

Summary
Human Vγ9Vδ2 T cells respond to tumour cells by sensing elevated levels of phosphorylated intermediates of the dysregulated mevalonate pathway, which is translated into activating signals by the ubiquitously expressed butyrophilin A1 (BTN3A1) through yet unknown mechanisms. Here, we developed an unbiased, genome-wide screening method that identified RhoB as a critical mediator of Vγ9Vδ2 TCR activation in tumour cells. Our results show that Vγ9Vδ2 TCR activation is modulated by the GTPase activity of RhoB and its redistribution to BTN3A1. This is associated with cytoskeletal changes that directly stabilize BTN3A1 in the membrane, and the subsequent dissociation of RhoB from BTN3A1. Furthermore, phosphoantigen accumulation induces a conformational change in BTN3A1, rendering its extracellular domains recognizable by Vγ9Vδ2TCRs. These complementary events provide further evidence for inside-out signaling as an essential step in the recognition of tumor cells by a Vγ9Vδ2TCR.
eTOC Blurb
Sebestyen et al. show that Vγ9Vδ2TCR activation is modulated by the GTPase activity of RhoB in tumour cells, and by the relocalization of RhoB to BTN3A1. Subsequently, a phosphoantigen-induced conformational change in BTN3A1 leads to its recognition by Vγ9Vδ2TCRs.
Introduction
γδT cells are unconventional T cells with strong reactivity towards a broad spectrum of tumours of diverse tissue origin. γδT cells combine potent anti-tumour effector functions with the recognition of broadly expressed tumour-associated molecules, and these features have put γδT cells in the spotlight for clinical application in cancer immunotherapy. Activation of γδT cells involves the sensing of metabolic changes in cancer cells that result in the expression of generic stress molecules. These molecules are upregulated upon transformation or distress (Bonneville et al., 2010, Vantourout and Hayday, 2013). However, progress in the clinical application of γδT cells for cancer treatment is hampered by conflicting published data from various labs that describe contradicting molecular requirements for γδT cell activation (Scheper et al., 2014, Vavassori et al., 2013a, Sandstrom et al., 2014), as well as by a lack of prognostic markers to assess which patients may benefit from such therapy.
Vγ9Vδ2 T cells, the major γδT cell subset in human peripheral blood, express γδT cell receptors (TCR) composed of Vγ9 and Vδ2 chains, and are specifically activated by intermediates of the mammalian mevalonate pathway (Gober et al., 2003, Constant et al., 1994), such as isopentenyl pyrophosphate (IPP), or by the microbial 2-C-methyl-D-erythritol 4-phosphate (MEP) pathway (Morita et al., 2007). Intracellular phosphoantigen (pAg) levels accumulate in tumour cells due to dysregulation of the mevalonate pathway or upon microbial infection, allowing the targeting of transformed or infected cells by Vγ9Vδ2 T cells. Similarly, intracellular phosphoantigen levels can be pharmaceutically increased by treating cells with mevalonate pathway inhibitors such as aminobisphosphonates (ABP), thereby sensitizing cells towards recognition by Vγ9Vδ2 T cells. Although the involvement of the Vγ9Vδ2 TCR in detecting elevated phosphoantigen levels was demonstrated as early as the 1990’s (Bukowski et al., 1998, Davodeau et al., 1993, Wang et al., 2010), the molecular determinants required for activation of Vγ9Vδ2 TCRs on target cells have long remained elusive.
Although recent breakthrough studies have identified the membrane-expressed butyrophilin BTN3A1 (CD277) as a key molecule in phosphoantigen-induced activation of Vγ9Vδ2 T cells, (Harly et al., 2012, Sandstrom et al., 2014, Vavassori et al., 2013b) they describe conflicting molecular roles for BTN3A1 in the Vγ9Vδ2 T cell activation process. While Vavassori and colleagues (Vavassori et al., 2013a) suggest that BTN3A1 as antigen presenting molecule triggers Vγ9Vδ2 T cell activation, data from (Sandstrom et al., 2014, Harly et al., 2012) support an inside-out signaling mechanism for BTN3A1, where phosphonatigens bind to its intracellular region and in which model immobilization of BTN3A1 at the cell surface may contribute to an extracellular cue for recognition by Vγ9Vδ2 TCRs. Since BTN3A1 is expressed on both transformed and healthy human cells (Compte et al., 2004), additional molecules are likely involved in mediating selective recognition of tumour targets by Vγ9Vδ2 T cells, including e.g. the cytoskeletal adaptor protein Periplakin (Rhodes et al., 2015). However, it is unclear how these different proposed molecules and mechanisms are linked. Here, we developed an unbiased, genome-wide screening method, SNP-Associated comPutational Pathway Hunt Including shRNA Evaluation (SAPPHIRE), and identified important molecular requirements for Vγ9Vδ2 T cell-mediated recognition of cancer (stem) cells. We propose a two-component mechanism occurring in a tumour cell, which allows Vγ9Vδ2 T cells to specifically sense cellular transformation.
Results
Identification of genetic loci associated with Vγ9Vδ2 TCR-mediated target cell recognition by SNP-Associated comPutational Pathway Hunt Including shRNA Evaluation (SAPPHIRE)
We hypothesized that differences in the genetic backgrounds of tumour cells affect recognition of these cells by Vγ9Vδ2 TCR engineered T cells. Therefore, we utilized the library of cell lines from the Centre d’Etude du Polymorphisme Humain (CEPH), which contains a large collection of EBV-transformed B cell lines (EBV-LCLs) obtained from several family pedigrees (Dausset et al., 1990) and genotyped for millions of SNPs (International HapMap, 2003). CD4+ αβT cells engineered to express one defined Vγ9Vδ2 TCR were utilized for the functional screening in order to eliminate fluctuations in recognition by a diverse γδTCR repertoire (Gründer et al., 2012) and varying expression of NK receptors (Scheper et al., 2014, Correia et al., 2013). The recognition phenotypes of EBV-LCLs by Vγ9Vδ2 TCR T cells were assessed by means of IFNγ production and showed an activating phenotype for 33 EBV-LCLs, while 7 were non-activating (Figure S1A and S1B). Importantly, in the zygocity analysis, EBV-LCLs with a non-activating phenotype represent more power than those with an activating phenotype analyzed with SAPPHIRE. Hypothetical zygosities for candidate genetic loci were deduced using classical Mendelian inheritance patterns within CEPH family trios, where the influence of candidate alleles on Vγ9Vδ2 TCR-mediated recognition was assumed to be dominant. The resulting recognition phenotypes combined with family pedigrees of the CEPH cell lines overcame the need to screen large numbers of LCL lines (Spaapen et al., 2008), and allowed the precise prediction of zygosities of candidate loci for 12 CEPH individuals (Figure 1A). The hypothetical loci zygosities were then correlated with available genotype information of SNPs within the study’s population (Spaapen et al., 2008), resulting in the identification of 17 SNPs whose genotypes correlated perfectly (100%) with predicted zygosities (Figure 1B). Since none of these 17 SNPs, nor their proxy SNPs within high linkage disequilibrium (r2 > 0.8), directly affected genes by causing changes in protein coding sequences, we speculated that rather than playing direct roles, the SNPs we identified could represent surrogate markers for genetic regions associated with susceptibility to Vγ9Vδ2 TCR+ T cell recognition. In addition, we queried the genomic vicinity of the 17 SNPs for neighboring candidate genes (Figure 1C).
Figure 1. SNP-Associated comPutational Pathway Hunt Including shRNA Evaluation (SAPPHIRE) to identify genetic loci associated with Vγ9Vδ2 TCR stimulation.
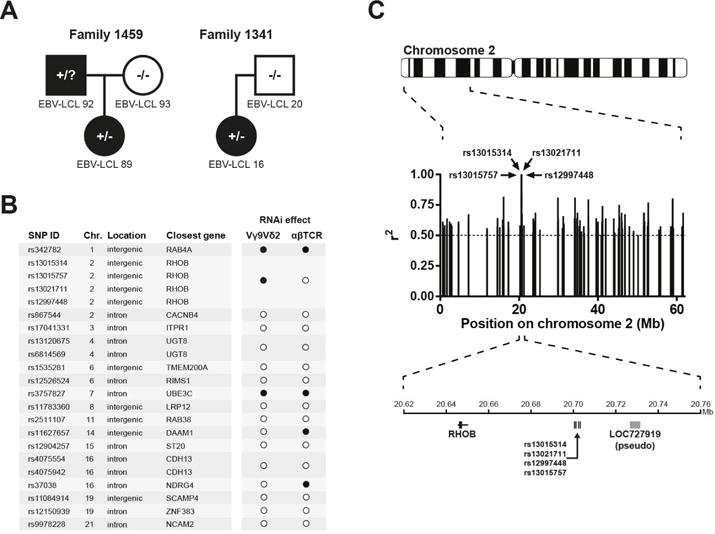
(A) The recognition of CEPH EBV-LCL lines by Vγ9Vδ2 TCR+ T cells provided the basis for deducing hypothetical zygosities of candidate loci in each cell line (black: recognized; white: not recognized; square: male; circle: female; +/−: heterozygous; −/−: homozygous negative; +/?: undetermined). Members of two CEPH families are shown as examples. For CEPH ID numbers of cell lines, see Figure S1A. (B) Genetic association analysis revealed 17 SNPs of which genotypes correlated 100% (r2 = 1) with predicted zygosities of cell lines. Locations and nearest neighboring genes of SNPs are indicated. The effect of knocking down candidate genes on recognition of EBV-LCL 48 by T cells transduced with either Vγ9Vδ2 TCR clone G115 or an HLA-A*0201-restricted WT1126–134-specific αβTCR are indicated by black circles (significant effect on T cell activation) and white circles (no effect). For testing recognition by WT1 αβTCR+ T cells, the EBV-LCL 48 line was pulsed with WT1126–134 peptide. (C) Associating SNPs resulting from association analysis with candidate genes. The genetic region of the SNPs neighboring RhoB is shown as an example. Each bar represents one SNP and r2 values represent correlation between predicted zygosities and SNP genotypes.
