

Abstract
The platinum drugs cisplatin, carboplatin, and oxaliplatin are highly utilized in the clinic and as a consequence have been extensively studied in the laboratory setting, sometimes by generating fluorophore-tagged analogs. Here, we synthesized two Pt(II) complexes containing ethane-1,2-diamine ligands linked to a BODIPY fluorophore, and compared their biological activity with previously reported Pt(II) complexes conjugated to carboxyfluorescein and carboxyfluorescein diacetate. The cytotoxicity and DNA-damage capacity of Pt-fluorophore complexes was compared to cisplatin, and the Pt-BODIPY complexes were found to be more cytotoxic with reduced cytotoxicity in cisplatin-resistant cells. Microscopy revealed a predominately cytosolic localization, with nuclear distribution at higher concentrations. Spheroids grown from parent and resistant cells revealed penetration of Pt-BODIPY into spheroids, and retention of the cisplatin-resistant spheroid phenotype. While most activity profiles were retained for the Pt-BODIPY complexes, accumulation in resistant cells was only slightly affected, suggesting that some aspects of Pt-fluorophore cellular pharmacology deviate from cisplatin.
Keywords: accumulation, trafficking, platinum, resistance
Introduction
Cisplatin (1, cis-[PtCl2(NH3)2]), carboplatin ([Pt(O,O′-cdbca)(NH3)2], cbdca = cyclobutane-1,1-dicarboxylate, Figure 1) and oxaliplatin ([Pt(ethanedioato-O,O′)(chxn)], chxn = 1R,2R-cyclohexane-1,2-diamine) are used in combination with other agents to treat a range of cancers. However, with the exception of most cases of testicular cancer, the treatment does not effectively eliminate tumors because they develop resistance to the drug. After entering cells, the platinum drugs exert their toxicity by binding to DNA and inducing apoptosis. Resistance is conferred through multiple cellular changes, including reduced uptake, reduced apoptotic signaling, up-regulated DNA damage repair mechanisms, altered cell cycle checkpoints, and disrupted assembly of the cytoskeleton [1]. Although the pleiotropic mechanisms underlying cisplatin resistance are well-documented, some mechanisms are still poorly understood and the clinical relevance of many mechanisms of cisplatin resistance remain unknown. There remains the need for further studies of mechanisms of action of platinum drugs, both alone and in combination with other therapeutics, to provide an understanding of why some new platinum agents such as BBR3464, picoplatin and satraplatin, have not succeeded in clinical trials [2].
Figure 1.

Structures of the compounds used in this study. For each platinum-dye complex, the Boc-protected dye was used as a control for comparison.
A persisting challenge when studying Pt-based drugs in biology is that there are few spectroscopic handles available for monitoring them, as there are for organic-based drugs [3]. For example, none of the Pt drugs are fluorescent in a way that is amenable to microscopy. In addition, radiolabeling with [3H] or [14C] is not feasible because the 6 protons on the ammine ligands of cisplatin exchange rapidly in water, and it contains no carbon. Carboplatin can be labeled on the ‘leaving’ cbdca ligand, limiting its utility [4, 5], but oxaliplatin’s chxn ligand can be labeled [6]. While isotopes of 15N and 195Pt are useful for mechanistic studies in solution (for example, by HSQC NMR) [7], these are not amenable to cellular studies. The main technique employed is an atomic detection technique such as GF-AAS or ICP-MS to measure bulk levels of Pt in cells because Pt is not endogenous to biology. However, little information about speciation and cellular localization of compounds can be gained. X-ray absorption spectroscopies such as X-ray absorption near-edge spectroscopy (XANES, for oxidation state) and X-ray fluorescence (XRF or SRIXE for cell localization) [8, 9], and nano-scale secondary ion mass spectrometry (NanoSIMS) are possible [10]. Bierbach and colleagues have reported a post-labeling system for determining sub-cellular localization of modified Pt complexes [11], and fluorescent sensors that can be applied to Pt-treated cells to image Pt species [12–14] have also emerged. However, these techniques require sample fixation, and therefore live-cell temporal imaging is not possible.
To address these challenges, a number of Pt complexes containing tethered fluorophores that retained the traditional structure-activity requirements for Pt(II) anticancer complexes (square planar complexes with cis leaving groups and amines) have been generated [15]. Reedijk and coworkers reported the synthesis of a Pt(II) complex conjugated to carboxyfluorescein diacetate (CFDA) through an ethane-1,2-diamine (en) ligand, in which the CFDA became fluorescent upon hydrolysis of the acetate groups to carboxyfluorescein (CF) [16]. Howell and co-workers subsequently used this complex (termed FDDP, fluorescent cisplatin, numbered 5 in the present paper) to show that the molecule was trafficking to vesicular compartments, with little detectable nuclear localization [17, 18]. Farrell and co-workers reported a cis-ammine/amine cisplatin analog tethered to N-(7-nitro-2,1,3-benzoxadiazol-4-yl)ethane-1,2-diamine (NBD), which undergoes lysosomal localization and was shown to be cytotoxic, but about 10-fold less so than cisplatin [19]. There is also a commercial ‘universal linkage system’ (ULYSIS) for DNA labeling based on the affinity of platinum for nucleobases [20]. These complexes do not conform to platinum structure-activity rules and contain only a single leaving group, having the general formula [PtCl(en)(NH2R)]+, with the primary amine tethered to a fluorophore (R), of which a number have been produced including cyanines, fluorescein, and rhodamine [21]. While not biologically active, the ULYSIS AlexaFluor-488 dye has been used to demonstrate defective trafficking of Pt in cisplatin-resistant cell lines [22]. As examples of more recent work, complexes tethered to BODIPY (described below) have been reported [23], including a cationic Pt complex for imaging mitochondrial localization [24]. Platts and co-workers reported platinum trimethyl bipyridyl thiolates that could emit in the near infrared for cellular imaging [25].
An obvious limitation to adding a fluorophore to a small inorganic complex is that it may significantly alter the physicochemical properties and biological activity of the complex. There has been no systematic assessment of the pharmacology of these analogs, and it is not clear whether the fluorescent analogs are an appropriate model for the cellular behavior of cisplatin and its congeners. For instance, none of the Pt-fluorophore complexes studied have reported significant nuclear localization.
A significant number of studies exist reporting the synthesis of Pt complexes conjugated to a cytotoxic ligand, such as a DNA intercalator that happens to also be fluorescent, in an attempt to generate a more toxic or DNA-specific experimental therapeutic. These constructs are by definition not appropriate for studying Pt localization as the fluorophore itself is not ‘innocent’ by design, and the fluorophore may strongly influence the cellular localization of the Pt center [3, 26]. Therefore, we synthesized several complexes reported in the literature, along with two new boron-dipyrromethene (BODIPY) -conjugated complexes that differ in the linker employed (amide and propanamide). It should be emphasized that probe molecules such as those described in this paper are not candidate experimental therapeutics, though there is also a rich literature describing Pt complexes tethered to intercalators [26]. The inherent fluorescence of many intercalators means that the cellular fate of these complexes can be visualized, but they are designed to modify the biological behavior of the Pt and as such do not represent candidate probes for the fate of Pt [3].
In this study, we assessed these complexes for solvent stability in dimethylsulfoxide (DMSO) and dimethylformamide (DMF), lipophilicity (distribution coefficient, logD), cytotoxicity against parental and cisplatin-resistant cancer cell lines, and cellular accumulation. Cellular localization was assessed using live-cell confocal microscopy at 1 and 24 h, and DNA damage was inferred by assessing phosphorylation of the histone H2A family member H2A.X in fixed cells. Cisplatin and a BODIPY-conjugated complex were also assessed for their activity and distribution in three dimensional (3D) culture in Matrigel, a gelatinous mixture of proteins derived from mouse sarcoma cells which is often used as a basement membrane matrix in 3D cell culture [27]. While this manuscript was in preparation, Miller et al. reported the synthesis of other BODIPY-conjugated cisplatin and carboplatin analogs, and reported their cytotoxicity and DNA-damage activities. A comparison with the activity for complexes synthesized in this study is made, and we qualify the conclusions in the studies of Miller et al by reporting that the activity of BODIPY-Pt is inactivated by the solvent DMSO [23]. By evaluating the efficacy of these fluorophore-conjugated Pt complexes as analogs for cisplatin, this study holds implications for further in vivo studies of platinum-based compounds for tumor treatment.
