). Allometric scaling was included unconditionally in all models, including the basic model absent of other covariates, with respect to the 2 clearance parameters by multiplying each parameter by [weight/reference weight]0.75 and, for the 2 volume terms, by multiplying each parameter by [weight/reference weight]. The reference weight was taken to be the median, 9.0 kg, except for the final model, in which it was taken to be that of a 1-year-old child, 8.43 kg. Interindividual variability and interoccasion variability, modeled as log-normal distributions, were estimated for each parameter except Q/F and V2/F, for which interoccasion variability was fixed at 50%, and ka, which was fixed at 0. The covariates sex, weight, age, weight-for-age z scores (calculated using World Health Organization standards [25]), hemoglobin level, parasite density (log transformed), HIV status, ART use, breast-fed status, and TMP-SMZ prophylaxis (Table 1) were evaluated for their influence on PK (Supplement).
Table 1.
Characteristics of Patients in the Population Pharmacokinetics (PK) and Outcomes Data Sets
| Parameter | Population PK Data Set (n = 101) | PK Outcomes Data Set (n = 100) |
|---|---|---|
| Malaria episodes, no. | ||
| Overall | 207 | 222 |
| Per child | ||
| 1 | 101 | 100 |
| 2 | 56 | 62 |
| 3 | 34 | 35 |
| 4 | 12 | 19 |
| 5 | 4 | 6 |
| Malaria episodes per child, no., median (range) | 1.9 (1–5) | 2.0 (1–5) |
| Episodes in male children, % | 47.3 | 47.8 |
| Weight, kg, median (range) | 9.1 (6.1–13.0) | 9.0 (6.1–13.3) |
| Total lumefantrine dose per treatment course, mg/kg, median (range) | 81.1 (55.4–118.0) | 80.0 (54.1–118.0) |
| Age, mo, median (range) | 14.4 (6.6–22.2) | 14.9 (6.6–24.2) |
| 6 to <12 | 50 | 49 |
| 12–18 | 120 | 126 |
| >18 to 24 | 37 | 47 |
| Hemoglobin level at diagnosis, g/dL, median (range) | 10.0 (5.6–15.9) | 10.0 (5.6–15.9) |
| Parasite density, parasites, geometric mean no./µL (95% CI) | 15 568 (128–159 393) | 17 603 (13 762–22 516) |
| HIV-infected children | ||
| Overall | 9 | 8 |
| Malaria episodes, no. | 16 | 18 |
| Episodes involving urban-resident children, no. (%) | Not included | 16 (7.2) |
| Episodes involving breast-feeding children, no. (%) | 95 | 101 (46) |
| Children receiving TMP-SMZ prophylaxis | ||
| Overall, no. | 47 | 43 |
| Malaria episodes, no. | 80 | 85 |
| Episodes in underweight children, no. (%) | 26 (12.6)a | 29 (13.1) |
Abbreviations: CI, confidence interval; HIV, human immunodeficiency virus; IQR, interquartile range; TMP-SMZ, trimethoprim-sulfamethoxazole.
a Median z score in the population PK data set was −0.74 (IQR, −3.63–2.08).
Association Analysis Between Day 7 Lumefantrine Concentration and Recurrent Malaria
Only P. falciparum infection episodes were included in the PD analysis. The risk of recurrent malaria was analyzed using Cox proportional hazard regression with a robust sandwich estimator, which accounts for repeated episodes and censoring of patients with incomplete follow-up. Covariates evaluated were age group, parasite density (log transformed), residence (urban vs rural), hemoglobin level, TMP-SMZ use, HIV status, day 3 or day 7 capillary whole-blood lumefantrine concentration, and being underweight, based on a weight-for-age z score with a −2 cutoff [25]. For the PD analysis, PK values below the LOD were treated as 0, and those below the LLOQ were retained as measured. Statistical significance was defined as a 2-sided P value of <.05. Stata, version SE12.1 (StataCorp, College Station, TX), and SAS, version 9.4 (SAS Institute, Cary, NC), were used.
RESULTS
A total of 105 children participated in PK sampling and experienced 249 episodes of malaria subjected to PK sampling over the 6-month period. In 99.5% of treatments included in the analysis, the full course of 6 doses was taken.
Population Pharmacokinetic Data
For the population PK analysis, 207 episodes in 101 children were included and contributed 806 evaluable capillary whole-blood concentration-time data (Figure 1; Supplementary Materials). A summary of demographic and clinical characteristics of the participants is in Table 1. The median total body weight–adjusted lumefantrine dose was 84.7 mg/kg (range, 65.4–118.0 mg/kg) for children aged ≤12 months, 80.0 mg/kg (range, 55.4–116.1 mg/kg) for children aged 12–18 months, and 72 mg/kg (range, 54.1–90.0 mg/kg) for children aged ≥18 months. Underweight children received a higher median milligrams per kilogram dose than children with a z score greater than or equal to −2 (90.0 vs 77.4 mg/kg; P < .001).
