

Abstract
Background
Nonsteroidal anti-inflammatory drugs (NSAIDs) are one of the most commonly used classes of medications and have been associated with hepatotoxicity. Studies of liver injury from NSAIDs have been retrospective and prospective data are lacking that provide details of cases.
Aim
To report the presenting feature and outcomes of subjects with severe drug induced liver injury from NSAIDS.
Methods
The U.S. Drug Induced Liver Injury Network is a prospective registry of severe idiosyncratic drug hepatotoxicity. All patients are evaluated in a standard fashion and followed for at least 6 months after onset.
Results
Of 1,221 DILIN cases that were adjudicated, 30 cases were attributed to 8 different NSAIDs. The mean age was 52 years old, 24 (80%) were women, and 21 (70%) were Caucasian. The mean latency to onset of laboratory abnormalities was 67 days. Common signs and symptoms at presentation were nausea (73%), jaundice (67%), and dark urine (67%). Mean peak serum AST, ALT, total bilirubin, and alkaline phosphatase were 898 U/L, 1060 U/L, 12.2 mg/dL, and 326 U/L, respectively. The most common pattern of injury was hepatocellular (70%) and autoantibodies were detected in 33% of cases. Diclofenac, was the most frequently implicated NSAID (16/30 cases), and characterized by hepatocellular injury in all cases. Seventeen cases resulted in hospitalization or prolongation of hospitalization and one patient died from complications of Stevens-Johnson syndrome rather than liver failure due to diclofenac.
Conclusions
Hepatocellular injury is the most common pattern seen with NSAID hepatotoxicity and diclofenac is the most frequently implicated agent. Given the number of available NSAID alternatives, diclofenac use should be limited to patients who fail other NSAIDs and a high level of suspicion for hepatotoxicity should be maintained.
Introduction
Since the 1970s, NSAIDs have been a mainstay among analgesic and anti-inflammatory agents and are among the most widely used classes of medications. More than 70 million prescriptions for NSAIDs are filled in the United States each year including over 11 million prescriptions for celecoxib, the most popular COX-2 inhibitor (www.drugs.com). Additionally, more than 30 billion over-the-counter NSAID tablets are sold annually, largely ibuprofen and naproxen [1]. Investigators have estimated that more than 1% of the U.S. population uses NSAIDs on a daily basis [2].
Although gastrointestinal side effects are common, hepatotoxicity is the leading reason that several drugs in the NSAID class including bromfenac, ibufenac, and benoxaprofen have been withdrawn from the market [3]. In fact, in large case series, NSAIDs have ranked second only to antimicrobials as a cause of DILI [4]. The estimated incidence of liver injury induced by NSAIDs has ranged from 1 to 9 cases per 100,000 persons [5, 6]. Given the enormous prescription and over-the-counter use of these agents, the safety profile of currently approved NSAIDs seems excellent, but systematic prospective studies providing details of NSAID hepatotoxicity are lacking.
The U.S. Drug Induced Liver Injury Network (DILIN) is a multicenter prospective registry designed to identify and carefully assess suspected cases of idiosyncratic drug induced liver injury from medications or herbal and dietary supplements [7]. Data are collected to both further characterize the clinical features of DILI and collect samples for future mechanistic studies. In DILIN, potential competing causes of liver injury are systematically excluded and causality is adjudicated by expert review. The aim of the current study is to report the presenting features and outcomes of all cases of NSAID hepatotoxicity prospectively enrolled in the DILIN database over a 10-year period.
Patients and methods
DILIN began enrolling patients in a prospective registry in September 2004. Details have been previously published [7]. Subjects had to be enrolled within 6 months of onset of DILI and had to meet predefined eligibility criteria: either 1) alanine aminotransferase (ALT) or aspartate aminotransferase (AST) > 5 times the upper limit of normal or alkaline phosphatase > 2 times ULN (or pretreatment baseline if baseline level was abnormal) on 2 consecutive occasions at least 1 day apart; or 2) total serum bilirubin > 2.5 mg/dL along with elevated ALT, AST, or alkaline phosphatase; or 3) international normalized ratio (INR) > 1.5 with elevated ALT, AST, or alkaline phosphatase.