Materials and Methods
Materials
All reagents and solvents were obtained from commercial sources and used as received unless otherwise noted. 5(6)-Carboxyfluorescein N-succinimidyl ester (CF SE), cisplatin, dimethylsulfoxide (DMSO), dimethylformamide (DMF), and 1-octanol were purchased from Sigma-Aldrich. 6-Carboxyfluorescein diacetate N-succinimidyl ester (CFDA SE) was purchased from Berry&Associates, and BODIPY LF SE was obtained from Life Technologies. [PtCl2(en)] was purchased from Alpha Aesar. Analytical HPLC analysis was performed on an Agilent 1200 Series instruments equipped with multi wavelength detector and Evaporative Light Scattering Detector (ELSD) using an Agilent Eclipse Plus column (4.6×50mm, 3.5μm) with a flow rate of 1 mL/min. Solvent A was water, solvent B was ACN, and a linear gradient of 5 % B to 95 % B over 10 min was used. APCI mass spectrometry was performed on 6130 Quadrupole LC/MS Agilent Technologies instrument equipped with diode array detector. 1H-NMR spectra were recorded with a Varian spectrometer operating at 400MHz. Chemical shifts are reported in parts per million (δ) and are referenced to tetramethylsilane (TMS). 195Pt-NMR spectra were acquired at 86 MHz (400MHz 1H) on a Bruker AV III (Bruker-Biospin, Switzerland) spectrometer using a spin echo (SE) sequence with a 10us echo time and a 24 kHz B1 field. Except for increased spectral quality, there was no discernable difference between the SE and simple 90-pulse-aquire spectra. Chemical shifts are referenced to K2[PtCl4] in D2O.
Synthesis
Compounds 3–10 were examined for biological activity in this study. Compounds 3–9 were previously reported as outlined below, and 10 is novel. Figure 2 illustrates the method of synthesis of the three BODIPY-containing compounds 8 (BODIPY-Boc), 9 (BODIPY-Pt) and 10 (BODIPY-prop-Pt). The Pt-free Boc-protected fluorophore derivatives 4 (CFDA-Boc), 6 (CF-Boc) and 8 (BODIPY-Boc) were obtained in the reaction of N-Boc-ethylenediamine with the corresponding N-succinimidyl ester of dye in the presence of base. To synthesize the Pt-containing BODIPY compounds, the precursors 11 and 12 were synthesized as described previously [16, 28]. The [(2,3-Diaminopropionic acid)(dichloro)platinum(II)] complex 14 was synthesized in a one-step procedure [29]. NMR, MS and IR structural characterization are in agreement with the published data for all previously reported compounds. The BODIPY FL-Pt analog 9 (BODIPY-Pt) was obtained by the reaction of 12 with BODIPY FL succinimidyl ester in the presence of base. Compound 10 was prepared by reaction of 14 with an amine derivative of BODIPY FL (15) using a standard coupling procedure (Figure 2). The fluorescein-containing Pt complexes 5 (CFDA-Pt) and 7 (CF-Pt) were synthesized as previously reported [16, 17].
Figure 2.

Scheme of syntheses for the BODIPY-containing compounds 8 (BODIPY-Boc), 9 (BODIPY-Pt), and 10 (BODIPY-prop-Pt).
tert-Butyl (2-(3-(5,5-Difluoro-7,9-dimethyl-5H-4λ4,5λ4-dipyrrolo[1,2-c:2′,1′-f][1,3,2]diazaborinin-3-yl)propanamido)ethyl)carbamate; BODIPY-Boc (8)
To a solution of BODIPY FL-SE (30 mg, 0.077 mmol) in anhydrous dichloromethane (10 mL) N-Boc-ethylenediamine (14 uL, 0.084 mmol) was added followed by triethylamine (12 uL, 0.084 mmol). The reaction was allowed to stir for 20 min and then was transferred to a separatory funnel. The organic layer was washed with water (2 × 6 mL) and brine (1 × 6 mL). The solvent was evaporated yielding 28 mg (Y 84%) of dark orange solid. 1H-NMR (400 MHz, CDCl3): δ 7.07 (s, 1H), 6.87 (d, 1H, J = 3.91 Hz), 6.27 (d, 1H, J = 3.91 Hz), 6.18 (bs, 1H), 6.11 (s, 1H), 4.91 (bs, 1H), 3.23–3.31 (m, 4H), 2.60–2.65 (m, 4H), 2.55 (s, 3H), 2.23 (s, 3H), 1.41 (s, 9H). LC/MS (APCI) m/z: 415 (M+-F)+.
[(3-(5,5-difluoro-7,9-dimethyl-5H-4λ4,5λ4-dipyrrolo[1,2-c:2′,1′-f][1,3,2]diazaborinin-3-yl)propan)aminomethyl)-1,2-ethylenediamine}dichloroplatinum(II)]; BODIPY-Pt (9)
To a solution of 12 (35 mg, 8.9×10−2 mmol) in deionized water (3 mL) a solution of BODIPY FL SE (35 mg, 8.9×10−2 mmol) in DMF (6 mL) followed by triethylamine (25 μL, 0.178 mmol) was added dropwise at room temperature. The reaction mixture was stirred for 2 h. Progress of the reaction was followed by ELSD-HPLC. The reaction mixture was diluted with deionized water (50 mL) and freeze-dried. Ice-cold water (7 mL) was added to the resulting powder and the mixture was stirred for 5 min. The precipitate was filtered off and washed with ice-cold water (3 × 5 mL), ethanol (2 × 5 mL) and diethyl ether (2 × 5 mL) affording 1 as dark orange solid (40.5 mg, Y 72%). 1H-NMR (400 MHz, DMF-d7): δ 8.16 (t, 1H, J = 5.48 Hz), 7.72 (s, 1H), 7.14 (d, 1H, J = 3.91 Hz), 6.42 (d, 1H, J = 3.92 Hz), 6.33 (s, 1H), 5.61 (d, 1H, J = 6.65 Hz), 5.36 (bs, 2H), 5.09 (t, 1H, J = 9.39 Hz), 3.62–3.68 (m, 1H), 3.48–3.53 (m, 2H), 3.24 (t, 2H, J = 7.63 Hz), 3.07–3.08 (m, 1H), 2.65–2.70 (m, 3H), 2.54 (s, 3H), 2.31 (s, 3H). 195Pt-NMR (86 MHz, DMF-d7): δ-2274. HRMS calculated for C17H24BCl2F2N5OPt (M + H) 628.1, found 628.1.
[(2,3-Diamino-N-(2-(3-(5,5-difluoro-7,9-dimethyl-5H-4λ4,5λ4-dipyrrolo[1,2-c:2′,1′-f][1,3,2]diazaborinin-3-yl)propanamido)ethyl)propanamide)dichloroplatinum (II)]; BODIPY-prop-Pt (10)
A solution of BODIPY FL SE (5 mg, 1.28×10−2 mmol) in dichloromethane (1.5 mL) was added dropwise to a solution of ethylenediamine (1.28 μL, 1.92×10−2 mmol) in dichloromethane (1.5 mL) and the mixture was stirred for 30 min. Progress of the reaction was monitored by ELSD-HPLC. The solvent was removed under reduced pressure and the resulting dark orange recidue was dried under high vacuum for 36 hours. The crude product 15 (4.3 mg, quantitative) was used in the next step of the synthesis. 1H-NMR (400 MHz, DMF-d7): δ 7.94 (bs, 1H), 7.71 (s, 1H), 7.12 (d, 1H, J = 3.92 Hz), 6.42 (d, 1H, J = 3.92 Hz), 6.32 (s, 1H), 3.21–3.26 (m, 4H), 2.60–2.64 (m, 4H), 2.53 (s, 3H), 2.313 (s, 3H). To a solution of 15 (4.7 mg, 1.27×10−2 mmol) in DMF (0.7 mL), N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (2.4 mg, 1.27×10−2 mmol), 1-hydroxybenzotriazole hydrate (1.7 mg, 1.27×10−2 mmol) and triethylamine (4.5 μL, 3.2×10−2 mmol) were added, and the mixture was stirred for 15 min at room temperature. A solution of 10 (4.7 mg, 1.27×10−2 mmol) in DMF (1 mL) was then added and the reaction mixture was stirred for 2 h. The mixture was diluted with deionized water (30 mL) and freeze-dried. The resulting residue was then treated with ice-cold water (4 mL), stirred for 5 min and filtered off. The solid was subsequently washed with ice-cold water (3 × 2 mL), ethanol (2 × 1.5 mL) and diethyl ether (2 × 1.5 mL) affording 2 as dark orange solid (5.3 mg, Y 61%). 1H-NMR (400 MHz, DMF-d7): δ 8.44–8.56 (m, 1H), 7.97–7.99 (m, 1H), 7.72 (s, 1H), 7.14 (d, 1H, J = 3.99 Hz), 6.42 (d, 1H, J = 3.99 Hz), 6.33 (s, 1H), 5.87–5.90 (m, 1H), 5.45–5.51 (m, 2H), 4.95–5.01 (m, 1H), 3.66–3.67 (m, 1H), 3.20–3.38 (m, 5H), 2.79–2.82 (m, 2H), 2.59–2.63 (m, 3H), 2.54 (s, 3H), 2.32 (s, 3H). 195Pt-NMR (86 MHz, DMF-d7): δ-2269. HRMS calculated for C19H27BCl2F2N6O2Pt (M + H) 686.1, found 686.1.