Figure 1.

Capillary whole-blood lumefantrine concentration-time profiles in Ugandan children ages 6 months to 2 years after 3 days of twice-daily dosing of 120 mg in combination with 20 mg of artemether. Concentrations below the LOD are presented as gray stars at the LOD value (52 ng/mL) on days 14 and 7 (most are stacked on top of one another).
Population Pharmacokinetic Model
The final population PK model for capillary whole-blood lumefantrine concentrations was a 2-compartment model with first-order absorption. A 1-compartment model resulted in a significantly worse fit. Age was identified as having a significant influence on F despite modeling clearance and volume of distribution parameters with allometric scaling. The model for F is as follows: = [1*Agei/12]0.596, in which is the typical value of bioavailability, Agei is the age of the ith child, 12 is the reference age of 12 months, and 0.596 is an estimated value. The final population parameter estimates are provided in Table 2. The prediction of CL/F in a typical 12-month-old child is 2.19 L/h, with 8% (CV) interindividual variability and 9% interoccasion variability. The point estimate of residual error (standard deviation) was 37.8% (proportional) and 19.2 ng/mL (additive). Figure 2 shows the prediction intervals of capillary whole-blood lumefantrine concentration. Half of all children are predicted to reach the LOD (52 ng/mL) in approximately 10 days. Figure 3 demonstrates the goodness of fit of the final model. Supplementary Figure 2A–C shows individual data and predictions for the first 18 individuals and demonstrate that retaining data less than the LOD (although the exact value is unknown) informed the predictions.
Table 2.
Pharmacokinetics (PK) Parameter Estimates of Lumefantrine in Capillary Blood for Ugandan Children 6 Months to 2 Years of Age
| Variable | Point Estimate | RSE, % | IIV, % (RSE, %) | IOV (%) |
|---|---|---|---|---|
| Population PK parameter | ||||
| CL/F (L/h)a | 2.19 | 8 | 8 (174) | 9.3 (105) |
| V1/F (L/8.43 kg) | 83.2 | 7 | 15.2 (46) | 8.6 (133) |
| Q/F (L/h)a | 0.23 | 35 | 50c | 50c |
| V2/F (L/8.43 kg) | 441 | 74 | 50c | 50c |
| ka (h−1) | 0.45c | … | … | … |
| F | 1c | … | 34.9 (23) | 59.4 (8) |
| Age effectb | 0.596 | 38 | … | … |
| Residual variability | ||||
| Proportional (%) | 37.8 | 5 | … | … |
| Additive (ng/mL) | 19.2 | 18 | … | … |
| Secondary parameter | Point Estimate | IQR | 95% Prediction Interval | |
| AUC0-∞ (μg/mL•h)d | 347.8 | 243.6–510.9 | 94.7–940.9 | |
| Day 7 concentration (ng/mL)e | 202.4 | 143.1–321.6 | 55.0–589.5 | |
Abbreviations: AUC0-∞, area under the curve from 0 to ∞; CL, capillary whole-blood clearance; F, relative bioavailability; RSE, relative standard error; IIV, interindividual variability; IOV, interoccasion variability; IQR, interquartile range; Q, intercompartmental clearance; V1, central volume of distribution; V2, peripheral volume of distribution.
a Model: Pk•[WT/8.43]0.75, in which WT is total body weight, and 8.43 is the typical weight for a 12-month-old child.
b Covariate model: F•[age/12]θx.
c Fixed.
d Median of values calculated from individual estimates (modes) of CL and F.
e Median of predicted values (modes) for individuals in population analysis with day 7 measurement (n = 99).
Figure 2.

Distribution of the predicted lumefantrine capillary whole-blood lumefantrine concentration versus time (shaded area) for the final model superimposed on the data (points). The median is given by the center line, and the prediction intervals are indicated by the depth of shading, as shown on the scale to the right. With inclusion of the informatively censored data below the LOD, predictions are appropriately lower than the data at 14 days.
Figure 3.

Goodness of fit plots for the final model, illustrating individual weighted residuals IWRES, based on the conditional (cond.) mode versus time (top left) or individual predictions (bottom left) and the normalized prediction distribution error (NPDE) plots versus time (top right) or population prediction (bottom right). Data are represented by points (red for predicted concentrations whose actual values are below the LOD and blue for all other concentrations). Graphs patterns (absence of systematic relationships between x and y variables) suggest reasonable specification of the structural and error models.
Clinical Outcome Following Artemether-Lumefantrine Therapy
Of the 249 episodes enrolled in the PK/PD study, 222 episodes in 100 children were analyzed for 28-day outcome (Table 1). For the 137 episodes in which TMP-SMZ was not received, 59.1% (81), 19.7% (27), and 21.2% (29) were classified as adequate clinical and parasitological response (ACPR), late clinical failure (LCF), and late parasitological failure (LPF), respectively. Among the 85 in which TMP-SMZ was received, 65.9% (56) were classified as ACPR, 5.9% (5) as LPF, and 28.2% (24) as LCF. At 63 days, 65.8% of episodes (146 of 222) were followed by an episode of recurrent malaria (LCF), with 95.9% (139 of 145) classified as new infection by multilocus genotyping (genotyping was unsuccessful in 1 sample).