Briefly, after providing written informed consent, subjects provided a medical history and underwent physical examination. Information was collected on dates, doses, and indications for the suspect drug and other concomitant medications and herbal or dietary supplements. Laboratory, imaging, and histologic results were extracted from the medical record. In situations in which key laboratory results were not available, these were obtained at the time of enrollment. Serum, plasma, and DNA samples were collected and stored in a central repository. Patients were asked to return for a six-month follow up evaluation to assess whether recovery was complete. Patients who continued to have abnormalities in liver tests or imaging results were asked to return at 12 and 24 months to fully assess whether chronic liver injury was present. Liver biopsies were not required for enrollment into DILIN, but biopsies done as part of regular medical evaluation and care were retrieved and reviewed by the DILIN hepatopathologist (DEK) in a standardized fashion [7].
The method of causality assessment has been described [7, 8] and was determined by expert consensus of 3 members of the causality committee (clinical site principal investigator and 2 reviewers from other sites) after independent review of a prepared summary of key clinical, laboratory, and diagnostic data from the baseline visit and clinical narrative. Specific information was provided to help exclude hepatitis A, B, and C, extrahepatic biliary obstruction, autoimmune hepatitis, alcoholic liver disease, and ischemic hepatitis. Testing for IgG and IgM antibody to hepatitis E was performed on stored serum samples from the study repository. In cases in which anti-HCV and HCV RNA testing results were not available, stored serum samples were retrieved and tested centrally.
Cases selected for this analysis were those in which an NSAID was implicated and was judged to be the cause of the liver injury. In the formal causality assessment process, the NSAID had to be adjudicated as probable (causality score of 3, 50–75% likelihood), very likely (2, 75–95% likelihood), or definite (1, > 95% likelihood). In cases involving multiple drugs, the NSAID had to be the highest-ranking drug. Additionally, a Roussel Uclaf Causality Assessment Method (RUCAM) score was calculated.
All cases were categorized as either hepatocellular, cholestatic, or “mixed” based upon initial serum ALT and alkaline phosphatase values as defined in the RUCAM system, the R ratio being calculated as the ALT divided by the alkaline phosphatase levels both expressed as an upper limit of normal. R ratios > 5 were considered hepatocellular, < 2 as cholestatic, and between 2 and 5 as mixed. In most analyses, cholestatic and mixed cases were grouped together. Cases were also categorized as having immunoallergic or autoimmune features. Immunoallergic features were scored if there were at least two of the following clinical features: rash, fever, facial edema, lymphadenopathy, or eosinophilia. Autoimmune features were scored if there was either a positive ANA or ASMA of 1:160 or greater.
Standard descriptive statistics were used to summarize the data including means, medians, standard deviations, and percentiles for continuous variables, and frequencies and percentages for categorical variables. Chi-square tests (or Fisher exact test for categories with small frequencies) and nonparametric tests were used to test the difference between the different NSAIDS for categorical and continuous variables. A p < 0.05 was considered statistically significant. All calculations were performed using SAS version 9.2 (Cary, NC).
Results
Patient Population and Presenting Features
Between September 2004 and February 2014, 1,322 cases were enrolled in the DILIN registry, 1,221 of which completed six months of follow up and formal causality assessment by the time of this analysis. In 39 cases (3%), an NSAID was implicated and in 30 instances, the NSAID was judged to be definitely (n = 9), very likely (n = 14), or probably (n = 7) the cause (Figure 1). RUCAM scores on the same 30 cases ranged from 2–12: being considered highly probable (RUCAM scores of 9 to 12) in 8, probable (6 to 8) in 18, possible (3 to 5) in 3, and unlikely (< 2) in one. The NSAIDs implicated included diclofenac (n = 16, 3 with misoprostol), celecoxib (n = 3), meloxicam (n = 3), etodolac (n = 2), oxaprozin (n = 2), ibuprofen (n = 2), sulindac (n = 1), and valdecoxib (n = 1). In nine instances, a second agent was also mentioned as possibly contributory, including amlodipine, pregabalin, metformin, tizanidine, doxycycline, citalopram, ranitidine, beta interferon, and ibuprofen.
FIGURE 1.
