

Abstract
Supplemental Digital Content is available in the text.
Key Words: Alzheimer disease, tau-negative Alzheimer disease, β-amyloid, tau, chorein, VPS13A
The “amyloid cascade” model of Alzheimer disease (AD) considers β-amyloid accumulation as a primary event that triggers tau hyperphosphorylation, neurofibrillary tangle formation, and ultimately neuron loss. However, this view is challenged by occasional cases without extensive tau pathology (referred to as “tau-negative” or “plaque-only” AD). Originally described by Alzheimer himself, this rare AD variant was only recently better characterized. Existence of a genetic defect targeting selectively the amyloid but not tau cascade is strongly suspected, yet no relevant mutation has been identified to date.1
Here, we report a case of probable tau-negative early-onset AD, carrying a heterozygous deletion in chorein (VPS13A; vacuolar protein sorting-associated protein-13A), a gene not previously known to be involved in AD. Chorein is expressed in the brain and its homozygous deficiency leads to chorea-acanthocytosis (ChAC),2 a debilitating neurodegenerative disease manifested by choreiform movements. Although chorein function remains unclear, it is thought to control intracellular protein trafficking.3 We show that chorein is involved in amyloid processing, and its heterozygous deficiency is associated with severe dementia, profound cerebral hypometabolism, and high amyloid burden, in the absence of any signs of ChAC and tau pathology.
MATERIALS AND METHODS
Neuroimaging
All the analyses were performed at the University Hospitals of Geneva, except for positron-emission tomography with Pittsburgh compound-B (PET-PiB), performed at the University Hospital of Zurich. The tracers used were: Ioflupane (123I) for DaTScan, 99mTc-HMPAO for SPECT, 2-deoxy-2-(18F)fluoro-d-glucose for FDG-PET.
Genetic Analysis
DNA was extracted from blood. The copy number variant analysis (Agilent Human Genome comparative genomic hybridization Microarray-Kit-244K) and exome-wide sequencing (paired-end sequencing, Illumina GAIIx/HiSeq-2000) were performed according to the manufacturer’s protocols.
In Vitro Assays
Human neuroblastoma SH-SY5Y cells transduced with wild-type human APP were transfected using Lipofectamine RNAiMAX with chorein-targeting (#4392420) or control (#4390843) Silencer Select siRNA (Ambion). After 72 hours, the cells and supernatants were collected. Total RNA was extracted from the cells (RNeasy Mini-kit; Qiagen), cDNA was obtained (PrimeScript-kit; Clontech), and gene expression was assessed (TaqMan/SYBRgreen PCR). Three housekeeping genes were simultaneously used for normalization (δ-aminolevulinate synthase-1, β-glucuronidase, transferrin receptor protein-1). Concentrations of tau, phospho-tau (p-tau), and β-amyloid peptides were measured in supernatants or cerebrospinal fluid (CSF) by enzyme-linked immunosorbent assay.
RESULTS
The proband is a Brazilian woman presenting with progressing cognitive deterioration since her early 50s. At the age of 58, the deficit was already important, affecting memory, time orientation, visuoconstructive and executive functions, praxis, gnosis, calculation abilities, and attention. Her Mattis dementia rating scale (MDRS) score was significantly lower than the age-adapted norms [109/144 (25 to 75 percentile range: 134 to 141)], and it declined progressively (Fig. 1A). At 62 years, all of the MDRS subscores were low, with a total score of 58/144 (Fig. 1B, supplementary Table 1, Supplemental Digital Content 1, http://links.lww.com/WAD/A125). Clinical and laboratory check-up (electrolytes, vitamins, thyroid hormones, HIV, HBV, HCV, syphilis serology, autoimmune markers) revealed no abnormalities.
FIGURE 1.