Day 3 or 7 Lumefantrine Concentration and Clinical Outcomes
The median concentration of lumefantrine on day 3 (n = 187 samples) was 2777 ng/mL (interquartile range [IQR], 1672–4760 ng/mL) and on day 7 (n = 216 samples) was 216 ng/mL (IQR, 136–345 ng/mL). Concentrations of lumefantrine on day 7 differed significantly in patients receiving TMP-SMZ (median, 243 ng/mL; IQR, 158–424 ng/mL; n = 85) versus those not receiving TMP-SMZ (median, 206 ng/mL; IQR, 129–302 ng/mL; n = 131; P = .018). While the total milligrams per kilogram dose was slightly higher in children receiving TMP-SMZ as compared to those not receiving TMP-SMZ (mean, 82.4 vs 78.3 mg/kg, respectively; P = .01), the total milligrams per kilogram dose did not explain the difference in lumefantrine levels between these groups. Concentrations on day 3 did not differ statistically, based on TMP-SMZ status.
The hazard of recurrent malaria (recurrent parasitemia or parasitemia plus fever) by 28 days is described in Table 3. Our receiver operating characteristic (ROC) curve analysis suggested that a day 7 capillary whole-blood lumefantrine concentration of approximately 200 ng/mL was the optimal cutoff for predicting the risk of 28-day recurrence, particularly for children not receiving TMP-SMZ (area under the ROC curve, 0.684). In our multivariate analysis, significant interaction was present between TMP-SMZ use and lumefantrine concentration (P = .0005). For children not receiving TMP-SMZ prophylaxis, a day 7 concentration of lumefantrine of <200 ng/mL conferred a 3-fold higher hazard of recurrent malaria as compared to concentrations of ≥200 ng/mL (hazard ratio, 2.97; 95% confidence interval, 1.59–5.55; P = .0007). For children receiving TMP-SMZ prophylaxis, the 28-day risk of malaria did not significantly vary on the basis of this threshold (hazard ratio, 0.50; 95% confidence interval, .22–1.13; P = .10). The 28-day cumulative risk of recurrent parasitemia, stratified by lumefantrine concentration threshold of 200 ng/mL and TMP-SMZ use, is shown in Figure 4. The 63-day cumulative risk of recurrent malaria (LCF only; secondary outcome) is shown in Supplementary Figure 2.
Table 3.
Cox Proportional Hazards Regression on Recurrent Malaria by Day 28, Unadjusted and Adjusted for Covariates
| Covariate | Unadjusted HR (95% CI) | P Value | Adjusted HR (95% CI) | P Value |
|---|---|---|---|---|
| Day 7 lumefantrine concentration <200 ng/mL | ||||
| Yes | 1.62 (1.05–2.64) | .03 | 1.21 (.71–2.07) | .48 |
| No | 1.00 | |||
| TMP-SMZ use | ||||
| Yes | 0.78 (.49–1.24) | .29 | 0.75 (.43–1.29) | .30 |
| No | 1.00 | |||
| TMP-SMZ*day 7 interaction | .001 | .0005 | ||
| Without TMP-SMZ use | ||||
| Day 7 concentration <200 ng/mL | 2.95 (1.61–5.43) | .0005 | 2.97 (1.59–5.55) | .0007 |
| Day 7 concentration ≥200 ng/mL | 1.00 | 1.00 | ||
| With TMP-SMZ use | ||||
| Day 7 concentration <200 ng/mL | 0.51 (.22–1.18) | .12 | 0.50 (.22–1.13) | .10 |
| Day 7 concentration ≥200 ng/mL | 1.00 | 1.00 | ||
| Age group, mo | .19 | .29 | ||
| ≥18 | 1.58 (.72–3.48) | .26 | 1.53 (.64–3.63) | .34 |
| 12–18 | 1.91 (.94–3.91) | .08 | 1.87 (.83–4.22) | .13 |
| 6–12 | 1.00 | 1.00 | ||
| Residence | ||||
| Rural | 1.53 (.52–4.44) | .44 | 1.21 (.43–3.43) | .71 |
| Urban | 1.00 | 1.00 | ||
| Hemoglobin level (g/dL)a | 0.94 (.83–1.06) | .32 | 0.93 (.83–1.05) | .25 |
| Parasite density (parasites/µL)a | 1.02 (.92–1.12) | .72 | 1.04 (.95–1.14) | .35 |
| Underweight (dichotomized)b | ||||
| Yes | 1.11 (.53–2.30) | .79 | 1.04 (.47–2.31) | .93 |
| No | 1.00 | 1.00 |
Abbreviations: CI, confidence interval; HR, hazard ratio; TMP-SMZ, trimethoprim-sulfamethoxazole.
a Values on the day of diagnosis; log-transformed parasite densities were used in the analysis.
b Weight-for-age z score less than −2 or greater than or equal to −2.