Mass spectrometry
Solutions of BODIPY-Pt were prepared using DMSO or DMF at a concentration of 10 μM. Solutions were kept at room temperature in the dark for 48 h prior to analysis. Accurate mass data were obtained on a Waters Premiere LCT ESI time-of-flight mass spectrometer operated in the positive ion W-mode at 10 K resolution. The high-performance liquid chromatography solvent pump was operated at 200 μL/minute and the solvent composition was 50:40:10 water:methanol:acetonitrile. Samples were flow injected via a sample loop and ionized in the electrospray ion source. The electrospray capillary was operated at 3.5 kV and at 350 °C. The desolvation gas was purified nitrogen. All test solutions were made fresh and incubated at room temperature in the dark prior to analysis. The MS data were analyzed using Waters MassLynx 4.1 software.
Distribution coefficient (log D) measurement
Distribution coefficient measurements were performed by making 200 μM solutions of compounds in 3 mL PBS, layered with 3 mL 1-octanol, vortexed briefly, and were shaken in the dark for 4 h. Mixtures were then allowed to settle at room temperature. Fluorescence in the 1-octanol and aqueous layers was determined using an Ibidi optical-bottom 96-well plate fluorimeter. All samples were run in triplicate and logD was calculated as log([oct]/[aq]). Data is reported as mean ± SD.
Cell lines and cell culture
This study used the KB-3-1 human cervical carcinoma cell line (a subline of HeLa), and its cisplatin-resistant sublines KB-CP.5 and KB-CP20. Originally, KB-CP.5 and KB-CP20 cells were selected in a single step in 0.5 μg/mL (1.6 μM) and in multiple steps up to 20 μg/mL (0.103 mM) cisplatin, respectively, in our laboratory as described previously [30]. All cell lines were thawed immediately before experimentation, and cell lines are characterized by NCI using short tandem repeat profiling. The cisplatin stock solutions used for culturing CP.5 and CP20 cells were prepared in PBS. The cisplatin-resistant cells were maintained in the presence of cisplatin, which was removed from growth medium 3 days prior to all experiments. All cell lines were grown as monolayer cultures at 37°C in humidified 5% CO2, using DMEM with 4.5 g/L glucose (from Invitrogen), supplemented with L-glutamine, penicillin, streptomycin, and 10% FBS (BioWhittaker).
Cytotoxicity
Cell survival was measured by the Cell-Titer Glo (Promega) assay. Cells were seeded at a density of 1,000 cells per well in 96-well opaque plates and incubated at 37 °C in humidified 5% CO2 for 24 h. Compound was serially diluted 1:3 (DMEM used as diluent) and added to plates to achieve the intended final concentrations. Solvent (DMSO and DMF) tolerance testing up to 8.3% under identical conditions at 24, 48, and 72 h incubation time points confirmed growth of all cell lines was unaffected. Cells were then incubated an additional 72 h, and the CellTiter-Glo assay was performed according to the manufacturer’s instructions (Promega). Bioluminescence values were measured on a Victor3V spectrophotometer (PerkinElmer). All CellTiter-Glo assays were performed in triplicate. IC50 values were defined as the drug concentrations required to reduce cellular proliferation to 50% of the untreated control well. Statistical analysis was performed using Prism 6 Prism 6 (GraphPad Software). All data are expressed as mean ± SD.
Three dimensional cell culture
Each well of a white 96-well plate was coated with 40 μL of 5 mg/mL matrigel and allowed to gel for 30 min at 37 °C. Cells were then seeded over this base layer at a density of 1000 cells per well in 100 μL of 2.5 mg/mL Matrigel (Corning Matrigel Matrix) and incubated at 37 °C and 5% CO2 for one day to culture single-cell suspensions, or four days to allow spheroid formation. Cells were then treated with compound as previously described. After drug incubation cytotoxicity was determined using CellTiter-Glo assays as previously described and according to the manufacturer’s directions.
H2AX immunohistochemical staining
KB-3-1 cells were fixed and stained with Phospho-Histone H2A.X (Se139; 20E3) Rabbit mAb (Alexa Fluor 647 Conjugate; all components from Cell Signaling Technology) following the manufacturer’s instructions and as previously described [31]. Cells were seeded at 5 × 104 cells per chamber in 8-chamber coverslips and incubated for 24 h. Cells were then incubated with Pt complexes for 24 h, after which they were washed with PBS, fixed in 4% paraformaldehyde, and blocked with blocking buffer (PBS/5% normal horse serum/0.3% Triton X-100) for 1 h. Diluted antibody was then applied, and cells were incubated overnight at 4°C. Cells were then washed and stained with DAPI nuclear stain (2 μg/mL for 1 h) and imaged using a Zeiss LSM 710 NLO confocal microscope.
Flow cytometry
Cells were trypsinized and 200,000 cells were added to 5 mL FACS tubes. Cells were then centrifuged, washed twice with IMDM supplemented with 5% fetal bovine serum, and resuspended in compound diluted in IMDM supplemented with 5% fetal bovine serum (final concentration 25 μM, only fluorescent compounds were examined). Cells were incubated at 37°C for 45 min, then washed as above and resuspended in PBS. Tubes were immediately placed on ice and analyzed by flow cytometry. FL-1 fluorescence intensities were measured using a FACSCanto flow cytometer equipped with a 488 nm argon laser for a total of 10,000 events per sample. Gating analysis was performed using FlowJo flow cytometry software.
Confocal microscopy
For two-dimensional confocal localization imaging, KB-3-1 and KBCP.5 cells were seeded at a density of 5 × 104 cells per well in 8-well chamber slides and allowed to attach at 37°C in humidified 5% CO2 for 24 h. For three-dimensional confocal localization imaging, each chamber was coated with 200 μL of 2.5 mg/mL Matrigel. Then 2000 cells in 200 μL of 2.5 mg/mL Matrigel were seeded in each well and were cultured for eight days at 37°C in humidified 5% CO2. Compound at a concentration of 10 μM was added directly to each well and cells were incubated for either 1 or 24 h for two-dimensional culture and 1 h for three-dimensional culture. Hoechst nuclear stain was added to each two-dimensional culture well (1 μg/mL) 1 h prior to imaging. Media were aspirated and cells were rinsed with PBS before imaging on a Zeiss 710 NLO confocal microscope.
Results and discussion
Design and synthesis of platinum-fluorophore conjugate
Our aim was to synthesize Pt-fluorophore complexes and compare then with cisplatin (1) and [PtCl2(en)] (2) (all structures shown in Figure 1). This latter compound was particularly relevant as the ethane moiety serves as the point for functionalization (3) and conjugation of fluorophores (synthetic schemes for BODIPY compounds shown in Figure 2). Based on previous literature, we prepared Boc-protected versions of CFDA (termed here CFDA-Boc, 4) and CF (termed here CF-Boc, 6), as well as CFDA (CFDA-Pt, 5) and CF (CF-Pt, 7) conjugated to Pt via an amide linker [17, 18]. We synthesized a Boc-protected BODIPY fluorophore (BODIPY-Boc, 8) and two BOPDIPY-conjugated Pt complexes that differ in the linker used: one with an amide linker (BODIPY-Pt, 9) and one with a propanamide linker (BODIPY-prop-Pt, 10). While this manuscript was in preparation, BODIPY-Pt (9) was reported by Miller et al [23], using an identical synthetic strategy (Figure 2) that proceeded via reaction of BODIPY FL succinimidyl ester (BODIPY FL SE, Figure 2) conjugated to the terminal amine of 12. The complexation to Pt did not result in altered excitation and emission maxima (not shown), consistent with previous reports [23]. 195Pt NMR spectra of BODIPY-Pt and BODIPY-prop-Pt (Supplementary Figure 1B, 1C, respectively) revealed a single peak in the range consistent with a PtN2Cl2 coordination sphere reported in the literature [32, 33], and distinct from that of K2[PtCl4] (Supplementary Figure 1A). It should be noted that an earlier report of ‘bodipy-cisplatin’ exists (prior to Miller et al.) in the literature but no details of synthesis, characterization or structure were included in that report [34].
