

Abstract
Peptides containing C-terminal esters are an important class of bioactive molecules that includes a-factor, a farnesylated dodecapeptide, involved in the mating of S. cerevisiae. Here, results that expand the scope of solid phase peptide synthetic methodology that uses trityl side-chain anchoring for the preparation of peptides with C-terminal cysteine alkyl esters are described. In this method, Fmoc-protected C-terminal cysteine esters are anchored to trityl chloride resin and extended by standard solid phase procedures followed by acidolytic cleavage and HPLC purification. Analysis using a Gly-Phe-Cys-OMe model tripeptide, revealed minimal epimerization of the C-terminal cysteine residue under basic conditions used for Fmoc deprotection. 1H-NMR analysis of the unfarnesylated a-factor precursor peptide confirmed the absence of epimerization. The side-chain anchoring method was used to produce wild type a-factor that contains a C-terminal methyl ester along with ethyl-, isopropyl- and benzyl-ester analogs in good yield. Activity assays using a yeast-mating assay demonstrate that while the ethyl and isopropyl esters manifest near-wild type activity, the benzyl ester-containing analog is ca. 100-fold less active. This simple method opens the door to the synthesis of a variety of C-terminal ester modified peptides that should be useful in studies of protein prenylation and other structurally related biological processes.
Graphical Abstract
Introduction
The development of peptide chemistry continues to be an area of intense investigation since peptides play a significant role in biochemical applications including therapeutics1,2 and peptidomimetics3. Due to the common use of peptides to address biological questions, efficient techniques are needed to incorporate modifications into their basic structure that enhance their properties.4 Peptides containing esters at their C-terminus can be used for a variety of biological applications including drug discovery. Using an alkyl ester to mask a carboxylic acid increases the hydrophobicity of the molecule and improves its membrane permeability.5,6 Depending on the nature of the alkyl ester, such molecules can undergo hydrolysis upon cell entry and release a bioactive carboxylic acid.7 Peptides with a methyl ester on their C-terminal cysteine are useful for studying protein prenylation since they are present in fully processed, farnesylated and geranylgeranylated proteins.8 Research shows that the presence of the prenyl moiety and methyl ester group on specific proteins is necessary for membrane targeting and proper functioning.9 However, even though ester-modified peptides can be useful for a variety of applications, only a few reports in the literature describe their synthesis via solid phase methods.10–14
An ideal methodology for the synthesis of peptides containing a C-terminal cysteine circumvents common limitations of existing methods without compromising their primary features. Specifically, such methodologies should feature the following: 1) use of commercially available resins, 2) manifest broad scope to introduce a variety of alkyl esters, 3) easy release of the desired peptide from the resin containing the alkyl ester functionality, 4) Allow for the incorporation of a variety of related derivatives within the amino acid sequence, 5) minimize the occurrence of side-reactions (i.e. cysteine racemization and β-elimination) due to the presence of a C-terminal cysteine, 6) use mild conditions and short reaction times, and 7) produce the desired peptide in high yield.
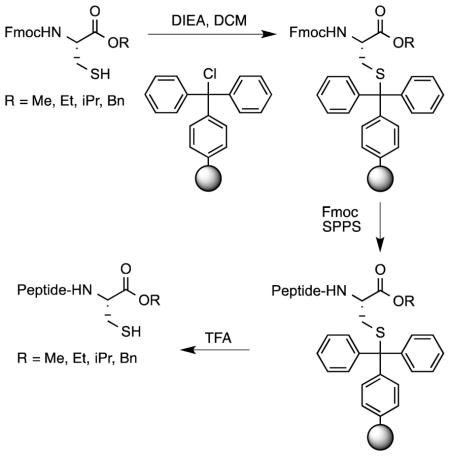
Here, we report new developments related to the trityl (Trt) side-chain anchoring method that expand the utility of that strategy published earlier by our group.15 The method reported here begins with the loading of an Fmoc-Cys-OR building block onto commercially available Trt-containing resin via its thiol functionality. The peptide sequence is then assembled using Fmoc-based solid phase peptide synthesis (SPPS). Since the linkage between the cysteinyl thiol and Trt resin is acid labile, the peptide can be cleaved from the resin upon treatment with mild acidic conditions (TFA-based solutions) along with simultaneous deprotection of the acid-labile amino acid side-chain protecting groups typically used in Fmoc SPPS. In our initial communication, the method was limited to the preparation of methyl esters; in the current study, the scope has been expanded to include ethyl, isopropyl and benzyl esters. An expanded analysis of cysteine racemization is also reported in both a tripeptide model system as well as a longer peptide. Finally, the new methodology has been used to produce analogs of a-factor, an important bioactive, farnesylated dodecapeptide, that incorporate different alkyl esters at the C-terminus. The preparation of different ester analogs of this peptide was not only useful to highlight the scope of the trityl-anchoring method but also allowed us to probe the effects of these ester modifications on the bioactivity of a-factor.
Results and Discussion
Loading of Fmoc-protected cysteine ester onto Trt-based resin via side-chain anchoring
In our initial report15, we established the use of Trt-Cl resin as the support of choice for SPPS through cysteine side-chain anchoring. This was achieved after coupling the resin to the free thiol of Fmoc-Cys-OMe. Because the Trt-Cl resin is commercially available, the only requirement needed to synthesize C-terminal methyl ester-containing peptides via this method is access to Fmoc-protected cysteine methyl ester. That material can be obtained in one step from Fmoc-Cys-OH. After effectively synthesizing peptides with methyl ester functionality on their C-termini15, we wanted to expand the scope of this methodology by investigating the synthesis of peptides with different alkyl esters at their C-termini.
The first step of SPPS via Trt-side chain anchoring is the attachment of an Fmoc-Cys-OR residue to Trt-Cl resin via its thiol functionality (Scheme 1). To obtain the requisite compounds, Fmoc-Cys-OH was treated with methanol, ethanol, 2-propanol or benzyl bromide to yield the corresponding methyl, ethyl, isopropyl and benzyl esters (1a–1e); those compounds were then used to prepare the desired resin-linked residues (2a–2e). As previously reported for the loading of Fmoc-Cys-OMe, the cysteine containing ethyl and isopropyl esters were loaded onto the resin after 24-hour incubations using two equivalents of the amino acid. The loading of amino acids onto the Trt-Cl resin (determined via subsequent Fmoc analysis) was >85%, 55%, and 46% for Fmoc-Cys-OMe, Fmoc-Cys-OEt, and Fmoc-Cys-OiPr, respectively. Efforts to increase the loading of Fmoc-Cys-OEt by treating the resin with fresh reagents for an additional 24 hours were not successful; the most likely explanation for this is that the resin may undergo some concomitant hydrolysis since the conditions were not rigorously anhydrous; given that reasonable loading was obtained, this was not further investigated. However, under the typical 24-hour reaction conditions, only 3% loading of Fmoc-Cys-OBn was obtained (Table 1). Overall, this shows that the presence of larger, hindered cysteine esters significantly decreases the efficiency of coupling between the resin-bound Trt group and the cysteine-derived thiol. In order to increase the loading of Fmoc-Cys-OBn on Trt resin, variations of the equivalents of added amino acid, reaction time, and temperature of coupling were made. The loading on the resin increased to 19% using a temperature of 37 °C and was further increased to ~50% when the temperature was raised to 50 °C. In general, the success of this reaction suggests that this method could be used to introduce a variety of alkyl esters with interesting functionalities including aromatic structures onto the C-terminus of Fmoc-protected cysteine for subsequent SPPS.
