

Abstract
To identify new pathways of estrogen action and novel estrogen receptor α (ERα) biomodulators, we performed high throughput screening and used follow on assays and bioinformatics to identify small molecule ERα inhibitors with a novel mode of action. These studies led to identification of rapid extranuclear activation of the endoplasmic reticulum stress sensor, the unfolded protein response (UPR), as a new pathway of estrogen-ERα action. Moreover, increasing evidence indicates that the mechanism underlying anticipatory activation of the UPR is shared among steroid and peptide hormones and is conserved from insects to humans. It is likely that this newly unveiled extranuclear pathway is used by diverse mitogenic hormones to prepare cells for the increased protein folding load that will occur during subsequent cell proliferation. Demonstrating biological relevance, elevated expression of a UPR gene signature in ERα positive breast cancer is a powerful new prognostic marker tightly correlated with subsequent resistance to tamoxifen, tumor recurrence and poor survival. In addition, overexpression of epidermal growth factor receptor and HER2/neu is positively correlated with increased UPR activation in breast cancer. This review describes recent research that demonstrates the importance of anticipatory UPR activation in therapy resistant tumors and discusses a promising small molecule biomodulator that inhibits tumor growth by tuning this UPR signaling pathway.
Keywords: Steroid Receptor, Breast cancer, Unfolded protein response, Cell death, Hormone action, Cancer
1. Introduction
The endoplasmic reticulum (EnR) plays a key role in synthesis, folding and transport of nascent peptides. Protein maturation in the EnR is a critical step in normal cell function and in cell survival. Modest changes in the cellular environment, such as changes in the intracellular Ca2+ level in the lumen of the EnR, nutrient availability, redox state, or in the rate of protein synthesis, can cause accumulation of misfolded or unfolded proteins. The resulting EnR stress [1, 2] leads to activation of the EnR stress response pathway, the unfolded protein response (UPR). The UPR consists of three main branches that together balance the synthesis of new proteins with the availability of chaperones and other proteins to help fold and transport proteins within cells. EnR stress activates autophosphorylation of the transmembrane kinase PERK (protein kinase RNA-like endoplasmic reticulum kinase) [2, 3]. Activated p-PERK phosphorylates eukaryotic initiation factor 2α (eIF2α), resulting in transient inhibition of most protein synthesis and increased translation of p58IPK and GADD34. If the stress is moderate, the p58IPK binds PERK, inhibiting PERK activation, and the GADD34 dephosphorylates eIF2α. This ultimately reverses PERK activation and protein synthesis is restored [4, 5]. The other arms of the UPR initiate with activation of the transcription factor ATF6α, leading to increased protein folding capacity and activation of the splicing factor IRE1α, which alternatively splices the transcription factor XBP1, leading to production of active spliced XBP1 (sp-XBP1), increased protein folding capacity and altered mRNA decay and translation (Fig. 1) [1–3].
Fig. 1.

Model for activation of the UPR by steroid or peptide hormones.
Diverse mitogenic hormones, acting via their respective receptors, stimulate cell proliferation and tumor growth [6–11]. Enhanced cell proliferation requires increased protein production, potentially leading to insufficient protein folding capacity and EnR stress. Although UPR activation has been described in multiple cancers [2, 12–15], until recently, it has not been a major research focus in hormone-dependent cancers. This review focuses on the pathophysiological importance of anticipatory UPR activation in hormone signaling as an early component of the cellular proliferation program and discusses the preclinical promises of targeting the UPR.
2. Steroid/peptide hormone activation of the UPR
Steroid and peptide hormones execute their biological functions through direct interaction with hormone-specific receptors [8, 9]. These include binding of mitogenic steroid hormones, 17β-estradiol (E2; estrogen), dihydrotestosterone (DHT; androgen) and ecdysone (Ec) to their respective nuclear receptors (ERα, AR and EcR) and of the peptide hormones epidermal growth factor (EGF) and vascular endothelial growth factor (VEGF) to their receptors (EGFR and VEGFR). Steroid hormones exert their molecular functions by regulating gene expression in the nucleus and cross-talking with diverse extranuclear signal transduction pathways. In the classical genomic action of steroid hormones, here illustrated using estrogen, estrogens bind to ERα; this results in receptor dimerization. Estrogen-ERα binds directly to genomic response elements and interacts with DNA indirectly through tethering to other proteins. This results in activation of a genomic program that alters the expression of thousands of genes and plays an important role in promoting the proliferation of ERα positive cancer cells [16–18].