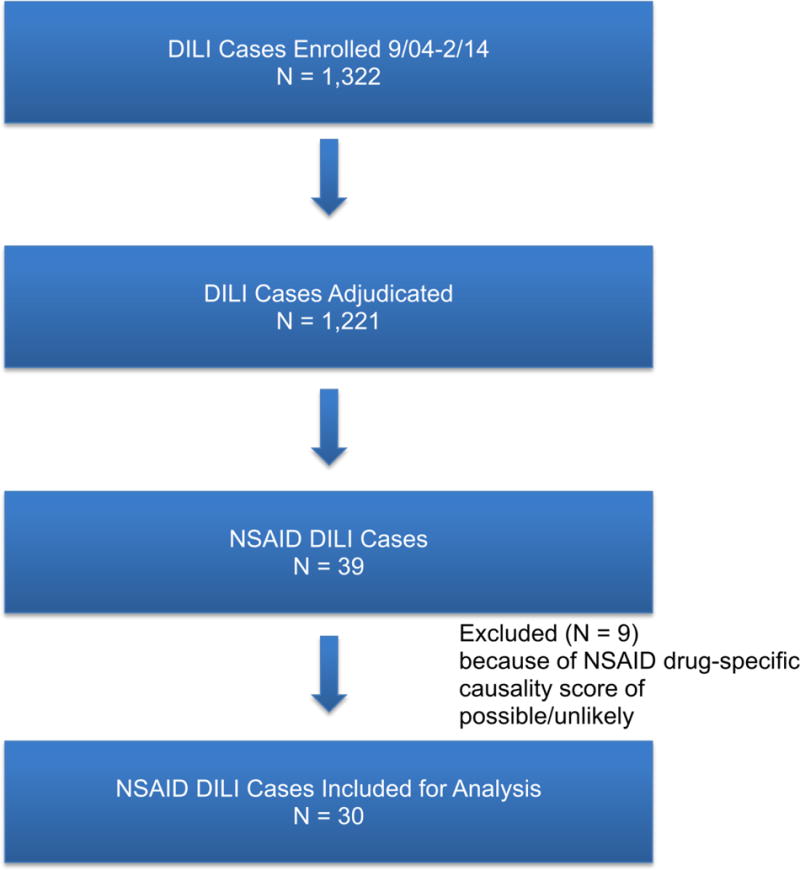
SELECTION OF NSAID LIVER INJURY CASES
Clinical features of the 30 cases are shown in Table 1. The mean age was 52 years and all were adults (age range 22–83 years); most were female (80%) and non-Hispanic whites (70%) (Table 1). The most common indication for NSAID prescriptions (19 cases) was chronic osteoarthritis and 15 patients gave a history of previous use of another NSAID (usually ibuprofen or naproxen). Diabetes mellitus was present in 10 cases (33%). One subject had pre-existing nonalcoholic fatty liver disease while another had chronic hepatitis C.
Table 1.
Demographic Data
| Mean age (range) in years | 52 (22–83) |
|
| |
| Female sex: n (%) | 24 (80%) |
|
| |
| Race and Ethnicity: n (%) | |
| Non-Hispanic White | 21 (70%) |
| African-American | 5 (17%) |
| Other | 3 (13%) |
| Hispanic/Latino | 1 (3%) |
|
| |
| Mean BMI (range) in kg/m2 | 30.7 (21.3–46.1) |
The most common symptoms at presentation were nausea (73%), dark urine (67%), and jaundice (67%). The time to onset ranged widely from 6 to 247 days (to first laboratory abnormality), but was less than 30 days in 17 subjects and was longer than 90 days in 9 (30%); all of them were receiving diclofenac or etodolac. Initial mean (and range) of liver test results were bilirubin 6.9 (0.3 to 33.4) mg/dL, ALT 946 (29–4640) U/L, AST 733 (45–2880) U/L, and alkaline phosphatase 240 (79–901) U/L. Peak bilirubin values averaged 12.2 mg/dL (range 0.4–38.3 mg/dL) and 20 (67%) were above 2.5 mg/dL, the criterion used to define icteric cases. INR values rose to 1.5 or above in 9 patients, 8 of whom received diclofenac. The severity of injury was scored as mild (bilirubin < 2.5 mg/dL) in 10 patients (33%), moderate in 11 (37%), and severe or fatal in 9 (30%). The only fatal case developed Stevens Johnson syndrome after receiving diclofenac, but died largely due to complications of the skin involvement and was recovering from the hepatitis at the time.
Clinical Phenotypes of Liver Injury
Not only the time to onset, but also the pattern of serum enzyme elevation (cholestatic, hepatocellular, or mixed) and severity of injury varied, but to some extent, fell into specific phenotypes based upon the implicated NSAID. The major clinical features for the five NSAIDs implicated in at least 2 cases are shown in Table 3 and Figure 2. A short description of the course and outcome of 6 cases is given in the Supplementary Appendix. In addition, some cases are described individually on the LiverTox website (http//livertox.nih.gov: in the subsection for each drug “Case Reports Submitted to LiverTox”).