Scheme 1.

Loading of Fmoc-Cys-OR onto trityl-based resin.
* is used to emphasize location of the stereogenic center of the cysteine α-carbon) producing a trityl side-chain anchored cysteine ester for SPPS.
Table 1.
Loading efficiency of Fmoc-Cys-OR onto Trt-Cl resin performed at 25 °C. The loading was determined by quantifying the Fmoc released after resin treatment with 10% piperidine in DMF.
| R group | Equiv of Fmoc-Cys-OR | Reaction time (h) | Loading (%) |
|---|---|---|---|
| Me (1a) | 2 | 24 | >85 |
| Et (1c) | 2 | 24 | 55 |
| Et (1c) | 2, 2a | 24, 24a | 57, 54a |
| iPr (1d) | 2 | 24 | 46 |
| Bn (1e) | 3 | 72 | 8 |
| Bn (1e) | 6 | 36b | 48 |
Trt-Cl resin was treated with 2 equiv of Fmoc-Cys-OEt and 4 equiv of DIEA for 24 h followed by retreatment with fresh reagents for another 24 h. The first value for the loading was obtained from a sample obtained after 24 h of reaction while the second value was obtained after the second treatment.
The temperature of the Fmoc-Cys-OBn reaction was increased to 37 °C (24 h) and further increased to 50 °C (12 h) to obtain a higher loading.
Epimerization studies of cysteine linked to Trt resin via side chain anchoring
Loss of chirality at C-terminal cysteine residues is a serious concern in Fmoc solid-phase peptide synthesis in two situations: 1) during Cys loading onto the resin as an ester and 2) during the amine deprotection cycles using basic conditions (piperidine-DMF). Since the methodology reported herein uses a side-chain anchoring strategy that does not require basic activation of the α-carboxyl of the Cys, the risk of racemization of the C-terminal Cys during resin loading is minimal.16 However, previous research suggests that repetitive treatment with piperidine can cause racemization under certain circumstances.17 In our original report, we investigated the extent of racemization of Cys in peptides prepared by side-chain anchoring using 2-Chlorotrityl resin. However, since we subsequently discovered that Trt-Cl resin give a higher yield of peptide due to lower peptide loss via β-elimination, we were interested in studying this question with a peptide prepared using this improved resin choice. To determine whether loss of chiral integrity of Cys occurs, model tripeptides 6a and 6b containing methyl esters at their C-termini were synthesized using the Trt side chain-anchoring method (Scheme 2). The C-terminal acid forms of these tripeptides have been previously reported to manifest baseline resolution when separated by HPLC.18 To determine the stage at which the cysteine on resin is more prone to racemization the exposure of resins 2a and 4a to 20% piperidine was prolonged for different periods of time (10 min, 2 h, 4 h and 16 h). Then, these resin samples were employed to synthesize the model tripeptide 6a. A D-Cys-containing peptide (6b) was synthesized as a standard for the determination of the retention time of the epimeric product in the HPLC chromatogram (Figure S6 and Figure S7). As expected, baseline resolution of these peptides was obtained but at a higher retention time due to the decrease in polarity resulting from modification of the C-terminus with a methyl ester (Rt 6a = 23.3 min vs. Rt 6b = 24.5 min). The extent of epimerization was quantified by HPLC peak integration (Table 2).
Scheme 2.

Synthesis of model tripeptides Gly-Phe-L-Cys-OMe and Gly-Phe-D-Cys-OMe.
Reaction conditions: a) 20% piperidine in DMF; b) Fmoc SPPS; c) 20% piperidine in DMF; d) 5% TFA in CH2Cl2.
Table 2.
Studies of L-Cys epimerization in samples of the model tripeptide 6a after piperidine treatment of a2a and b4a for different periods of time.
| Exposure Time | D-cysteine (%)a | D-cysteine (%)b |
|---|---|---|
| 10 min | 1.4 ± 0.2 | 0.5 ± 0.1 |
| 2 h | 3.3 ± 0.6 | 2.8 ± 0.3 |
| 4 h | 5.0 ± 0.4 | 5.5 ± 0.1 |
| 16 h | 4.6 ± 0.2 | 17.8 ± 0.3 |
For the first experiment (piperidine treatment of 2a), the results show that 1.4% of the undesired D-Cys-containing peptide forms when the tripeptide is synthesized using standard SPPS deprotection conditions (10 minute treatment). A modest and entirely acceptable level (~5%) of D-Cys-containing peptide was noted after 4 hours of treatment. This same extent of epimerization was obtained after exposing the amino acid-resin 2a for 16 hours. These results illustrate that the presence of a free amine on cysteine methyl ester linked trityl resin via its thiol supresses racemization. While prolonged exposure of the free amine form to basic deprotection conditions is unlikely to occur under typical SPPS conditions, the situation modeled by exposure of 4a to prolonged piperidine treatment is more representative since the C-terminal cysteine ester with an acylated amine will be subjected to every Fmoc deprotection step in the course of peptide synthesis. Interestingly, the extent of epimerization obtained after treating 2a or 4a from 10 min up to 4 hours is comparable (Table 2). A more pronounced level of epimerization was obtained when treating model peptide-resin 4a for 16 hours with piperidine-DMF (L:D ~8:2). This result is comparable to that reported for a different Cys side-chain anchoring strategy (8% epimerization in 24 hours) involving the use of a specialized xanthenyl-based resin.16 In general, these observations are consistent with the greater acidity of the cysteinyl α-proton when the amine is acylated compared to the free amine form. Given the modest level of epimerization of 4a observed after 4 hours of piperidine treatment (<6%), it appears this method can be safely employed to prepare peptides up to 24 residues in length without appreciable racemization.
