

Anomalous signal from sulfur atoms present in most proteins was used for de novo phasing of XFEL data and solving a GPCR structure.
Keywords: Crystallography, x-ray free electron laser, serial femtosecond crystallography, SAD, sulfur, GPCR, lipidic cubic phase, native phasing, de novo structure, protein
Abstract
Serial femtosecond crystallography (SFX) takes advantage of extremely bright and ultrashort pulses produced by x-ray free-electron lasers (XFELs), allowing for the collection of high-resolution diffraction intensities from micrometer-sized crystals at room temperature with minimal radiation damage, using the principle of “diffraction-before-destruction.” However, de novo structure factor phase determination using XFELs has been difficult so far. We demonstrate the ability to solve the crystallographic phase problem for SFX data collected with an XFEL using the anomalous signal from native sulfur atoms, leading to a bias-free room temperature structure of the human A2A adenosine receptor at 1.9 Å resolution. The advancement was made possible by recent improvements in SFX data analysis and the design of injectors and delivery media for streaming hydrated microcrystals. This general method should accelerate structural studies of novel difficult-to-crystallize macromolecules and their complexes.
INTRODUCTION
The recent development of x-ray free-electron lasers (XFELs) is in the process of transforming macromolecular crystallography for both structural analysis (1) and time-resolved molecular imaging (2). Extremely bright and ultrashort x-ray pulses enable high-resolution data collection from micrometer-sized crystals at room temperature with minimal radiation damage. Because each crystal is destroyed by a powerful XFEL pulse, crystallographic data sets are typically collected using the serial femtosecond crystallography (SFX) approach, in which microcrystals are delivered to the intersection with the pulsed XFEL beam in a continuous hydrated stream (3). A fast detector operating at the XFEL pulse repetition rate collects diffraction images from microcrystals intersecting the beam at random orientations. After identification of diffraction peaks and indexing, structure factor amplitudes are determined by averaging reflection intensities measured in each diffraction pattern over stochastic variables, such as the microcrystal size and the orientation of individual crystals (4).
Recently developed enhancements to the basic Monte Carlo integration method enable more accurate data to be obtained from fewer diffraction patterns (5, 6). Meanwhile, the development of viscous crystal delivery media and special injectors (7–10) has allowed a marked reduction in crystal consumption for both membrane and soluble proteins. One of the most successful approaches to date involves the application of the gel-like lipidic cubic phase (LCP) for growth and delivery of microcrystals across the pulsed x-ray beam (11–13). The use of microcrystal injectors bypasses the need for mounting the crystals, whereas femtosecond-duration x-ray pulses from the FEL obviate cryocooling and extensive crystal optimization for challenging membrane proteins and their complexes with soluble partners. Combined with the improvements in data processing, this enables the determination of accurate reflection intensities from much smaller amounts of protein than previously possible.
Most protein structures obtained by SFX so far have been determined using the molecular replacement (MR) method for solving the crystallographic phase problem. However, experimental phasing of XFEL data without using previous models is difficult because all existing approaches require a very high accuracy of structure factor amplitude measurements, as compared to MR. The first successful experimental phasing of SFX data was demonstrated with lysozyme crystals using single-wavelength anomalous dispersion (SAD) on gadolinium, which provides a very strong anomalous signal (14). Recent attempts to use SAD phasing of SFX data from another test soluble protein, luciferin-regenerating enzyme, with a more conventional mercury compound were initially unsuccessful, but the collection of additional data on native crystals and the use of the SIRAS (single isomorphous replacement with anomalous signal) method made it possible to solve the structure (15). Both of these methods rely on the incorporation of heavy atoms into protein crystals, which requires extensive screening of various compounds, many of which are toxic or suffer from poor solubility. Quite often, efficient incorporation is not attainable, and using native elements for phasing is therefore preferable. For example, native copper ions were used to phase SFX data for copper-nitride reductase (16); however, copper or other heavy atoms are not widespread in biological macromolecules.
A more general phasing method that uses anomalous signal from native sulfur atoms, which are ubiquitous in most proteins, was introduced more than 30 years ago (17) but has not been commonly used for a long time because of the extremely low level of the anomalous signal and the challenges associated with data collection and processing. During the past decade, the method was revisited and started gaining popularity because of the advances in synchrotron radiation sources and in the data processing software (18, 19). However, the conventional wisdom was that, for this method to be successful, one needs a nearly perfect, large crystal that diffracts to high resolution and withstands radiation damage. A few years ago, it was shown that sulfur SAD phasing could be achieved by averaging data from multiple weakly diffracting small crystals (20), and an optimized method for routine sulfur SAD phasing at synchrotron beamlines was published (21). Recently, successful reports of sulfur SAD phasing of SFX data for test soluble proteins, lysozyme (22) and thaumatin (23), have been published. Phasing SFX data from weakly diffracting crystals of membrane proteins and complexes represents a next level of difficulty. Here, we demonstrate the ability to automatically solve a macromolecular structure by native sulfur SAD using SFX data collected at room temperature from micrometer-sized crystals of the human A2A adenosine receptor (A2AAR), which belongs to the pharmaceutically important but difficult-to-crystallize superfamily of G protein–coupled receptors (GPCRs).
RESULTS
Anomalous SFX data were collected at the Coherent X-Ray Imaging (CXI) end station (24) of the Linac Coherent Light Source (LCLS), as previously described (11). Microcrystals (average size, 5 × 5 × 2 μm3; fig. S1) of the A2AAR with apocytochrome b562RIL (BRIL) fused into its third intracellular loop (A2AAR-BRIL) (25) in complex with the antagonist ZM241385 were grown (26) and delivered inside LCP using a viscous medium microinjector (7). An x-ray energy of 6 keV (wavelength, 2.07 Å) was used as a compromise between the strength of anomalous scattering from sulfur atoms (K-edge, 2.472 keV), the efficiencies of the Kirkpatrick-Baez mirrors and of the detector, as well as the detector-size and wavelength limits on resolution. The sample was injected within a vacuum chamber to minimize background scattering, and the XFEL beam was attenuated to ~14% of its full power (~170 μJ per pulse) to prevent oversaturation of the Cornell-SLAC (Stanford Linear Accelerator Center) pixel array detector (CSPAD). At this x-ray energy, the anomalous difference in structure factors is expected to be less than 1.5% (17), requiring a very high precision of collected data and therefore a very high multiplicity (many measurements of each reflection). Within ~17 hours, a total of 7,324,430 images were collected at 120 images/s, in which 1,797,503 crystal diffraction patterns were identified using the Cheetah hit finding software (27). We successfully indexed 593,996 of these hits using the CrystFEL software package (28). The final reflection list was obtained by merging data from 578,620 indexed patterns using iterative scaling and resulted in a data set at 2.5 Å resolution. This resolution was limited by the x-ray energy, detector size, and minimal achievable sample-to-detector distance. To further extend resolution, we collected additional data at an x-ray energy of 9.8 keV (wavelength, 1.27 Å). This high-resolution data set was assembled from 72,735 indexed patterns and was truncated at 1.9 Å resolution on the basis of the correlation coefficient (CC*) >0.5 criterion (table S1).