While the genomic actions of steroid hormones are initiated rapidly, they play out over many hours. A disparate set of rapid extranuclear actions of steroid receptors, often initiated at or near the plasma membrane, influence diverse cell functions and also play a pivotal role in modulating the receptors genomic program [19–21]. While much attention focused on rapid effects of steroid hormones on established signal transduction pathways, rapid effects of estrogen and other steroid hormones on activation of the UPR were largely unexplored. We recently showed that, within 1 minute, estrogens, acting via ERα, activate phospholipase C gamma (PLCγ), producing inositol triphosphate (IP3). The IP3 binds to and opens the EnR inositol triphosphate receptor (IP3R) calcium channels allowing rapid efflux of calcium from the lumen of the EnR into the cytosol (Fig. 1). This rapid calcium efflux activates the UPR, inducing chaperones (Fig. 1). Notably, inhibition or knockdown of pathway components strongly inhibits estrogen stimulated cell proliferation and nearly abolishes subsequent estrogen-ERα induction and repression of gene expression (Fig. 1) [22]. Moreover, analysis of data from approximately 1,000 ERα positive breast cancers shows that elevated expression of a UPR gene signature at diagnosis is a powerful new prognostic marker tightly correlated with subsequent resistance to tamoxifen, tumor recurrence and poor survival [22].
The well-studied oncogenic mitogen EGF, acting through EGF receptors, rapidly activates the ERK and AKT signaling pathways and alters gene expression. EGF-EGFR activation of these pathways promotes tumor growth and invasion, and is antiapoptotic [23–25]. Although EGF is a peptide hormone and EGFR is a plasma membrane receptor, EGF-EGFR and E2-ERα share the same general pathway for rapid anticipatory activation of the UPR (Fig. 1; see Section 2 above) [26]. Activation of the anticipatory UPR pathway is a newly described action of EGF, and it facilitates EGF stimulated cell cycle progression in different ways [26]. We find that EGF-EGFR activation of the anticipatory UPR pathway is required for EGF induced immediate early gene expression. Notably, while blocking UPR activation abolishes EGF regulated immediate early gene expression, it has no effect on EGF activation of the ERK and AKT pathways. This indicates that at early times, the anticipatory UPR pathway and the ERK pathway are independent regulators, which converge at the level of gene expression [26]. EGF induced chaperone production also contribute to cell proliferation; knocking down the chaperone producing arms of the UPR inhibits EGF stimulated cell proliferation [26].
Tumor growth and metastasis depends on new growth in the vascular network [27]. In endothelial cells, vascular endothelial growth factor (VEGF), acting through VEGF receptors, promotes cell viability and angiogenesis, the formation of new blood vessels [27–29]. Recently, anticipatory activation of the UPR has been identified as a new mode of action in VEGF signaling. In a notable difference between the estrogen and VEGF pathways, the activation mechanism is reported to be independent of the PLCγ-IP3-calcium pathway [30]. In the absence of EnR stresses, VEGF primes cells by activating the ATF6α and PERK arms of the UPR and anticipates the needs for subsequent VEGF-induced vascularization [30].