Table 3.
Comparison of NSAID Cases by Implicated Agent
| Feature | Diclofenac | Celecoxib | Meloxicam | Etodolac | Oxaprozin |
|---|---|---|---|---|---|
|
| |||||
| N | 16 | 3 | 3 | 2 | 2 |
|
| |||||
| Causality: | 1.9 | 2.0 | 2.3 | 2.0 | 1.5 |
| Definite | 4 | 1 | 0 | 1 | 1 |
| Very Likely | 9 | 1 | 2 | 0 | 1 |
| Probable | 3 | 1 | 1 | 1 | 0 |
|
| |||||
| Severity | 2.8 | 3.0 | 3 | 2 | 1 |
| Mild [1] | 6 | 0 | 0 | 0 | 2 |
| Mod [2,3] | 2 | 3 | 3 | 2 | 0 |
| Severe [4,5] | 8 | 0 | 0 | 0 | 0 |
|
| |||||
| Fatality | 6% | 0% | 0% | 0% | 0% |
|
| |||||
| Duration Rx | 90.1 | 58.7 | 11.0 | 211.5 | 18.0 |
|
| |||||
| Immunoallergic | 25% | 0% | 33% | 0% | 100% |
|
| |||||
| Autoimmune | 38% | 0% | 0% | 0% | 0% |
|
| |||||
| Prednisone | 38% | 33% | 33% | 0% | 50% |
|
| |||||
| ALT (initial) | 1386 | 656 | 360 | 492 | 478 |
|
| |||||
| Alk P (initial) | 182 | 319 | 901 | 235 | 228 |
|
| |||||
| Bilirubin (initial) | 8.0 | 7.4 | 12.3 | 3.9 | 0.7 |
|
| |||||
| Bilirubin (peak) | 14.8 | 11.1 | 12.3 | 4.0 | 1.4 |
|
| |||||
| INR (peak) | 1.5 | 1.1 | 1.1 | 1.1 | 1.2 |
|
| |||||
| Symptoms | 94% | 100% | 100% | 100% | 100% |
|
| |||||
| Jaundice | 69% | 100% | 100% | 100% | 50% |
|
| |||||
| Itching | 38% | 33% | 0% | 50% | 100% |
|
| |||||
| Rash | 31% | 0% | 33% | 50% | 100% |
|
| |||||
| Fever | 25% | 0% | 33% | 0% | 100% |
|
| |||||
| ANA | 31% | 0% | 0% | 0% | 50% |
|
| |||||
| ASMA | 25% | 0% | 0% | 0% | 0% |
FIGURE 2.
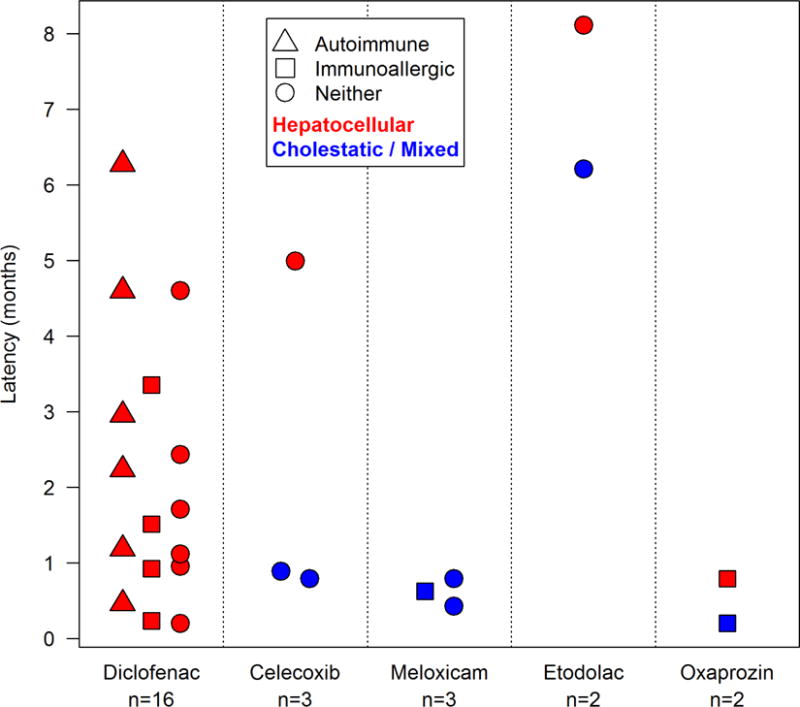
PATTERN OF INJURY AND LATENCY BY NSAID
Diclofenac was the most commonly implicated NSAID (16 cases, 53%). All cases were hepatocellular with marked elevations in mean initial serum ALT (mean 1508 ± 1171 U/L) and AST (mean 1284 ± 822 U/L) with modest increases in alkaline phosphatase (mean 209 ± 63 U/L). The latency to onset varied considerably from quite short (6 days) to long (191 days). Immunoallergic features (fever, rash, facial edema, eosinophilia) occurred in 4 subjects and autoimmune features in 6 (38%), including 3 who were treated with prednisone; in all three, however, corticosteroid was eventually stopped without recurrence of liver injury.