To test the relevance of these genes for Vγ9Vδ2 TCR+ T cell-mediated target recognition, we knocked down all 17 SNP-adjacent genes in the Vγ9Vδ2 TCR+ T cell-activating EBV-LCL line 48. We then assessed the effect of knockdown on activation of Vγ9Vδ2 TCR+ T cells by measuring IFNγ production, which was reduced upon the knockdown of three genes (RAB4A, RHOB and UBE3C) (Figure 1B). To ensure that potential knockdown effects pointed to genes that selectively affect Vγ9Vδ2 TCR-dependent activation, the three knock-down variants of EBV-LCL line 48 were pulsed with Wilm’s tumour 1 (WT1) peptide and tested for recognition with T cells engineered to express the cognate WT1-specific αβTCR (Kuball et al., 2007). The selective knockdown of the small GTPase RhoB significantly affected the activation of Vγ9Vδ2 TCR− but not αβTCR-engineered T cells (Figure 1B). Similar data were observed after partial knock down of RhoB in the prototypic Vγ9Vδ2 T cell target cell line Daudi (Figure S2B), as well as after CRISPR/Cas-mediated partial RhoB knock out in the renal carcinoma cell line MZ1851RC (Figure 2A). Interestingly, even complete knock out of RhoB in 293 HEK cells resulted only in partial depletion of target cell mediated Vγ9Vδ2 TCR T cell activation (Figure S2A). In addition, knock out of either RhoA or RhoC genes in 293 HEK cells did not significantly influence their ability to activate Vγ9Vδ2 TCR+ T cells (Figure 2B and Figure S2C), emphasizing that RhoB modulates the recognition of tumour cells by a defined Vγ9Vδ2 TCR in a non-redundant role.
Figure 2. RhoB activity correlates with target cell recognition by Vγ9Vδ2 TCR+ T cells.
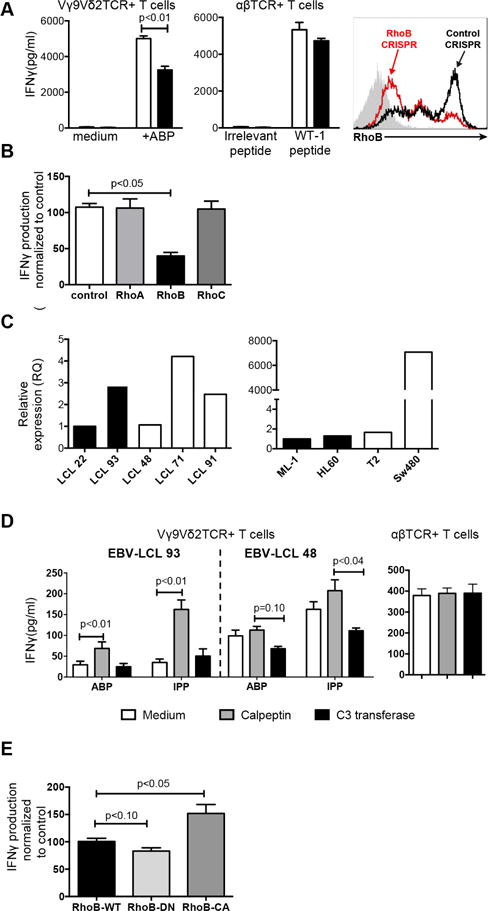
(A) RhoB was knocked out in the renal cancer cell line MZ1851RC using the CRISPR/Cas system. MZ1851RC cells were treated with either pamidronate or HLA-A*0201-restricted WT1126–134 peptide and the effect on target cell recognition by Vγ9Vδ2 TCR+ and WT1 αβTCR+ T cells, respectively, was determined by measuring IFNγ production. A guide RNA targeting an irrelevant sequence was used as control for knock out, while medium or irrelevant peptide loaded tumor cells were used as controls for T cell stimulation. Data show mean±S.E.M of three independent experiments, in duplicate samples. Level of knock out was determined by intracellular flow cytometry. (B) The effect of knock-out of RhoA, B and C in 293 HEK cells on recognition by Vγ9Vδ2 TCR+ T cells was assessed by measuring IFNγ production. A guide RNA targeting an irrelevant sequence was used as control. Figure shows IFNγ production normalized to irrelevant knock out samples of three independent experiments, in duplicate samples. (C) RNA expression of RhoB was measured by qPCR in either non-recognized (black bars) or recognized (white bars) EBV-LCLs and tumour cell lines. Data is representative of 2 repeated experiments. (D) The non-recognized EBV-LCL line 93 or the recognized EBV-LCL 48 were pretreated with either calpeptin or C3 transferase in combination with pamidronate or IPP and the effect on stimulation of Vγ9Vδ2 TCR+ T cells was assessed by measuring IFNγ. The effect of Rho-modulating compounds on recognition of WT1126–134 peptide-pulsed EBV-LCL 48 cells by WT1 αβTCR+ T cells was measured in parallel. Data shows mean +/− S.E.M of at least three independent experiments. (E) HEK 293 cells were transfected with dominant-negative (RhoB-DN), constitutively active (RhoB-CA) or wild type RhoB (RhoB-WT) and the effect of activity variants on target cell recognition by Vγ9Vδ2 TCR+ T cells in the presence of pamidronate was determined by measuring IFNγ. Figure shows IFNγ production normalized to wild type RhoB samples of three independent experiments. Significance of data has been analyzed by Mann-Whitney test on figure A, B and D, and by Kruskal-Wallis test and Dunn’s multiple comparison test on figure E.
Target cell recognition by Vγ9Vδ2 TCRs depends on Rho GTPase activity
Genomic sequencing of RhoB and associated promoter regions did not identify genetic variations associated with our marker SNP (data not shown), suggesting that RhoB is indirectly regulated by other gene loci influencing expression or activity (Pombo and Dillon, 2015). Our primary observations however confirmed that neither RhoB RNA nor protein levels in various recognized and two non-recognized tumour cell lines correlated with their ability to activate Vγ9Vδ2 TCR+ T cells (Figure 2C, S2D and S2E). We therefore assessed the impact of RhoB GTPase activity by pretreating different tumour cell lines with either the Rho GTPase activator calpeptin or inhibitor C3 transferase (Aktories et al., 1992, Schoenwaelder and Burridge, 1999). Pretreatment with calpeptin significantly sensitized EBV-LCL 93 cells for recognition by Vγ9Vδ2 TCR+ T cells while, conversely, inhibition of Rho GTPase with C3 transferase resulted in significantly reduced activation of Vγ9Vδ2 TCR+ T cells by LCL 48 cells (Figure 2D). Importantly, modulation of Rho GTPase activity did not affect the recognition of WT1 peptide-pulsed EBV-LCL 48 cells by WT1 αβTCR-transduced T cells. To specify that enzymatic activity of RhoB regulates tumour cell recognition by Vγ9Vδ2 TCR+ T cells, we transfected HEK 293 cells with wildtype or dominant negative form corresponding to the GDP-bound state (RhoB-DN), or with the constitutively active form corresponding to the GTP-bound form (RhoB-CA) of RhoB (Kamon et al., 2006) and used them as target cells for Vγ9Vδ2 TCR+ T cells (Figure 2E). HEK cells overexpressing RhoB-CA mutants were able to trigger significantly stronger Vγ9Vδ2 TCR-specific responses than cells expressing wild type RhoB, while RhoB-DN-transfected HEK cells showed a significantly reduced ability to stimulate Vγ9Vδ2 TCR+ T cells compared to cells expressing wildtype RhoB. Thus, these data suggest that the biochemical activity of RhoB GTPases is a critical factor modulating the recognition of cancer cells by Vγ9Vδ2 TCR+ T cells.