The structure was solved with a two-stage sulfur SAD phasing procedure using the A2A_S-SAD anomalous data set collected at the x-ray energy of 6 keV. In the first stage, SHELXD (19) was used to determine the sulfur atom substructure. The A2AR-BRIL construct contains 24 sulfurs (15 Cys and 9 Met) per 447 residues (Cys+Met residue content, ~5.4%), including 8 sulfurs engaged in four disulfide bonds. The resolution cutoff was the most critical parameter for the successful sulfur atom search, with optimal results obtained at 3.5 Å resolution (fig. S2A). A scatter plot of SHELXD correlation coefficients between the observed and calculated E values (CCall/CCweak; fig. S2B) showed the main cluster of random solutions with a few strong ones, which were well separated from the main cluster. A sharp drop in the occupancy of sulfur atoms from 0.72 to 0.42 (fig. S2C) was used as an indicator for distinguishing 16 correct sulfur atoms (Fig. 1) from five incorrect ones in the found solution, with CCall/CCweak = 32/12. In the second stage, the partial sulfur substructure solution found with SHELXD was used as an input for substructure refinement, log-likelihood gradient map substructure completion, and phasing with Phaser EP (29). The resulting phases were improved by density modification and phase extension using Resolve (30). Although density modification clearly improved the maps, it was not used at its full potential because of the absence of noncrystallographic symmetry and a relatively low solvent content (53%). The model was traced automatically with phenix.autobuild (31) to 59% completeness. The resulting model contained all eight receptor helices, with only the BRIL fusion and some loops missing. The electron density maps at the different stages of the phasing process are shown in Fig. 2. Density for the ligand ZM241385 became clear after automatic tracing (fig. 2C), validating the correct structure solution. Subsequently, we tried running the same diffraction data through an automated structure-determination pipeline, X2DF, which explores a wide range of settings (32). Several additional combinations of different parameters were found to yield structure solutions (fig. S3). The success in the phasing of SFX S-SAD data has been made possible by recent advances in data processing software. In particular, the new scaling algorithm introduced in CrystFEL version 0.6.1 appeared to be critical for improving data quality and solving the structure. Partiality correction was not necessary to achieve this result, and in fact appeared to slightly decrease the data quality (fig. S4), and did not lead to structure determination.
Fig. 1. Sulfur peaks in the anomalous difference A2AAR Fourier map.
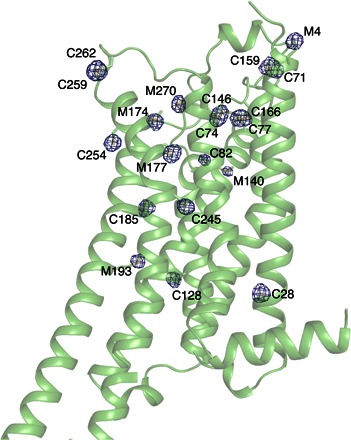
Sulfur density is contoured at 3 σ and overlaid on the A2AAR crystal structure. Twenty sulfur atoms could be identified from the map. BRIL fusion moiety containing one ordered sulfur atom (M1033) is not shown. Three sulfurs (M-24, C-13, and M1058) are disordered and do not have electron density.
Fig. 2. Improvements in electron density at different stages of the phasing process.
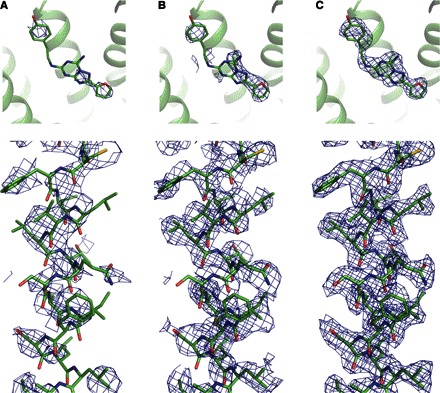
(A) Phaser EP map. (B) Resolve density modified map. (C) Autobuild autotraced map. Omit electron density around the ligand is shown on the top panels. 2mFo-DFc electron density map for helix III is shown on the bottom panels. All maps are contoured at 1.0 σ.
The final data set used for the structure solution contained 578,620 indexed patterns. To find the minimum number of indexed patterns necessary for successful structure determination, we performed phasing, density modification, and autotracing using data ranging from 100,000 to 550,000 indexed patterns in 50,000 pattern increments (fig. S5). All phasing attempts with a number of patterns lower than 500,000 have failed, even when the correct sulfur atom positions were used, highlighting the requirement of collecting data with high multiplicity for accurate calculation of anomalous differences from weak anomalous sulfur scattering. With 550,000 indexed patterns, structure determination was straightforward, and autotracing yielded a 46% complete model with 19 cycles in phenix.autobuild. However, with 500,000 indexed patterns, autotracing produced a 36% complete model with 19 cycles and required twice as many cycles (38 cycles) to produce a similar model with all receptor helices (53% complete) as the one obtained from 550,000 patterns.
Reducing the number of indexed patterns also had a negative impact on the initial sulfur atom search using SHELXD. With 550,000 patterns, 15 sulfur atoms were found with the same parameters as for the full data set. With 500,000 and 450,000 patterns, 14 sulfur atoms were found, but this required optimization of the search parameters in SHELXD (such as the resolution cutoff) and increase in the number of trials from 1000 to 5000. With 400,000 patterns, only six sulfur atoms were found after an extensive search, and with 300,000 patterns and lower, the substructure could not be found.
Because the sulfur content in proteins varies, to analyze the effect of the number of sulfur atoms in the substructure on the success of phasing, we consecutively removed the 10 weakest sulfur atoms from the complete 21-atom substructure and performed phasing, density modification, and autotracing using the complete data set. With nine sulfur atoms removed, the phasing was successful with a figure of merit (FOM) of 0.371 and with 45% of the structure built automatically. However, the removal of 10 sulfur atoms carried a negative effect, and no model was produced with 36 cycles of autotracing. Therefore, in this case, it was possible to solve the structure starting from as low as 12 sulfur atoms per 447 residues (2.7%).
After solving and refining the structure at 2.5 Å with the A2A_S-SAD data set, the resolution was extended to 1.9 Å using the additional A2A_High-Res data set collected at 9.8 keV. The final, 1.9 Å room temperature A2AAR-BRIL structure (A2A_S-SAD_1.9) contains 396 of 447 residues (excluding disordered N- and C-terminal tags and one BRIL loop 1045–1055), the ligand ZM241385, 3 cholesterols, 21 lipids, 1 sodium ion, 1 polyethylene glycol (PEG), and 105 water molecules (table S2 and fig. S6). Alternatively, we solved the structure using MR with the A2A_High-Res data set truncated at 1.9 and 2.5 Å. Comparison of the A2A_S-SAD_2.5 and A2A_MR_2.5 structures (fig. S7) showed minimal differences as expected, with a root mean square deviation (RMSD) of 0.43 Å for Cα atoms of all resolved residues in the protein (0.60 Å for all atoms). Discrepancies were mostly observed in the solvent-exposed regions on the protein surface, where side chains of bulky residues adopted alternative conformations and could not be unambiguously modeled. Also, the B factor distribution showed no substantial deviations of one structure from the other (fig. S8). Functionally important protein regions, such as the ligand-binding pocket and the sodium ion binding site (25), have very similar quality of electron density maps (fig. S9).