In addition, cases of diclofenac hepatotoxicity were often severe; 8 of the 16 cases developed coagulopathy (INR ≥ 1.5) with jaundice. As mentioned above, only one patient died, and the cause of death was considered septicemia and complications of Stevens Johnson syndrome rather than hepatic failure. Six patients had serum enzyme elevations without jaundice, including two who presented with a clinical picture resembling chronic hepatitis. In both patients, diclofenac had been continued despite identification of serum aminotransferase elevations, and the abnormalities did not improve until diclofenac was stopped. None of the 16 patients developed evidence of chronic liver injury that persisted after diclofenac was stopped; a single patient with an abnormal alkaline phosphatase at 6 months had normal values at 12 and 24 months after onset. Three cases of diclofenac liver injury are included in the supplemental material, two with severe, acute icteric hepatitis (one short and one long latency) and one with an anicteric, chronic hepatitis-like presentation.
Etodolac was implicated in two cases of DILI, both with a long latency (166 and 216 days), a mixed and hepatocellular pattern of serum enzyme elevations (R values 2.8 and 19.3), moderate severity in that both were jaundiced, but peak bilirubin levels were 3.8 and 4.0 mg/dL and neither patient was hospitalized or had INR elevations. Thus, etodolac cases were similar to but perhaps milder than those associated with diclofenac, another proprionic acid derivative. Neither case had immunoallergic or autoimmune features.
Celecoxib was implicated in three cases, two with short latency (16 and 23 days), usually with a mixed pattern of liver enzyme elevations and without immunoallergic features. One patient developed a severe cholestatic hepatitis and ultimately recovered clinically, but had continued evidence of cholestatic injury 6, 12, and 24 months after onset, compatible with some degree of vanishing bile duct syndrome.
Meloxicam was implicated in three cases, all 3 with a short latency (13–24 days) and cholestatic pattern of liver enzymes (R values 1.1–1.5) along with jaundice and pruritus. One case had mild immunoallergic features. One case appeared to have persistent serum alkaline phosphatase elevations, but all values ultimately returned to normal when she was seen at 12 and 24 months after onset.
Oxaprozin was implicated in only two cases, but both were similar in having a short latency (6 and 24 days), prominent ALT elevations (713 and 775 U/L), immunoallergic features (fever and rash), and a mild, self-limited course.
The other 4 cases (2 from ibuprofen and 1 each from sulindac and valdecoxib) had variable courses, latencies, and severity and no clear-cut predominance of immunoallergic or autoimmune features. The two cases of ibuprofen induced liver injury were both considered “probable” and both occurred in patients with suspected pre-existing liver disease (one with both hepatitis C and alcoholic liver disease and one with autoimmune hepatitis).
Hepatitis E testing was done on all but 2 cases. Four samples were positive for IgG anti-HEV, but none of the four were positive for IgM anti-HEV.
Liver Histology
Ten cases had liver biopsies available for central review (Table 2). An acute hepatitis with a hepatocellular pattern of injury was identified in 5 cases, 3 of which were due to diclofenac and 2 to oxaprozin. The remaining 5 cases showed cholestatic hepatitis and one case each was attributed to celecoxib, ibuprofen, meloxicam, diclofenac, and valdecoxib. None of the cases showed histologic changes indicative of autoimmune hepatitis.
Table 2.