Synthesis of a-factor and a-factor ester analogs
a-Factor is a bioactive farnesylated dodecapeptide containing a C-terminal methyl ester involved in the mating of yeast. This molecule has attracted considerable interest in the field of protein prenylation since its biogenesis involves the same sequence of enzymatic reactions (S-prenylation, proteolysis and methylation) observed with larger proteins.19 Protein prenylation is an area of intense interest since inhibition of this process has important therapeutic applications.20,21 During the mating of yeast, this molecule is secreted and stimulates the cell surface receptors on an opposite cell type, which then results in a cellular mating response between two different cells.22–24 Studies have shown that absence of the methyl ester functionality on this peptide decreases the bioactivity ~100 fold relative to the wild type peptide indicating an important relationship between structure and activity.25
The a-factor sequence was assembled using Fmoc-based SPPS starting with Fmoc-Cys-OMe anchored on trityl resin (2a) to obtain the peptide-resin 7a (Scheme 3). Since the linkage between the cysteinyl thiol and Trt resin is acid labile, the peptide was cleaved from the resin upon treatment with Reagent K (a cocktail of TFA containing cation scavengers)26 along with simultaneous deprotection of the acid-labile side-chain protecting groups. Using this concise procedure, it was possible to obtain the a-factor precursor peptide 8a in good yield (82%) with a crude purity of 81% (Figure 1A). The epimeric a-factor precursor peptide, 8b, containing a D-Cys residue was prepared in an analogous fashion (Figure 1B).
Scheme 3.

Synthesis of a-factor (9a) and a-factor C-terminal ester analogs (9c–e) using the trityl side-chain anchoring strategy.
Figure 1.

HPLC chromatograms of crude peptides (8a–8e) after cleavage from the trityl resin. A) 8a; B) 8b; C) 8c; D) 8d; E) 8e.
Having demonstrated the success of this simple method for the preparation of the a-factor precursor, we next undertook the synthesis of a-factor analogs containing different alkyl esters at the C-terminus. The syntheses of the a-factor precursor peptides (8c–e) were initiated on resin loaded with Fmoc-protected cysteine esters including an ethyl ester (2c), isopropyl ester (2d) or benzyl ester (2e). These alkyl groups were selected due to their small size and their stability as C-terminal esters under SPPS acidic/basic conditions. Importantly, studies have shown that the use of large modifications at the C-terminus of a-factor significantly affects the bioactivity of the pheromone.25 Apart from exploring the versatility of the trityl side-chain anchoring methodology, the ability to prepare a-factor analogs with incremental C-terminal modifications allows the functional consequences of such changes to be studied in a systematic manner.
The a-factor ester analogs were synthesized using the same methodology employed for the synthesis of the wild type a-factor (Scheme 3). Different Fmoc-Cys-OR residues attached to Trt-Cl resin (2c–e) were used to initiate peptide assembly via SPPS. Deprotection of side-chains and cleavage of the peptide from the resin were achieved simultaneously in the presence of Reagent K to give peptides 8c–e with crude purity higher than 70% as shown in Table 3 and Figure 1C–D. Farnesylation of 8a and 8c–e was performed using farnesyl bromide in the presence of Zn(OAc)2 under acidic conditions as described by Naider and coworkers11 and previously reported by Mullen et. al27 for a-factor synthesis to yield 9a and 9c–e.
Table 3.
Yield and purity of crude peptides 8a–e synthesized using the trityl side-chain anchoring method.
Purity of peptides 8a–e in the crude mixture after deprotection from the resin.
Yield of peptides in the crude mixture based on weight and purity of crude material.
1H-NMR analysis of a-factor precursor peptide
While the experiments with the model tripeptide described above provide compelling evidence that Cys racemization is not a significant problem when using the side-chain anchoring approach reported here, we wanted to confirm that this was true for a larger, biologically relevant peptide. Unfortunately, efforts to separate an epimeric mixture of 8a and 8b via HPLC were not successful (Figure S8). Both peptides exhibited essentially identical retention times, suggesting that it would not be possible to employ chromatographic methods to investigate possible Cys epimerization in the context of this larger peptide. Since NMR spectroscopy has also been used to analyze the stereochemical composition of mixtures of diastereomeric peptides28 and complexes of D,L-amino acids,29 that technique was employed here. Inspection of the fingerprint region of a 2D 1H-1H TOCSY NMR spectrum obtained using 8a (Figure 2A) shows a single doublet due to Hα-HN coupling. Since the absence of a second doublet (that would indicate the presence of the D-Cys-containing epimer 8b) does not unambiguously prove that D-Cys is not present, we elected to analyze an authentic sample of 8b. Importantly, the fingerprint region of the 2D NMR spectrum obtained using that material (Figure 2C) shows a clear difference in the Cys-related protons. In the L-Cys-containing epimer (8a), the Hα-HN and Hβ-HN cross peaks localize near 8.0 ppm while in the D-Cys-containing epimer (8b), they are shifted upfield near 7.9 ppm. A color-coded expanded view of the superposition of the two individual spectra (from Figure 2A and Figure 2C) highlights these variations (Figure 2B; 8a in black, 8b in red). These differences were also readily seen in the NMR spectrum of an equimolar mixture of 8a and 8b (Figure S13). However, in the spectrum of purified 8a (Figure 2A), no evidence of the D-Cys-containing epimer (8b) was observed. Based on the signal to noise level in the 2D spectrum of 8a (Figure 2A), we estimate that the amount of 8b that could be present must be less than 5%. That low level of epimerization is consistent with the results noted above obtained with the tripeptide model. Overall, the absence of stereochemical erosion in this dodecapeptide provides a compelling case for the utility of the side-chain anchoring strategy reported here.
Figure 2.

Fingerprint region of 700-MHz 2D 1H–1H TOCSY NMR spectrum of a-factor precursor peptides. A) Spectrum of L-Cys containing a-factor precursor peptide (8a); B) Superposition of the spectra of 8a (black) and 8b (red); C) D-Cys containing a-factor precursor peptide (8b) in DMSO-d6. Assignments are indicated adjacent to each cross peak.
Structural analysis of a-factor (9a) produced via side chain anchoring
While the mass of 9a produced by the side chain anchoring method was established to be correct using ESI-MS, 1H-NMR was employed to confirm the site of prenylation. Accordingly, a 2D 1H-1H TOCSY NMR spectrum was obtained for 9a and a portion of the spectrum from the fingerprint region is shown in Figure 3. The newly acquired spectrum shows excellent agreement when compared to previously reported NMR data obtained from a-factor (prepared via two different routes employing Boc/Bzl protection11 or Fmoc SPPS using a hydrazinobenzoyl specialized linker27). Importantly, in the fingerprint region, the only significant difference between the spectrum of 9a and the precursor peptide, 8a (see Figure 2A) is a shift in the Cys-related Hα-HN and Hβ-HN cross peaks which is consistent with regioselective alkylation on the cysteinyl thiol. Thus, this data provides unambiguous confirmation that the side chain anchoring methodology presented here can be successfully used to prepare a-factor.
Figure 3.

Fingerprint region of 700-MHz 2D 1H-1H TOCSY NMR spectrum of a-factor (9a) in DMSO-d6. Assignments are labeled adjacent to each cross peak.