The intracellular distribution of RhoB in cancer (stem) cells marks susceptibility for Vγ9Vδ2 TCR-mediated recognition
Rho proteins have been shown to translocate within cellular compartments upon activation (Kranenburg et al., 1997). Therefore, we hypothesized an impact on subcellular localization of RhoB in recognized versus non-recognized cells. Additionally, ABP treatment of cells have been demonstrated to increase the GTP-bound forms of Rho GTPases (Dunford et al., 2006) that could provide a possible link between dysregulation of mevalonate pathway in tumor cells and RhoB cellular compartmentalization. This hypothesis was supported by confocal imaging which revealed that distribution of RhoB patches that were primarily reported to locate to membranes of intracellular vesicles such as endosomes (Adamson et al., 1992, Michaelson et al., 2001), segregated strongly with the susceptibility of different target tumour cell lines to Vγ9Vδ2 TCR-mediated recognition. RhoB was selectively excluded from nuclear areas in cells that are able to activate Vγ9Vδ2 TCR+ T cells (Figure 3A, B, S3A and S3E). Crucially, re-localization of RhoB from the nucleus, or from the nuclear membrane to extranuclear sites was induced by ABP as well as by soluble phosphoantigen IPP in cell lines (Figure 3C and D), emphasizing that this process is dependent on accumulation of intracellular phosphoantigen. Expressing transgenic RhoB-GFP in either recognized or non-recognized tumour cell lines followed intracellular distribution of the endogenous RhoB (Figure S3B), suggesting post-translational regulation of RhoB. The homogenous intracellular distribution of other GTPases such as RhoA did not change upon ABP treatment (Figure S3C), supporting the observation that RhoA knock down did not impact Vγ9Vδ2 TCR-mediated recognition (Figure 2B). We found that RhoB was excluded from the nucleus in human, but not in mouse dendritic cells selectively treated with ABP, even though RhoB protein sequences are identical in both species (Figure 3E and S3D). This is consistent with our previous observations that human, but not mouse monocyte-derived dendritic cells are able to activate Vγ9Vδ2 TCR+ T cells (Marcu-Malina et al., 2011). In order to test whether redistribution of RhoB occurs upon ABP treatment in leukemic cancer stem cells also, we sorted leukemic blasts, cancer stem cells and healthy stem cells from the very same donor based on flow markers (Terwijn et al., 2014) and we quantified distribution of RhoB. Indeed, in primary blasts from an acute myeloid leukemia patient/donor, RhoB localization correlated with the recognition by Vγ9Vδ2 TCR-engineered T cells (Figure 3B and S3A) including leukemic stem cells, but not healthy stem cells from the same donor (Figure 3F). This supports our previous observation that Vγ9Vδ2 TCR-mediated recognition selectively targets the leukemic but not the healthy stem cell pool (Marcu-Malina et al., 2011). Taken together, the intracellular distribution of the small GTPase RhoB strongly correlates with recognition of diverse tumour targets, including cancer stem cells, and exclusion of RhoB from the nucleus is a signature of tumour cells that are susceptible for targeting by Vγ9Vδ2 TCR+ T cells.
Figure 3. Intracellular distribution of RhoB correlates with the recognition of target cells by Vγ9Vδ2 TCR+ T cells.
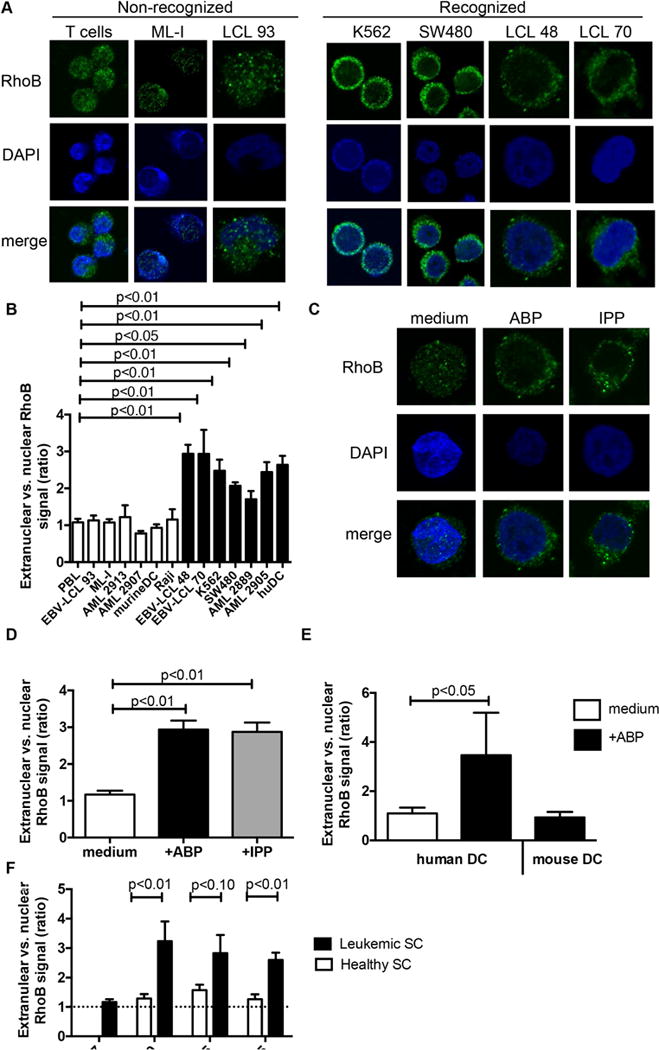
(A) Non-recognized healthy T cells, leukemic cell line ML-I and EBV-LCL 93 cells, and recognized leukemia cell line K562, colon carcinoma cell line SW480, EBV-LCL 48 and EBV-LCL 70 cells were treated with pamidronate and loaded onto poly-L-lysine-coated coverslips. Attached cells were fixed and permeabilized, and stained using RhoB-specific antibody followed by an Alexa Fluor 488-conjugated secondary antibody. RhoB distribution was subsequently analyzed by confocal microscopy and representative images are shown (green: RhoB; blue: nucleus [DAPI]). (B) T cells from healthy donors, EBV-LCL line 93, 48 and 70, tumour cell lines ML-I, K562, SW480, Raji, primary AML blasts AML2913, AML2907, AML2889, AML2905, and murine and human DCs were treated with pamidronate and analysed for the intracellular distribution of RhoB in confocal microscopy. White bars represent target cells with a non-activating phenotype, while black bars indicate target cells that are able to activate Vγ9Vδ2 TCR+ T cells. The RhoB signal ratio between nuclear and extranuclear cellular compartments was measured using ImageJ image analysis software. Graphs show average ratios of at least 10 different cells ±S.E.M. Statistical significance compared to PBL was determined by using Kruskal-Wallis test and Dunn’s multiple comparison test. (C) The intracellular RhoB distribution and (D) extranuclear/nuclear RhoB signal ratios with ABP pamidronate and soluble IPP sensitization for recognized, ABP/IPP-sensitive EBV-LCL 48 were analyzed as in (A and B). Graphs show average ratios of at least 10 different cells ±S.E.M. Statistical significance compared to untreated EBV-LCL 48 was determined by using Mann-Whitney test. (E) The intracellular RhoB distribution in the presence or absence of ABP pamidronate was determined in monocyte derived human dendritic cells from two different donors. Bone marrow derived mouse dendritic cells (>95% CD11c+) were treated with ABP pamidronate and used for intracellular labeling of RhoB. Graphs show average ratios of at least 10 different cells ±S.E.M. Statistical significance compared to LPS treated human DCs was determined by using Mann-Whitney test. (F) CD34+CD38− leukemic stem cells were sorted from four patients of which leukemic blasts were recognized (AML 2889, AML 1665, AML 2575) and non-recognized (AML 2907), respectively, and the ratios between extranuclear and nuclear RhoB signal was measured. Graphs show average ratios of at least 10 different cells ±S.E.M. Statistical significance compared to CD34+CD38− healthy stem cells was determined by using Mann-Whitney test.
RhoB regulates membrane mobility of BTN3A1 on cancer cells
Decreased mobility of the transmembrane protein BTN3A1 on target tumor cells has recently been identified as a critical determinant of Vγ9Vδ2 T cell activation (Harly et al., 2012, Sandstrom et al., 2014, Vavassori et al., 2013b). However, the mechanisms linking intracellular accumulation of phosphoantigens to changes in BTN3A1 membrane mobility are unclear. Therefore, the impact of ABP and RhoB, an important player in the cytoskeletal reorganization and formation of actin stress fibers (Prendergast, 2001), on mobility of BTN3A1 was tested. Consistent with previous reports (Harly et al., 2012, Sandstrom et al., 2014), treating 293 HEK cells with the ABP zoledronate resulted in decreased BTN3A1 membrane mobility. Strikingly, treatment with calpeptin induced immobilization of BTN3A1 to similar levels as those of ABP treatment alone, while C3-transferase counteracted this ABP-effect (Figure 4A). This points to the possibility that Rho GTPase activity acts on BTNA1 membrane immobilization upstream of the mevalonate pathway. Selective depletion of RhoB by CRISPR/Cas inhibited the ABP-induced immobilization of BTN3A1 to levels comparable to those of medium controls, suggesting that ABP-mediated changes in BTN3A1 mobility depend on RhoB.
Figure 4. RhoB activity modulates BTN3A1 membrane mobility and its association with the actin cytoskeleton.
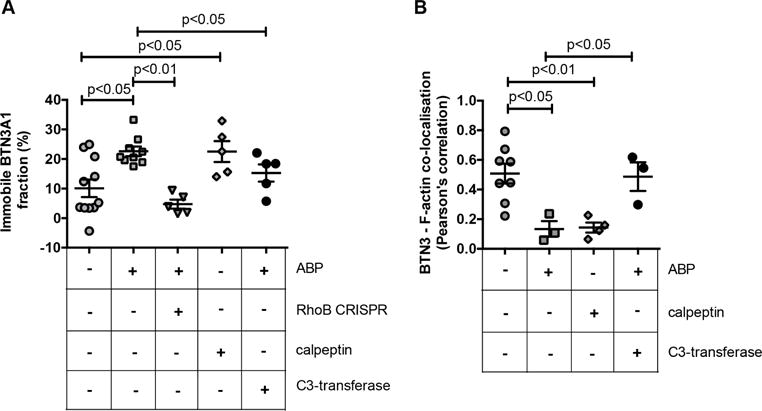
(A) HEK 293 cells were transfected with BTN3A1-emGFP fusion constructs and treated with medium, ABP zoledronate, or calpeptin. Zoledronate treatment was also applied to HEK 293 BTN3A1-emGFP+ cells in which RhoB was knocked out by CRISPR/Cas or in cells that were also treated with C3-transferase. BTN3A1 mobility was measured within selected membrane regions of interest by subsequent photobleaching and FRAP measurement as described in the Materials & Methods section. The figure shows the percentage of BTN3A1 immobile fraction upon treatments applied (+). Symbols represent single cell measurements, of 2 experiments. (B) HEK 293 cells were pretreated with pamidronate or with calpeptin and BTN3 molecules and filamentous actin (F-actin) were stained using a fluorescently labeled anti-BTN3 antibody and fluorescent phalloidin, respectively. The colocalization of BTN3 and F-actin was subsequently assessed by determining the localization correlation of both signals, as described in the Materials & Methods section. Symbols represent single cell measurements, of a single experiment. The center line and error bars represent average and S.E.M., p values indicate significance analyzed by using Mann-Whitney test.