Lipophilicity of fluorescent compounds was assessed by distribution coefficient ([octanol]:[phosphate-buffered saline, pH 7.2] partition, log D) using fluorescence measurements and compared to literature data for the parent Pt complexes cisplatin (1) and [PtCl2(en)] (2) (Table 1) [35]. The CFDA-Pt and CF-Pt complexes partitioned preferentially to the aqueous layer (both logD values ~ −2), similar to 1 and 2. The Pt complexes with BODIPY both had positive logD values (BODIPY-Pt logD = 0.98, BODIPY-prop-Pt logD = 0.66). The slightly lower logD of BODIPY-prop-Pt can be accounted for based on the second hydrophilic amide incorporated into the propanamide linker. Miller et al. reported a logP value of 0.58 for BODIPY-Pt, which is slightly lower than our report, but as their partition was into unbuffered water (which they defined as logP) our discrepancy is not unreasonable.
Table 1.
Cytotoxicity (IC50, μM) of compounds against parental KB-3-1 and cisplatin-resistant KB CP.5 and KB CP20 cell lines. Distribution coefficients (logD) of each compound and percent increase in molecular weight compared with cisplatin shown for Pt complexes.
| Compound Solvent | Percent mass increase | LogD | IC50 (μM) | ||
|---|---|---|---|---|---|
|
| |||||
| KB 3-1 | KB CP.5 | KB CP20 | |||
| Cisplatin (1) | |||||
| Saline | - | [−2.28 ± 0.046]1 | 2.5 ± 0.2 | 25.1 ± 3.4 | >100 |
| DMF | 1.8 ± 0.1 | 23.9 ± 5.3 | 124.7 ± 31.5 | ||
|
| |||||
| [PtCl2(en)] (2) | |||||
| Saline | - | [−2.30 ± 0.019]1 | 2.7 ± 1.9 | 94.9 ± 8.3 | >100 |
| DMF | 1.0 ± 0.6 | 150.0 ± 11.8 | >500 | ||
|
| |||||
| Pt-Boc (3) | |||||
| DMF | 133% | ND | 16.7 ± 0.9 | 172.4 ± 28.7 | 431.9 ± 96.9 |
|
| |||||
| CFDA-Boc (4) | |||||
| DMF | - | −0.063± 0.014 | 116.1 ± 8.9 | 104.1 ± 2.4 | 131.6 ± 5.7 |
|
| |||||
| CFDA-Pt (5) | |||||
| DMF | 308% | −2.09 ± 0.011 | 71.6 ± 29.5 | 276.1 ± 38.4 | 440.3 ± 40.9 |
|
| |||||
| CF-Boc (6) | |||||
| DMF | - | −2.35 ± 0.037 | >500 | >500 | >500 |
|
| |||||
| CF-Pt (7) | |||||
| DMF | 265% | −2.66 ± 0.009 | >500 | >500 | >500 |
|
| |||||
| BODIPY-Boc (8) | |||||
| DMF | – | 2.15 ± 0.023 | 86.7 ± 0.8 | 86.9 ± 4.4 | 53.6 ± 2.9 |
|
| |||||
| BODIPY-Pt (9) | |||||
| DMF | 222% | 0.98 ± 0.04 | 35.7 ± 1.5 | 103.0 ± 17.0 | 226.5 ± 47.2 |
| DMSO | 391.7 ± 35.2 | 459.7 ± 113.2 | >500 | ||
|
| |||||
| BODIPY-prop-Pt (10) | |||||
| DMF | 251% | 0.66 ± 0.011 | 21.5 ± 5.8 | 44.6 ± 2.7 | 192.5 ± 50.3 |
Hall, M.D.; Amjadi, S.; Zhang, M.; Beale, P.J.; Hambley T.W. J. Inorg. Biochem. 2004, 98, 1614
Excipient interactions and utility
Given the limited aqueous solubility of the platinum-fluorophore complexes, an organic solvent was required to create stock solutions of agents. We recently reported the deleterious effects of DMSO on the biological activity of platinum(II) complexes [31], caused by ligand replacement and coordination to DMSO via its sulfur [36]. A number of alternative solvents have been utilized in the literature for drug dissolution for biological experiments, one of which is DMF. There is surprisingly little information on the inherent cytotoxicity of DMF. The highest percentage DMF utilized in cytotoxicity assays is 0.25% in cell culture media, with subsequent 1:3 serial dilutions of drug reducing it further (see Materials and Methods). We have previously shown that DMF had no deleterious effect on cisplatin cytotoxicity when used as the stock solution solvent, but that DMF did cause cell death at higher concentrations than would be utilized in in vitro assays (3%) [31]. To determine whether DMF could be utilized as a general solvent for Pt complexes in biological assays, its effect on cellular viability was examined to ensure solvent toxicity would not exclude its use. KB-3-1 cells were seeded, then exposed to varying concentrations of DMF from 8.3 – 0.001 % (1.1 mM – 164 μM). After 4, 24, 48 and 72 h, cellular viability was determined, with longer incubation times producing toxicity at higher percentages (Supplementary Figure 2). No effect on viability occurred at 4 h, and at 24 and 48 h concentrations lower than 1% did not affect viability. After 72 h, 0.34% DMF had little effect on cell viability, with 1% DMF inhibiting cell growth almost completely. This indicates that at the percentages of DMF utilized in biological assays, there should be no contributing effect from DMF.
To assess DMF’s utility, we examined whether it modified Pt-Boc. This was examined in two ways. First, Pt-Boc (3) was dissolved in DMSO-d6 and DMF-d7 and stored in the dark at room temperature. 1H NMR spectra of the solutions were collected over time (0 h to 1 week, Figure 3).
Figure 3.

Timecourse 1H NMR spectra of Pt-Boc dissolved in (a) DMF and (b) DMSO. Stability of Pt-Boc in solution was measured at 0 h, 24 h, 48 h, 72 h, 1 week, and in the case of DMF 10 weeks. The NH region of Pt-Boc is highlighted in the spectra by a red bar.
As expected based on previous work, DMSO modified Pt-Boc. The 0 h sample spectrum was collected immediately after dissolution, and examination of peaks in the NH region (highlighted in Figure 3, en ligand at 5.3 and Boc-protected amine at 6.7) revealed minor peaks already appearing upfield of the parent peaks due to reaction with DMSO. The reaction with DMSO appeared to be complete at 24 h because there were no evident subsequent changes to the spectrum, and at least two products existed in solution, with little to no parent compound remaining. In contrast, DMF resulted in no noticeable spectral changes after storage at room temperature for 1 or 10 weeks. Second, these observations were supported by ESI-MS (positive mode) spectra of DMF and DMSO solutions of Pt-Boc (10 μM) collected after 48 h. In DMF, the parent peak corresponded to unmodified Pt-Boc (m/z = 647.1, Pt-Boc+NH4+), and no peaks could be assigned to DMF-related adducts (Supplementary Figure 3). The mass spectrum of Pt-Boc in DMSO revealed a base peak corresponding to the replacement of a Cl ligand by DMSO (m/z = 671.1, [PtCl(DMSO)(enBoc)]+), consistent with the reaction products of cisplatin and other platinum(II) complexes previously reported [31, 37, 38].
We compared the cytotoxicity of cisplatin and [PtCl2(en)] dissolved in DMF and in saline (consistent with the clinically formulated solution) towards KB-3-1 cells. This confirmed that DMF did not deactivate either complex (Table 1). Furthermore, cisplatin-resistant KB CP.5 cells demonstrated the same degree of cross-resistance to both complexes in either solvent, but IC50 values were not reached against the highly resistant KB CP20 cells. The maximum concentration tested was lower, as the saline stock solutions are prepared to 3 mM, while DMF stock solutions can be prepared at higher concentrations. As expected, [PtCl2(en)] was less cytotoxic than cisplatin [39].