DILI Cases with Histology for Central Review
| Drug Name | Case Number | Biopsy Findings | DILI Onset to Biopsy (days) |
|---|---|---|---|
| celecoxib | 2 | cholestatic hepatitis | 7 |
| diclofenac-misoprostol | 5 | mild chronic hepatitis, eosinophils | −200* |
| diclofenac | 12 | zone 3 necrosis with hepatitis, plasma cells, eosinophils | 23 |
| diclofenac | 14 | acute cholestatic hepatitis, eosinophils, plasma cells | 7 |
| diclofenac | 17 | acute hepatitis, zone 3 necrosis, eosinophils | 7 |
| ibuprofen | 23 | cholestatic hepatitis, plasma cells, eosinophils | 12 |
| meloxicam | 25 | cholestatic hepatitis, eosinophils | 0 |
| oxaprozin | 27 | acute hepatitis, zone 3 necrosis, plasma cells, eosinophils | 12 |
| oxaprozin | 28 | zone 3 necrosis with hepatitis, eosinophils, plasma cells | −6* |
| valdecoxib | 30 | cholestatic hepatitis, mild zone 3 necrosis | 43 |
The minus sign denotes that the biopsy was performed prior to the recognized DILI onset (presumably liver tests were abnormal at the time of the biopsy).
Discussion
Since NSAIDs are readily available over the counter or are commonly prescribed, details about presentation and recovery from drug induced liver injury caused by them should be useful for clinicians. In the U.S., 6% of adults report using NSAIDs in a month and 24% of this group used nonprescription ibuprofen [9]. Although other studies have reported hepatotoxicity from NSAIDS, details on dosing, duration of therapy, and time to recovery are frequently lacking, as is the systematic exclusion of other liver diseases [10–12].
Studies from other countries are consistent with results from DILIN and hepatotoxicity from NSAIDS accounts for a substantial number of cases in DILI registries from Iceland, Sweden, and Spain [10–12]. Furthermore, similar to our findings, diclofenac is the most common NSAID identified causing liver injury. A population based study from Iceland reported that diclofenac was the only NSAID implicated in drug induced liver injury with an estimated incidence of 11 per 100,000 [10]. DILIN is not a population-based study, however in 2007 approximately 7 million prescriptions were written for diclofenac-misoprostol (Arthrotec®) or generic diclofenac compared to 24 million prescriptions for generic ibuprofen, suggesting that in the United States the incidence of hepatotoxicity from diclofenac is much higher than ibuprofen. (www.drugtopics.modernmedicine.com).
A striking finding of our study is the wide spectrum of liver injury associated with NSAIDs as a class. Diclofenac presented only as hepatocellular injury with or without autoimmune or immunoallergic features. Latency was much shorter for those presenting with an immunoallergic phenotype compared to the other diclofenac phenotypes, which had a variable latency. In contrast, the three cases of meloxicam were all cholestatic/mixed and had a relative short latency of less than a month. The two etolodac cases presented with a long latency while the two oxaprozin cases presented with immunoallergic features and short latency. There were only two ibuprofen cases; one presented with hepatocellular injury and the other with a bland cholestatic injury. Thus, as a class NSAIDS have a wide spectrum of liver injury, but each individual NSAID seems to have its own signature phenotype.
Diclofenac was approved for use in the United States in 1988 for the treatment of osteoarthritis, rheumatoid arthritis, and ankylosing spondylitis. It was recognized as causing elevations in aminotransferases. In a randomized trial of 17,289 subjects, aminotransferases more than three times the upper limit of normal were seen in 3.1% of subjects, mostly within the first 6 months of therapy [13]. In the current series, the latency was variable and half of the diclofenac subjects had severe injury requiring hospitalization. Serious liver injury from diclofenac has been reported to occur in 6.3 per 100,000 diclofenac users [14]. Therefore, clinicians should consider alternative agents to diclofenac, but if it is prescribed and liver tests are elevated, it should be discontinued.
Of note, some common NSAIDs were not reported in this study including naproxen, ketoprofen, indomethacin, and nabumetone. This is likely because some of these agents are now rarely used (e.g. indomethacin, nabumetone). However, others are commonly used and can be found over-the-counter. Ibuprofen was implicated in two cases that were only rated as probable. One case was a rechallenge with ibuprofen in a patient who previously had diclofenac injury; this may be a case of autoimmune hepatitis. In the other ibuprofen case, the subject had a history of hepatitis C and alcohol use. Given the widespread use of over the counter and prescription ibuprofen, it seems reasonable to conclude that liver toxicity from ibuprofen is an exceedingly rare event. Nevertheless, the safety of NSAIDs in patients with established liver disease deserves to be prospectively and more critically assessed.