Furthermore, we compared the room temperature A2A_S-SAD_1.9 structure with the previously solved structure of the same protein at a synchrotron under cryo-conditions [Protein Data Bank (PDB) ID: 4EIY (25)]. Both structures overlay very well in the receptor part with an RMSD of 0.24 Å for all resolved Cα atoms, excluding the BRIL fusion protein (fig. S10). Most differences are observed in the loop regions of the receptor and in BRIL, which is tilted out from the receptor, accounting for the larger unit cell dimensions in the room temperature structure. As expected, the average B factor of the room temperature structure is ~20 Å2 higher than that of the cryocooled structure; nevertheless, the relative distribution of B factors in both structures is very similar, with higher B factor values in the loop regions and on the protein termini (fig. S11). Similar to the previous results (11), we observed slight improvements in the strength of most interactions that involve charged side chains in the room temperature structure (table S3), even though B factors for the interacting atoms usually increased. The current room temperature XFEL structure also provides important insights into water and ion binding to the receptor under close to physiological conditions (Fig. 3). A total of 101 ordered waters that interact with the receptor are observed in the XFEL structure (compared to 166 waters in PDB: 4EIY), of which 88 waters are located at the same (within 1 Å distance) positions. Most of the waters preserved at room temperature have relatively low B factors in both structures and form multiple polar contacts with the protein residues. At the same time, most high–B-factor waters are lost in the XFEL structure, suggesting that they do not have well-defined bound conformations at room temperature. Whereas most of the mobile waters are located in the intra- and extracellular loop regions of the receptor, tightly bound waters form two contiguous clusters. The first cluster is defined by close proximity (5 Å) to the ligand, supporting the notion that ligand binding is strongly mediated by a network of water interactions (33). The second cluster of waters, highly conserved in class A GPCRs, fills the binding pocket of the sodium ion, which itself has an identical position in both structures, emphasizing the stability of the extensive network of ionic and polar interactions around the sodium ion, which plays a key role in the receptor activation mechanism (34).
Fig. 3. Comparison of resolved water molecules between the room temperature XFEL structure (A2A_S-SAD_1.9) and the cryocooled synchrotron structure (PDB: 4EIY).
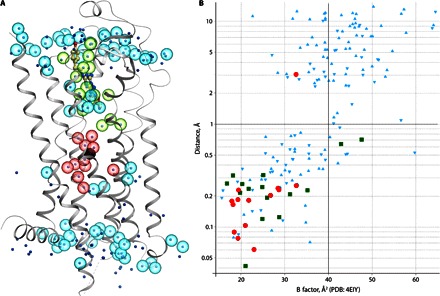
(A) Cartoon representation of the XFEL structure with overlaid waters. Water molecules from the XFEL structure are shown as semitransparent spheres, whereas waters from PDB: 4EYI are shown as dots, colored by location: green, close proximity to ligand (<5 Å); red, sodium ion pocket (<10 Å); cyan, other regions. (B) Conservation of the water positions between PDB: 4EIY and XFEL structures. For each water molecule in PDB: 4EIY, the distance to the closest water in the XFEL structure is shown on the y axis, whereas its B factor is shown on the x axis. Data points are colored the same way as in (A). Positions of water molecules can be considered as conserved if the distance between corresponding water molecules in two structures is less than 1 Å.
DISCUSSION
Compared to the previously reported S-SAD phasing of SFX data for the soluble proteins lysozyme (22) and thaumatin (23), phasing of A2AAR data required approximately four times more indexed patterns (table S4). In addition to lower crystal symmetry and lower sulfur content, the diffraction power of A2AAR microcrystals is substantially (one to two orders of magnitude) lower compared to lysozyme crystals of comparable size. At the same time, the background scattering from an LCP stream 50 μm in diameter, in which A2AAR microcrystals were delivered, is substantially higher (8) than the background from a liquid stream 5 μm in diameter used to deliver thaumatin crystals. These factors, together with potentially lower isomorphicity of A2AAR microcrystals, contribute to the challenge of native sulfur phasing of SFX data for difficult membrane proteins. Here, protein consumption required for de novo phasing was very reasonable (~2.7 mg) due to the very efficient operation of the LCP injector (7). Our results that ~600,000 indexed patterns are potentially sufficient to phase GPCR data starting with 12 ordered sulfur atoms per 447 residues (2.7%) can be placed in perspective with the fact that over 88% of all human proteins have Cys and Met residue content higher than 2.7% (fig. S12). Thus, our report provides an important reference point reassuring that most human proteins could be phased by S-SAD for de novo structure determination with XFELs.
Although this experiment took only two shifts (24 hours) of LCLS beam time, including experiment and sample changeover time, the scarcity of beam time is definitely currently a limiting factor for all XFEL experiments including de novo phasing and time-resolved studies. Future XFEL sources will have higher pulse repetition rates, enabling the acquisition of similar amounts of data in much less time. New XFEL facilities are coming online in the next few years (European XFEL, SwissFEL, PAL, and LCLS-II), providing additional capacity through an increased number of beamlines to choose from. New detectors will also have higher dynamic range, improving the quality of data. Further developments of the data processing software should result in determination of more accurate structure factors from fewer diffraction patterns. Therefore, we expect that within a few years, native sulfur phasing with XFELs will become a routine exercise.
MATERIALS AND METHODS
Protein expression and purification
Expression and purification of A2AAR construct engineered for crystallization, containing BRIL fusion protein in the third intracellular loop (A2AAR-BRIL), were done as previously described (25). Briefly, A2AAR-BRIL was expressed in Spodoptera frugiperda (Sf9) insect cells for 48 hours at 27°C using recombinant baculovirus at a multiplicity of infection of 5. Cells were harvested by centrifugation and stored at −80°C until use.
Frozen insect cell pellets were thawed on ice and disrupted by dounce homogenization in a hypotonic buffer containing 10 mM Hepes (pH 7.5), 10 mM MgCl2, 20 mM KCl, and EDTA-free cOmplete protease inhibitor cocktail (Roche). Insect cell membranes were collected by centrifugation at 150,000g for 45 min. Extensive washing of the isolated raw membranes was performed by repeated dounce homogenization and centrifugation in a high osmotic buffer containing 1.0 M NaCl, 10 mM Hepes (pH 7.5), 10 mM MgCl2, and 20 mM KCl (three times) to remove soluble and membrane associated proteins. Purified membranes were resuspended in a storage buffer containing 10 mM Hepes (pH 7.5), 10 mM MgCl2, 20 mM KCl, and 20% glycerol, flash-frozen in liquid nitrogen, and stored at −80°C until further use.