Prostate cancer is a leading cause of cancer in men. Androgens, such as testosterone and dihydrotestosterone (DHT), exert their biological functions through the androgen receptor and play a pivotal role in proliferation of prostate cancer [31, 32]. At early time points, DHT did not rapidly induce sp-XBP1 and BiP mRNA (Fig. 2). As a control, in the AR-positive LNCaP and LAPC4 human prostate cancer cell lines, prostate-specific antigen (PSA), which is a classic DHT inducible gene, is rapidly and robustly induced (8 hours; Fig. 2). Thus, rapid UPR activation is not part of DHT signaling pathway. However, activation of the UPR was observed at later times (e.g. 24 to 48 hours) [33]. Together, these data suggest DHT may activate the UPR through a reactive UPR mechanism, in which UPR activation is stimulated by accumulation of misfolded and unfolded proteins as the cells proliferate, or through a nuclear gene expression program that induces UPR-related mRNAs. Although the exact molecular mechanism underlying DHT activation of the UPR requires further exploration, pharmacological inhibition of IRE1α significantly reduces prostate tumor growth [33]. This supports the biological significance of activation of the UPR pathway in prostate cancer progression.
Fig. 2.
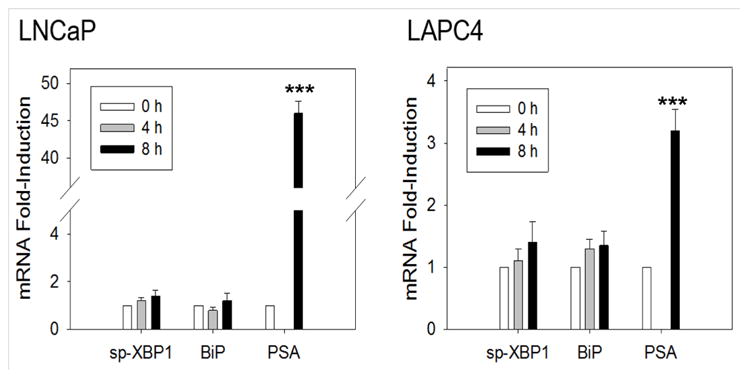
DHT does not induce rapid activation of the IRE1α and ATF6α arms of the UPR in prostate cancer cells. qRT-PCR time course comparing the effect of DHT on DHT-AR induction of spliced-XBP1 (sp-XBP1), BiP and prostate-specific antigen (PSA) in AR positive LNCaP and LAPC4 cells. The qRT-PCR procedures have been described previously [22, 39]. Briefly, the cells were plated in RPMI growth medium with 10% charcoal stripped fetal bovine serum for three days prior to treating the cells with 10 nM DHT at the indicated times. (n=3; 0 h set to 1). Data are mean ± s.e.m. ***P < 0.001.
The transformation of insects from larvae to adults is commonly known as metamorphosis. Neuronal remodeling is a crucial process for development of both vertebrates and invertebrates but is particularly critical for metamorphosis. During this process, axons and dendrites undergo a precisely controlled program of pruning via cell death followed by regrowth [34]. This remodeling of axons and dendrite events ultimately depends on the molecular function of Ec-EcR [34]. As a part of rapid extranuclear action of Ec-EcR, Ec regulates the cytosolic calcium level through a PLCγ-IP3-calcium dependent pathway, resulting in phosphorylation and activation of protein kinase C that modulates transcriptional activity of ultraspircle in the lepidopteran insect Helicoverpa armigera [35]. Depletion of PLCγ leads to metamorphosis defects and blocks induction of Ec regulated genes [35].
Although the mechanisms underlying anticipatory UPR activation vary among different hormone-mediated signaling pathways, an important consequence of anticipatory UPR activation is the induction of molecular chaperones that primes cells to mitigate damage due to future cell stress that may occur during proliferation, or under various physiological conditions. Activation of the UPR before the cellular stress and before the accumulation of unfolded protein is the key feature that distinguishes the anticipatory UPR pathway from the well-studied reactive UPR pathway.