To next assess a role for RhoB-induced cytoskeletal rearrangements in mediating the observed changes in BTN3A1 mobility, the relation between BTN3 molecules and F-actin was investigated by colocalization experiments. HEK 293 cells were stained with fluorescently labeled anti-BTN3 and phalloidin, and colocalization coefficients were determined in response to treatment with ABP and calpeptin. In cells in a culture medium, a variable but considerable colocalization between BTN3 and F-actin was observed, and was markedly reduced by ABP treatment (Figure 4B). Strikingly, and similar to its effect on BTN3A1 membrane mobility, calpeptin reduced colocalization between BTN3 and F-actin to comparable levels observed with ABP treatment. This reduction suggests that both phosphoantigen accumulation and Rho activation may induce the formation of membrane domains surrounded by cytoskeleton, where BTN3 molecules could be trapped and immobilized, as recently described for FcεRI (Andrews et al., 2008). Importantly, C3-transferase prevented ABP-induced BTN3-actin segregation, indicating again the crucial involvement of active Rho in this process. Together, these data suggest that RhoB activity contributes to target recognition by Vγ9Vδ2 TCR+ T cells by modulating BTN3A1 membrane mobility through cytoskeletal rearrangements.
RhoB interacts with BTN3A1 homodimers in cancer cells recognized by Vγ9Vδ2 TCRs
Given the strong requirement for RhoB activity in the membrane immobilization of BTN3A1, we questioned whether regulation of BTN3A1 involved direct interactions with RhoB. Using in situ proximity ligation assay (PLA), RhoB and BTN3 were observed to be in close proximity in recognized EBV-LCL 48 cells only when pretreated with the ABP (Figure 5A). Importantly, PLA signals were typically excluded from the nuclear area and distributed close to the plasma membrane, in line with our data that RhoB is involved in Vγ9Vδ2 TCR+ T cell recognition by regulating membrane-expressed BTN3A1.
Figure 5. RhoB interacts with BTN3 molecules and dissociates after phosphoantigen treatment.
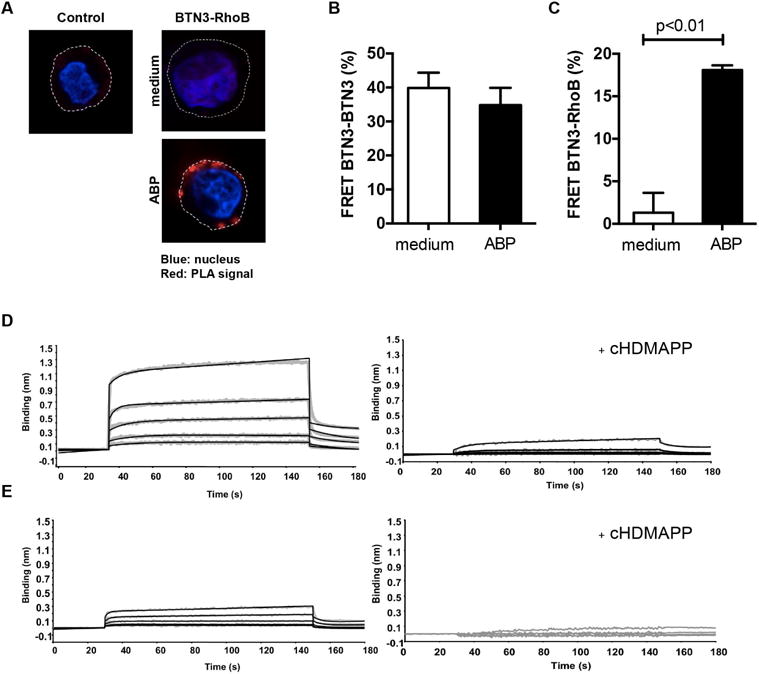
(A) EBV-LCL 48 cells were treated either with medium or ABP pamidronate, loaded onto poly-L-lysine-coated coverslips and permeabilized. The interaction between RhoB and BTN3 was subsequently assessed by Duolink PLA using anti-RhoB and anti-CD277 antibodies. Duolink PLA without antibodies against RhoB and BTN3 served as negative control (red: PLA signal; blue: nucleus [DAPI]; dotted line: cell membrane). Figures are representative of two independent experiments. (B) HEK 293 cells were treated with either medium or pamidronate and co-stained with equal amount of anti-CD277-PE (donor) as well as anti-CD277-DyLight 680 (acceptor) antibodies and FRET efficiency in cells was measured as described in Materials and Methods. Data shown is mean±S.E.M. of three independent experiments, in triplicate samples, where Mann-Whitney test was used to analyze statistical significance. (C) HEK 293 cells were pretreated either with medium or pamidronate, trypsinized, permeabilized and stained with anti-RhoB-Alexa Fluor 488 (FRET donor) and anti-CD277-DyLight 680 (FRET acceptor) antibodies. FRET efficiency was subsequently measured by flow cytometry as described in Materials and Methods. Data show mean±S.E.M of three independent experiments, in triplicate samples, where Mann-Whitney test was used to analyze statistical significance. (D) Concentration dependent binding of the full-length BTN3A1 intracellular domain (BFI) with RhoGTPase in the presence or absence of the phosphoantigen cHDMAPP. Binding of BFI to RhoGTPase was measured using Biolayer Interferometry (BLI) either in the absence of cHDMAPP (left panel) or presence of cHDMAPP (1:1) (right panel). Concentrations of BTN3A1 BFI shown in the upper panel are 6.25, 12.5, 25, 50 and 100uM shown in grey. The kinetics fitting curves are shown as black. In the lower panel, concentrations of BTN3A1 BFI shown are 3.75, 7.5, 15, 30 and 60uM shown in grey. The kinetics fitting curves are shown as black. (E) Same experimental setup but with recombinant BTN3A1 B30.2 domain, lacking the N terminal region connector to the transmembrane domain. In the left panel, the interaction was measured without cHDMAPP. Concentrations of BTN3A1 B30.2 shown were 12.5, 25, 50, 100 and 200uM shown in grey. The kinetics fitting curves are shown as black. In the lower panel, the interaction was measured with cHDMAPP (1:1). Concentrations of B30.2 domain shown are 3.75, 7.5, 15, 30 and 60uM shown in grey. Data show one representative experiment.
To determine whether BTN3A1 exists as a homodimer when expressed in a cellular context, as suggested from crystallization studies (Palakodeti et al., 2012), and to study RhoB-BTN3A1 interactions at even higher resolution, we examined close interactions by utilizing fluorescence resonance energy transfer (FRET). Flow cytometry FRET measurements were performed on ABP-sensitive HEK 293 cells (Harly et al., 2012), by either overexpressing FRET compatible fusion proteins or labeling endogenous proteins with antibodies coupled to FRET-compatible fluorochromes. These experiments showed that BTN3A1 molecules are expressed as homodimers on the cell surface of cancer cells (Figure 5B), however the pairing of BTN3A1 molecules was insensitive to ABP-induced phosphoantigen accumulation. Close association between RhoB and BTN3A1 was undetectable in ABP-untreated HEK cells, but increased markedly after treating cells with the ABP (Figure 5C). We used Biolayer Interferometry (BLI) to formally define a possible docking site for RhoB on the intracellular domain of BTN3A1. RhoB binding was detected with recombinant full length BTN3A1 intracellular domain (BFI) (Figure 5D left panel and Table S1), yet was significantly reduced when using a recombinant BTN3A B30.2 domain, lacking the N terminal region connector to the transmembrane domain (Figure 5E left panel). These data indicate an important role for the membrane proximal region of the BTN3A1 intracellular domain in binding to RhoB GTPase. Interestingly, RhoB binding to BFI was almost completely abolished in the presence of soluble phosphoantigen cHDMAPP (Figure 5D right panel). When in the same experiment, the physiologically more relevant, but much lower affinity pAg IPP was applied, BLI was unable to resolve pAg-induced dissociation of RhoB from BFI (Figure S4C), very likely due to the technical limitation of the assay. In summary, our data indicate that RhoB and BTN3 molecules closely interact at the surface membrane selectively in a Vγ9Vδ2-stimulatory context and that the physical interaction is regulated by phosphoantigens.