Biological assay of synthetic a-factor analogs incorporating different C-terminal esters
Since the side chain anchoring route reported here provided facile access to a variety of a-factor analogs that incorporate different ester groups, we decided to analyze the activity of these analogs to obtain insight into the relationship between the C-terminal a-factor structure and biological activity. This was accomplished using a yeast growth arrest assay in which cells expressing the Ste3p receptor (MATα cells) undergo growth arrest in the presence of a-factor. For these experiments, a plate containing a lawn of MATα S. cerevisiae cells (strain RC757) was treated with different amounts of a-factor (Figure 4, panels A, C, E, G, and I). The presence of active a-factor causes a zone of growth inhibition to develop in the region where the compound is applied with the endpoint given by the lowest amount of a-factor applied that produces a distinct clear zone (compared with partial clearing at lower amounts). Parallel experiments with cells that do not express the Ste3p receptor (MATa cells, strain LM102) were performed to establish that growth inhibition was not due to nonspecific toxicity (Figure 4, panels B, D, F, H, and J). Both wild type a-factor (Figure 4A) and the synthetic material produced here gave endpoints of 0.12 ng. That value is in good agreement with data obtained using wild type a-factor in previous studies or from samples prepared via other synthetic routes11,25,30,31 indicating that the new method reported here provides material that manifests full biological activity.
Figure 4.

Biological assay of pheromones. Growth arrest in response to pheromone was determined for the a-factor responsive strain RC757 (left column; A, C, E, G, I) or the α-factor responsive strain LM102 (right column: B, D, F, H, J). A, B - wild type a-factor (9a), C, D – ethyl ester a-factor (9c), E, F – isopropyl ester a-factor (9d), G, H, I, J – benzyl ester a-factor (9e). The templates in the middle indicate the amount of synthetic a-factor and its analogs spotted on the plates. The benzyl ester a-factor analog was applied to the plates in two different sets of concentrations as indicated in the corresponding middle templates. At the top of each plate 2.5 μL of cells secreting α-factor (MATα) (on the left) or a-factor (MATa) were applied to the lawn as indicated in the templates.
Next the biological activity of the a-factor analogs was evaluated using the same assay. The observed endpoint for the ethyl ester analog 9c (Figure 4C) was half (0.06 ng) that of the wild-type a-factor indicating that the substitution of an ethyl group in place of the naturally occurring methyl ester has a minimal effect on biological activity. Similarly, an end-point of 0.25 ng (Figure 4E) was observed for the isopropyl ester analog showing that it has comparable activity when compared with the natural pheromone. More striking results were observed with the benzyl ester, which showed no activity in the standard growth arrest assay using a range of 2.0 – 0.015 ng of compound (Figure 4G). To quantify the reduced activity, an additional experiment employing larger amounts of material (20 – 0.15 ng) was conducted (Figure 4I). That data gave an end-point of 10 ng indicating that replacement of the methyl ester moiety present in wild type a-factor with a benzyl ester group results in an approximately 100-fold reduction in activity. A summary of the biological data is provided in Table 4. Overall, these results demonstrate that small changes in the size of the C-terminal alkoxy group have minimal effects on the bioactivity of a-factor. However, when the size of the ester group exceeds 100 Å3, there is a precipitous drop in activity25,32 suggesting that there is a defined binding pocket within the receptor that interacts with the C-terminal region of a-factor.
Table 4.
End-point concentration of a-factor and a-factor analogs as determined in a growth arrest assay. Alcohol group volumes are the molecular volumes calculated for the parent alcohols CH3OH (MeOH), CH3CH2OH (EtOH), (CH3)2CHOH (iPrOH) and C6H5CH2OH (BnOH).
| a-factor analog | End-point concentration (ng) | C-terminal alkoxy group | Alcohol group volume (Å3)33 |
|---|---|---|---|
| 9a (WT)a | 0.12 | CH3O | 37.2 |
| 9ab | 0.12 | CH3O | 37.2 |
| 9c | 0.06 | CH3CH2O | 54.0 |
| 9d | 0.25 | (CH3)2CHO | 70.6 |
| 9e | 10 | C6H5CH2O | 109 |
Data for wild-type a-factor.
Data acquired for synthetic a-factor was published previously by our group.15
Conclusion
In summary, a versatile method has been developed for the synthesis of peptides containing C-terminal cysteine esters that is based on side-chain anchoring via the thiol group of cysteine. A model tripeptide was synthesized using this strategy and analyzed via HPLC to investigate possible epimerization; under conditions that would be typically used for the synthesis of peptides less than 24 residues (4 hours of total piperidine exposure), no significant cysteine racemization was observed. The absence of epimerization was also directly established in a longer, dodecameric peptide via 1H-NMR analysis; for that work, the same route was used to prepare the epimeric peptide containing a D-Cys residue. 1H-NMR was also used to confirm the selectivity of the subsequent thiol alkylation that yielded a-factor, a bioactive farnesylated peptide with a C-terminal methyl ester. The method described herein was then used to efficiently synthesize a-factor ester analogs in good yield and high purity. The bioactivities of the synthesized a-factor analogs were analyzed using a growth arrest assay. While the activities of the ethyl and isopropyl ester analogs were similar to that of the wild type methyl ester, the benzyl ester analog was almost 100-fold less active suggesting that the C-terminal ester of a-factor is an important structural element recognized by its cognate receptor. The success of this simple method for the synthesis of peptides containing C-terminal cysteine alkyl esters using commercially available trityl resin opens the door to the synthesis of a wide variety of C-terminal ester modified peptides functionalized with modified isoprenoids34–36 that should be useful in studies of protein prenylation and other structurally related biological processes.
Experimental Section
General Methods
All reagents and solvents were used as received. All the solvents were HPLC grade. DIEA (N,N-Diisopropylethylamine) and TFA were of Sequalog/peptide synthesis grade. Standard Fmoc/HCTU chemistry was used for SPPS of peptides. Fmoc-based SPPS was performed on a Protein Technologies PS3 automated peptide synthesizer. Additional steps performed on resin (after coupling/deprotection of N-terminus) were carried out using a polypropylene syringe equipped with a porous polypropylene disc at the bottom. Peptide synthesis and other transformations were performed at 25 °C unless otherwise indicated. Loading of first amino acids on resin was determined using Fmoc quantitative analysis26 and the Fmoc solutions were analyzed at 301 nm on a UV-Visible spectrophotometer. The peptides were cleaved off the resin using Reagent K. (Preparation of Reagent K: The specific amount of compounds used to do the mixture were: 0.5 g phenol, 0.5 mL of thioanisole, 0.5 mL of H2O, 0.25 mL ethanedithiol and 8.25 mL of TFA. Then N2(g) was bubbled into the solution to remove any oxygen dissolved in the mixture.) The peptides were analyzed using Reverse Phase High Performance Liquid Chromatography (RP-HPLC) and the products were observed at 220 nm. The analytical column used was a C18 Agilent Microsorb-MV 100–5 (4.6 × 250 mm) and the preparative column was a Phenomenex C18 (10 μm, 10.00 × 250 mm). The buffer solutions used for HPLC analysis and purifications were: Buffer A (0.1% TFA in H2O) and Buffer B (0.1% TFA in CH3CN). The retention times of the a-factor analogs are based on a flow rate of 1 mL/min and on a gradient increase of 1% Buffer B/min after equilibration of the column with 1% Buffer B. The solutions containing purified peptides were lyophilized and analyzed by ESI-MS. The retention times of the model tripeptides are based on a flow rate of 1 mL/min and a gradient increase of 1.6% Buffer B/min after equilibration of the column with 5% Buffer B.