Figure 4.
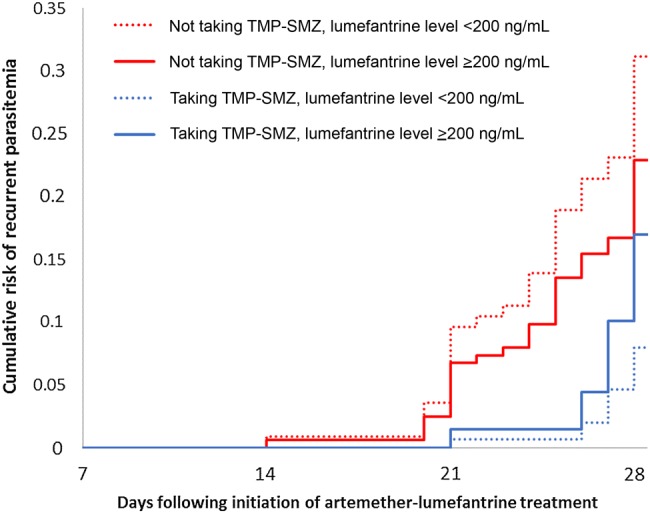
Cumulative risk of recurrent parasitemia by day 28 following treatment with artemether-lumefantrine, stratified by trimethoprim-sulfamethoxazole (TMP-SMZ) status and lumefantrine concentration of 200 ng/mL. Cox proportional hazards risk was adjusted for repeated measures, age, residence, underweight status, parasite density, and hemoglobin level on the day of diagnosis and was stratified by TMP-SMZ status and capillary whole-blood lumefantrine threshold of 200 ng/mL.
We also examined the association of day 3 lumefantrine concentration with 28-day outcomes in 182 children who had both day 3 and day 7 concentrations available. In our multivariate analysis, for children not receiving TMP-SMZ, each 1-unit increase in natural log–transformed day 3 lumefantrine concentration was associated with a 49% reduced hazard of 28-day recurrent malaria (P = .002). In comparison, each 1-unit increase in the natural log–transformed day 7 lumefantrine concentration was associated with a 20% reduced hazard of 28-day recurrent malaria (P = .002).
DISCUSSION
Dosing guidelines for AL, the most widely used ACT, are based on ranges of body weights, with the ratio of artemether to lumefantrine held fixed at 1:6, although the PK/PD of this regimen has not been evaluated thoroughly in the most vulnerable populations [17]. We report the single largest population PK/PD data set of lumefantrine in infants and very young children to-date. A 2-compartment model with first-order absorption best described our PK data, consistent with previous reports [26–29]. The most notable finding of our PK evaluation was that, within the age range studied, older children had a significantly larger bioavailability than younger children, after accounting for allometric scaling of clearances and volumes of distribution. Our model predicts a point estimate of relative bioavailability of 0.7 for a 6-month-old child as compared to 1.5 in a 2-year-old child, with a value of 1 for the reference 1-year-old population. We identified a previous report of age-dependent lumefantrine bioavailability in children aged 3 months to 12 years [30]. In this study, in the presence of concomitant food intake, a doubling of age was associated with a 22% increase in lumefantrine bioavailability.
Based on the pathways of elimination and transport of lumefantrine, a decrease in bioavailability, rather than an increase, would be expected with maturation. Lumefantrine is eliminated primarily via metabolism by cytochrome p450 3A4 (CYP3A4) and is also a substrate of P-glycoprotein [31, 32]. Both are expressed in the gut wall, mature over the first years of life, and act in concert to limit oral bioavailability. Alternative factors may explain our observed age-dependent increase in bioavailability. One explanation may relate to immature biliary function and reduced bile salt formation (approximately 50% of adults) [33]. Ionization and the partitioning of lumefantrine, a highly lipophilic drug, would be expected to exhibit a slower dissolution rate and lower permeability in the diminished intra-duodenal bile salt environment [21, 34]. Other age-related changes include alterations in protein binding and changes in the gastrointestinal tract, such as maturation of intestinal mucosa, gastric emptying, and intestinal transit [35–37]. Being able to understand the cause of lower average concentrations in the youngest of children is critical. For instance, no dose adjustment may be required if changes are related to protein binding, while adjustment may be warranted if differences are due to the amount absorbed.