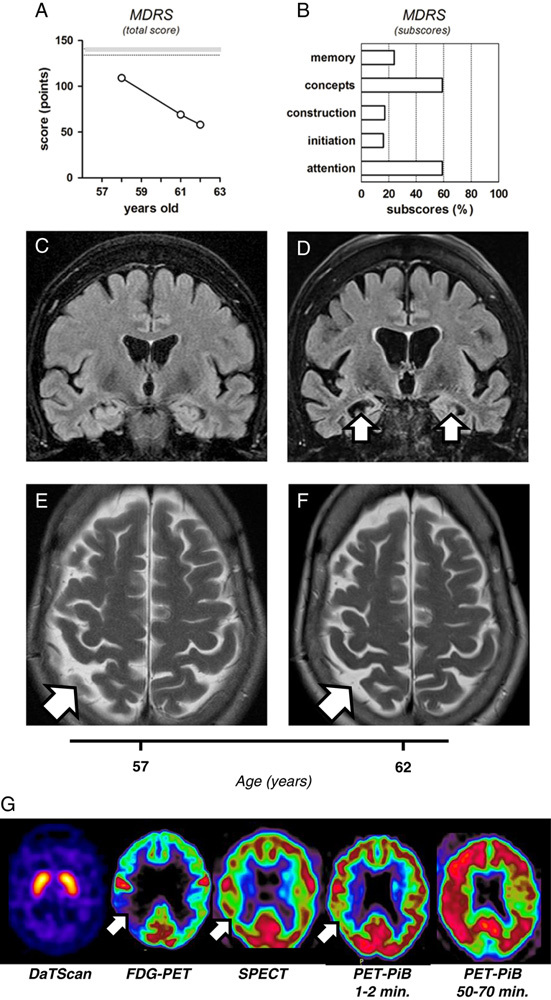
Neurocognitive and brain structural and metabolic assessment of the reported patient. A, Changes in the total score of Mattis dementia rating scale (MDRS), indicating progressive cognitive decline (109/144, 69/144, and 58/144, at the age of 58, 61, and 62, respectively). Shadowed range indicated on the panel represents a 25 to 75 percentile interval, previously established for the population younger than 70 years old. B, Specific MDRS subscores collected at the age of 62, and expressed as a percentage of the maximal possible value for each subscore. C-D, The coronal FLAIR imaging, demonstrating a clear progression of the hippocampal atrophy (white arrows) within 5 years of follow-up, from the age of 57 (C) to 62 (D). Note the right hemispheric dominance of the hippocampal atrophy and consecutive dilation of the temporal horns of the lateral ventricles. E-F, The axial T2 shows a mild parietal atrophy (white arrows), again with right hemispheric predominance, which only slowly progressed between the age to 57 (E) and 62 (F). Note the absence of microvascular white matter lesions. G, Metabolic neuroimaging. DaTScan imaging shows normal uptake of the tracer in the caudate and putamen. FDG-PET indicates global decrease in cerebral metabolism, particularly severe in the right temporo-parietal region (white arrow). SPECT reveals generalized hypoperfusion, slightly predominant on the right side (white arrow). Positron-emission tomography with Pittsburgh compound-B (PET-PiB) shows decreased tracer activity after 1 to 2 minutes, especially in the right temporo-parietal region (white arrow), and massive and widespread retention of PiB after 50 to 70 minutes, again with right predominance.
Despite major neuropsychological deficits, brain magnetic resonance imaging (MRI) revealed no abnormalities at the age of 57 (Fig. 1C), and mild atrophy at 62 (Fig. 1D), with no white matter hyperintensities in T2-weighted images (Figs. 1E). However, the progression of cerebral atrophy over this 5-year period was accelerated, particularly in the right hippocampus (Figs. 1C, D; Scheltens MTA score progression from 1 to 2). Moreover, functional imaging at the age of 62 showed extensive deficits compatible with advanced AD. In fMRI sequences, ASL perfusion and CO2BOLD reserve were decreased in the temporal and parietal lobes (not shown). FDG-PET showed profound temporo-parietal hypometabolism, with right-side predominance (Fig. 1G). SPECT revealed marked hypoperfusion, with similar regional distribution (Fig. 1G). Electroencephalography was abnormal, displaying generalized intermittent theta-waves, with temporal predominance (not shown). Only DaTScan appeared normal, indicating that dopaminergic circuits were not primarily affected (Fig. 1G). Most importantly, PET-PiB revealed massive amyloid deposition with a distribution pattern typical for AD (Fig. 1G), concordant with a parallel decrease in CSF Aβ42 level (320 pg/mL). Notwithstanding already advanced dementia, the CSF tau/p-tau concentrations remained low within a 2-year interval (244.5±44.5 and 52.5±15.5 pg/mL, respectively).