Synthesis of Fmoc-L-Cys-OMe (1a)
A solution of Fmoc-Cys-OH • H2O (0.5 g, 1.38 mmol) in CH3OH (~10 mL) was prepared and 6 drops of 37% HCl were added to this solution. The mixture was left stirring overnight at rt. A wet white solid (with excess of CH3OH) was obtained. The material was dissolved in acetone and the mixture of solvents was evaporated under reduced pressure to obtain the product as a white solid (0.470 g, 97%). No further purification was needed. Rf 0.5 (silica gel, hexane/EtOAc, 1:1, v/v). 1H NMR (300 MHz, CDCl3): δ 1.37 (t, 1H, SH, J=8.6 Hz), 3.01 (s (broad), 2H), 3.81 (s, 3H), 4.24 (t, 1H, J=7.0), 4.44 (d, 2H, J=6.3), 4.68 (s (broad), 1H), 5.69 (s (broad), NH), 7.33 (t, 2H, J=7.3), 7.42 (t, 2H, J=7.3), 7.62 (d, 2H, J=7.3), 7.78 (d, 2H, J=7.3). 13C NMR (75.0 MHz, CDCl3): δ 27.2, 47.2, 52.9, 55.2, 67.1, 120.0, 125.1, 127.1, 127.8, 141.3, 143.6, 155.6, 170.4. HRMS (ESI-TOF) m/z: [M+Na]+ Calcd for C19H19NO4S 380.0933; Found 380.0905.
Synthesis of Fmoc-D-Cys-OMe (1b)
Compound 1b was prepared via the same procedure described above for 1a using Fmoc-D-Cys-OH as starting material.
Synthesis of Fmoc-Cys-OEt (1c)
A solution of Fmoc-Cys-OH • H2O (0.10 g, 0.26 mmol) in EtOH (~10 mL) was prepared and 6 drops of 37% HCl were added to this solution. The mixture was left stirring overnight at reflux (80 °C). A wet white solid (with excess of EtOH) was obtained. The material was dissolved in acetone and the mixture of solvents was evaporated under reduced pressure to obtain the product as a white solid (0.109 g, 99%). No further purification was needed. Rf 0.7 (silica gel, hexane/EtOAc, 1:3, v/v). 1H NMR (300 MHz, CDCl3): δ 1.34 (m, 3H, SH), 3.01 (s (broad), 2H), 4.25 (m, 3H), 4.43 (d, 2H, J=6.9), 4.65 (s (broad), 1H), 5.72 (s (broad), NH), 7.33 (t, 2H, J=7.4), 7.41 (t, 2H, J=7.4), 7.62 (d, 2H, J=7.4), 7.78 (d, 2H, J=7.4). 13C NMR (75.0 MHz, CDCl3): δ 14.3, 27.2, 47.2, 55.2, 62.1, 67.1, 120.0, 125.1, 127.1, 127.8, 141.3, 143.6, 155.6, 169.9. HRMS (ESI-TOF) m/z: [M+Na]+ Calcd for C20H21NO4S 394.1089; Found 394.1085.
Synthesis of Fmoc-Cys-OiPr (1d)
A solution of Fmoc-Cys-OH • H2O (0.1 g, 0.26 mmol) in iPrOH (~10 mL) was prepared and 12 drops of 37% HCl were added to this solution. The mixture was left stirring overnight at reflux (83 °C). The material was dissolved in acetone and the mixture of solvents was evaporated under reduced pressure to obtain a solution that was purified via column chromatography (hexanes/EtOAc, 9:1). The product was obtained as a white solid (57 mg, 51%). Rf 0.71 (silica gel, hexane/EtOAc, 7:3, v/v). 1H NMR (300 MHz, CDCl3): δ 1.32 (m, 6H, SH), 3.00 (s (broad), 2H), 4.23 (t, 1H, J=3.4), 4.43 (d, 2H, J=6.6), 4.61 (s (broad), 1H), 5.79 (s (broad), NH), 7.32 (t, 2H, J=7.5), 7.41 (t, 2H, J=7.5), 7.61 (d, 2H, J=7.5), 7.77 (d, 2H, J=7.5). 13C NMR (75.0 MHz, CDCl3): δ 21.8, 27.2, 47.2, 50.8, 55.2, 67.1, 120.0, 125.1, 127.1, 127.8, 141.3, 143.7, 155.7, 169.5. HRMS (ESI-TOF) m/z: [M+Na]+ Calcd for C21H23NO4S 408.1246; Found 408.1258.
Synthesis of Fmoc-Cys-OBn (1e)
A solution of Fmoc-Cys(Trt)-OH (0.5 g, 0.85 mmol) in DMF (6 mL) was prepared and benzyl bromide (112 uL, 0.94 mmol) and DIEA (148 uL, 0.85 mmol) were added to this solution. The mixture was left stirring for 2 h on a reflux at 60 °C. The salt byproduct was removed by crystallization with Et2O. The product remained in the filtrate and the DMF was removed by lyophilization. The material was used without further purification. Fmoc-Cys(Trt)-OBn (0.3 g, 0.44 mmol, 1 equiv) was dissolved in 25 mL of 1% TFA solution in CH2Cl2. Triisopropylsilane (0.9 mL, 4.4 mmol, 10 equiv) was added to this solution resulting in no immediate color change. TLC showed 95% conversion after 2 h of constant stirring. A solution of 1% TEA in CH2Cl2 (28 mL) was added slowly to the reaction mixture to neutralize the TFA. 20 mL of water were added to the mixture and the CH2Cl2 was evaporated under reduced pressure. CH2Cl2 (25 mL) was added to the solution and an extraction with water (20 mL - 2×) was performed. The product (CH2Cl2 solution) was dried with MgSO4 and filtered. The solvents were removed under reduced pressure and the product was purified by flash chromatography (hexanes/EtOAc, 9.5:5). Rf 0.48 (silica gel, hexane/EtOAc, 7:3, v/v). 1H NMR (300 MHz, CDCl3): δ 1.27 (t, 1H, SH, J=8.9 Hz), 3.01 (s (broad), 2H), 4.23 (t, 1H, J=7.0), 4.43 (d, 2H, J=6.8), 4.72 (s (broad), 1H), 5.23 (q, 2H, J=12.8), 5.74 (s (broad), NH), 7.37 (m (arom), 9H), 7.61 (d, 2H, J=7.6), 7.78 (d, 2H, J=7.6). 13C NMR (75.0 MHz, CDCl3): δ 27.2, 47.2, 55.2, 67.2, 67.8, 120.0, 125.1, 127.1, 127.8, 128.6, 128.7, 129.5, 135.0, 141.3, 143.78, 155.6, 169.9. HRMS (ESI-TOF) m/z: [M+Na]+ Calcd for C25H23NO4S 456.1246; Found 456.1248.