A direct comparison of our PK results to those of previous studies is limited by differences in sampling methods (whole-blood and use of filter paper vs plasma), matrices (capillary vs venous) and assay methods (liquid chromatography tandem mass spectrometry vs high-performance liquid chromatography). In a study of children similar in weight (5–14.9 kg) to those in our study, the average area-under-the-curve from 0 to infinity (AUC0-∞) was 441–577 µg/mL•h, and the average peak concentration (Cmax) was 5.2–6.1 µg/mL, both of which are generally higher than our findings, although this prior study used venous plasma, which may contain higher concentrations than recovered from capillary whole-blood [8, 11]. In studies of adult patients with malaria, the AUC in venous plasma has ranged from 252 to 758 µg/mL•h [12, 21, 27, 38], and in studies of older African children (up to 13 years old) treated under supervision and in whom lumefantrine was measured for up to at least 14 days, the average AUC0-∞ in venous plasma ranged from 460 to 704 µg/mL•h, compared with 345 µg/mL•h in our study [11, 24, 30, 39]. The Cmax in these studies of older children, and that by Mwesigwa et al, ranged from 5.6 to 9.4 µg/mL, compared with approximately 3.9 µg/mL (90% prediction interval, 1.5–9.8 µg/mL) in our study [9].
In contrast to AUC data, more information is available to compare day 7 lumefantrine concentrations across different age groups. Our data reveal that children <2 years of age have a median day 7 capillary whole-blood concentration of 216 ng/mL. Two previous studies using the same sampling and matrix reported a median day 7 concentration of 304 ng/mL (IQR, 191–523 ng/mL) in children aged 6 months to 5 years [40] and a median of 430 ng/mL (IQR 282, 634) ng/mL in children aged 6 months to 15 years [41]. A recent WWARN meta-analysis, which included the above studies, suggested that children aged <3 years had the lowest day 7 exposure to lumefantrine, particularly those with a low weight-for-age z score [8].
Our study also revealed a potential PK interaction between TMP-SMZ and AL use, an interesting finding as TMP-SMZ use is recommended by the World Health Organization in all HIV-infected and HIV-exposed children until HIV can be definitively excluded [42]. Children receiving TMP-SMZ had higher day 7 lumefantrine concentrations than children not receiving TMP-SMZ (median, 243 ng/mL vs 206 ng/mL; P = .018), a finding that was not explained by the slightly higher milligrams per kilogram dose received by children taking TMP-SMZ. To our knowledge, the only other study to assess the PK of AL in the context of TMP-SMZ also found a higher level of lumefantrine in children taking TMP-SMZ [43]. It should be noted, however, that TMP-SMZ use was not identified as a significant covariate in our population PK analysis. While our current understanding of the metabolism of lumefantrine [31], TMP, and SMZ [44, 45] is unable to explain this finding, it is likely incomplete, as evidenced by 2 recent studies. A study of pregnant women receiving TMP-SMZ and mefloquine found that SMZ concentrations were reduced by approximately 50% in those receiving mefloquine [46], and a study of TMP-SMZ and rifampin, a potent CYP-inducer, reported significant reductions in TMP-SMZ concentrations in those receiving rifampin [47].
TMP-SMZ use, which itself provides protection against malaria, also influenced lumefantrine PK/PD. Among those not receiving TMP-SMZ, there was a nearly 3-fold higher risk of 28-day recurrence if the concentration of lumefantrine was <200 ng/mL (P = .0007). For children receiving TMP-SMZ, lumefantrine concentrations were not associated with the risk of recrudescence, likely because of the independent protective effects of TMP-SMZ against malaria. In these children, there was a trend towards an opposite interaction (a higher risk of recurrence with higher lumefantrine concentrations; Supplementary Figure 3), these findings should be interpreted with caution. One potential explanation could be an attenuation of the protective TMP-SMZ effect seen at higher lumefantrine levels, although the mechanism behind such an effect remains unclear. Assessment of TMP-SMZ levels and the potential impact of TMP-SMZ on lumefantrine absorption in future studies may be informative.
Finally, we evaluated the comparative predictive value of day 3 lumefantrine concentrations in those for whom both day 3 and day 7 concentrations were available. Our analysis revealed that day 3 concentrations were stronger predictors of 28-day recurrence than day 7 concentrations (HR, 0.51 and 0.80, respectively; P=.002 for both). These findings are supported by our earlier results, which suggest that AUC and day 3 lumefantrine exposure are highly correlated (r2 = 0.94; P < .0001) [9]. If day 3 concentrations prove to be equivalent or improved predictors of treatment outcomes than day 7 concentrations, it may be logistically easier to sample at this earlier time point in future PK/PD studies.
Our analysis has several limitations. Owing to its simplicity and convenience, capillary sampling on treated filter papers was used. While the use of dried blood spots is advantageous in field operations because it does not require plasma sample preparation and freeze shipping, its usefulness is limited because of the need for extraction of samples in a bioanalytical laboratory, as well as its decreased sensitivity and increased variability [8, 22]. In addition, the correlation between capillary whole-blood and venous plasma remains unclear. However, while average concentrations were lower in capillary whole-blood on filter paper than in venous plasma, a recent meta-analysis did not find an association of assay matrix and day 7 concentrations in a multivariable analysis [8].