Family history revealed subjective memory impairment in the proband’s daughter (aged 44), and psychotic episodes in the proband and at least 2 of her children (Fig. 2A). Copy number variant analysis showed in the proband and her daughter a heterozygous deletion in chromosome 9q21.2, eliminating chorein exons 70 to 73 (Fig. 2B; other family members not available). Exome-wide sequencing revealed no mutation in chorein, APP, PSEN1, PSEN2, APOE, nicastrin, Aph-1, CD147, TMP-21, clusterin, GAB2, ATXN1, CD3, PCDH11X, CR1, PICALM, BIN-1, EXOC3L2, MTHFD1L, SORL1, EPHA1, ABCA7, and CD2AP (not shown). The proband’s ApoE genotype was ε3/ε4.
FIGURE 2.

Chorein deficiency and its impact on amyloid processing. A, Family tree, with the proband indicated by black arrow. The proband and 2 of 3 of her children presented with neuropsychiatric disorders (black symbols; clinical details in the text). Clinical profile of other family members remained undetermined (white symbols). B, Illustration of the results of genetic analysis. Deleted exons in chorein are marked with gray asterisk. The alternative splicing site is marked with black asterisk, and the exon 69 (skipped in the canonical form of chorein) is shadowed. C–F, In vitro chorein knockdown system. The neuronal SH-SY5Y cells were transfected with specific, chorein-targeting siRNA (white bars) or negative control siRNA (black bars). Expression of chorein (C), as well as APP, PSEN1, PSEN2, and SORL1 (E) was assessed by quantitative PCR and expressed as a percentage of its respective negative control; chorein knockdown had no impact on the viability/proliferation of SH-SY5Y cells (D); concentration of Aβ40, but not Aβ42, was significantly decreased in supernatants from SH-SY5Y cells upon chorein knockdown (F); concentration of Aβ40 was low in the cerebrospinal fluid of the proband, as compared with mean reference values previously reported for healthy controls (black arrow and dashed line) and demented patients carrying PSEN1 pathogenic mutations (gray arrow and dashed line) (G). For quantitative PCR analysis 7 (n=7; C) or 4 (n=4; E) independent experiments were performed. For cell counting (D) and measurement of Aβ concentrations (F), 5 independent experiments were performed (n=5). All of the values in the panels (C–E) are presented as percentages of the respective control groups (means±SEM). The significance of the differences (chorein knockdown vs. control) was tested by the 2-tailed Mann-Whitney nonparametric test (E), or by the 2-tailed paired t test for the sets of data following normal distribution (C, D, F; normal distribution confirmed by Kolmogorov-Smirnov test). The difference between the means were considered significant if P<0.05. NS indicates statistically not significant; **P<0.01.
Chorein expression in SH-SY5Y cells was decreased by 50% (Fig. 2C), with no impact on viability/proliferation (Fig. 2D), total protein synthesis (not shown), or expression of APP, PSEN1, PSEN2, and SORL1 (Fig. 2E). Expression of the constituents of α- (ADAM-10, ADAM-17), β- (BACE-1), and γ- (Nicastrin, Aph-1, Pen-2) secretases, and of γ-secretase modulators (TMP21, CD147), was also unaffected (not shown). In contrast, the concentration of Aβ40 (but not Aβ42) in culture supernatants was significantly decreased upon chorein knockdown (470±70 vs. 579±63 ng/mL; Fig. 2F), and low Aβ40 level (50 pg/mL) was found in the proband’s CSF (Fig. 2G).