General Procedure for the Loading of Fmoc-Cys-OR (R = Me, Et, iPr) to Trt-Cl resin
Trityl chloride (Trt-Cl) resin, with a loading of 1.40 mmol/g, (1 equiv) was placed into a fritted syringe and was washed with DMF (3×). The Fmoc-Cys-OR (2 equiv) was added into a separate test tube and dissolved with CH2Cl2 (3 mL). This solution was added to the syringe containing the resin followed by the addition of DIEA (4 equiv). The mixture was left to react overnight while mixing with a rotisserie. The next morning, CH3OH (50 μL) was added to the solution and allowed to react for 5 min in order to cap any unreacted Trt-Cl groups on the resin. Then, the reagents were drained from the syringe and the resin was washed with DMF (3×) and CH2Cl2 (3×). Loading of Fmoc-Cys-OR on the resin was calculated via Fmoc-quantitative analysis (see Table 1).26
Procedure for the Loading of Fmoc-Cys-OBn to Trt-Cl resin
The trityl chloride (Trt-Cl) resin, with a loading of 1.40 mmol/g, (1 equiv, 40 mg) was placed into a round bottom flask and was suspended in CH2Cl2. Fmoc-Cys-OBn (6 equiv, 144 mg, 0.333 mmol) was added into a separate test tube and dissolved with CH2Cl2 (3 mL). This solution was added to the flask before adding DIEA (8 equiv, 77.5 μL, 0.45 mmol). The mixture was left to react 24 h at 37 °C with occasional manual stirring. The next day, the reaction was refluxed at 50 °C for 2 h at which point 50 μL of CH3OH were added to the solution (at rt) and reacted for 5 min in order to cap any remaining Trt-Cl groups on the resin. Then, the resin was transferred to a fritted syringe and the reagents were drained off. The resin was washed with DMF (3×) and CH2Cl2 (3×). Loading of Fmoc-Cys-OBn on the resin (0.680 mmol/g, 39.5 mg resin, 48.6% loading) was calculated via Fmoc-quantitative analysis26 (Table 1).
Synthesis of the model tripeptide Gly-Phe-L-Cys-OMe (6a)
Fmoc-based SPPS was used to couple Fmoc-Phe-OH and Fmoc-Gly-OH onto resin loaded with Fmoc-Cys-OCH3 (2a). The peptide 6a was obtained after treatment of the resin with 2 mL of 1% TFA/CH2Cl2 for 2 min (Repeated 10×). The TFA/original was evaporated to 5% of its volume and the peptide was precipitated with 5 mL of Et2O. The suspension was centrifuged to form a pellet that was rinsed twice with Et2O and dissolved in 2 mL of DMF. ESI-MS showed the mass of an oxidized peptide (disulfide bond) that was reduced with 4 equiv of DTT in DIEA (4 equiv) to obtain the desired peptide. ESI-MS: for C15H21N3O4S [M+H+]+; calcd 340.1333, found 340.1383 [M+Na+]+; calcd 362.12, found 362.10.
Synthesis of model tripeptide Gly-Phe-D-Cys-OMe (6b)
Fmoc-based SPPS was used to prepare 6b as described above for 6a. As noted for 6a, ESI-MS showed the mass of an oxidized peptide (disulfide bond) that was reduced with 4 equiv of DTT in DIEA (4 equiv) to obtain the desired peptide. ESI-MS: for C15H21N3O4S [M+H+]+; calcd 340.1333, found 340.1310 [M+Na+]+; calcd 362.12, found 362.12.
Synthesis of YIIKGVFWDPAC-OMe (8a)
The resin 2a, (1 equiv, 64 mg, 0.81 mmol/g) containing the side-chain Fmoc-Cys-OCH3 residue was placed in reaction vessel for an automated peptide synthesizer together with 4 equivalents of each Fmoc-protected amino acid and 4 equivalents of the activating agent HCTU. After the peptide was synthesized, the resin was transferred to a syringe, washed 3× with CH2Cl2 and dried in vacuum. Then, the peptide was deprotected and cleaved from the resin after treatment with 10 mL of Reagent K for 1 h followed by Et2O precipitation. The peptide was centrifuged to form a pellet that was rinsed twice with Et2O. The product was obtained as a white solid, and this crude product was used in the subsequent alkylation step without purification (75 mg, 81% purity of crude, 82% yield). MS (ESI-TOF) m/z: [M+2H+]2+ Calcd for C70H100N14O16S 713.3661; Found 713.3055.
Synthesis of YIIKGVFWDPAC-OMe containing D-Cys (8b)
Peptide 8b was prepared using resin 2b (1 equiv, 30 mg, 0.80 mmol/g) via the same procedure described above for 8a (40 mg, 75% purity of crude, 85% yield). MS (ESI-TOF) m/z: [M+2H+]2+ Calcd for C70H100N14O16S 713.3661; Found 713.4649
Synthesis of YIIKGVFWDPAC-OEt (8c)
Peptide 8c was prepared using resin 2c (1 equiv, 120 mg, 0.75 mmol/g) via the same procedure described above for 8a. The product was obtained as a white solid, and this crude product was used in the next step without purification (147.36 mg, 76% purity of crude, 84% yield). MS (ESI-TOF) m/z: [M+2H+]2+ Calcd for C71H102N14O16S 720.3740; Found 720.3745.
Synthesis of YIIKGVFWDPAC-OiPr (8d)
Peptide 8d was prepared using resin 2d (1 equiv, 38 mg, 0.64 mmol/g) via the same procedure described above for 8a. The product was obtained as a white solid, and this crude product was used in the next step without purification (39 mg, 72% purity of crude, 80% yield). MS (ESI-TOF) m/z: [M+2H+]2+ Calcd for C72H104N14O16S 727.3818; Found 727.3869.