An additional limitation was the lack of sufficient sampling times during the absorption phase, making it difficult to detect an absorption delay and related kinetics in our study. We were able to overcome these limitations by fixing the absorption rate constant (ka) to a prior reported value of 0.45 h−1. Additionally, since a limited amount of material were available, we were not able to determine the levels of desbutyl-lumefantrine (DBL) in this investigation. Measurement of DBL levels may have been of interest for its contribution to the therapeutic efficacy of lumefantrine, and as a secondary point, DBL levels are of value for the assessment of the rate and extent of CYP3A4 metabolism in this age group and its implication on the oral bioavailability of lumefantrine. Finally, the number of underweight children and children taking ART limited our ability to detect an impact of these variables on PK exposure.
In summary, a population PK approach has been used to characterize the PK of lumefantrine in young children age 6 months to 2 years. Lumefantrine's PK were best described by a 2-compartment open model with first-order absorption and linear elimination. Of the covariates studied, age was identified as the variable significantly affecting lumefantrine bioavailability. Exposure on day 7 in our patient population was generally lower than those previously reported in older children and adults despite higher actual milligrams per kilogram doses in this group, though at least some of the difference may be due to a difference in sampling methods. In addition, TMP-SMZ use was found to modulate the association between lumefantrine exposure and recurrent parasitemia. Further investigation into the mechanisms of altered AL exposure in young children, as well as optimization of dosing in this age group, should be done to improve exposure and reduce the risk of recurrent malaria in this vulnerable population.
Supplementary Data
Supplementary materials are available at http://jid.oxfordjournals.org. Consisting of data provided by the author to benefit the reader, the posted materials are not copyedited and are the sole responsibility of the author, so questions or comments should be addressed to the author.
Notes
Acknowledgments. We thank the clinical study team and our administrative staff; the children who participated in this study and their parents and guardians; Dr Grant Dorsey and Moses Kamya, for their support with the primary drug efficacy study; and Fangyong Li, from the Yale Center for Analytic Sciences, for help with the statistical analysis.
Disclaimer. The findings and conclusions of this report are those of the authors and do not necessarily represent the official position of the Centers for Disease Control and Prevention. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Financial support. This work was supported by the Doris Duke Charitable Foundation (clinical scientist development award to S. P. [grant 2007055]); Novartis Pharmaceuticals, through a clinical services agreement (study number COA566A 0940005); the President′s Emergency Plan for AIDS Relief, through the Department of Health and Human Services/Centers for Disease Control and Prevention (U62P024421); the National Center for HIV, Viral Hepatitis, STD, and TB Prevention; the National Institute of Child Health and Human Development (award R01HD068174); and the Global AIDS Program.
Potential conflicts of interest. All authors: No reported conflicts. All authors have submitted the ICMJE Form for Disclosure of Potential Conflicts of Interest. Conflicts that the editors consider relevant to the content of the manuscript have been disclosed.
References
- 1.World Health Organization. World malaria report 2015. Geneva: WHO, 2015. [Google Scholar]
- 2.White NJ, van Vugt M, Ezzet F. Clinical pharmacokinetics and pharmacodynamics and pharmacodynamics of artemether-lumefantrine. Clin Pharmacokinet 1999; 37:105–25. [DOI] [PubMed] [Google Scholar]
- 3.Amaratunga C, Lim P, Suon S et al. . Dihydroartemisinin-piperaquine resistance in Plasmodium falciparum malaria in Cambodia: a multisite prospective cohort study. Lancet Infect Dis 2016; 16:357–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Takala-Harrison S, Jacob CG, Arze C et al. . Independent emergence of artemisinin resistance mutations among Plasmodium falciparum in Southeast Asia. J Infect Dis 2015; 211:670–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Saunders DL, Vanachayangkul P, Lon C et al. . Dihydroartemisinin-piperaquine failure in Cambodia. N Engl J Med 2014; 371:484–5. [DOI] [PubMed] [Google Scholar]