DISCUSSION
Both structural and functional imaging data indicate that early-onset dementia in this patient can be attributed to a primary neurodegenerative process. First, there were no MRI signs of vascular changes at 57 and 62 years. Second, the rate of hippocampal atrophy was largely increased, indicating that the medial temporal lobe was preferentially affected. Third, functional imaging revealed marked hypoperfusion and hypometabolism, particularly in temporo-parietal regions. This pattern is consistent with AD, and PET-PiB confirmed extensive amyloid deposition. Admittedly, some Parkinson disease-related dementing disorders can present with concomitant AD-type Aβ pathology; however, this is unlikely considering the patient’s preserved dopaminergic circuits (normal neurological status and DaTScan, no sensitivity to antipsychotics), and the diagnosis of early-onset AD seems highly probable. Although neuropathological verification was not available here, CSF markers correlate well with Braak staging and the observed sharp divergence between the amyloid and tau markers indicates that this is a tau-negative variant.
The genetic background of tau-negative AD remains unknown.1 In our patient, exome-wide analysis revealed a heterozygous deletion of chorein exons 70 to 73, indispensable for the protein's function.4 The well-established neuroprotective role of chorein makes it an attractive pathogenic candidate here. Importantly, many ChAC patients develop early dementia,5 indicating that chorein is involved in preservation of cognitive functions. Even though ChAC shows recessive inheritance pattern, chorein hemizygosity is not clinically silent, and reduction in chorein expression has a detrimental impact on different tissues.6 Interestingly, 2 siblings with heterozygous 495+1G>A mutation were recently reported, presenting with early-onset cognitive impairment.7 The cognitive deficit in these patients was not characterized in detail, but a dramatic loss of parvalbumin-positive cortical interneurons was found in the temporal lobe. Given involvement of these neurons in the pathogenesis of AD, it seemed conceivable that the heterozygous chorein deletion might be relevant to the proband’s AD-type dementia. Although the scarcity of available family members precluded association analysis, we attempted to address chorein’s implication from a functional perspective. Chorein belongs to the VPS13 family, and is abundantly expressed in the neocortex and hippocampus. The members of other VPS families as well as some functionally related proteins are known to control intracellular APP trafficking, and they are increasingly reported as risk or causative factors in neurodegenerative diseases, including AD.8 We therefore studied a possible link between chorein and amyloid in vitro, showing that reduction in chorein expression selectively decreases secretion of Aβ40, but not Aβ42 peptide. The reason of such selectivity is unclear. Aβ peptides of different length are generated in distinct cellular compartments. In the secretory pathways, Aβ42 is generated mainly in the ER, whereas Aβ40 in the trans-Golgi network.9 Thus, it can be speculated that being located in the Golgi compartment,3 chorein might preferentially influence Aβ40 generation and/or trafficking, and its deficiency could interfere with this process, resulting in an imbalance between Aβ40 and Aβ42 in favor of the latter. Increase in Aβ42/Aβ40 ratio is one of the principal pathomechanisms in AD, the ratio being probably more relevant than the total Aβ amount. At first glance, the 20% decrease in Aβ40 secretion upon chorein knockdown appears mild, but some PSEN mutations decrease Aβ40 to a similar extent, still being pathogenic.10 Moreover, the very low Aβ40 level in the proband’s CSF indicates that the impact of chorein knockdown on Aβ40 secretion might be underestimated in our in vitro experiments.
In conclusion, our findings suggest that heterozygous chorein deficiency affects amyloid processing and might be implicated in tau-negative AD, although its penetrance remains unknown. This hypothesis could not be assessed in this single-case study, and although cognitive impairment in the carriers of heterozygous chorein mutations was reported,7 the incidence of dementia in such individuals was never systematically investigated. Clinical manifestation of heterozygous chorein deficiency and the age of symptoms onset would be very likely modulated by additional (epi)genetic factors, and ApoE4 (a major AD risk factor) might have played a precipitating role in the reported case. Although our exome-wide screening did not disclose any other factors that could potentially synergize with chorein deletion, their presence cannot be formally excluded.