Before solubilization, purified membranes were thawed on ice in the presence of 4 mM theophylline (Sigma), iodoacetamide (2.0 mg/ml; Sigma), and EDTA-free cOmplete protease inhibitor cocktail (Roche). After incubation for 30 min at 4°C, membranes were solubilized by incubation in a buffer containing 50 mM Hepes (pH 7.5), 800 mM NaCl, 1% (w/v) n-dodecyl-β-d-maltopyranoside (DDM) (Anatrace), and 0.2% (w/v) cholesteryl hemisuccinate (CHS) (Sigma) for 3 hours at 4°C. The unsolubilized material was removed by centrifugation at 250,000g for 45 min. The supernatant was incubated overnight with TALON immobilized metal affinity chromatography resin (1 ml of resin per 1 liter of expression culture; Takara-Clontech) in the presence of 20 mM imidazole. After overnight binding, the resin was washed with 10 column volumes of 50 mM Hepes (pH 7.5), 800 mM NaCl, 10% (v/v) glycerol, 25 mM imidazole, 0.1% (w/v) DDM, 0.02% (w/v) CHS, 10 mM MgCl2, 8 mM adenosine 5′-triphosphate (Sigma), and 100 μM ZM241385 (Tocris; prepared as 100 mM stock in dimethyl sulfoxide), followed by 5 column volumes of 50 mM Hepes (pH 7.5), 800 mM NaCl, 10% (v/v) glycerol, 50 mM imidazole, 0.05% (w/v) DDM, 0.01% (w/v) CHS, and 100 μM ZM241385. The receptor was eluted with 3 ml of elution buffer containing 25 mM Hepes (pH 7.5), 800 mM NaCl, 10% (v/v) glycerol, 220 mM imidazole, 0.01% (w/v) DDM, 0.002% (w/v) CHS, and 100 μM ZM241385. Purified receptor was concentrated to ~60 mg/ml with a 100-kDa molecular weight cutoff Amicon concentrator (Millipore). Receptor purity and monodispersity were analyzed by SDS–polyacrylamide gel electrophoresis and analytical size exclusion chromatography.
Sample preparation
Concentrated protein samples of A2AAR-BRIL in complex with ZM241385 were reconstituted into LCP by mixing with molten lipid using a syringe mixer (35). The protein-LCP mixture contained 40% (w/w) protein solution, 54% (w/w) monoolein (Sigma), and 6% (w/w) cholesterol (Sigma). Crystals for SFX data collection were obtained in Hamilton gas-tight syringes using the following procedure (26). Approximately 6 μl of protein-laden LCP was injected into a 100-μl syringe filled with 60 μl of precipitant solution [28% (v/v) PEG-400, 40 mM sodium thiocyanate, and 100 mM sodium citrate (pH 5.0)] and incubated for 24 hours at 20°C. After crystals had formed, excess precipitant solution was carefully removed, followed by the addition of ~3 μl of 7.9 MAG (monoacylglycerol) (36). The 7.9 MAG was used to prevent the appearance of a lipidic lamellar crystal phase due to rapid dehydration and cooling upon injection of LCP into vacuum (10−4 torr). The microcrystal samples were characterized on site at LCLS by optical and ultraviolet fluorescence imaging. The average microcrystal size was 5 × 5 × 2 μm3 (fig. S1).
Anomalous SFX data collection and processing
Experiments were performed using the CXI instrument (24) at the LCLS at SLAC National Accelerator Laboratory. The LCLS was operated at a wavelength of 2.07 Å (6.0 keV), delivering individual x-ray pulses of 45-fs pulse duration and ~1.7 × 1011 photons per pulse focused into a spot size of approximately 1.5 μm in diameter using a pair of Kirkpatrick-Baez mirrors. Microcrystals of A2AAR-BRIL/ZM241385 were delivered in the LCP medium using a microextrusion injector (7) with 50-μm nozzle running at a flow rate of ~220 nl/min. Diffraction images were recorded at a rate of 7200 patterns/min (120 Hz) with the 2.3-megapixel CSPAD (64 independent detectors of 194 pixels × 184 pixels with a pixel size of 110 × 110 μm2 each tiled to cover an area of 200 × 200 mm2) (37). Background subtraction and detector correction were performed with the Cheetah software (27). Pedestal signal arising from the detector was removed by subtracting an average dark image from each frame and using unbonded pixels as a shot-to-shot dark reference for common mode corrections. Hot pixels were identified and masked. The software was also used to discriminate the patterns containing crystal diffraction, which were named “hits,” from the rest of the blank shots by locating pixel clusters that lie above a given threshold. These images were processed with the CrystFEL software package (version 0.6.1) (28). The unit cell parameters were first determined using a subset of the collected data. Subsequent indexing was performed by comparing the resulted unit cell parameters to the determined ones, allowing a tolerance of 15% in reciprocal space axis lengths and 3° in reciprocal space angles. The detector geometry and the sample-to-detector distance were first optimized using a virtual powder pattern from lysozyme crystals, which were collected at the beginning of the experiment, and further refined at this stage using geoptimiser (38). Multiple indexing runs were performed, each by using finer detector geometry corrections to get the final stream of processed data. The complete set of scattered intensities was obtained by merging (treating Friedel pairs as separate reflections) and iteratively scaling all the reflections using partialator (CrystFEL package) without partiality correction. Specifically, the algorithm first generates a reference set, averaging the reflections from single diffraction patterns and applying polarization corrections, followed by applying linear and Debye-Waller scaling of the diffraction patterns using least-squares minimization of residuals on a logarithmic scale, similar to the method previously described (39). Three cycles of scaling were used to generate the final data set. Through these scaling cycles, 15,376 crystals were rejected because they had either not enough common reflections with the merged data set to permit scaling or relative Debye-Waller factors greater than 100 Å2 or lower than −100 Å2. The final A2A_S-SAD data set was truncated at 2.5 Å resolution using the CC* >0.7 criterion (fig. S4A and table S1).
High-resolution SFX data collection and processing
The high-resolution data set (A2A_High-Res) was collected using a similar setup as for the A2A_S-SAD set, except for the x-ray energy of 9.8 keV (1.27 Å), and the flux of ~6.4 × 1010 photons per pulse. A total of 948,961 images were collected, 232,283 of which were identified as hits with the Cheetah program (24.5% hit rate). Of these hits, 72,735 were successfully indexed and merged using the standard CrystFEL pipeline of Monte Carlo averaging. The merging procedure was performed using per-pattern resolution cutoff with the “pushres 1.8” option and without an additional scaling step. Scaling and partiality refinement were tested but did not result in any significant improvement of data quality. The final A2A_High-Res data set was truncated at 1.9 Å resolution using the CC* >0.5 criterion (table S1).