Synthesis of YIIKGVFWDPAC-OBn (8e)
Peptide 8e was prepared using resin 2e (1 equiv, 39 mg, 0.67 mmol/g) via the same procedure described above for 8a. The product was obtained as a white solid, and this crude product was used in the next step without purification (51.4 mg, 76% purity of crude, 90% yield). MS (ESI-TOF) m/z: [M+2H+]2+ Calcd for C76H104N14O16S 751.3818; Found 751.3798.
Synthesis of YIIKGVFWDPAC(Far)-OMe (9a)
The a-factor precursor peptide containing the methyl ester at the C-terminus (8a) (1 equiv, 3.9 mg, 3 μmol) was dissolved in DMF/BuOH/0.1% aq TFA (10 mL of a 4:2:1 mixture) together with farnesyl bromide (5 equiv, 3 μL, 15 μmol). In a separate test tube, Zn(OAc)2•2H2O (5 equiv, 2.3 mg, 15 μmol) was dissolved in 0.50 mL of 0.1% aq TFA. The Zn(OAc)2 solution was added to the peptide solution and the reaction was left stirring for 1.5 h. The reaction mixture was then filtered and purified by preparative HPLC. The product was obtained as a white solid (4 mg, 96% purity, 91% yield). MS (ESI-TOF) m/z: [M+2H+]2+ Calcd for C85H124N14O16S 815.4600; Found 815.4186.
Synthesis of YIIKGVFWDPAC(Far)-OEt (9c)
The a-factor precursor peptide containing the ethyl ester at the C-terminus (8c) (1 equiv, 20 mg, 14 μmol) was used to prepare the farnesylated form 9c using the procedure outlined for 9a. The product was obtained as a white solid (7 mg, 97% purity, 31% yield). MS (ESI-TOF) m/z: [M+2H+]2+ Calcd for C86H126N14O16S 822.4679; Found 822.4301.
Synthesis of YIIKGVFWDPAC(Far)-OiPr (9d)
The a-factor precursor peptide containing the isopropyl ester at the C-terminus (8d) (1 equiv, 28 mg, 19 μmol) was used to prepare the farnesylated form 9d using the procedure outlined for 9a. The product was obtained as a white solid (5 mg, 92 % purity, 15% yield). MS (ESI-TOF) m/z: [M+2H+]2+ Calcd for C87H128N14O16S 829.4757; Found 829.4845.
Synthesis of YIIKGVFWDPAC(Far)-OBn (9e)
The a-factor-OBn precursor peptide containing the benzyl ester at the C-terminus (8e) (1 equiv, 7.15 mg, 3 μmol) was used to prepare the farnesylated form 9e using the procedure outlined for 9a. The product was obtained as a white solid (6.5 mg, 93% purity, 80% yield). ESI-MS: for C91H128N14O16S [M+2H+]2+; calcd 853.48, found 853.44. MS (ESI-TOF) m/z: [M+2H+]2+ Calcd for C91H128N14O16S 853.4780; Found 853.4424.
1H-NMR analysis of peptides 8a and 9a
For NMR analysis, the a-factor precursor peptide (8a) was dissolved in DMSO-d6 to a concentration of 700 μM. The a-factor peptide (9a) was dissolved in DMSO-d6 to a concentration of 2.4 mM. NMR data was acquired on a Bruker Avance 700-MHz spectrometer with a 5-mm TXI cryoprobe, and processed using TopSpin software. 1D 1H spectra were acquired with 64 scans using 1 s recycle delay, 30° pulse, 3.1 s acquisition time with a sweep width of 15 ppm. Data was Fourier transformed with 0.3 Hz line broadening applied and manually phase-corrected. 1H-1H-TOCSY spectra were acquired using 32 scans/increment, spectral widths of 15 ppm and a 60-ms mixing time. 6300 × 256 points were collected in the directly and indirectly detected dimensions, respectively. Data was zero-filled and Fourier-transformed with a shifted sine-bell squared function. 2× linear prediction was applied in the indirect dimension.
Epimerization Studies – Piperidine Treatment of Fmoc-Cys(Trt-resin)-OMe
Resin (2a, 100 mg) containing Fmoc-Cys-OMe attached via its thiol functionality was separated in four batches of 25 mg and treated with 20% piperidine/DMF for 10 min, 2 h, 4h, and 16 h. Then, the resin samples were washed with DMF and CH2Cl2 and dried in vacuum. Next they were each elongated by SPPS to produce samples of the tripeptide Gly-Phe-L-Cys-OMe (6a, see synthesis of 6a described above) that were subsequently analyzed by reversed-phase HPLC to determine the extent of Cys epimerization.
Epimerization Studies – Piperidine Treatment of Fmoc-Gly-Phe-Cys(Trt-resin)-OMe
A batch of resin (4a, 100 mg) prepared via standard SPPS (see synthesis of 6a described above) was separated into four 25 mg portions. The resin samples were treated with 20% piperidine/DMF for 10 min, 2 h, 4h, and 16 h after peptide synthesis. Then, the resins were washed with DMF and CH2Cl2 and dried in vacuum before peptide cleavage.
Growth Arrest Assay
RC757 cells were cultured in YEPD (1% yeast extract, 2% peptone, 2% dextrose) and LM102 cells were cultured in MLT medium to ensure plasmid maintenance.15 Cells were grown overnight at 30 °C with shaking in liquid medium. For use in the growth arrest assay, cells were harvested by centrifugation (1000 × g), washed twice with sterile H2O, and resuspended to a final concentration of 1 × 106 cells/mL in H2O. The cell suspension (1 mL) was combined with 3 mL of Noble agar (1.1% in H2O) and overlaid onto solid medium (YEPD or MLT containing 2% agar). The peptides were dissolved in CH3OH (10 ng/μL) and diluted in 0.5% bovine serum albumin (BSA) to a final concentration of 6.4 ng/μL, then serially diluted in 0.5% BSA to generate solutions of the desired concentrations. 2.5 μL of each dilution was spotted onto the overlay containing RC757 or LM102 cells. The plates were spotted in triplicate, and incubated 17 h at 30 °C. The experiment was repeated in quadruplicate with similar results. The endpoint of the assay was determined to be the lowest concentration at which a clear zone of inhibition, which indicates growth arrest, was observed.
Supplementary Material
Acknowledgments
This research was supported by the National Institute of Health Grants GM008700, GM084152 and GM058842 (M.D.D.), 5T32GM008347-22 (W.H.) and GM22087 (J.M.B.)