- 6.Barnes KI, Watkins WM, White NJ. Antimalarial dosing regimens and drug resistance. Trends Parasitol 2008; 24:127–34. [DOI] [PubMed] [Google Scholar]
- 7.Worldwide Antimalarial Resistance Network: A. L. Dose Impact Study Group. The effect of dose on the antimalarial efficacy of artemether-lumefantrine: a systematic review and pooled analysis of individual patient data. Lancet Infect Dis 2015; 15:692–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.WorldWide Antimalarial Resistance Network Lumefantrine: P. K. P. D. Study Group. Artemether-lumefantrine treatment of uncomplicated Plasmodium falciparum malaria: a systematic review and meta-analysis of day 7 lumefantrine concentrations and therapeutic response using individual patient data. BMC Med 2015; 13:227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mwesigwa J, Parikh S, McGee B et al. . Pharmacokinetics of artemether-lumefantrine and artesunate-amodiaquine in children in Kampala, Uganda. Antimicrob Agents Chemother 2010; 54:52–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sambol NC, Yan L, Creek DJ et al. . Population Pharmacokinetics of Piperaquine in Young Ugandan Children Treated With Dihydroartemisinin-Piperaquine for Uncomplicated Malaria. Clin Pharmacol Ther 2015; 98:87–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Djimde AA, Tekete M, Abdulla S et al. . Pharmacokinetic and pharmacodynamic characteristics of a new pediatric formulation of artemether-lumefantrine in African children with uncomplicated Plasmodium falciparum malaria. Antimicrob Agents Chemother 2011; 55:3994–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hietala SF, Martensson A, Ngasala B et al. . Population pharmacokinetics and pharmacodynamics of artemether and lumefantrine during combination treatment in children with uncomplicated falciparum malaria in Tanzania. Antimicrob Agents Chemother 2010; 54:4780–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Staehli Hodel EM, Guidi M, Zanolari B et al. . Population pharmacokinetics of mefloquine, piperaquine and artemether-lumefantrine in Cambodian and Tanzanian malaria patients. Malar J 2013; 12:235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Homsy J, Dorsey G, Arinaitwe E et al. . Protective efficacy of prolonged co-trimoxazole prophylaxis in HIV-exposed children up to age 4 years for the prevention of malaria in Uganda: a randomised controlled open-label trial. Lancet Glob Health 2014; 2:e727–36. [DOI] [PubMed] [Google Scholar]
- 15.Manyando C, Njunju EM, D'Alessandro U, Van Geertruyden JP. Safety and efficacy of co-trimoxazole for treatment and prevention of Plasmodium falciparum malaria: a systematic review. PLoS One 2013; 8:e56916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Okello PE, Van Bortel W, Byaruhanga AM et al. . Variation in malaria transmission intensity in seven sites throughout Uganda. Am J Trop Med Hyg 2006; 75:219–25. [PubMed] [Google Scholar]
- 17.World Health Organization. Guidelines for the treatment of malaria. 3rd ed. Geneva, Switzerland, 2015. [Google Scholar]
- 18.Arinaitwe E, Sandison TG, Wanzira H et al. . Artemether-lumefantrine versus dihydroartemisinin-piperaquine for falciparum malaria: a longitudinal, randomized trial in young Ugandan children. Clin Infect Dis 2009; 49:1629–37. [DOI] [PubMed] [Google Scholar]
- 19.Wanzira H, Kakuru A, Arinaitwe E et al. . Longitudinal outcomes in a cohort of Ugandan children randomized to artemether-lumefantrine versus dihydroartemisinin-piperaquine for the treatment of malaria. Clin Infect Dis 2014; 59:509–16. [DOI] [PubMed] [Google Scholar]
- 20.Greenhouse B, Myrick A, Dokomajilar C et al. . Validation of microsatellite markers for use in genotyping polyclonal Plasmodium falciparum infections. Am J Trop Med Hyg 2006; 75:836–42. [PMC free article] [PubMed] [Google Scholar]
- 21.Ashley EA, Stepniewska K, Lindegardh N et al. . How much fat is necessary to optimize lumefantrine oral bioavailability? Trop Med Int Health 2007; 12:195–200. [DOI] [PubMed] [Google Scholar]
- 22.Blessborn D, Romsing S, Annerberg A et al. . Development and validation of an automated solid-phase extraction and liquid chromatographic method for determination of lumefantrine in capillary blood on sampling paper. J Pharm Biomed Anal 2007; 45:282–7. [DOI] [PubMed] [Google Scholar]
- 23.Monolix. Monolix users guide. http://www.lixoft.eu/products/monolix/documentation/ Accessed 2 November 2015.