Activity of platinum-fluorophore complexes
Compounds were tested for their inherent cytotoxicity towards parent KB-3-1 cells, and two cisplatin-resistant sub-lines, KB CP.5 and KB CP20, which show low- and high-grade resistance respectively. The Boc-protected dyes were also assessed as controls to ascertain whether the dyes possessed biological activity independent of Pt. Cisplatin and [PtCl2(en)] were tested as positive controls. Consistent with previous results for cisplatin, CP.5 cells showed resistance to cisplatin (10-fold) or [PtCl2(en)] (40-fold). Against CP20 cells, [PtCl2(en)] did not achieve an IC50 (up to 500 μM), and were 60-fold resistant to cisplatin. Pt-Boc was less cytotoxic than its analog [PtCl2(en)] (KB-3-1 IC50 = 16.7 ± 0.9 μM), but cross-resistance profiles were similar (CP.5 cells 10-fold resistant, CP20 cells 26-fold resistant), indicating that the conjugation to the ethane-1,2-diamine backbone did not inactivate the complex.
The BODIPY-Pt complex showed activity against KB-3-1 cells (IC50 = 35.7 ± 1.5 μM), and CP.5 and CP20 cells showed cross-resistance. Miller et al. reported the IC50 of this complex against two ovarian cancer cell lines to be >700 μM, however they dissolved the complex in DMSO [23]. When we dissolved BODIPY-Pt in DMSO, the IC50 against KB-3-1 was reduced by an order of magnitude to 391.7 ± 35.2 μM, confirming that the lack of activity reported by Miller et al. was probably due to inactivation by DMSO, rather than the inherent inactivity of the complex. The BODIPY-prop-Pt complex showed the greatest cytotoxicity of any Pt-dye complex against KB-3-1 cells (IC50 = 35.7 ± 1.5 μM), with cross-resistance to CP.5 (2-fold) and CP20 (10-fold) cells.
The CFDA-Pt complex showed low-level activity against KB-3-1 cells (IC50 = 71.6 ± 29.5 μM), and both CP.5 (4-fold) and CP20 (6-fold) cells showed cross-resistance. The Boc-protected dye CFDA-Boc also demonstrated cytotoxicity towards cells (IC50 = 116.1 ± 8.9 μM), weaker than that of the Pt-CFDA complex, but with equal activity against the cisplatin-resistant cells. The CF-Pt complex, representing the hydrolyzed dye form of CFDA-Pt, did not demonstrate cytotoxicity up to 500 μM, nor did CF-Boc (Table 1). This is likely due to the negative charge of the complex conferred by the organic acid groups, and is reflected in its slightly lower logD value (−2.66).
DNA damage potential
To assess whether the Pt-dye complexes could elicit DNA damage and cell killing normally elicited by cisplatin [2], we examined phosphorylation of the histone H2A family member H2A.X. H2A.X is phosphorylated at serine 139 (termed γH2A.X) as part of the cellular response to DNA damage, recruiting DNA-repair proteins, and is also phosphorylated during apoptosis induced by DNA damage [40]. H2A.X has been shown to be phosphorylated in cells exposed to cisplatin, carboplatin, and oxaliplatin [41, 42], and to correlate with the cytotoxic potency of platinum complexes [31]. Cells were treated with compounds, and γH2A.X foci were evaluated by immunofluorescence microscopy after 24 h treatment (Figure 4, magenta). The antibody absorbed and fluoresced at different wavelengths to the compounds to ensure fluorescence spillover was not responsible for false-positive signals. Cells treated with cisplatin, [PtCl2(en)], and Pt-Boc elicited a strong DNA damage response compared with control (untreated) cells.
Figure 4.

Assessment of the ability of dyes and platinum-dye conjugates to elicit DNA damage. (a) Control, (b) Cisplatin (5 μM), (c) [PtCl2(en)] (25 μM), and (d) Pt-Boc (25 μM) are non-fluorescent compounds. Fluorescent compounds are shown in (e) CFDA-Pt (100 μM), (f) CF-Pt (100 μM), (g) BODIPY-Pt (50 μM), and (h) BODIPY-prop-Pt (50 μM). Each image shows the fluorescent compound (green), H2A.X (magenta), and a merge with the DAPI nuclear stain (blue).
While CF-Pt and CFDA-Pt did not produce γH2A.X foci even at high concentrations (100 μM), BODIPY-Pt and BODIPY-prop-Pt did so at 50 μM. This is supported by Miller’s observation that fluorescently-labelled 53BP1 protein localized to the nucleus in cells treated with BODIPY-Pt (50 μM). We also examined the ULYSIS 546 platinum complex at 100 μM (Supplementary Figure 4). The concentration of ULYSIS 546 reagent provided by the vendor is not disclosed, but using absorption spectrometry a standard curve was generated with AlexaFluor 546 dye to determine the ULYSIS 546 reagent concentration to be 100 μM. No γH2A.X foci were evident in ULYSIS 546-treated cells. None of the fluorophore-Boc compounds produced foci associated with γH2A.X, indicating that while they were weakly cytotoxic, the dyes could not induce DNA damage independent of platinum (Supplementary Figure 5).
Cellular distribution of Pt-fluorophore complexes
One feature of many cisplatin-resistant cells is diminished accumulation compared with parent cells [43]. We examined the cellular accumulation of fluorophore-Boc and Pt-fluorophore complexes in KB-3-1 and CP20 cells using flow cytometry, to examine whether resistant cells accumulate lower levels of Pt-dye consistent with previous observations for Pt drugs [22]. Cells were incubated with compounds for 45 min, then cellular fluorescence was measured, and values normalized against accumulation in parental cells (Figure 5). CP20 cells accumulated 25% less BODIPY-Pt than parent cells, while BODIPY-Boc accumulation was unaffected in resistant cells.
Figure 5.
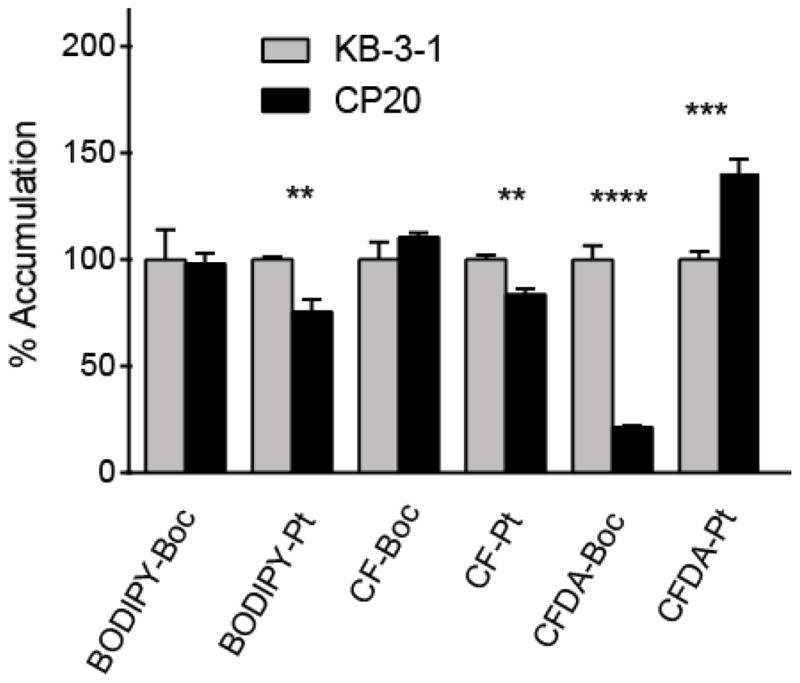
Cellular accumulation of the dyes and dye-platinum conjugates at 25 μM for 45 minutes in KB-3-1 and CP.20 cells assessed by flow cytometry. Values shown are an average of a triplicate and error bars represent standard deviation, and for each compound uptake is normalized to florescence in KB-3-1 cells. A Student’s T test was performed for each compound between the two cell lines. Comparisons that are statistically significant are marked (p value <0.05 * <0.01 ** <0.001 *** <0.0001 ****).
The Pt-CF also showed a small but statistically significant 17% reduction in accumulation, while the CF-Boc control was unaffected. In contrast, the control CFDA-Boc dye showed a large 80% reduction in accumulation in resistant cells, whereas Pt-CFDA accumulation actually increased in CP20 cells. The absolute fluorescence measured in CFDA-treated KB-3-1 cells was two orders of magnitude greater than the CF-treated cells, consistent with the hydrolytic trapping of CFDA dyes in cells, and the poor cellular permeability of CF dyes. The weakly-resistant CP.5 cell line does not show reduced levels of Pt compared with KB-3-1 cells, and this was the case for the Pt-dye complexes examined here (not shown).