Phosphoantigen accumulation associates with conformational changes of BTN3A1 dimers
Conformational changes of BTN3A1 in response to elevated levels of phosphoantigens have been proposed to serve as a stimulatory signature for Vγ9Vδ2 TCRs (Palakodeti et al., 2012, Harly et al., 2012, Sandstrom et al., 2014), however no experimental data have so far been reported to substantiate this hypothesis. The crystal structures of the extracellular domain of BTN3A1 in complex with the functionally well-characterized BTN3-specific antibodies 20.1 and 103.2 (Harly et al., 2012) have recently been resolved, and have revealed that both antibodies bind to different epitopes on the membrane-distal Ig-V domains of BTN3A1 dimers (Figure 6A) (Palakodeti et al., 2012). To study BTN3A1 conformational changes in response to increased phosphoantigen levels, surface membranes of either unstimulated or ABP-stimulated HEK 293 cells were labeled with the fluorescent lipid conjugate BODIPY FL (donor), and subsequently stained with acceptor dye-labeled BTN3-specific antibodies on ice, in order to prevent conformational changes that could be driven by these antibodies under physiological circumstances (Gaspar et al., 2001). Without ABP stimulation, potent FRET efficiencies between stained membrane and both antibodies were observed (Figure 6B), suggesting that the BTN3 Ig-V domain is in close proximity to the cell membrane. Strikingly however, treatment of cells with ABP resulted in a marked reduction in FRET signals (Figure 6B), demonstrating that intracellular phosphoantigen accumulation associates with a conformational change of BTN3 molecules. This change involves a pronounced distancing of the Ig-V domain from the cell membrane. Importantly, Rho-inhibitor C3-transferase treatment (Figure S2F) of cells did not prevent ABP-driven conformational changes of the BTN3A1 homodimer (Figure 6B) indicating that this conformational change is independent of the enzymatic activity of RhoB. These data provide support for the hypothesis that increases in intracellular phosphoantigen levels can induce extracellular changes in BTN3A1 dimers that may act as, or contribute to a molecular signature recognized by Vγ9Vδ2 TCRs.
Figure 6. Intracellular phosphoantigen accumulation induces extracellular conformational changes in BTN3.
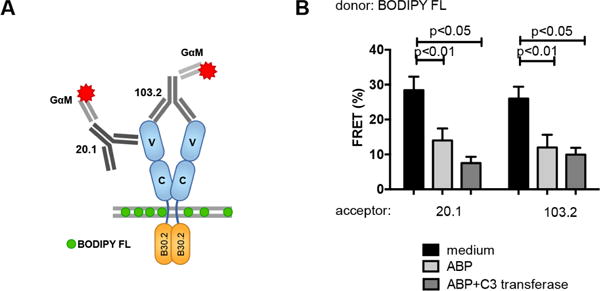
HEK 293 cells were pretreated with medium, C3 transferase and/or pamidronate and the surface membrane of cells was subsequently stained with the fluorescent lipid conjugate BODIPY FL (FRET donor) and BTN3 molecules were labeled with mouse anti-CD277 mAbs originating either from clone 20.1 or from the clone 103.2 followed by staining with secondary Alexa Fluor 594-conjugated Fab fragment (GαM) (FRET acceptor). FRET efficiency was measured by flow cytometry and data represent mean±S.E.M of at least three independent experiments in triplicate samples. Statistical significance of data was analyzed by Mann-Whitney test.
Discussion
The translation of tumour-reactive Vγ9Vδ2 T cells to clinical applications has proven challenging, due to a limited understanding of the molecular mechanisms by which Vγ9Vδ2 T cells recognize their targets. Important progress has been made in this area by recent elegant reports identifying BTN3A1 as a phosphoantigen sensor that plays a key role in the activation of Vγ9Vδ2 T cells (Vavassori et al., 2013b, Harly et al., 2012, Sandstrom et al., 2014). These studies suggest, however, nearly opposing mechanisms for the exact role of BTN3A1 in this process. Unfortunately, this contradiction makes it unclear how intracellular phosphoantigen accumulation translates into extracellular signatures that can be recognized by Vγ9Vδ2 TCRs, and acts as a barrier to the clinical development of Vγ9Vδ2 T cell-based therapies. In this study, we provide insights to support a two-component mechanism necessary to fully activate Vγ9Vδ2 TCR+ T cells. We describe the critical role of the small GTPase RhoB in spatial redistribution of BTN3A1, as well as conformational changes in BTN3A1 at the tumour cell surface. We propose that ABP stimulation induces activation of RhoB and its compartmentalization to membrane proximal areas, where it can directly interact with BTN3A1, as has been demonstrated for several other Rho GTPases (Dunford et al., 2006, Kranenburg et al., 1997). While biochemical activity of RhoB induces a close vicinity of RhoB with BTN3A1 and membrane rearrangements of BTN3A1 dimers, phosphoantigens allow for a dissociation of RhoB from BTN3A1 and conformational changes in BTN3A1 at the cell membrane, which are then independent of RhoB activity. These findings, based on the model of Vγ9Vδ2TCR engineered CD4+ αβT cells, are most likely transferrable to the recognition of Vγ9Vδ2 T cells, as both populations have comparable activity against defined target cells as we recently demonstrated (Marcu-Malina et al., 2011). However, we cannot exclude that the modulating effect of RhoB for the recognition of tumor cells by Vγ9Vδ2 T cells can be partially overcome by utilizing Vγ9Vδ2TCRs of higher affinity, or additional co-stimulatory molecules (Gründer et al., 2012). Together, these data suggest that RhoB is a key actor in the cellular mechanisms that couple phosphoantigen accumulation to BTN3A1-mediated triggering of Vγ9Vδ2 TCR+ T cell responses.
The intrinsic GTPase activity of RhoB that cycles between biologically active GTP-bound and inactive GDP-bound conformations enables the differential association with downstream signaling pathways. As such, RhoB functions as a molecular switch in diverse cellular processes, ranging from gene transcription to the regulation of cytoskeletal changes and vesicle transport (Prendergast, 2001). Our data provide several lines of evidence that support a direct link between the mevalonate pathway-dependent changes in RhoB activity in tumor cells, cytoskeletal reorganization, and tumor cell induced activation of Vγ9Vδ2 TCR+ T cells. However, the biological activity of RhoB seems to be partially genetically predetermined, as suggested by our initial screening strategy, we could not identify a direct genetic link of the surrogate marker SNP with RhoB activity. It is possible that differential SNPs found in recognized tumour cells, with the frequency of around 70% in the normal population, act as enhancer of a distant genomic region through chromatin looping (Pombo and Dillon, 2015), and indirectly influence RhoB activity. The importance of RhoB expression in the studied cancer cell lines was further emphasized by the loss in proliferative activity of complete RhoB knock-downs. Although we encountered a genetic block in a small subset of patients who were largely non-responsive to Vγ9Vδ2 TCR+ T cell mediated tumour control, our results identified in RhoB a crucial link connecting intracellular changes to the recognition of target cells by Vγ9Vδ2 TCR+ T cells (Gober et al., 2003). These changes are induced by the accumulation of phosphoantigens occurring in the majority of cancer cells.
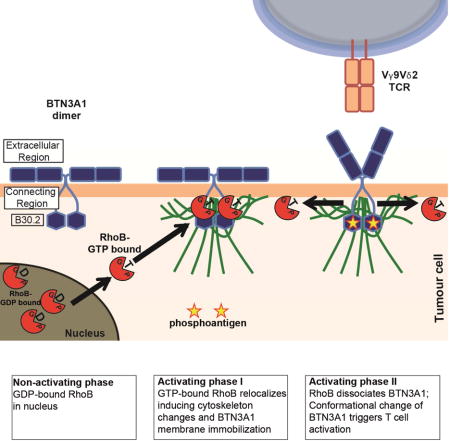
Our data suggest that pAgs act differently in separate cell compartments and support a two-component model. A dysregulated mevalonate pathway leads to the activation and relocation of RhoB to the membrane-proximal connector region of the BTN3A1 cytoplasmic tail, through a process requiring GTPase activity (Figure 7). B30.2 domains have been reported to function as scaffold modules by spatiotemporally sequestering signaling proteins to distinct subcellular locations (Perfetto et al., 2013). This interaction favors an enhanced submembranous cytoskeleton arrangement, which leads to the observed immobilization of BTN3A1 at the cell surface, similar to its reported role in modulating membrane structures such as focal adhesions (Allal et al., 2002, Vega et al., 2012). Moreover, our data suggest a sequential, second cascade of events in the transmembrane compartment, which is necessary for recognition by a Vγ9Vδ2 TCR+ T cell: RhoB dissociates from immobilized BTN3A1 most likely after phosphoantigen binding to B30.2 and the conformational change in the extracellular domain of BTN3A1, which is then independent of RhoB GTPase activity. Our data furthermore most likely implicate the involvement of an additional protein in this molecular complex which is required for the recognition by Vγ9Vδ2 TCR, as others (Gu et al., 2014) have suggested. This observation corroborates data from crystallographic experiments using soluble BTN3A1 domains, which revealed a pronounced flexibility of BTN3A1 dimers correlating with treatment with the agonist 20.1 antibody which can mimic phosphoantigen-induced stimulation of Vγ9Vδ2 T cells (Palakodeti et al., 2012). Our proposed two-component model supports an “inside-out” mechanism of phosphoantigen signaling to Vγ9Vδ2 TCR+ T cells as others have suggested (Palakodeti et al., 2012, Sandstrom et al., 2014), whereby intracellular phosphoantigen accumulation is translated into surface changes of BTN3A1 by RhoB.
Figure 7. Two-component mechanism in tumour cells that leads to activation of Vγ9Vδ2 TCR T cells.