- 24.Salman S, Page-Sharp M, Griffin S et al. . Population pharmacokinetics of artemether, lumefantrine, and their respective metabolites in Papua New Guinean children with uncomplicated malaria. Antimicrob Agents Chemother 2011; 55:5306–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.World Health Organization. WHO Multicentre Growth Reference Study Group 2006. WHO child growth standards. Length/height-for-age, weight-for-age, weight-for-length, weight-for-height and body mass index-for-age: methods and development. Geneva: World Health Organization, 2006. [Google Scholar]
- 26.Ezzet F, van Vugt M, Nosten F, Looareesuwan S, White NJ. Pharmacokinetics and pharmacodynamics of lumefantrine (benflumetol) in acute falciparum malaria. Antimicrob Agents Chemother 2000; 44:697–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kloprogge F, McGready R, Hanpithakpong W et al. . Lumefantrine and desbutyl-lumefantrine population pharmacokinetic-pharmacodynamic relationships in pregnant women with uncomplicated Plasmodium falciparum malaria on the Thailand-Myanmar border. Antimicrob Agents Chemother 2015; 59:6375–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hoglund RM, Byakika-Kibwika P, Lamorde M et al. . Artemether-lumefantrine coadministration with antiretrovirals; population pharmacokinetics and dosing implications. Br J Clin Pharmacol 2015; 79:636–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Maganda BA, Ngaimisi E, Kamuhabwa AA, Aklillu E, Minzi OM. The influence of nevirapine and efavirenz-based anti-retroviral therapy on the pharmacokinetics of lumefantrine and anti-malarial dose recommendation in HIV-malaria co-treatment. Malar J 2015; 14:179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Borrmann S, Sallas WM, Machevo S et al. . The effect of food consumption on lumefantrine bioavailability in African children receiving artemether-lumefantrine crushed or dispersible tablets (Coartem) for acute uncomplicated Plasmodium falciparum malaria. Trop Med Int Health 2010; 15:434–41. [DOI] [PubMed] [Google Scholar]
- 31.Lefevre G, Thomsen M. Clinical pharmacokinetics of artemether and lumefantrine (Riamet®). Clin Drug Investig 1999; 18:467–80. [Google Scholar]
- 32.Wahajuddin, Raju KS, Singh SP, Taneja I. Investigation of the functional role of P-glycoprotein in limiting the oral bioavailability of lumefantrine. Antimicrob Agents Chemother 2014; 58:489–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bartelink IH, Rademaker CM, Schobben AF, van den Anker JN. Guidelines on paediatric dosing on the basis of developmental physiology and pharmacokinetic considerations. Clin Pharmacokinet 2006; 45:1077–97. [DOI] [PubMed] [Google Scholar]
- 34.Heimann G. Enteral absorption and bioavailability in children in relation to age. Eur J Clin Pharmacol 1980; 18:43–50. [DOI] [PubMed] [Google Scholar]
- 35.Fernandez E, Perez R, Hernandez A, Tejada P, Arteta M, Ramos JT. Factors and mechanisms for pharmacokinetic differences between pediatric population and adults. Pharmaceutics 2011; 3:53–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rakhmanina NY, van den Anker JN. Pharmacological research in pediatrics: from neonates to adolescents. Adv Drug Deliv Rev 2006; 58:4–14. [DOI] [PubMed] [Google Scholar]
- 37.Colussi D, Parisot C, Legay F, Lefevre G. Binding of artemether and lumefantrine to plasma proteins and erythrocytes. Eur J Pharm Sci 1999; 9:9–16. [DOI] [PubMed] [Google Scholar]
- 38.Mosha D, Guidi M, Mwingira F et al. . Population pharmacokinetics and clinical response for artemether-lumefantrine in pregnant and nonpregnant women with uncomplicated Plasmodium falciparum malaria in Tanzania. Antimicrob Agents Chemother 2014; 58:4583–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Abdulla S, Sagara I, Borrmann S et al. . Efficacy and safety of artemether-lumefantrine dispersible tablets compared with crushed commercial tablets in African infants and children with uncomplicated malaria: a randomised, single-blind, multicentre trial. Lancet 2008; 372:1819–27. [DOI] [PubMed] [Google Scholar]
- 40.Ngasala BE, Malmberg M, Carlsson AM et al. . Efficacy and effectiveness of artemether-lumefantrine after initial and repeated treatment in children <5 years of age with acute uncomplicated Plasmodium falciparum malaria in rural Tanzania: a randomized trial. Clin Infect Dis 2011; 52:873–82. [DOI] [PubMed] [Google Scholar]
- 41.Ursing J, Kofoed PE, Rodrigues A et al. . Similar efficacy and tolerability of double-dose chloroquine and artemether-lumefantrine for treatment of Plasmodium falciparum infection in Guinea-Bissau: a randomized trial. J Infect Dis 2011; 203:109–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.World Health Organization. Guidelines on co-trimoxazole prophylaxis for HIV-related infections among children, adolescents and adults: Recommendations for a public health approach, 2006. http://www.who.int/hiv/pub/plhiv/ctx/en/ Accessed 1 March 2016.
- 43.Kredo T, Mauff K, Van der Walt JS et al. . Interaction between artemether-lumefantrine and nevirapine-based antiretroviral therapy in HIV-1-infected patients. Antimicrob Agents Chemother 2011; 55:5616–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Soeters HM, von Gottberg A, Cohen C, Quan V, Klugman KP. Trimethoprim-sulfamethoxazole prophylaxis and antibiotic nonsusceptibility in invasive pneumococcal disease. Antimicrob Agents Chemother 2012; 56:1602–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Goldman JL, Leeder JS, Van Haandel L, Pearce RE. In vitro hepatic oxidative biotransformation of trimethoprim. Drug Metab Dispos 2015; 43:1372–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Green M, Otieno K, Katana A et al. . Pharmacokinetics of mefloquine and its effect on sulfamethoxazole and trimethoprim steady-state blood levels in intermittent preventive treatment (IPTp) of pregnant HIV-infected women in Kenya. Malar J 2016; 15:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ribera E, Pou L, Fernandez-Sola A et al. . Rifampin reduces concentrations of trimethoprim and sulfamethoxazole in serum in human immunodeficiency virus-infected patients. Antimicrob Agents Chemother 2001; 45:3238–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}