We next examined the cellular localization of fluorophore-Boc and Pt-fluorophore complexes (10 μM) in KB-3-1 cells using live-cell confocal microscopy following incubation for 1 or 24 h (Figure 6). CF-Boc showed very low cellular signal, whereas CFDA-Boc, CF-Pt and CFDA-Pt localized to punctae and ring-like structures at the cell periphery consistent with previous reports of endocytosis and vesicular localization of FDDP [17, 18]. CFDA-Pt was distributed evenly through the cell at 1 h, but at 24 h it was not observed except in occasional cells that appeared to be dead or dying (rounded). The same was true for the Boc-protected dye, suggesting elimination or metabolism of the dye in cells over time. Both BODIPY-Pt and BODIPY-prop-Pt showed stronger cellular signal at 1 h compared with 24 h, with diffuse signal observed in the perinuclear space, and no discernible co-localization with the DNA dye Hoechst. The ULYSIS 546 platinum complex appeared in KB-3-1 cells diffusely distributed in the perinuclear space at 1 h, and as large vesicular structures at 24 h (Supplementary Figure 6). In the resistant CP.5 cells, compounds were strongly localized in large vesicles at 1 h, and in most cases these were largely absent from cells after 24 h (Supplementary Figure 7). This is consistent with previous observations that resistant cells sequester platinum complexes in vesicular structures [44].
Figure 6.

Confocal images of live KB-3-1 cells in culture after incubation with dye or dye-platinum conjugate at 10 μM for either 1 h or 24 h: (a) CF-Boc 1 h, (b) CF-Boc 24 h, (c) CF-Pt 1 h, (d) CF-Pt 24 h, (e) CFDA-Boc 1 h, (f) CFDA-Boc 24 h, (g) CFDA-Pt 1 h, (h) CFDA-Pt 24 h, (i) BODIPY-Boc 1 h, (j) BODIPY-Boc 24 h, (k) BODIPY-Pt 1 h, (l) BODIPY-Pt 24 h, (m) BODIPY-prop-Pt 1 h, and (n) BODIPY-prop-Pt 24 h. Each image represents a merge of the Hoechst nuclear stain (1 μg/mL blue) and the fluorescent compound (green).
The H2A.X microscopy of fixed cells pre-treated with compounds also allowed examination of fluorescence, as the H2A.X antibody (AlexaFluor 647-conjugated, excitation 633 nm) spectral properties do not overlap with the fluorophores conjugated to Pt (excitation 488 nm) (Figure 4). Cells were treated with 50 μM compound (compared with 10 μM for the live microscopy described above). At this higher concentration, both BODIPY platinum complexes demonstrated cytosolic and nuclear localization, as did the CFDA-Pt complex, while CF-Pt did not produce any signal (Figure 4). This is consistent with observations using XRF to examine cisplatin distribution in A2780 ovarian carcinoma cells, which showed nuclear and cytotsolic pools of cisplatin [45]. At the same concentration, the dye controls produced minimal cellular signal (Supplementary Figure 5).
Pt-fluorophore complexes in multicellular systems
We also examined the permeability and activity of BODIPY-Pt in 3-dimensional (3D) culture. There is surprisingly little literature on the activity of platinum complexes in 3D culture compared with 2D and no literature we could identify examining the behavior of cisplatin-resistant cell lines grown in 3D [46, 47], despite the fact that cells growing in multicellular structures have been shown to have different response patterns to cytotoxic agents [48]. These differences stem from factors such as high interstitial pressure, hypoxia, a large proportion of cells being quiescent (not dividing) and high metabolite concentrations leading to extracellular acidity [49]. We established KB-3-1 and KB-CP.5 cells as spheroids by seeding them in Matrigel and allowing growth for 1 week (until spheroid diameter was approximately 100 μm). Spheroids were incubated with compounds for 1 h, and then confocal microscopy was used to examine the distribution (Figure 7). The BODIPY-Boc control demonstrated higher accumulation at the periphery of the spheroid, supported by the intensity histogram generated from a cross-section (Figure 7C,D). This is consistent with observations for many small molecules, with limited diffusion into spheroids for a range of reasons including lysosomal and other trapping mechanisms [50, 51]. In contrast, BODIPY-Pt did not appear to accumulate at the periphery, with little accumulation occurring in the outer leaflet of the spheroids but strong fluorescence in the center. This unusual accumulation feature was not related to cell death, as spheroids were treated with a low concentration (10 μM for 1 h). The CF dyes showed little to no accumulation in spheroids, and while the CFDA-Boc demonstrated a ring-like fluorescence at the spheroid periphery, CFDA-Pt demonstrated little to no accumulation in spheroids, in contrast with monolayer (2D) cells. The CP.5 spheroids demonstrated similar cell distribution patterns to KB-3-1 spheroids (Supplementary Figure 8).
Figure 7.

Distribution of BODIPY-Boc and BODIPY-Pt at 10 μM in KB-3-1 three-dimensional spheroids grown in Matrigel. Confocal images show (a) BODIPY-Pt and (b) BODIPY-Box distribution. The optical slice of each image is 4.0 μM. Each image shows the fluorescent compound (green) merged with the bright field image. Graphical representation of fluorescence intensity plotted as a factor of distance across spheroid for (c) BODIPY-Pt and (d) BODIPY-Boc. Arrows signify the boundaries of the spheroids.
To determine whether the cisplatin-resistant cells maintained their phenotype in 3D, we examined the activity of cisplatin, [PtCl2(en)] and BODIPY-Pt against cells grown in Matrigel. One common strategy for assessing response to cytotoxins in 3D is to grow spheroids on a non-adherent surface such as agarose, and assess cellular response based on changes in diameter (and therefore spheroid volume) or by a biochemical read-out [52]. Cells can also grow as spheroids in protein matrices such as Matrigel, and while viability assays are complicated by 3D architecture for a number of reasons [53], we utilized CellTiter Glo viability assays (which assay ATP content) with a prolonged incubation period (60 min) to ensure all cells were lysed [54].
Two Matrigel growth conditions were examined. Cells were plated in Matrigel, and allowed to establish for 24 h or 4 days before addition of compounds. At 24 h, cells were considered to be single cells, whereas at 4 days multicellular spheroids were apparent in Matrigel (observed by transmission microscopy, not shown). We examined these Matrigel conditions for two reasons. First, we wanted to assess if differences in drug response were due to multicellularity versus an effect conferred simply by the presence of Matrigel. For example, it has been shown that Matrigel, as a dense protein solution, participates in drug-protein binding [55], though the extent of cisplatin binding to Matrigel is not known. Second, we wondered whether the single-cell suspension of cells in Matrigel responded to cisplatin in a similar manner to 2D cells, which also have minimal cell-cell contacts.
As anticipated based on general literature evidence, the spheroids (3D 4 day) were less sensitive to cisplatin than monolayer (2D) cells (Table 2, 2D IC50 = 2.5 ± 0.1 μM, 3D 4 day IC50 = 9.4 ± 0.7 μM). The same pattern was observed for [PtCl2(en)]. Spheroids formed from CP.5 cells retained a resistant phenotype towards cisplatin (3D 4 day: 3.4-fold resistant vs. 2D: 8.4-fold resistant) and [PtCl2(en)], though the degree of relative resistance was diminished. The sensitivity of single-cell Matrigel suspension (3D 1 day) was intermediate between 2D cells and 3D spheroids. In most cases CP.5 cells grown in 2D and 3D for 1 day showed statistically equivalent sensitivity. The BODIPY-Pt complex also showed reduced efficacy against 3D 4-day spheroids (Table 2, 2D IC50 = 36.9 ± 1.3 μM, 3D 4 day IC50 = 140.9 ± 6.6 μM), spheroids were cross-resistant.
Table 2.