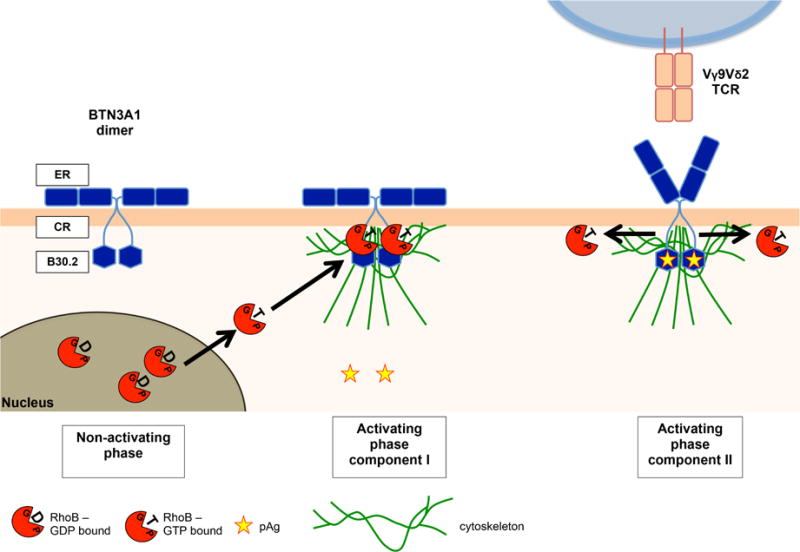
Non-activating phase: no accumulation of phosphoantigens (pAg, yellow stars, which retains GDP-bound RhoB (red circles+GDP) from extranuclear areas. Activating phase component I: Accumulation of phosphoantigens is followed by more GTP-bound RhoB formation (red circles+GTP). GTP-bound RhoB undergoes subcellular re-compartmentalization (black arrows) accumulating at extranuclear areas and facilitates spatial redistribution of BTN3A1 by promoting cytoskeletal trapping (green lines) in the plasma membrane binding to the B30.2 domain proximal connector region (CR) of BTN3A1 (blue hexagon). Activating phase component II: GTP-bound RhoB dissociates (black arrow) from while pAg binds to the B30.2 domain of BTN3A1, which triggers a conformational change of the extracellular region (ER) of BTN3A1 leading to Vγ9Vδ2 TCR T cell activation.
Consistent with our observations, other recent data also suggest that additional factors are likely to be involved in the recognition of tumour cells by Vγ9Vδ2 TCR+ T cells. In particular, transfer of human BTN3A1 alone to rodent cells, which are not recognized by Vγ9Vδ2 T cells (Morita et al., 2007), was insufficient to sensitize cells towards recognition by Vγ9Vδ2 T cells (Sandstrom et al., 2014). Instead, this sensitization required co-transfer of a substantial proportion of the human chromosome 6 (Riano et al., 2014, Vavassori et al., 2013b), suggesting that the combined presence of BTN3A1 and additional human genes located on this chromosome is essential for facilitating target cell recognition by Vγ9Vδ2 T cells. The human RhoB gene locates to chromosome 2, and moreover, protein sequences of RhoB are fully conserved between human and rodents. Thus, other human-specific genes that mediate activation of Vγ9Vδ2 T cells are yet to be identified. These might include GTPase activating proteins (GAPs) and guanine nucleotide exchange factors (GEFs); both of which coordinate actions that regulate the balance between the active, GTP-bound state and inactive, GDP-bound state of Rho GTPases (Rossman et al., 2005, Moon and Zheng, 2003). However, none of the known regulators of Rho GTPase signaling locate to the region on chromosome 6 required to transfer Vγ9Vδ2 T cell susceptibility to rodent cells. Identification of these missing factors linking RhoB and BTN3A1 to target cell recognition by Vγ9Vδ2 TCR+ T cells is therefore an important challenge for further elucidating the molecular activation mechanisms of Vγ9Vδ2 TCR+ T cells.
The identification of RhoB as a mechanistic link between transformation-associated dysregulation of the mevalonate pathway and BTN3A1-dependent activation of Vγ9Vδ2 TCR+ T cells has important implications for cancer immunotherapeutic concepts using Vγ9Vδ2 T cells or their TCRs. Our observations strongly suggest the potential of Vγ9Vδ2 TCR+ T cell-mediated immune interventions to target selectively the leukemic but not the healthy stem cell fraction. Thereby, the divergent intracellular distribution of RhoB among recognized and non-recognized tumour cells may be used as a biological surrogate marker for the identification of patients susceptible to Vγ9Vδ2 TCR+ T cell-mediated tumour cell killing. Secondly, although we have not been able to provide a direct genetic link between the marker SNP and the function of RhoB, our data strongly argue for a genetic predisposition, which most likely acts via post-translational regulation of RhoB, and partially explains why some patients do not benefit from Vγ9Vδ2 TCR+ T cell-mediated tumour control. Thirdly, our data put RhoB forward as a key therapeutic target for improving immunotherapy using Vγ9Vδ2 TCR+ T cells. In contrast to BTN3A1, which is ubiquitously expressed on healthy as well as malignant cells, the data presented here and elsewhere (Prendergast, 2001) demonstrate that RhoB possesses transformation-selective characteristics, such as spatial distribution, and could therefore represent a more selective therapeutic target compared to BNT3A1. Moreover, the mobilization of Vγ9Vδ2 T cells in cancer patients using in vivo or ex vivo stimulation protocols with ABPs such as zoledronate (Fournie et al., 2013, Fisher et al., 2014, Scheper et al., 2014) is likely limited by the poor pharmacokinetic profile of ABPs (Lin, 1996) and their toxicity to T cells upon prolonged exposure (Wang et al., 2011). Agents modulating RhoB GTPase activity could raise attractive opportunities to complement currently pursued Vγ9Vδ2 TCR+ T cell-based therapies. In summary, with RhoB we provide a key molecule that regulates the ability of cancer cells to activate Vγ9Vδ2 TCR+ T cells, by orchestrating BTN3A1 in the plasma membrane,. This knowledge will enable the pre-selection of patients susceptible to Vγ9Vδ2 T cell based therapies, and will enhance the efficacy of such therapies.
Experimental procedures
Retroviral transduction of TCRs
The Vγ9Vδ2TCR genes were transduced into αβT cells using retroviral supernatant from Phoenix-Ampho packaging cells that were transfected with gag-pol, env and pBullet retroviral constructs containing TCR genes.
Functional T cell assays
IFNγ ELISPOT was performed as15,000 T cells and 50,000 target cells were cocultured in nitrocellulose plates and the number of spots was quantified using ELISPOT Analysis Software.
Zygosity/SNP correlation analysis
Recognition of CEPH EBV-LCL lines by Vγ9Vδ2 TCR+ T cells was defined in IFNγ ELIspots. Hypothetical zygosities for candidate genetic loci were deduced using classical Mendelian inheritance patterns within CEPH family pedigrees, where the influence of candidate alleles on Vγ9Vδ2 TCR-mediated recognition was assumed to be dominant. Correlations of predicted zygosities with Hapmap SNP genotypes of CEPH individuals were calculated with the software tool ssSNPer.
Confocal microscopy and data analysis
Cells were allowed to attach onto poly-L-lysine-precoated coverslips and were permeabilized and stained with anti-RhoB antibody. Images were acquired using a Zeiss confocal laser scanning microscope LSM 700. Ratios between nuclear and extranuclear signal of RhoB was determined using ImageJ software, using DAPI staining to mark nuclei.
Statistical analysis
All experiments were repeated at least three times unless otherwise indicated. All data were shown as mean ± SEM. Statistical significance was analysed by either Mann-Whitney or Kruskal-Wallis test and Dunn’s multiple comparison test.
Supplementary Material
Highlights.
Identification of SNPs near RhoB are associated with poor Vg9Vd2 T cell activation
RhoB activity and distribution in tumor cells modulates Vg9Vd2 T cell activation
Relocalization of RhoB induces membrane immobility of BTN3A1
BTN3A1-dependent tumour recognition by a Vg9Vd2 TCR
Acknowledgments
Funding for this study was provided by ZonMW 43400003 and VIDI-ZonMW 917.11.337, KWF UU 2010-4669, UU 2013-6426, UU 2014-6790 and UU 2015-7601, Vrienden van het UMCU, AICR 10-0736 & 15-0049 to J.K. NIH R01 AI115471 to EJA & Lady Tata Memorial Trust to ZS. RJL was supported by Dutch Cancer Society (KWF) grant UU 2012-5667.
We acknowledge A.N. Snel (Department of Hematology, VU University Medical Center, Amsterdam, The Netherlands) for his technical assistance during FACSsorting of leukemic end healthy stem cell fractions, and Paul Coffer (Department of Cell Biology, University Medical Center Utrecht, the Netherlands) and Jeanette Leusen (Laboratory of Translational Immunology, University Medical Center Utrecht, the Netherlands) for their critical reading and discussions.
J. Kuball is co-founder and chief scientific officer of gadeta (www.gadeta.nl).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Author contributions
Z.S., W.S. and J.K. designed the study and wrote the manuscript. Z.S., W.S., R.O. and T.M. designed and performed the computational SNP analysis. S.G. and E.A. designed and performed in vitro protein expression and purification, and BLI assays. Z.R., Z.S. generated T cells and performed EBV-LCL screening. A.C., A.V., Z.R. and Z.S. designed and performed Rho activity assays. M.S., M.N. and A.V. performed mRNA analysis. R.J.L. designed and produced CRISPR/Cas constructs and viruses. C.M.P., E.S. and Z.S. designed and performed FRAP analysis. Z.S. and C.M.P. performed FRET analysis. C.C., A.V. and Z.S. performed confocal analysis. G.J.S. designed and carried out FACS sorting of leukemia stem cells. D.O. produced and purified CD277 antibodies.