3. The UPR acts as a double-edged sword to control cell fate
Anticipatory UPR activation is protective; deletion or inhibition of UPR components is an emerging therapeutic strategy that reduces tumor growth and increases susceptibility of cancer cells to therapeutic agents [33, 36, 37]. Recently, we described a novel strategy to target cancer cells, not by inhibiting the UPR, but by toxic hyperactivation of the UPR. Through high throughput screening follow on assays and bioinformatics, we identified a clinically promising small molecule ERα biomodulator, BHPI [38, 39]. BHPI binds non-competitively to ERα and distorts and exaggerates the normal estrogen-ERα pathway for anticipatory activation of the UPR [39]. Comparing the cytotoxic actions of BHPI to the protective anticipatory activation of the UPR by estrogen, BHPI induces hyperactivation of PLCγ, which leads to greatly increased production of IP3 and a massive efflux of calcium stored in the lumen of EnR into the cytosol. Because of this massive release of calcium into the cytosol, the magnitude and duration of UPR activation by BHPI is much larger than what is seen with estrogens [22, 39]. This leads to strong activation of the PERK arm of the UPR, resulting in extensive eIF2α phosphorylation and near quantitative inhibition of protein synthesis [39]. Cytosolic calcium levels are tightly regulated because high levels of calcium in the cytosol are toxic [40–43]. To maintain cellular calcium homeostasis, the cell activates sarco/endoplasmic reticulum Ca2+-ATPase (SERCA) pumps, which catalyze ATP-dependent transfer of calcium from the cytosol into the lumen of the EnR [44–46]. Since the IP3R calcium channels are still open, the calcium pumped into the EnR leaks back out to cytosol creating a futile cycle that rapidly depletes cellular ATP. This ATP depletion results in increased AMP, which activates the metabolic sensor AMPK [39]. Together, high levels of cytosolic calcium and AMPK activation activate the Ca2+/calmodulin-dependent kinase, eukaryotic elongation factor 2 kinase (CAMKIII/eEF2K). This results in phosphorylation and inactivation of eukaryotic elongation factor 2 (eEF2), inhibiting protein synthesis at a second site [39, 47, 48]. Anticipatory UPR activation by estrogen is weak and transient because the induced expression of UPR molecular chaperones helps resolve UPR stress and ultimately reverses UPR activation [1, 4, 12]. In contrast, since BHPI blocks global protein synthesis including synthesis of UPR-induced molecular chaperones such as p58IPK, GADD34, and BiP, BHPI activation of UPR is unresolvable.
BHPI is exceptionally effective as a new preclinical anticancer drug. It works in ERα positive cancer cells that do not require estrogens or ERα for growth and also in cells that are therapy-resistant [39]. Importantly, at nanomolar concentrations, BHPI does not only stop cancer cell growth, but also kills many breast and endometrial cancer cell lines. In the mouse xenograft model of breast cancer in which BHPI treatment was initiated when tumors reached about 45 mm2, control and BHPI treated tumors were both continuously exposed to estrogen. BHPI, at the reasonable dose of 15 mg/kg daily for 10 days, rapidly stopped tumor growth and induced substantial regression of 48 out of 52 tumors [39]. Moreover, BHPI was well tolerated by the mice [39].
An intriguing question to ask is, since BHPI hyperactivates the UPR converting it from protective to toxic, why is BHPI not toxic to normal ERα positive cells? The UPR is nearly off in most normal cells. We hypothesize that since UPR expression is already elevated as part of the mechanism that protects cancer cells [39], it is actually easier for BHPI hyperactivation of the UPR to push the already activated UPR from cytoprotective to cytotoxic in a cancer cell than it is in a normal cell that starts with a much lower level of UPR activation.
In summary, BHPI is the first example of an ERα biomodulator that targets the UPR pathway and converts it from cytoprotective to cytotoxic.
4. Conclusion
Steroid and peptide hormones play key roles in normal cell physiology and in pathology. The ability to tolerate various cellular stresses is crucial for cell survival, especially in the tumor microenvironment. Anticipatory UPR activation is an emerging rapid extranuclear signaling pathway that is activated by different mitogenic hormones to resolve future cellular stresses. These rapid responses and early events can also lead to downstream genomic effects and are important in cross-talk, leading to reciprocal regulation of the nuclear and extranuclear pathways. Identification of the players that communicate molecular messages, especially elevated intracellular calcium and increased chaperone levels from the UPR pathway to the nucleus is essential to understanding how the UPR pathway influences the gene regulation network. Moreover, disruption or strong enhancement of the UPR offers a new approach to cancer therapy and to overcoming resistance to current therapies.