Cytotoxicity (IC50, μM) of compounds against parental KB-3-1 and cisplatin-resistant B CP.5 cell lines. Cells were analyzed in 2D culture and 3D culture grown for either 1 or 4 days.
| IC50 (μM) | |||
|---|---|---|---|
|
| |||
| Culture method | 2D | 3D 1 Day | 3D 4 Day |
| Cisplatin (1) | |||
| KB-3-1 | 2.5 ± 0.1 | 4.5 ± 0.7 | 9.4 ± 0.7 |
| CP.5 | 21.0 ± 2.8 | 22.9 ± 4.6 | 32.1 ± 6.9 |
|
| |||
| [PtCl2(en)] (2) | |||
| KB-3-1 | 4.1 ± 0.2 | 21.4 ± 1.0 | 150* |
| CP.5 | 107.4 ± 10.9 | 297.7 ± 21.8 | >500 |
|
| |||
| BODIPY-Pt (9) | |||
| KB-3-1 | 36.9 ± 1.3 | 53.8 ± 7.1 | 140.9 ± 6.6 |
| CP.5 | 141.5 ± 12.5 | 185.7 ± 47.2 | 299.5 ± 92.2 |
single data point
Conclusions
In this study, we examined several fluorophore-conjugated platinum complexes and assessed them in the context of the mechanism of action of cisplatin. The BODIPY-tethered platinum complexes demonstrated elevated cytotoxicity compared with the CF/CFDA-conjugation complexes, cross-resistance to cisplatin-resistant cells and phosphorylation of H2A.X. At higher concentrations, nuclear localization of the BODIPY platinum complexes was observed, along with cytosolic/perinuclear staining that could be detected at lower concentrations. This observation is consistent with results from other techniques examining Pt distribution in cells, as diverse as subcellular fractionation [56, 57] and XRF/SRIXE [45]. Our observations of activity are consistent with that of Miller et al. who used a localization assay to demonstrate that 53BP1 relocalized to the nucleus following BODIPY-Pt treatment, with the qualification that DMSO as a solvent inactivates these complexes as demonstrated here. In other words, while DNA binding correlates with cytotoxicity of Pt complexes, the majority of Pt in a cell is not bound to DNA. The CF and CFDA platinum complexes demonstrated poor cytotoxicity, and DNA localization and H2A.X phosphorylation was not observed.
The main discrepancy appears to be related to uptake defects in cisplatin-resistant cells. While cisplatin-resistant cell lines (CP.5 and CP20) showed cross-resistance to Pt-fluorophore complexes indicating, the reduction in accumulation measured by flow cytometry (Figure 5) for BODIPY-Pt and CF-Pt is less than that observed for cells exposed to agents such as [14C]carboplatin, cisplatin, and dyes used as markers of endocytosis (50–75% reduction)[22, 58]. This result suggests that the platinum-fluorophore complexes must enter the cell through pathways somewhat different from those used by the compounds listed above. Exactly what the pathway of these BODIPY-platinum compounds is within the cell to the nucleus remains to be determined, but since the cisplatin-resistant cells that we examined are resistant to these compounds, and their defect is in uptake and intracellular trafficking of platinum derivatives, it appears likely that they share intracellular trafficking pathways with clinically used cytotoxic platinum compounds. We believe these BODIPY complexes demonstrate improved potential for understanding the intracellular trafficking of platinum compounds in vitro and in vivo.
Supplementary Material
Acknowledgments
This research was supported by the Intramural Research Program of the National Institutes of Health, National Cancer Institute. We thank George Leiman for editorial assistance, and Robert O’Connor (NIDDK) for assistance with NMR acquisition.
Footnotes
Conflicts of interest: None declared
Contributor Information
Justin C. Jagodinsky, Laboratory of Cell Biology, National Cancer Institute, Center for Cancer Research, National Institutes of Health, Bethesda, MD 20892, Phone: 301-496-1530, Fax: 301-402-0450.
Agnieszka Sulima, Imaging Probe Development Center, National Institutes of Health, Rockville, MD.
Yiqi Cao, Laboratory of Cell Biology, National Cancer Institute, Center for Cancer Research, National Institutes of Health, Bethesda, MD 20892, Phone: 301-496-1530, Fax: 301-402-0450.
Joanna E. Poprawski, Laboratory of Cell Biology, National Cancer Institute, Center for Cancer Research, National Institutes of Health, Bethesda, MD 20892, Phone: 301-496-1530, Fax: 301-402-0450
Burchelle N. Blackman, Imaging Probe Development Center, National Institutes of Health, Rockville, MD
John R. Lloyd, Advanced Mass Spectrometry Facility, National Institute of Diabetes & Digestive & Kidney Diseases, National Institutes of Health, Bethesda, MD 20892
Rolf E. Swenson, Imaging Probe Development Center, National Institutes of Health, Rockville, MD
Michael M. Gottesman, Email: mgottesman@nih.gov, Laboratory of Cell Biology, National Cancer Institute, Center for Cancer Research, National Institutes of Health, Bethesda, MD 20892, Phone: 301-496-1530, Fax: 301-402-0450
Matthew D. Hall, Laboratory of Cell Biology, National Cancer Institute, Center for Cancer Research, National Institutes of Health, Bethesda, MD 20892, Phone: 301-496-1530, Fax: 301-402-0450
References
- 1.Shen DW, Pouliot LM, Hall MD, Gottesman MM. Pharmacol Rev. 2012;64:706–721. doi: 10.1124/pr.111.005637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kelland L. Nat Rev Cancer. 2007;7:573–584. doi: 10.1038/nrc2167. [DOI] [PubMed] [Google Scholar]
- 3.Klein AV, Hambley TW. Chem Rev. 2009;109:4911–4920. doi: 10.1021/cr9001066. [DOI] [PubMed] [Google Scholar]
- 4.Hah SS, Stivers KM, de Vere White RW, Henderson PT. Chem Res Toxicol. 2006;19:622–626. doi: 10.1021/tx060058c. [DOI] [PubMed] [Google Scholar]
- 5.Shen DW, Liang XJ, Gawinowicz MA, Gottesman MM. Mol Pharmacol. 2004;66:789–793. doi: 10.1124/mol.66.4.. [DOI] [PubMed] [Google Scholar]
- 6.Jong NN, Nakanishi T, Liu JJ, Tamai I, McKeage MJ. J Pharmacol Exp Ther. 2011;338:537–547. doi: 10.1124/jpet.111.181297. [DOI] [PubMed] [Google Scholar]
- 7.Berners-Price SJ, Ronconi L, Sadler PJ. Prog Nucl Magn Reson Spectrosc. 2006;49:65–98. [Google Scholar]
- 8.McRae R, Bagchi P, Sumalekshmy S, Fahrni CJ. Chem Rev. 2009;109:4780–4827. doi: 10.1021/cr900223a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hall MD, Foran GJ, Zhang M, Beale PJ, Hambley TW. J Am Chem Soc. 2003;125:7524–7525. doi: 10.1021/ja0354770. [DOI] [PubMed] [Google Scholar]
- 10.Wedlock LE, Kilburn MR, Liu R, Shaw JA, Berners-Price SJ, Farrell NP. Chem Commun (Camb) 2013;49:6944–6946. doi: 10.1039/c3cc42098a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ding S, Qiao X, Suryadi J, Marrs GS, Kucera GL, Bierbach U. Angew Chem Int Ed Engl. 2013;52:3350–3354. doi: 10.1002/anie.201210079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Qiao X, Ding S, Liu F, Kucera GL, Bierbach U. J Biol Inorg Chem. 2014;19:415–426. doi: 10.1007/s00775-013-1086-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shen C, Harris BD, Dawson LJ, Charles KA, Hambley TW, New EJ. Chem Commun (Camb) 2015;51:6312–6314. doi: 10.1039/c4cc08077g. [DOI] [PubMed] [Google Scholar]