Structure determination
Integrated and scaled intensities of the A2A_S-SAD data set in CrysFEL format were converted to the XSCALE (40) format, followed by conversion to the CCP4 mtz and SHELX formats. Anomalous signal strength was analyzed with phenix.xtriage (31) and was found to extend to ~3.6 Å (fig. S2A). The substructure was found with SHELXD (19) (OS X version 2013/2) using the graphical user interface hkl2map (41), varying the resolution cutoff from 3.0 to 3.6 Å in 0.1 Å increments, the Emin from 1.5 to 1.2, the number of sites from 10 to 20, the number of disulfides from 0 to 4, and the number of trials from 1000 to 5000. A clear drop in occupancy from 0.72 to 0.42 at the 3.5 Å resolution cutoff (fig. S2C) was used as an indicator for the correct solution. After removing the five low-occupancy atoms, the substructure was used as an input in Phaser EP (29) for log-likelihood gradient map substructure completion and phasing. The resulting phases for both enantiomorphs with an FOM of 0.418 and a log-likelihood gain of 505 were subjected to density modification and phase extension using Resolve (30) with the mask type “Wang.” The correct hand was identified by success in autotracing. The model was traced automatically with phenix.autobuild (31), and it was possible to build 264 of 447 residues (59%) with R/Rfree = 0.31/0.34.
Alternatively, we applied the automated crystal structure determination pipeline X2DF for the structure determination. X2DF automatically performs heavy atom search, S-SAD phasing, phase extension, density modification, and automated model building using different programs based on the “parameter-space screening” strategy (32). In the current version of X2DF pipeline, the parameters are different crystallography programs, such as phenix.autosol or SHELXC/D/E for heavy atom searching and phasing; DM or Parrot for density modification; phenix.autobuild, ARP/wARP, or BUCCANEER for model building; and high-resolution cutoff values and incremental step values for heavy atom search, phasing, phase extension, number of heavy atoms, solvent content, space groups, etc. For difficult-to-solve structures, such as S-SAD phasing, only a limited number of combinations of parameters may lead to successful structure solutions. Taking advantage of high-performance computer cluster’s parallel computing power, X2DF pipeline explores a much larger multidimensional parameter space than what a human can do manually.
In this case, the 2.5 Å data set was subjected to the following steps of structure solution determination using the X2DF pipeline. The pipeline was configured to use SHELXC/D for sulfur atom substructure determination, Phaser EP for phasing, DM for phase extension, and phenix.autobuild for model building. Three-dimensional parameter-space screening was performed. The three screening parameters were (i) number of heavy atoms (screening range, 1 to 24 based on the sequence), (ii) high-resolution limits for heavy atom search (screening range, 2.5 to 4.0 Å; step, 0.1 Å), and (iii) high-resolution limits for phasing (screening range, 2.5 to 4.0 Å; step, 0.1 Å). A total of 7843 computing jobs were created, of which 200 jobs returned the correct structure solutions. The best combination resulted in a structure solution with 294 of 447 residues (66%) automatically traced, an R value of 28.7%, and an Rfree value of 30.2%. The best number of sulfur sites in the search was 10; however, SHELX yielded 18 sites, from which six low-occupancy sites were removed automatically before phase calculation. We observed that multiple combinations of high-resolution limits for sulfur site search and phasing could lead to structure solutions (fig. S3). The best high-resolution limits for sulfur site search and phasing were 3.60 and 3.25 Å, respectively. This result demonstrates that the X2DF pipeline could be used as a powerful tool for S-SAD structure determination with SFX data.
Structure refinement
The structure was further built and refined by repetitive cycling with phenix.refine (42) using experimental phase restraints with MLHL (phased maximum likelihood) target function followed by manual examination and rebuilding of the refined coordinates with Coot (43). The final model (A2A_S-SAD_2.5), refined to R/Rfree = 17.4/22.8 against the 2.5 Å A2A_S-SAD data using isotropic ADPs and three TLS groups consisting of residues −2 to 208, 1001 to 1106, and 219 to 308, contains the complete A2AAR-BRIL construct sequence, except for an unresolved gap (1045–1055) in one of the BRIL loops, ligand ZM241385, 3 cholesterol molecules, 20 lipids, and 70 waters. The overall structure had a good stereochemistry with no Ramachandran outliers (99.0% in favored and 1.0% in allowed regions), as determined with MolProbity (44).
Resolution was then further extended to 1.9 Å by refining against the A2A_High-Res data set while using the same Rfree set, extended to 1.9 Å, and experimental phase restraints from the 2.5 Å A2A_S-SAD data. The final A2A_S-SAD_1.9 model, refined to R/Rfree = 17.3/20.8 using isotropic ADPs and three TLS groups consisting of residues −2 to 208, 1001 to 1106, and 219 to 308, contains the complete A2AAR-BRIL construct sequence except for an unresolved gap (1045–1055) in one of the BRIL loops, ligand ZM241385, 3 cholesterol molecules, 21 lipids, and 105 waters. The overall structure had a good stereochemistry with no Ramachandran outliers (99.0% in favored and 1.0% in allowed regions), as determined with MolProbity (44).
In parallel, we applied MR using the previously obtained A2AAR structure (PDB: 4EIY; without BRIL, ligands, and waters) as a search model, using the A2A_High-Res data truncated at 1.9 and 2.5 Å resolution. The MR structures were refined by repetitive cycling with phenix.refine (42) using the same Rfree set as for the S-SAD structures, followed by manual examination and rebuilding of the refined coordinates with Coot (43). The final refinement was done with three TLS groups, consisting of residues −2 to 208, 1001 to 1106, and 219 to 308. The final model was fully completed with no gaps except missing residues 1044–1055 in BRIL. The overall structure contained a good stereochemistry with no Ramachandran outliers (99.0% in favored and 1.0% in allowed regions), as determined by MolProbity (44). The crystallographic refinement statistics are shown in table S2.