Acknowledgments
Supported by Carter (to XZ), Westcott and Carter (to NA) and NSF (to MZ) predoctoral fellowships, and NIH RO1 DK 071909 and DOD BCRP BC13187 (to DS).
Abbreviations
- AR
androgen receptor
- CAMKIII/eEF2K
eukaryotic elongation factor 2 kinase
- Ec
ecdysone
- EcR
ecdysone receptor
- EGF
epidermal growth factor
- EGFR
epidermal growth factor receptor
- eIF2α
eukaryotic initiation factor 2 α
- EnR
endoplasmic reticulum
- ERα
estrogen receptor α
- IP3
inositol triphosphate
- IP3R
inositol triphosphate receptor
- PERK
protein kinase RNA-like endoplasmic reticulum kinase
- PLCγ
phospholipase C gamma
- SERCA
sarco/endoplasmic reticulum Ca2+-ATPase
- UPR
unfolded protein response
- VEGF
vascular endothelial growth factor
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Ron D, Walter P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat Rev Mol Cell Biol. 2007;8:519–29. doi: 10.1038/nrm2199. [DOI] [PubMed] [Google Scholar]
- 2.Wang M, Kaufman RJ. The impact of the endoplasmic reticulum protein-folding environment on cancer development. Nat Rev Cancer. 2014;14:581–97. doi: 10.1038/nrc3800. [DOI] [PubMed] [Google Scholar]
- 3.Korennykh A, Walter P. Structural basis of the unfolded protein response. Annu Rev Cell Dev Biol. 2012;28:251–77. doi: 10.1146/annurev-cellbio-101011-155826. [DOI] [PubMed] [Google Scholar]
- 4.Novoa I, Zeng H, Harding HP, Ron D. Feedback inhibition of the unfolded protein response by GADD34-mediated dephosphorylation of eIF2alpha. J Cell Biol. 2001;153:1011–22. doi: 10.1083/jcb.153.5.1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yan W, Frank CL, Korth MJ, Sopher BL, Novoa I, Ron D, et al. Control of PERK eIF2alpha kinase activity by the endoplasmic reticulum stress-induced molecular chaperone P58IPK. Proc Natl Acad Sci U S A. 2002;99:15920–5. doi: 10.1073/pnas.252341799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Deroo BJ, Korach KS. Estrogen receptors and human disease. J Clin Invest. 2006;116:561–70. doi: 10.1172/JCI27987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Evans RM. The steroid and thyroid hormone receptor superfamily. Science. 1988;240:889–95. doi: 10.1126/science.3283939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Huang P, Chandra V, Rastinejad F. Structural overview of the nuclear receptor superfamily: insights into physiology and therapeutics. Annu Rev Physiol. 2010;72:247–72. doi: 10.1146/annurev-physiol-021909-135917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Katzenellenbogen BS. Dynamics of steroid hormone receptor action. Annu Rev Physiol. 1980;42:17–35. doi: 10.1146/annurev.ph.42.030180.000313. [DOI] [PubMed] [Google Scholar]
- 10.Musgrove EA, Sutherland RL. Biological determinants of endocrine resistance in breast cancer. Nat Rev Cancer. 2009;9:631–43. doi: 10.1038/nrc2713. [DOI] [PubMed] [Google Scholar]
- 11.Tyson JJ, Baumann WT, Chen C, Verdugo A, Tavassoly I, Wang Y, et al. Dynamic modelling of oestrogen signalling and cell fate in breast cancer cells. Nat Rev Cancer. 2011;11:523–32. doi: 10.1038/nrc3081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bravo R, Parra V, Gatica D, Rodriguez AE, Torrealba N, Paredes F, et al. Endoplasmic reticulum and the unfolded protein response: dynamics and metabolic integration. Int Rev Cell Mol Biol. 2013;301:215–90. doi: 10.1016/B978-0-12-407704-1.00005-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gao Y, Sartori DJ, Li C, Yu QC, Kushner JA, Simon MC, et al. PERK is required in the adult pancreas and is essential for maintenance of glucose homeostasis. Mol Cell Biol. 2012;32:5129–39. doi: 10.1128/MCB.01009-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hotamisligil GS. Endoplasmic reticulum stress and the inflammatory basis of metabolic disease. Cell. 2010;140:900–17. doi: 10.1016/j.cell.2010.02.