Footnotes
1H-NMR spectra, HPLC chromatograms and Mass Spectrometry Data. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Kaspar AA, Reichert JM. Drug Discovery Today. 2013;18:807. doi: 10.1016/j.drudis.2013.05.011. [DOI] [PubMed] [Google Scholar]
- 2.Medina SH, Schneider JP. J Controlled Release. 2015;209:317. doi: 10.1016/j.jconrel.2015.05.267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Grauer A, Koenig B. Eur J Org Chem. 2009:5099. [Google Scholar]
- 4.Gongora-Benitez M, Tulla-Puche J, Albericio F. ACS Comb Sci. 2013;15:217. doi: 10.1021/co300153c. [DOI] [PubMed] [Google Scholar]
- 5.Zablocki JA, Tjoeng FS, Bovy PR, Miyano M, Garland RB, Williams K, Schretzman L, Zupec ME, Rico JG, Lindmark RJ, Toth MV, Mcmackins DE, Adams SP, Panzerknodle SG, Nicholson NS, Taite BB, Salyers AK, King LW, Campion JG, Feigen LP. Biorg Med Chem. 1995;3:539. doi: 10.1016/0968-0896(95)00045-i. [DOI] [PubMed] [Google Scholar]
- 6.Doh HJ, Cho WJ, Yong CS, Choi HG, Kim JS, Lee CH, Kim DD. J Pharm Sci. 2003;92:1008. doi: 10.1002/jps.10353. [DOI] [PubMed] [Google Scholar]
- 7.Meijler MM, Arad-Yellin R, Cabantchik ZI, Shanzer A. J Am Chem Soc. 2002;124:12666. doi: 10.1021/ja027013s. [DOI] [PubMed] [Google Scholar]
- 8.Zhang FL, Casey PJ. Annu Rev Biochem. 1996;65:241. doi: 10.1146/annurev.bi.65.070196.001325. [DOI] [PubMed] [Google Scholar]
- 9.Glomset JA, Gelb MH, Farnsworth CC. Trends Biochem Sci. 1990;15:139. doi: 10.1016/0968-0004(90)90213-u. [DOI] [PubMed] [Google Scholar]
- 10.Turner RA, Weber RJ, Lokey RS. Org Lett. 2010;12:1852. doi: 10.1021/ol100471k. [DOI] [PubMed] [Google Scholar]
- 11.Xue CB, Caldwell GA, Becker JM, Naider F. Biochem Bioph Res Co. 1989;162:253. doi: 10.1016/0006-291x(89)91989-x. [DOI] [PubMed] [Google Scholar]
- 12.Millington CR, Quarrell R, Lowe G. Tetrahedron Lett. 1998;39:7201. [Google Scholar]
- 13.Ludolph B, Eisele F, Waldmann H. J Am Chem Soc. 2002;124:5954. doi: 10.1021/ja025768t. [DOI] [PubMed] [Google Scholar]
- 14.O’Reilly N, Charbin A, Lopez-Serra L, Uhlmann F. Yeast. 2012;29:233. doi: 10.1002/yea.2906. [DOI] [PubMed] [Google Scholar]
- 15.Diaz-Rodriguez V, Mullen DG, Ganusova E, Becker JM, Distefano MD. Org Lett. 2012;14:5648. doi: 10.1021/ol302592v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Barany G, Han YX, Hargittai B, Liu RQ, Varkey JT. Biopolymers. 2003;71:652. doi: 10.1002/bip.10593. [DOI] [PubMed] [Google Scholar]
- 17.Han YX, Albericio F, Barany G. J Org Chem. 1997;62:4307. doi: 10.1021/jo9622744. [DOI] [PubMed] [Google Scholar]
- 18.Huang Z, Derksen DJ, Vederas JC. Org Lett. 2010;12:2282. doi: 10.1021/ol100645t. [DOI] [PubMed] [Google Scholar]
- 19.Michaelis S, Barrowman J. Microbiol Mol Biol Rev. 2012;76:626. doi: 10.1128/MMBR.00010-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Palsuledesai CC, Distefano MD. ACS Chem Biol. 2015;10:51. doi: 10.1021/cb500791f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ochocki JD, Distefano MD. Medchemcomm. 2013;4:476. doi: 10.1039/C2MD20299A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Anderegg RJ, Betz R, Carr SA, Crabb JW, Duntze W. J Biol Chem. 1988;263:18236. [PubMed] [Google Scholar]
- 23.Xie H, Becker JM, Gibbs RA, Naider F. J Pept Res. 2000;55:372. doi: 10.1034/j.1399-3011.2000.00705.x. [DOI] [PubMed] [Google Scholar]
- 24.Khouri O, Sherrill C, Roise D. Biochemistry. 1996;35:14553. doi: 10.1021/bi961594e. [DOI] [PubMed] [Google Scholar]
- 25.Marcus S, Caldwell GA, Miller D, Xue CB, Naider F, Becker JM. Mol Cell Biol. 1991;11:3603. doi: 10.1128/mcb.11.7.3603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chan WC, White PD. In: Fmoc Solid Phase Peptide Synthesis: A Practical Approach. Hames BD, editor. Oxford University Press; Oxford, New York: 2000. p. 41. [Google Scholar]
- 27.Mullen DG, Kyro K, Hauser M, Gustavsson M, Veglia G, Becker JM, Naider F, Distefano MD. Biorg Med Chem. 2011;19:490. doi: 10.1016/j.bmc.2010.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Torres AM, Menz I, Alewood PF, Bansal P, Lahnstein J, Gallagher CH, Kuchel PW. FEBS Lett. 2002;524:172. doi: 10.1016/s0014-5793(02)03050-8. [DOI] [PubMed] [Google Scholar]
- 29.Kumari D, Bandyopadhyay P, Suryaprakash N. J Org Chem. 2013;78:2373. doi: 10.1021/jo3025016. [DOI] [PubMed] [Google Scholar]
- 30.Xue CB, Ewenson A, Becker JM, Naider F. Int J Pept Prot Res. 1990;36 doi: 10.1111/j.1399-3011.1990.tb01295.x. [DOI] [PubMed] [Google Scholar]
- 31.Ewenson A, Marcus S, Becker JM, Naider F. Int J Pept Prot Res. 1990;35 doi: 10.1111/j.1399-3011.1990.tb00944.x. [DOI] [PubMed] [Google Scholar]
- 32.Xue CB, Becker JM, Naider F. Int J Pept Protein Res. 1991;37:476. doi: 10.1111/j.1399-3011.1991.tb00764.x. [DOI] [PubMed] [Google Scholar]
- 33.Molinspiration 2015; Vol. 2015, p Interactive property calculator
- 34.Wollack JW, Zeliadt NA, Mullen DG, Amundson G, Geier S, Falkum S, Wattenberg EV, Barany G, Distefano MD. J Am Chem Soc. 2009;131:7293. doi: 10.1021/ja805174z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rose MW, Xu J, Kale TA, O’Doherty G, Barany G, Distefano MD. Biopolymers. 2005;80:164. doi: 10.1002/bip.20239. [DOI] [PubMed] [Google Scholar]
- 36.Kale TA, Hsieh S-aJ, Rose MW, Distefano MD. Curr Top Med Chem. 2003;3:1043. doi: 10.2174/1568026033452087. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.