Supplementary Material
Acknowledgments
We thank A. Walker for assistance with manuscript preparation. A. Batyuk thanks A. Plückthun for helpful discussions. Funding: This work was supported by NIH grants R01 GM108635 (V.C.), U54 GM094618 (V.C., V.K., and R.C.S.), U54 GM094599 (P.F.), and R01 GM095583 (P.F.); by the Ministry of Science and Technology of China grant 2014CB910400 (Z.-j.L.); and by the NSF Science and Technology Center award 1231306 (J.C.H.S, U.W., W.L., and P.F.). C.G. thanks the PIER Helmholtz Graduate School and, together with L.G., A. Barty, and T.A.W., the Helmholtz Association for financial support through project-oriented funds. P.A.P. and V.C. acknowledge support from the Russian Ministry of Education and Science project 5-100. Parts of the sample delivery system used at LCLS for this research was funded by the NIH grant P41 GM103393, formerly P41 RR001209. Use of LCLS at Stanford Linear Accelerator Center (SLAC) National Accelerator Laboratory is supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences under contract number DE-AC02-76SF00515. Author contributions: A. Batyuk prepared the samples, participated in data collection, solved the structure, analyzed the data, and wrote the paper; L.G. processed and analyzed the data and contributed to the writing of the paper; C.G. participated in data collection and processed and analyzed the data; A.I., B.S., M.-Y.L., and W.L. prepared the samples, participated in data collection, and analyzed the data; G.W.H. performed structure refinement; M.P. and Z.-j.L. analyzed the data and contributed to the writing of the paper; G.N., D.J., C.L., and Y.Z. operated the LCP injector; U.W. designed and operated the LCP injector and contributed to the writing of the paper; P.A.P. and V.K. analyzed the results and contributed to the writing of the paper; S.B., M.S.H., A.A., and M.L. set up the CXI beamline controls and data acquisition, operated the beamline, and performed the data collection; P.F. assisted with sample characterization and data collection; A. Barty and T.A.W. wrote the data processing software and contributed to data processing and the writing of the paper; J.C.H.S. contributed to data analysis and to the writing of the paper; R.C.S. supervised GPCR production; V.C. conceived the project, designed the experiments, supervised the data collection, analyzed the results, and wrote the paper. Competing interests: U.W. and J.C.H.S. have filed a patent application (US 20160051995) for the LCP injector. All other authors declare that they have no competing interests. Data and materials availability: Coordinates and the structure factors have been deposited in the Protein Data Bank under accession codes 5K2C (A2A S-SAD_1.9 data set), 5K2D (A2A MR_1.9 data set), 5K2A (A2A S-SAD_2.5 data set), and 5K2B (A2A MR_2.5 data set). Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/2/9/e1600292/DC1
fig. S1. A2A-BRIL/ZM241385 microcrystals used for data collection.
fig. S2. Strength of anomalous signal and sulfur atom search.
fig. S3. Parameter-space screening results for S-SAD phasing using the X2DF pipeline.
fig. S4. Effect of different data processing methods on data merging metrics.
fig. S5. Dependence of anomalous signal measurability on the number of indexed patterns.
fig. S6. Final 1.9 Å XFEL room temperature A2AAR-BRIL structure (A2A_S-SAD_1.9).
fig. S7. Structure-factor amplitude difference Fourier map between A2A_S-SAD_2.5 and A2A_MR_2.5 structures.
fig. S8. B factor comparison between A2A_S-SAD_2.5 and A2A_MR_2.5 structures.
fig. S9. Comparison of 2mFo-DFc electron density maps for the ligand- and sodium-binding pockets obtained by S-SAD and MR phasing.
fig. S10. Cα-Cα difference distance matrix between A2A_S-SAD_1.9 and previously determined A2AAR structure (PDB: 4EIY).
fig. S11. B factor comparison between room temperature A2A_S-SAD_1.9 and previously determined cryocooled A2AAR structure (PDB: 4EIY).
fig. S12. Distribution of Cys and Met residues in human proteins.
table S1. Data collection statistics.
table S2. Data refinement statistics.
table S3. Comparison of interactions involving charged residues between PDB: 4EIY and A2A_S-SAD_1.9 structures.
table S4. Comparison of protein and data collection parameters for successful S-SAD phasing of XFEL data.
REFERENCES AND NOTES
- 1.Schlichting I., Serial femtosecond crystallography: The first five years. IUCrJ 2, 246–255 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tenboer J., Basu S., Zatsepin N., Pande K., Milathianaki D., Frank M., Hunter M., Boutet S., Williams G. J., Koglin J. E., Oberthuer D., Heymann M., Kupitz C., Conrad C., Coe J., Roy-Chowdhury S., Weierstall U., James D., Wang D., Grant T., Barty A., Yefanov O., Scales J., Gati C., Seuring C., Srajer V., Henning R., Schwander P., Fromme R., Ourmazd A., Moffat K., Van Thor J. J., Spence J. C. H., Fromme P., Chapman H. N., Schmidt M., Time-resolved serial crystallography captures high-resolution intermediates of photoactive yellow protein. Science 346, 1242–1246 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chapman H. N., Fromme P., Barty A., White T. A., Kirian R. A., Aquila A., Hunter M. S., Schulz J., DePonte D. P., Weierstall U., Doak R. B., Maia F. R. N. C., Martin A. V., Schlichting I., Lomb L., Coppola N., Shoeman R. L., Epp S. W., Hartmann R., Rolles D., Rudenko A., Foucar L., Kimmel N., Weidenspointner G., Holl P., Liang M., Barthelmess M., Caleman C., Boutet S., Bogan M. J., Krzywinski J., Bostedt C., Bajt S., Gumprecht L., Rudek B., Erk B., Schmidt C., Hömke A., Reich C., Pietschner D., Strüder L., Hauser G., Gorke H., Ullrich J., Herrmann S., Schaller G., Schopper F., Soltau H., Kühnel K.-U., Messerschmidt M., Bozek J. D., Hau-Riege S. P., Frank M., Hampton C. Y., Sierra R. G., Starodub D., Williams G. J., Hajdu J., Timneanu N., Seibert M. M., Andreasson J., Rocker A., Jönsson O., Svenda M., Stern S., Nass K., Andritschke R., Schröter C.-D., Krasniqi F., Bott M., Schmidt K. E., Wang X., Grotjohann I., Holton J. M., Barends T. R. M., Neutze R., Marchesini S., Fromme R., Schorb S., Rupp D., Adolph M., Gorkhover T., Andersson I., Hirsemann H., Potdevin G., Graafsma H., Nilsson B., Spence J. C. H., Femtosecond X-ray protein nanocrystallography. Nature 470, 73–77 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kirian R. A., White T. A., Holton J. M., Chapman H. N., Fromme P., Barty A., Lomb L., Aquila A., Maia F. R. N. C., Martin A. V., Fromme R., Wang X., Hunter M. S., Schmidt K. E., Spence J. C. H., Structure-factor analysis of femtosecond microdiffraction patterns from protein nanocrystals. Acta Crystallogr. A A67, 131–140 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.White T. A., Post-refinement method for snapshot serial crystallography. Philos. Trans. R. Soc. Lond. B Biol. Sci. 369, 20130330 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Uervirojnangkoorn M., Zeldin O. B., Lyubimov A. Y., Hattne J., Brewster A. S., Sauter N. K., Brunger A. T., Weis W. I., Enabling X-ray free electron laser crystallography for challenging biological systems from a limited number of crystals. Elife 4, e05421 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Weierstall U., James D., Wang C., White T. A., Wang D., Liu W., Spence J. C. H., Doak R. B., Nelson G., Fromme P., Fromme R., Grotjohann I., Kupitz C., Zatsepin N. A., Liu H., Basu S., Wacker D., Han G. W., Katritch V., Boutet S., Messerschmidt M., Williams G. J., Koglin J. E., Seibert M. M., Klinker M., Gati C., Shoeman R. L., Barty A., Chapman H. N., Kirian R. A., Beyerlein K. R., Stevens R. C., Li D., Shah S. T. A., Howe N., Caffrey M., Cherezov V., Lipidic cubic phase injector facilitates membrane protein serial femtosecond crystallography. Nat. Commun. 