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Malhi H, Kaufman RJ. Endoplasmic reticulum stress in liver disease. J Hepatol. 2011;54:795–809. doi: 10.1016/j.jhep.2010.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Carroll JS, Liu XS, Brodsky AS, Li W, Meyer CA, Szary AJ, et al. Chromosome-wide mapping of estrogen receptor binding reveals long-range regulation requiring the forkhead protein FoxA1. Cell. 2005;122:33–43. doi: 10.1016/j.cell.2005.05.008. [DOI] [PubMed] [Google Scholar]
- 17.Hah N, Danko CG, Core L, Waterfall JJ, Siepel A, Lis JT, et al. A rapid, extensive, and transient transcriptional response to estrogen signaling in breast cancer cells. Cell. 2011;145:622–34. doi: 10.1016/j.cell.2011.03.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.York B, O’Malley BW. Steroid receptor coactivator (SRC) family: masters of systems biology. J Biol Chem. 2010;285:38743–50. doi: 10.1074/jbc.R110.193367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hammes SR, Davis PJ. Overlapping nongenomic and genomic actions of thyroid hormone and steroids. Best Pract Res Clin Endocrinol Metab. 2015;29:581–93. doi: 10.1016/j.beem.2015.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Levin ER. Plasma membrane estrogen receptors. Trends Endocrinol Metab. 2009;20:477–82. doi: 10.1016/j.tem.2009.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Song RX, Santen RJ. Membrane initiated estrogen signaling in breast cancer. Biol Reprod. 2006;75:9–16. doi: 10.1095/biolreprod.105.050070. [DOI] [PubMed] [Google Scholar]
- 22.Andruska N, Zheng X, Yang X, Helferich WG, Shapiro DJ. Anticipatory estrogen activation of the unfolded protein response is linked to cell proliferation and poor survival in estrogen receptor alpha-positive breast cancer. Oncogene. 2015;34:3760–9. doi: 10.1038/onc.2014.292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Masuda H, Zhang D, Bartholomeusz C, Doihara H, Hortobagyi GN, Ueno NT. Role of epidermal growth factor receptor in breast cancer. Breast Cancer Res Treat. 2012;136:331–45. doi: 10.1007/s10549-012-2289-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sainsbury JR, Farndon JR, Needham GK, Malcolm AJ, Harris AL. Epidermal-growth-factor receptor status as predictor of early recurrence of and death from breast cancer. Lancet. 1987;1:1398–402. doi: 10.1016/s0140-6736(87)90593-9. [DOI] [PubMed] [Google Scholar]
- 25.Salomon DS, Brandt R, Ciardiello F, Normanno N. Epidermal growth factor-related peptides and their receptors in human malignancies. Crit Rev Oncol Hematol. 1995;19:183–232. doi: 10.1016/1040-8428(94)00144-i. [DOI] [PubMed] [Google Scholar]
- 26.Yu L, Andruska N, Zheng X, Shapiro DJ. Anticipatory activation of the unfolded protein response by epidermal growth factor is required for immediate early gene expression and cell proliferation. Mol Cell Endocrinol. 2015 doi: 10.1016/j.mce.2015.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Folkman J. Tumor angiogenesis: therapeutic implications. N Engl J Med. 1971;285:1182–6. doi: 10.1056/NEJM197111182852108. [DOI] [PubMed] [Google Scholar]