References
- ADAMSON P, PATERSON HF, HALL A. Intracellular localization of the P21rho proteins. J Cell Biol. 1992;119:617–27. doi: 10.1083/jcb.119.3.617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- AKTORIES K, MOHR C, KOCH G. Clostridium botulinum C3 ADP-ribosyltransferase. Curr Top Microbiol Immunol. 1992;175:115–31. doi: 10.1007/978-3-642-76966-5_6. [DOI] [PubMed] [Google Scholar]
- ALLAL C, PRADINES A, HAMILTON AD, SEBTI SM, FAVRE G. Farnesylated RhoB prevents cell cycle arrest and actin cytoskeleton disruption caused by the geranylgeranyltransferase I inhibitor GGTI-298. Cell Cycle. 2002;1:430–7. doi: 10.4161/cc.1.6.272. [DOI] [PubMed] [Google Scholar]
- ALLISON TJ, WINTER CC, FOURNIE JJ, BONNEVILLE M, GARBOCZI DN. Structure of a human gammadelta T-cell antigen receptor. Nature. 2001;411:820–824. doi: 10.1038/35081115. [DOI] [PubMed] [Google Scholar]
- ANDREWS NL, LIDKE KA, PFEIFFER JR, BURNS AR, WILSON BS, OLIVER JM, LIDKE DS. Actin restricts FcepsilonRI diffusion and facilitates antigen-induced receptor immobilization. Nat Cell Biol. 2008;10:955–63. doi: 10.1038/ncb1755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BONNEVILLE M, O’BRIEN RL, BORN WK. Gammadelta T cell effector functions: a blend of innate programming and acquired plasticity. Nat Rev Immunol. 2010;10:467–78. doi: 10.1038/nri2781. [DOI] [PubMed] [Google Scholar]
- BUKOWSKI JF, MORITA CT, BAND H, BRENNER MB. Crucial role of TCR gamma chain junctional region in prenyl pyrophosphate antigen recognition by gamma delta T cells. J Immunol. 1998;161:286–93. [PubMed] [Google Scholar]
- COMPTE E, PONTAROTTI P, COLLETTE Y, LOPEZ M, OLIVE D. Frontline: Characterization of BT3 molecules belonging to the B7 family expressed on immune cells. Eur J Immunol. 2004;34:2089–99. doi: 10.1002/eji.200425227. [DOI] [PubMed] [Google Scholar]
- CONSTANT P, DAVODEAU F, PEYRAT MA, POQUET Y, PUZO G, BONNEVILLE M, FOURNIE JJ. Stimulation of human gamma delta T cells by nonpeptidic mycobacterial ligands. Science. 1994;264:267–270. doi: 10.1126/science.8146660. [DOI] [PubMed] [Google Scholar]
- CORREIA DV, LOPES A, SILVA-SANTOS B. Tumor cell recognition by gammadelta T lymphocytes: T-cell receptor vs. NK-cell receptors. Oncoimmunology. 2013;2:e22892. doi: 10.4161/onci.22892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DAUSSET J, CANN H, COHEN D, LATHROP M, LALOUEL JM, WHITE R. Centre d’etude dupolymorphisme humain (CEPH): collaborative genetic mapping of the human genome. Genomics. 1990;6:575–7. doi: 10.1016/0888-7543(90)90491-c. [DOI] [PubMed] [Google Scholar]
- DAVODEAU F, PEYRAT MA, HALLET MM, GASCHET J, HOUDE I, VIVIEN R, VIE H, BONNEVILLE M. Close correlation between Daudi and mycobacterial antigen recognition by human gamma delta T cells and expression of V9JPC1 gamma/V2DJC delta-encoded T cell receptors. J Immunol. 1993;151:1214–23. [PubMed] [Google Scholar]
- DUNFORD JE, ROGERS MJ, EBETINO FH, PHIPPS RJ, COXON FP. Inhibition of protein prenylation by bisphosphonates causes sustained activation of Rac, Cdc42, and Rho GTPases. J Bone Miner Res. 2006;21:684–94. doi: 10.1359/jbmr.060118. [DOI] [PubMed] [Google Scholar]
- FISHER JP, HEUIJERJANS J, YAN M, GUSTAFSSON K, ANDERSON J. gammadelta T cells for cancer immunotherapy: A systematic review of clinical trials. Oncoimmunology. 2014;3:e27572. doi: 10.4161/onci.27572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FOURNIE JJ, SICARD H, POUPOT M, BEZOMBES C, BLANC A, ROMAGNE F, YSEBAERT L, LAURENT G. What lessons can be learned from gammadelta T cell-based cancer immunotherapy trials? Cell Mol Immunol. 2013;10:35–41. doi: 10.1038/cmi.2012.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GASPAR R, JR, BAGOSSI P, BENE L, MATKO J, SZOLLOSI J, TOZSER J, FESUS L, WALDMANN TA, DAMJANOVICH S. Clustering of class I HLA oligomers with CD8 and TCR: three-dimensional models based on fluorescence resonance energy transfer and crystallographic data. J Immunol. 2001;166:5078–86. doi: 10.4049/jimmunol.166.8.5078. [DOI] [PubMed] [Google Scholar]
- GOBER HJ, KISTOWSKA M, ANGMAN L, JENO P, MORI L, DE LIBERO G. Human T cell receptor gammadelta cells recognize endogenous mevalonate metabolites in tumor cells. J Exp Med. 2003;197:163–8. doi: 10.1084/jem.20021500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GRÜNDER C, VAN DORP S, HOL S, DRENT E, STRAETEMANS T, HEIJHUURS S, SCHOLTEN K, SCHEPER W, SEBESTYEN Z, MARTENS A, STRONG R, KUBALL J. gamma9 and delta2CDR3 domains regulate functional avidity of T cells harboring gamma9delta2TCRs. Blood. 2012;120:5153–62. doi: 10.1182/blood-2012-05-432427. [DOI] [PubMed] [Google Scholar]
- GU S, NAWROCKA W, ADAMS EJ. Sensing of Pyrophosphate Metabolites by Vgamma9Vdelta2 T Cells. Front Immunol. 2014;5:688. doi: 10.3389/fimmu.2014.00688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HARLY C, GUILLAUME Y, NEDELLEC S, PEIGNE CM, MONKKONEN H, MONKKONEN J, LI J, KUBALL J, ADAMS EJ, NETZER S, DECHANET-MERVILLE J, LEGER A, HERRMANN T, BREATHNACH R, OLIVE D, BONNEVILLE M, SCOTET E. Key implication of CD277/butyrophilin-3 (BTN3A) in cellular stress sensing by a major human gammadelta T-cell subset. Blood. 2012;120:2269–79. doi: 10.1182/blood-2012-05-430470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- INTERNATIONAL HAPMAP, C. The International HapMap Project. Nature. 2003;426:789–96. doi: 10.1038/nature02168. [DOI] [PubMed] [Google Scholar]
- JOHNSON AD, HANDSAKER RE, PULIT SL, NIZZARI MM, O’DONNELL CJ, DE BAKKER PI. SNAP: a web-based tool for identification and annotation of proxy SNPs using HapMap. Bioinformatics. 2008;24:2938–9. doi: 10.1093/bioinformatics/btn564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KAMON H, KAWABE T, KITAMURA H, LEE J, KAMIMURA D, KAISHO T, AKIRA S, IWAMATSU A, KOGA H, MURAKAMI M, HIRANO T. TRIF-GEFH1-RhoB pathway is involved in MHCII expression on dendritic cells that is critical for CD4 T-cell activation. EMBO J. 2006;25:4108–19. doi: 10.1038/sj.emboj.7601286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KRANENBURG O, POLAND M, GEBBINK M, OOMEN L, MOOLENAAR WH. Dissociation of LPA-induced cytoskeletal contraction from stress fiber formation by differential localization of RhoA. J Cell Sci. 1997;110(Pt 19):2417–27. doi: 10.1242/jcs.110.19.2417. [DOI] [PubMed] [Google Scholar]
- KUBALL J, DOSSETT ML, WOLFL M, HO WY, VOSS RH, FOWLER C, GREENBERG PD. Facilitating matched pairing and expression of TCR chains introduced into human T cells. Blood. 2007;109:2331–8. doi: 10.1182/blood-2006-05-023069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LIN JH. Bisphosphonates: a review of their pharmacokinetic properties. Bone. 1996;18:75–85. doi: 10.1016/8756-3282(95)00445-9. [DOI] [PubMed] [Google Scholar]
- MARCU-MALINA V, HEIJHUURS S, VAN BUUREN M, HARTKAMP L, STRAND S, SEBESTYEN Z, SCHOLTEN K, MARTENS A, KUBALL J. Redirecting alphabeta T cells against cancer cells by transfer of a broadly tumor-reactive gammadeltaT-cell receptor. Blood. 2011;118:50–9. doi: 10.1182/blood-2010-12-325993. [DOI] [PubMed] [Google Scholar]