- 14.White JD, Osborn MF, Moghaddam AD, Guzman LE, Haley MM, DeRose VJ. J Am Chem Soc. 2013;135:11680–11683. doi: 10.1021/ja402453k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hambley TW. Coordin Chem Rev. 1997;166:181–223. [Google Scholar]
- 16.Molenaar C, Teuben JM, Heetebrij RJ, Tanke HJ, Reedijk J. J Biol Inorg Chem. 2000;5:655–665. doi: 10.1007/s007750000153. [DOI] [PubMed] [Google Scholar]
- 17.Katano K, Safaei R, Samimi G, Holzer A, Tomioka M, Goodman M, Howell SB. Clin Cancer Res. 2004;10:4578–4588. doi: 10.1158/1078-0432.CCR-03-0689. [DOI] [PubMed] [Google Scholar]
- 18.Safaei R, Katano K, Larson BJ, Samimi G, Holzer AK, Naerdemann W, Tomioka M, Goodman M, Howell SB. Clin Cancer Res. 2005;11:756–767. [PubMed] [Google Scholar]
- 19.Benedetti BT, Peterson EJ, Kabolizadeh P, Martínez A, Kipping R, Farrell NP. Mol Pharm. 2011;8:940–948. doi: 10.1021/mp2000583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.van Belkum A, Linkels E, Jelsma T, Houthoff HJ, van den Berg F, Quint W. J Virol Methods. 1993;45:189–200. doi: 10.1016/0166-0934(93)90103-x. [DOI] [PubMed] [Google Scholar]
- 21.Heetebrij RJ, Talman EG, v Velzen MA, van Gijlswijk RP, Snoeijers SS, Schalk M, Wiegant J, vd Rijke F, Kerkhoven RM, Raap AK, Tanke HJ, Reedijk J, Houthoff HJ. Chembiochem. 2003;4:573–583. doi: 10.1002/cbic.200200498. [DOI] [PubMed] [Google Scholar]
- 22.Liang XJ, Shen DW, Chen KG, Wincovitch SM, Garfield SH, Gottesman MM. J Cell Physiol. 2005;202:635–641. doi: 10.1002/jcp.20253. [DOI] [PubMed] [Google Scholar]
- 23.Miller MA, Askevold B, Yang KS, Kohler RH, Weissleder R. ChemMedChem. 2014;9:1131–1135. doi: 10.1002/cmdc.201300502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sun T, Guan X, Zheng M, Jing X, Xie Z. ACS Med Chem Lett. 2015;6:430–433. doi: 10.1021/acsmedchemlett.5b00041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Steel HL, Allinson SL, Andre J, Coogan MP, Platts JA. Chem Commun (Camb) 2015;51:11441–11444. doi: 10.1039/c5cc04003e. [DOI] [PubMed] [Google Scholar]
- 26.Baruah H, Barry CG, Bierbach U. Curr Top Med Chem. 2004;4:1537–1549. doi: 10.2174/1568026043387313. [DOI] [PubMed] [Google Scholar]
- 27.Hughes CS, Postovit LM, Lajoie GA. Proteomics. 2010;10:1886–1890. doi: 10.1002/pmic.200900758. [DOI] [PubMed] [Google Scholar]
- 28.Benoist E, Loussouarn A, Remaud P, Chatal J-F, Gestin J-F. Synthesis. 1998;8:1113–1118. [Google Scholar]
- 29.Altman J, Wilchek M. Inorganica Chimica Acta. 1985;101:171–173. [Google Scholar]
- 30.Shen DW, Akiyama S, Schoenlein P, Pastan I, Gottesman MM. Br J Cancer. 1995;71:676–683. doi: 10.1038/bjc.1995.134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hall MD, Telma KA, Chang KE, Lee TD, Madigan JP, Lloyd JR, Goldlust IS, Hoeschele JD, Gottesman MM. Cancer Res. 2014;74:3913–3922. doi: 10.1158/0008-5472.CAN-14-0247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bancroft DP, Lepre CA, Lippard SJ. J Am Chem Soc. 1990;112:6880–6871. [Google Scholar]
- 33.Yu C, Guo Z, Sadler PJ. In: Cisplatin: Chemistry and biochemistry of a leading anticancer drug. Lippert B, editor. Wiley Online Library; Zurich, Switzerland: 2006. [Google Scholar]
- 34.Huang Z, Huang Y. Cancer Invest. 2005;23:26–32. [PubMed] [Google Scholar]
- 35.Hall MD, Amjadi S, Zhang M, Beale PJ, Hambley TW. J Inorg Biochem. 2004;98:1614–1624. doi: 10.1016/j.jinorgbio.2004.05.017. [DOI] [PubMed] [Google Scholar]
- 36.Kerrison SJS, Sadler PJ. J Chem Soc Chem Commun. 1977;23:861–863. [Google Scholar]
- 37.Zenda M, Yasui H, Oishi S, Masuda R, Fujii N, Koide T. Chem Biol Drug Des. 2015;85:519–526. doi: 10.1111/cbdd.12450. [DOI] [PubMed] [Google Scholar]
- 38.Fischer SJ, Benson LM, Fauq A, Naylor S, Windebank AJ. Neurotoxicology. 2008;29:444–452. doi: 10.1016/j.neuro.2008.02.010. [DOI] [PubMed] [Google Scholar]
- 39.Ellis LT, Er HM, Hambley TW. Aust J Chem. 1995;48:793–806. [Google Scholar]
- 40.Rogakou EP, Nieves-Neira W, Boon C, Pommier Y, Bonner WM. J Biol Chem. 2000;275:9390–9395. doi: 10.1074/jbc.275.13.9390. [DOI] [PubMed] [Google Scholar]
- 41.Chiu SJ, Lee YJ, Hsu TS, Chen WS. Chem Biol Interact. 2009;182:173–182. doi: 10.1016/j.cbi.2009.08.019. [DOI] [PubMed] [Google Scholar]
- 42.Cruet-Hennequart S, Villalan S, Kaczmarczyk A, O’Meara E, Sokol AM, Carty MP. Cell Cycle. 2009;8:3039–3050. [PubMed] [Google Scholar]
- 43.Hall MD, Okabe M, Shen DW, Liang XJ, Gottesman MM. Annu Rev Pharmacol Toxicol. 2008;48:495–535. doi: 10.1146/annurev.pharmtox.48.080907.180426. [DOI] [PubMed] [Google Scholar]
- 44.Liang XJ, Mukherjee S, Shen DW, Maxfield FR, Gottesman MM. Cancer Res. 2006;66:2346–2353. doi: 10.1158/0008-5472.CAN-05-3436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hall MD, Dillon CT, Zhang M, Beale P, Cai Z, Lai B, Stampfl APJ, Hambley TW. J Biol Inorg Chem. 2003;8:726–732. doi: 10.1007/s00775-003-0471-6. [DOI] [PubMed] [Google Scholar]
- 46.Jones AC, Stratford IJ, Wilson PA, Peckham MJ. Br J Cancer. 1982;46:870–879. doi: 10.1038/bjc.1982.296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Erlichman C, Vidgen D, Wu A. J Natl Cancer Inst. 1985;75:499–505. [PubMed] [Google Scholar]
- 48.Friedrich J, Ebner R, Kunz-Schughart LA. Int J Radiat Biol. 2007;83:849–871. doi: 10.1080/09553000701727531. [DOI] [PubMed] [Google Scholar]
- 49.Tredan O, Galmarini CM, Patel K, Tannock IF. J Natl Cancer Inst. 2007;99:1441–1454. doi: 10.1093/jnci/djm135. [DOI] [PubMed] [Google Scholar]
- 50.Minchinton AI, Tannock IF. Nat Rev Cancer. 2006;6:583–592. doi: 10.1038/nrc1893. [DOI] [PubMed] [Google Scholar]
- 51.Durand RE. Cancer Chemother Pharmacol. 1990;26:198–204. doi: 10.1007/BF02897199. [DOI] [PubMed] [Google Scholar]
- 52.Friedrich J, Seidel C, Ebner R, Kunz-Schughart LA. Nat Protoc. 2009;4:309–324. doi: 10.1038/nprot.2008.226. [DOI] [PubMed] [Google Scholar]
- 53.Vinci M, Gowan S, Boxall F, Patterson L, Zimmermann M, Court W, Lomas C, Mendiola M, Hardisson D, Eccles SA. BMC Biol. 2012;10:29. doi: 10.1186/1741-7007-10-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Chen X, Thibeault SL. Acta Biomater. 2010;6:2940–2948. doi: 10.1016/j.actbio.2010.01.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zhang Y, Lukacova V, Reindl K, Balaz S. J Biochem Biophys Methods. 2006;67:107–122. doi: 10.1016/j.jbbm.2006.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sharma RP, Edwards IR. Biochem Pharmacol. 1983;32:2665–2669. doi: 10.1016/0006-2952(83)90073-4. [DOI] [PubMed] [Google Scholar]
- 57.Sancho-Martinez SM, Prieto-Garcia L, Prieto M, Lopez-Novoa JM, Lopez-Hernandez FJ. Pharmacol Ther. 2012;136:35–55. doi: 10.1016/j.pharmthera.2012.07.003. [DOI] [PubMed] [Google Scholar]
- 58.Gottesman MM, Hall MD, Liang X-J, Shen D-w. In: Platinum and other heavy metal compounds in cancer chemotherapy: Molecular mechanisms and clinical applications. Bonetti A, Leone R, Muggia F, Howell SB, editors. Springer-Verlag; New York: 2009. pp. 83–88. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.