The mechanism of liver injury from NSAIDs is not well understood and it is been proposed that acidic moiety of NSAIDs or reactive adducts of NSAID metabolites may bind to host proteins and cause cellular injury in susceptible individuals [15, 16]. Aithal and Day proposed a multistep theory for diclofenac-induced liver and the liver injury was dose dependent and seen mostly at doses of 150 mg or higher [17]. Our results support this contention because daily doses of 150 mg or higher were reported in 12 of 16 diclofenac cases in our series. Although polymorphisms in UGT2B7, CYP2C8 and ABCC2 have been associated with diclofenac hepatotoxicity, this association has not been consistently demonstrated [18, 19].
In summary, NSAIDs are commonly used and effective non-narcotic pain medications that are rarely, but consistently reported to cause hepatotoxicity. Diclofenac is the most frequent offender and presents with a hepatocellular injury (often severe) with or without autoimmune or immunoallergic features. Given the plethora of NSAIDs available, it seems reasonable to avoid prescribing diclofenac or consider it a second or third line agent.
Supplementary Material
Key Points.
-
✓
NSAIDs are one of the most commonly used classes of medications and have been associated with hepatotoxicity.
-
✓
Hepatocellular injury is the common pattern seen with NSAID hepatotoxicity
-
✓
Diclofenac is the most frequently implicated agent.
-
✓
Diclofenac use should be limited to patients who fail other NSAIDs and a high level of suspicion for hepatotoxicity should be maintained.
Acknowledgments
Study Support: NIDDK, NIH. See DILIN website https://dilin.dcri.duke.edu/publications-1 for a complete listing of funding sources, sites, investigators, co-investigators, coordinators, and staff.
List of abbreviations
- NSAIDs
nonsteroidal anti-inflammatory drugs
- DILIN
Drug Induced Liver Injury Network
- AST
aspartate aminotransferase
- ALT
alanine aminotransferase
- U/L
units per liter
- COX-2
cyclooxygenase-2
- DILI
drug induced liver injury
- ULN
upper limit of normal
- mg/dL
milligrams per deciliter
- INR
international normalized ratio
- DNA
deoxyribonucleic acid
- IgG
immunoglobulin G
- IgM
immunoglobulin M
- Anti-HCV
antibody to hepatitis C virus
- HCV RNA
hepatitis C virus ribonucleic acid
- RUCAM
Roussel Uclaf Causality Assessment Method
- ANA
antinuclear antibody
- ASMA
antismooth muscle antibody
Footnotes
Conflicts of interest: None
References
- 1.Wolfe M, Lichtenstein D, Singh G. Gastrointestinal toxicity of nonsteroidal anti-inflammatory drugs. N Engl J Med. 1999;340:1888–1899. doi: 10.1056/NEJM199906173402407. [DOI] [PubMed] [Google Scholar]
- 2.Lichtenstein D, Sygal S, Wolfe M. Nonsteroidal anti-inflammatory drugs and the gastrointestinal tract: the double-edged sword. Arthritis Rheum. 1995;38:5–18. doi: 10.1002/art.1780380103. [DOI] [PubMed] [Google Scholar]
- 3.Goldkind L, Laine L. A systematic review of NSAIDs withdrawn from the market due to hepatotoxicity: lessons learned from the bromfenac experience. Pharmacoepidemiol Drug Saf. 2006;15(4):213–220. doi: 10.1002/pds.1207. [DOI] [PubMed] [Google Scholar]
- 4.Bjornsson E. Review article: drug-induced liver injury in clinical practice. Aliment Pharmacol Ther. 2010;32:3–13. doi: 10.1111/j.1365-2036.2010.04320.x. [DOI] [PubMed] [Google Scholar]
- 5.Rostom A, Goldkind L, Laine L. Nonsteroidal anti-inflammatory drugs and hepatic toxicity: a systematic review of randomized controlled trials in arthritis patients. Clin Gastroenterol Hepatol. 2005;3:489–498. doi: 10.1016/s1542-3565(04)00777-3. [DOI] [PubMed] [Google Scholar]