- MICHAELSON D, SILLETTI J, MURPHY G, D’EUSTACHIO P, RUSH M, PHILIPS MR. Differential localization of Rho GTPases in live cells: regulation by hypervariable regions and RhoGDI binding. J Cell Biol. 2001;152:111–26. doi: 10.1083/jcb.152.1.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MOON SY, ZHENG Y. Rho GTPase-activating proteins in cell regulation. Trends Cell Biol. 2003;13:13–22. doi: 10.1016/s0962-8924(02)00004-1. [DOI] [PubMed] [Google Scholar]
- MORITA CT, JIN C, SARIKONDA G, WANG H. Nonpeptide antigens, presentation mechanisms, and immunological memory of human Vgamma2Vdelta2 T cells: discriminating friend from foe through the recognition of prenyl pyrophosphate antigens. Immunol Rev. 2007;215:59–76. doi: 10.1111/j.1600-065X.2006.00479.x. [DOI] [PubMed] [Google Scholar]
- PALAKODETI A, SANDSTROM A, SUNDARESAN L, HARLY C, NEDELLEC S, OLIVE D, SCOTET E, BONNEVILLE M, ADAMS EJ. The molecular basis for modulation of human Vgamma9Vdelta2 T cell responses by CD277/butyrophilin-3 (BTN3A)-specific antibodies. J Biol Chem. 2012;287:32780–90. doi: 10.1074/jbc.M112.384354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PERFETTO L, GHERARDINI PF, DAVEY NE, DIELLA F, HELMER-CITTERICH M, CESARENI G. Exploring the diversity of SPRY/B30.2-mediated interactions. Trends Biochem Sci. 2013;38:38–46. doi: 10.1016/j.tibs.2012.10.001. [DOI] [PubMed] [Google Scholar]
- POMBO A, DILLON N. Three-dimensional genome architecture: players and mechanisms. Nat Rev Mol Cell Biol. 2015;16:245–57. doi: 10.1038/nrm3965. [DOI] [PubMed] [Google Scholar]
- PRENDERGAST GC. Actin’ up: RhoB in cancer and apoptosis. Nat Rev Cancer. 2001;1:162–8. doi: 10.1038/35101096. [DOI] [PubMed] [Google Scholar]
- RHODES DA, CHEN HC, PRICE AJ, KEEBLE AH, DAVEY MS, JAMES LC, EBERL M, TROWSDALE J. Activation of Human gammadelta T Cells by Cytosolic Interactions of BTN3A1 with Soluble Phosphoantigens and the Cytoskeletal Adaptor Periplakin. J Immunol. 2015;194:2390–8. doi: 10.4049/jimmunol.1401064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RIANO F, KARUNAKARAN MM, STARICK L, LI J, SCHOLZ CJ, KUNZMANN V, OLIVE D, AMSLINGER S, HERRMANN T. Vgamma9Vdelta2 TCR-activation by phosphorylated antigens requires butyrophilin 3 A1 (BTN3A1) and additional genes on human chromosome 6. Eur J Immunol. 2014;44:2571–6. doi: 10.1002/eji.201444712. [DOI] [PubMed] [Google Scholar]
- ROSSMAN KL, DER CJ, SONDEK J. GEF means go: turning on RHO GTPases with guanine nucleotide-exchange factors. Nat Rev Mol Cell Biol. 2005;6:167–80. doi: 10.1038/nrm1587. [DOI] [PubMed] [Google Scholar]
- SANDSTROM A, PEIGNE CM, LEGER A, CROOKS JE, KONCZAK F, GESNEL MC, BREATHNACH R, BONNEVILLE M, SCOTET E, ADAMS EJ. The intracellular B30.2 domain of butyrophilin 3A1 binds phosphoantigens to mediate activation of human Vgamma9Vdelta2 T cells. Immunity. 2014;40:490–500. doi: 10.1016/j.immuni.2014.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SCHEPER W, GRUNDER C, STRAETEMANS T, SEBESTYEN Z, KUBALL J. Hunting for clinical translation with innate-like immune cells and their receptors. Leukemia. 2014;28:1181–90. doi: 10.1038/leu.2013.378. [DOI] [PubMed] [Google Scholar]
- SCHEPER W, VAN DORP S, KERSTING S, PIETERSMA F, LINDEMANS C, HOL S, HEIJHUURS S, SEBESTYEN Z, GRUNDER C, MARCU-MALINA V, MARCHANT A, DONNER C, PLACHTER B, VERMIJLEN D, VAN BAARLE D, KUBALL J. gammadeltaT cells elicited by CMV reactivation after allo-SCT cross-recognize CMV and leukemia. Leukemia. 2013;27:1328–38. doi: 10.1038/leu.2012.374. [DOI] [PubMed] [Google Scholar]
- SCHOENWAELDER SM, BURRIDGE K. Evidence for a calpeptin-sensitive protein-tyrosine phosphatase upstream of the small GTPase Rho. A novel role for the calpain inhibitor calpeptin in the inhibition of protein-tyrosine phosphatases. J Biol Chem. 1999;274:14359–67. doi: 10.1074/jbc.274.20.14359. [DOI] [PubMed] [Google Scholar]
- SEBESTYEN Z, NAGY P, HORVATH G, VAMOSI G, DEBETS R, GRATAMA JW, ALEXANDER DR, SZOLLOSI J. Long wavelength fluorophores and cell-by-cell correction for autofluorescence significantly improves the accuracy of flow cytometric energy transfer measurements on a dual-laser benchtop flow cytometer. Cytometry. 2002;48:124–35. doi: 10.1002/cyto.10121. [DOI] [PubMed] [Google Scholar]
- SPAAPEN RM, LOKHORST HM, VAN DEN OUDENALDER K, OTTERUD BE, DOLSTRA H, LEPPERT MF, MINNEMA MC, BLOEM AC, MUTIS T. Toward targeting B cell cancers with CD4+ CTLs: identification of a CD19-encoded minor histocompatibility antigen using a novel genome-wide analysis. J Exp Med. 2008;205:2863–72. doi: 10.1084/jem.20080713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- STANISLAWSKI T, VOSS RH, LOTZ C, SADOVNIKOVA E, WILLEMSEN RA, KUBALL J, RUPPERT T, BOLHUIS RL, MELIEF CJ, HUBER C, STAUSS HJ, THEOBALD M. Circumventing tolerance to a human MDM2-derived tumor antigen by TCR gene transfer. Nat Immunol. 2001;2:962–970. doi: 10.1038/ni1001-962. [DOI] [PubMed] [Google Scholar]
- TERWIJN M, ZEIJLEMAKER W, KELDER A, RUTTEN AP, SNEL AN, SCHOLTEN WJ, PABST T, VERHOEF G, LOWENBERG B, ZWEEGMAN S, OSSENKOPPELE GJ, SCHUURHUIS GJ. Leukemic stem cell frequency: a strong biomarker for clinical outcome in acute myeloid leukemia. PLoS One. 2014;9:e107587. doi: 10.1371/journal.pone.0107587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VAN DE WEIJER ML, BASSIK MC, LUTEIJN RD, VOORBURG CM, LOHUIS MA, KREMMER E, HOEBEN RC, LEPROUST EM, CHEN S, HOELEN H, RESSING ME, PATENA W, WEISSMAN JS, MCMANUS MT, WIERTZ EJ, LEBBINK RJ. A high-coverage shRNA screen identifies TMEM129 as an E3 ligase involved in ER-associated protein degradation. Nat Commun. 2014;5:3832. doi: 10.1038/ncomms4832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VANTOUROUT P, HAYDAY A. Six-of-the-best: unique contributions of gammadelta T cells to immunology. Nat Rev Immunol. 2013;13:88–100. doi: 10.1038/nri3384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VAVASSORI S, KUMAR A, WAN GS, RAMANJANEYULU GS, CAVALLARI M, EL DS, BEDDOE T, THEODOSSIS A, WILLIAMS NK, GOSTICK E, PRICE DA, SOUDAMINI DU, VOON KK, OLIVO M, ROSSJOHN J, MORI L, DE LG. Butyrophilin 3A1 binds phosphorylated antigens and stimulates human gammadelta T cells. Nat Immunol. 2013b doi: 10.1038/ni.2665. [DOI] [PubMed] [Google Scholar]
- VEGA FM, COLOMBA A, REYMOND N, THOMAS M, RIDLEY AJ. RhoB regulates cell migration through altered focal adhesion dynamics. Open Biol. 2012;2:120076. doi: 10.1098/rsob.120076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WANG H, FANG Z, MORITA CT. Vgamma2Vdelta2 T Cell Receptor recognition of prenyl pyrophosphates is dependent on all CDRs. J Immunol. 2010;184:6209–22. doi: 10.4049/jimmunol.1000231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WANG H, SARIKONDA G, PUAN KJ, TANAKA Y, FENG J, GINER JL, CAO R, MONKKONEN J, OLDFIELD E, MORITA CT. Indirect stimulation of human Vgamma2Vdelta2 T cells through alterations in isoprenoid metabolism. J Immunol. 2011;187:5099–113. doi: 10.4049/jimmunol.1002697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- YANG TP, BEAZLEY C, MONTGOMERY SB, DIMAS AS, GUTIERREZ-ARCELUS M, STRANGER BE, DELOUKAS P, DERMITZAKIS ET. Genevar: a database and Java application for the analysis and visualization of SNP-gene associations in eQTL studies. Bioinformatics. 2010;26:2474–6. doi: 10.1093/bioinformatics/btq452. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.