5, 3309 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Conrad C. E., Basu S., James D., Wang D., Schaffer A., Roy-Chowdhury S., Zatsepin N. A., Aquila A., Coe J., Gati C., Hunter M. S., Koglin J. E., Kupitz C., Nelson G., Subramanian G., White T. A., Zhao Y., Zook J., Boutet S., Cherezov V., Spence J. C. H., Fromme R., Weierstall U., Fromme P., A novel inert crystal delivery medium for serial femtosecond crystallography. IUCrJ 2, 421–430 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fromme R., Ishchenko A., Metz M., Chowdhury S. R., Basu S., Boutet S., Fromme P., White T. A., Barty A., Spence J. C. H., Weierstall U., Liu W., Cherezov V., Serial femtosecond crystallography of soluble proteins in lipidic cubic phase. IUCrJ 2, 545–551 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sugahara M., Mizohata E., Nango E., Suzuki M., Tanaka T., Masuda T., Tanaka R., Shimamura T., Tanaka Y., Suno C., Ihara K., Pan D., Kakinouchi K., Sugiyama S., Murata M., Inoue T., Tono K., Song C., Park J., Kameshima T., Hatsui T., Joti Y., Yabashi M., Iwata S., Grease matrix as a versatile carrier of proteins for serial crystallography. Nat. Methods 12, 61–63 (2015). [DOI] [PubMed] [Google Scholar]
- 11.Liu W., Wacker D., Gati C., Han G. W., James D., Wang D., Nelson G., Weierstall U., Katritch V., Barty A., Zatsepin N. A., Li D., Messerschmidt M., Boutet S., Williams G. J., Koglin J. E., Seibert M. M., Wang C., Shah S. T. A., Basu S., Fromme R., Kupitz C., Rendek K. N., Grotjohann I., Fromme P., Kirian R. A., Beyerlein K. R., White T. A., Chapman H. N., Caffrey M., Spence J. C. H., Stevens R. C., Cherezov V., Serial femtosecond crystallography of G protein–coupled receptors. Science 342, 1521–1524 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang H., Unal H., Gati C., Han G. W., Liu W., Zatsepin N. A., James D., Wang D., Nelson G., Weierstall U., Sawaya M. R., Xu Q., Messerschmidt M., Williams G. J., Boutet S., Yefanov O. M., White T. A., Wang C., Ishchenko A., Tirupula K. C., Desnoyer R., Coe J., Conrad C. E., Fromme P., Stevens R. C., Katritch V., Karnik S. S., Cherezov V., Structure of the angiotensin receptor revealed by serial femtosecond crystallography. Cell 161, 833–844 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kang Y., Zhou X. E., Gao X., He Y., Liu W., Ishchenko A., Barty A., White T. A., Yefanov O., Han G. W., Xu Q., de Waal P. W., Ke J., Tan M. H. E., Zhang C., Moeller A., West G. M., Pascal B. D., Van Eps N., Caro L. N., Vishnivetskiy S. A., Lee R. J., Suino-Powell K. M., Gu X., Pal K., Ma J., Zhi X., Boutet S., Williams G. J., Messerschmidt M., Gati C., Zatsepin N. A., Wang D., James D., Basu S., Roy-Chowdhury S., Conrad C. E., Coe J., Liu H., Lisova S., Kupitz C., Grotjohann I., Fromme R., Jiang Y., Tan M., Yang H., Li J., Wang M., Zheng Z., Li D., Howe N., Zhao Y., Standfuss J., Diederichs K., Dong Y., Potter C. S., Carragher B., Caffrey M., Jiang H., Chapman H. N., Spence J. C. H., Fromme P., Weierstall U., Ernst O. P., Katritch V., Gurevich V. V., Griffin P. R., Hubbell W. L., Stevens R. C., Cherezov V., Melcher K., Xu H. E., Crystal structure of rhodopsin bound to arrestin by femtosecond X-ray laser. Nature 523, 561–567 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Barends T. R. M., Foucar L., Botha S., Doak R. B., Shoeman R. L., Nass K., Koglin J. E., Williams G. J., Boutet S., Messerschmidt M., Schlichting I., De novo protein crystal structure determination from X-ray free-electron laser data. Nature 505, 244–247 (2014). [DOI] [PubMed] [Google Scholar]
- 15.Yamashita K., Pan D., Okuda T., Sugahara M., Kodan A., Yamaguchi T., Murai T., Gomi K., Kajiyama N., Mizohata E., Suzuki M., Nango E., Tono K., Joti Y., Kameshima T., Park J., Song C., Hatsui T., Yabashi M., Iwata S., Kato H., Ago H., Yamamoto M., Nakatsu T., An isomorphous replacement method for efficient de novo phasing for serial femtosecond crystallography. Sci. Rep. 5, 14017 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fukuda Y., Tse K. M., Nakane T., Nakatsu T., Suzuki M., Sugahara M., Inoue S., Masuda T., Yumoto F., Matsugaki N., Nango E., Tono K., Joti Y., Kameshima T., Song C., Hatsui T., Yabashi M., Nureki O., Murphy M. E. P., Inoue T., Iwata S., Mizohata E., Redox-coupled proton transfer mechanism in nitrite reductase revealed by femtosecond crystallography. Proc. Natl. Acad. Sci. U.S.A. 113, 2928–2933 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hendrickson W. A., Teeter M. M., Structure of the hydrophobic protein crambin determined directly from the anomalous scattering of sulphur. Nature 290, 107–113 (1981). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ramagopal U. A., Dauter M., Dauter Z., Phasing on anomalous signal of sulfurs: What is the limit?. Acta Crystallogr. D Biol. Crystallogr. D59, 1020–1027 (2003). [DOI] [PubMed] [Google Scholar]
- 19.Sheldrick G. M., Experimental phasing with SHELXC/D/E: Combining chain tracing with density modification. Acta Crystallogr. D Biol. Crystallogr. D66, 479–485 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liu Q., Dahmane T., Zhang Z., Assur Z., Brasch J., Shapiro L., Mancia F., Hendrickson W. A., Structures from anomalous diffraction of native biological macromolecules. Science 336, 1033–1037 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Weinert T., Olieric V., Waltersperger S., Panepucci E., Chen L., Zhang H., Zhou D., Rose J., Ebihara A., Kuramitsu S., Li D., Howe N., Schnapp G., Pautsch A., Bargsten K., Prota A. E., Surana P., Kottur J., Nair D. T., Basilico F., Cecatiello V., Pasqualato S., Boland A., Weichenrieder O., Wang B.-C., Steinmetz M. O., Caffrey M., Wang M., Fast native-SAD phasing for routine macromolecular structure determination. Nat. Methods 12, 131–133 (2015). [DOI] [PubMed] [Google Scholar]