Supplementary Material
Supplemental Digital Content is available for this article. Direct URL citations appear in the printed text and are provided in the HTML and PDF versions of this article on the journal's Website, www.alzheimerjournal.com.
ACKNOWLEDGMENTS
The authors thank Professor Alfred Buck from the University Hospital of Zurich for performing PET-PiB analysis, and Terese Escarra, Eladia Ballmann-Croisier, Julien Prados, and Rafael Fernandez from the University Hospitals of Geneva for their excellent technical assistance. They are grateful to Professor Patrick R. Hof from the Fishberg Department of Neuroscience, Icahn School of Medicine at Mount Sinai, in New York, for his critical comments on the manuscript.
Footnotes
M.J.L. is the recipient of scientific fellowships from the University Hospitals of Geneva and the Vachoux Foundation.
The authors declare no conflicts of interest.
REFERENCES
- 1.Klünemann HH, Fronhöfer W, Werner-Füchtenbusch D, et al. Characterization of the kindred of Alois Alzheimer’s patient with plaque-only dementia. Alzheimer Dis Assoc Disord. 2006;20:291–294. [DOI] [PubMed] [Google Scholar]
- 2.Rampoldi L, Dobson-Stone C, Rubio JP, et al. A conserved sorting-associated protein is mutant in chorea-acanthocytosis. Nat Genet. 2001;28:119–120. [DOI] [PubMed] [Google Scholar]
- 3.Hayashi T, Kishida M, Nishizawa Y, et al. Subcellular localization and putative role of VPS13A/chorein in dopaminergic neuronal cells. Biochem Biophys Res Commun. 2012;419:511–516. [DOI] [PubMed] [Google Scholar]
- 4.Dobson-Stone C, Danek A, Rampoldi L, et al. Mutational spectrum of the CHAC gene in patients with chorea-acanthocytosis. Eur J Hum Genet. 2002;10:773–781. [DOI] [PubMed] [Google Scholar]
- 5.Tomiyasu A, Nakamura M, Ichiba M, et al. Novel pathogenic mutations and copy number variations in the VPS13A gene in patients with chorea-acanthocytosis. Am J Med Genet B Neuropsychiatr Genet. 2011;156B:620–631. [DOI] [PubMed] [Google Scholar]
- 6.Ichiba M, Nakamura M, Kusumoto A, et al. Clinical and molecular genetic assessment of a chorea-acanthocytosis pedigree. J Neurol Sci. 2007;263:124–132. [DOI] [PubMed] [Google Scholar]
- 7.Connolly B, Hazrati LN, Lang AE. Neuropathological findings in chorea-acanthocytosis: new insights into mechanisms underlying parkinsonism and seizures. Acta Neuropathol. 2014;127:613–615. [DOI] [PubMed] [Google Scholar]
- 8.Pottier C, Hannequin D, Coutant S, et al. High frequency of potentially pathogenic SORL1 mutations in autosomal dominant early-onset Alzheimer disease. Mol Psychiatry. 2012;17:875–879. [DOI] [PubMed] [Google Scholar]
- 9.Hartmann T, Bieger SC, Brühl B, et al. Distinct sites of intracellular production for Alzheimer’s disease Aβ40/42 amyloid peptides. Nat Med. 1997;3:1016–1020. [DOI] [PubMed] [Google Scholar]
- 10.Chávez-Gutiérrez L, Bammens L, Benilova I, et al. The mechanism of γ-secretase dysfunction in familial Alzheimer disease. EMBO J. 2012;31:2261–2274. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Digital Content is available for this article. Direct URL citations appear in the printed text and are provided in the HTML and PDF versions of this article on the journal's Website, www.alzheimerjournal.com.