- 6.Garcia Rodriguez L, Perez Gutthann S, Walker AM, Lueck L. The role of non-steroidal anti-inflammatory drugs in acute liver injury. BMJ. 1992;305:865–868. doi: 10.1136/bmj.305.6858.865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fontana RJ, Watkins PB, Bonkovsky HL, Chalasani N, Davern T, Serrano J, et al. Drug-Induced Liver Injury Network (DILIN) prospective study: rationale, design and conduct. Drug Saf. 2009;32:55–68. doi: 10.2165/00002018-200932010-00005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chalasani N, Fontana RJ, Bonkovsky HL, Watkins PB, Davern T, Serrano J, et al. Causes, clinical features, and outcomes from a prospective study of drug-induced liver injury in the United States. Gastroenterology. 2008;135:1924–1934. 1934.e1–e4. doi: 10.1053/j.gastro.2008.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Paulose-Ram R, Hirsch R, Dillon C, Gu Q. Prescription and non-prescription analgesic use among the U.S. adult population: results from the third national health and nutritional examination survey (NHANES III) Pharmacoepidemiol Drug Saf. 2003;12:315–326. doi: 10.1002/pds.755. [DOI] [PubMed] [Google Scholar]
- 10.Bjornsson ES, Bergmann OM, Bjornsson HK, Kvaran RB, Olafsson S. Incidence, presentation, and outcomes in patients with drug-induced liver injury in the general population of Iceland. Gastroenterology. 2013;144:1419–1425. doi: 10.1053/j.gastro.2013.02.006. [DOI] [PubMed] [Google Scholar]
- 11.Andrade RJ, Lucena MI, Fernandez MC, Pelaez G, Pachkoria A, Garcia-Ruiz E. Drug-Induced Liver Injury: an analysis of 461 incidences submitted to the Spanish registry over a 10-year period. Gastroenterology. 2005;129:512–521. doi: 10.1053/j.gastro.2005.05.006. [DOI] [PubMed] [Google Scholar]
- 12.Bjornsson E, Jerlstad P, Bergqvist A, Olsson R. Fulminant drug-induced hepatic failure leading to death or liver transplantation in Sweden. Scand J Gastroenterol. 2005;40:1095–1101. doi: 10.1080/00365520510023846. [DOI] [PubMed] [Google Scholar]
- 13.Laine L, Goldkind L, Curtis SP, Connors LG, Yanqiong Z, Cannon CP. How common is diclofenac-associated liver injury? Analysis of 17,289 arthritis patients in a long-term prospective clinical trial. AJG. 2009;104:356–362. doi: 10.1038/ajg.2008.149. [DOI] [PubMed] [Google Scholar]
- 14.deAbajo FJ, Montero D, Madurga M, Garcia Rodriguez LA. Acute and clinically relevant drug-induced liver injury: a population based case-control study. Br J Clin Pharmacol. 2004;58:71–80. doi: 10.1111/j.1365-2125.2004.02133.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bessone F. Nonsteroidal anti-inflammatory drugs: what is the actual risk of liver damage. World J Gastroenterol. 2010;16(45):5651–5661. doi: 10.3748/wjg.v16.i45.5651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kishida T, Onozato T, Kanazawa T, Tanaka S, Kuroda J. Increase in covalent binding of 5-hydroxydiclofenac to hepatic tissues in rats co-treated with lipopolysaccharide and diclofenac: involvement in the onset of diclofenac-induced idiosyncratic hepatotoxicity. J Toxicol Sci. 2012;37(6):1143–1156. doi: 10.2131/jts.37.1143. [DOI] [PubMed] [Google Scholar]
- 17.Aithal G, Day C. Nonsteroidal anti-inflammatory drug-induced hepatotoxicity. Clin Liver Dis. 2007;11:563–575. doi: 10.1016/j.cld.2007.06.004. [DOI] [PubMed] [Google Scholar]
- 18.Daly AK, Aithal GP, Leathart JB, Swainsbury RA, Dang TS, Day CP. Genetic susceptibility to diclofenac-induced hepatotoxicity: contribution of UGT2B7, CYP2C8, and ABCC2 genotypes. Gastroenterology. 2007;132:272–281. doi: 10.1053/j.gastro.2006.11.023. [DOI] [PubMed] [Google Scholar]
- 19.Urban TJ, Shen Y, Stolz A, Chalasani N, Fontana RJ, Rochon J. Limited contribution of common genetic variants to risk for liver injury due to a variety of drugs. Pharmacogenet Genomics. 2012;22(11):784–795. doi: 10.1097/FPC.0b013e3283589a76. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.