- 22.Nakane T., Song C., Suzuki M., Nango E., Kobayashi J., Masuda T., Inoue S., Mizohata E., Nakatsu T., Tanaka T., Tanaka R., Shimamura T., Tono K., Joti Y., Kameshima T., Hatsui T., Yabashi M., Nureki O., Iwata S., Sugahara M., Native sulfur/chlorine SAD phasing for serial femtosecond crystallography. Acta Crystallogr. D Biol. Crystallogr. D71, 2519–2525 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nass K., Meinhart A., Barends T. R. M., Foucar L., Gorel A., Aquila A., Botha S., Doak R. B., Koglin J., Liang M., Shoeman R. L., Williams G., Boutet S., Schlichting I., Protein structure determination by single-wavelength anomalous diffraction phasing of X-ray free-electron laser data. IUCrJ 3, 180–191 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liang M., Williams G. J., Messerschmidt M., Seibert M. M., Montanez P. A., Hayes M., Milathianaki D., Aquila A., Hunter M. S., Koglin J. E., Schafer D. W., Guillet S., Busse A., Bergan R., Olson W., Fox K., Stewart N., Curtis R., Miahnahri A. A., Boutet S., The Coherent X-ray Imaging instrument at the Linac Coherent Light Source. J. Synchrotron Radiat. 22, 514–519 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liu W., Chun E., Thompson A. A., Chubukov P., Xu F., Katritch V., Han G. W., Roth C. B., Heitman L. H., IJzerman A. P., Cherezov V., Stevens R. C., Structural basis for allosteric regulation of GPCRs by sodium ions. Science 337, 232–236 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liu W., Ishchenko A., Cherezov V., Preparation of microcrystals in lipidic cubic phase for serial femtosecond crystallography. Nat. Protoc. 9, 2123–2134 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Barty A., Kirian R. A., Maia F. R. N. C., Hantke M., Yoon C. H., White T. A., Chapman H., Cheetah: Software for high-throughput reduction and analysis of serial femtosecond X-ray diffraction data. J. Appl. Crystallogr. 47, 1118–1131 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.White T. A., Kirian R. A., Martin A. V., Aquila A., Nass K., Barty A., Chapman H. N., CrystFEL: A software suite for snapshot serial crystallography. J. Appl. Crystallogr. 45, 335–341 (2012). [Google Scholar]
- 29.McCoy A. J., Grosse-Kunstleve R. W., Adams P. D., Winn M. D., Storoni L. C., Read R. J., Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Terwilliger T. C., Maximum-likelihood density modification. Acta Crystallogr. D Biol. Crystallogr. D56, 965–972 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L.-W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., Richardson J. S., Terwilliger T. C., Zwart P. H., PHENIX: A comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. D66, 213–221 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liu Z.-J., Lin D., Tempel W., Praissman J. L., Rose J. P., Wang B.-C., Parameter-space screening: A powerful tool for high-throughput crystal structure determination. Acta Crystallogr. D Biol. Crystallogr. D61, 520–527 (2005). [DOI] [PubMed] [Google Scholar]
- 33.Mason J. S., Bortolato A., Congreve M., Marshall F. H., New insights from structural biology into the druggability of G protein-coupled receptors. Trends Pharmacol. Sci. 33, 249–260 (2012). [DOI] [PubMed] [Google Scholar]
- 34.Katritch V., Fenalti G., Abola E. E., Roth B. L., Cherezov V., Stevens R. C., Allosteric sodium in class A GPCR signaling. Trends Biochem. Sci. 39, 233–244 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Caffrey M., Cherezov V., Crystallizing membrane proteins using lipidic mesophases. Nat. Protoc. 4, 706–731 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Misquitta Y., Cherezov V., Havas F., Patterson S., Mohan J. M., Wells A. J., Hart D. J., Caffrey M., Rational design of lipid for membrane protein crystallization. J. Struct. Biol. 148, 169–175 (2004). [DOI] [PubMed] [Google Scholar]
- 37.Blaj G., Caragiulo P., Carini G., Carron S., Dragone A., Freytag D., Haller G., Hart P., Hasi J., Herbst R., Herrmann S., Kenney C., Markovic B., Nishimura K., Osier S., Pines J., Reese B., Segal J., Tomada A., Weaver M., X-ray detectors at the Linac Coherent Light Source. J. Synchrotron Radiat. 22, 577–583 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yefanov O., Mariani V., Gati C., White T. A., Chapman H. N., Barty A., Accurate determination of segmented X-ray detector geometry. Opt. Express 23, 28459–28470 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kabsch W., Processing of X-ray snapshots from crystals in random orientations. Acta Crystallogr. D Biol. Crystallogr. D70, 2204–2216 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kabsch W., XDS. Acta Crystallogr. D Biol. Crystallogr. D66, 125–132 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pape T., Schneider T. R., HKL2MAP: A graphical user interface for macromolecular phasing with SHELX programs. J. Appl. Crystallogr. 37, 843–844 (2004). [Google Scholar]
- 42.Afonine P. V., Grosse-Kunstleve R. W., Echols N., Headd J. J., Moriarty N. W., Mustyakimov M., Terwilliger T. C., Urzhumtsev A., Zwart P. H., Adams P. D., Towards automated crystallographic structure refinement with phenix.refine. Acta Crystallogr. D Biol. Crystallogr. D68, 352–367 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Emsley P., Lohkamp B., Scott W. G., Cowtan K., Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. D66, 486–501 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chen V. B., Arendall W. B. III, Headd J. J., Keedy D. A., Immormino R. M., Kapral G. J., Murray L. W., Richardson J. S., Richardson D. C., MolProbity: All-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. D66, 12–21 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/2/9/e1600292/DC1
fig. S1. A2A-BRIL/ZM241385 microcrystals used for data collection.
fig. S2. Strength of anomalous signal and sulfur atom search.
fig. S3. Parameter-space screening results for S-SAD phasing using the X2DF pipeline.
fig. S4. Effect of different data processing methods on data merging metrics.
fig. S5. Dependence of anomalous signal measurability on the number of indexed patterns.
fig. S6. Final 1.9 Å XFEL room temperature A2AAR-BRIL structure (A2A_S-SAD_1.9).
fig. S7. Structure-factor amplitude difference Fourier map between A2A_S-SAD_2.5 and A2A_MR_2.5 structures.
fig. S8. B factor comparison between A2A_S-SAD_2.5 and A2A_MR_2.5 structures.
fig. S9. Comparison of 2mFo-DFc electron density maps for the ligand- and sodium-binding pockets obtained by S-SAD and MR phasing.
fig. S10. Cα-Cα difference distance matrix between A2A_S-SAD_1.9 and previously determined A2AAR structure (PDB: 4EIY).
fig. S11. B factor comparison between room temperature A2A_S-SAD_1.9 and previously determined cryocooled A2AAR structure (PDB: 4EIY).
fig. S12. Distribution of Cys and Met residues in human proteins.
table S1. Data collection statistics.
table S2. Data refinement statistics.
table S3. Comparison of interactions involving charged residues between PDB: 4EIY and A2A_S-SAD_1.9 structures.
table S4. Comparison of protein and data collection parameters for successful S-SAD phasing of XFEL data.