- 28.Kieran MW, Kalluri R, Cho YJ. The VEGF pathway in cancer and disease: responses, resistance, and the path forward. Cold Spring Harb Perspect Med. 2012;2:a006593. doi: 10.1101/cshperspect.a006593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nishida N, Yano H, Nishida T, Kamura T, Kojiro M. Angiogenesis in cancer. Vasc Health Risk Manag. 2006;2:213–9. doi: 10.2147/vhrm.2006.2.3.213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Karali E, Bellou S, Stellas D, Klinakis A, Murphy C, Fotsis T. VEGF Signals through ATF6 and PERK to promote endothelial cell survival and angiogenesis in the absence of ER stress. Mol Cell. 2014;54:559–72. doi: 10.1016/j.molcel.2014.03.022. [DOI] [PubMed] [Google Scholar]
- 31.Perner S, Cronauer MV, Schrader AJ, Klocker H, Culig Z, Baniahmad A. Adaptive responses of androgen receptor signaling in castration-resistant prostate cancer. Oncotarget. 2015;6:35542–55. doi: 10.18632/oncotarget.4689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Watson PA, Arora VK, Sawyers CL. Emerging mechanisms of resistance to androgen receptor inhibitors in prostate cancer. Nat Rev Cancer. 2015;15:701–11. doi: 10.1038/nrc4016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sheng X, Arnoldussen YJ, Storm M, Tesikova M, Nenseth HZ, Zhao S, et al. Divergent androgen regulation of unfolded protein response pathways drives prostate cancer. EMBO Mol Med. 2015;7:788–801. doi: 10.15252/emmm.201404509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Boulanger A, Dura JM. Nuclear receptors and Drosophila neuronal remodeling. Biochim Biophys Acta. 2015;1849:187–95. doi: 10.1016/j.bbagrm.2014.05.024. [DOI] [PubMed] [Google Scholar]
- 35.Liu W, Cai MJ, Zheng CC, Wang JX, Zhao XF. Phospholipase Cgamma1 connects the cell membrane pathway to the nuclear receptor pathway in insect steroid hormone signaling. J Biol Chem. 2014;289:13026–41. doi: 10.1074/jbc.M113.547018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Das I, Krzyzosiak A, Schneider K, Wrabetz L, D’Antonio M, Barry N, et al. Preventing proteostasis diseases by selective inhibition of a phosphatase regulatory subunit. Science. 2015;348:239–42. doi: 10.1126/science.aaa4484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Harding HP, Zyryanova AF, Ron D. Uncoupling proteostasis and development in vitro with a small molecule inhibitor of the pancreatic endoplasmic reticulum kinase, PERK. J Biol Chem. 2012;287:44338–44. doi: 10.1074/jbc.M112.428987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Andruska N, Mao C, Cherian M, Zhang C, Shapiro DJ. Evaluation of a luciferase-based reporter assay as a screen for inhibitors of estrogen-ERalpha-induced proliferation of breast cancer cells. J Biomol Screen. 2012;17:921–32. doi: 10.1177/1087057112442960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Andruska ND, Zheng X, Yang X, Mao C, Cherian MM, Mahapatra L, et al. Estrogen receptor alpha inhibitor activates the unfolded protein response, blocks protein synthesis, and induces tumor regression. Proc Natl Acad Sci U S A. 2015;112:4737–42. doi: 10.1073/pnas.1403685112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Monteith GR, McAndrew D, Faddy HM, Roberts-Thomson SJ. Calcium and cancer: targeting Ca2+ transport. Nat Rev Cancer. 2007;7:519–30. doi: 10.1038/nrc2171. [DOI] [PubMed] [Google Scholar]
- 41.Orrenius S, Zhivotovsky B, Nicotera P. Regulation of cell death: the calcium-apoptosis link. Nat Rev Mol Cell Biol. 2003;4:552–65. doi: 10.1038/nrm1150. [DOI] [PubMed] [Google Scholar]
- 42.Papp B, Brouland JP. Altered Endoplasmic Reticulum Calcium Pump Expression during Breast Tumorigenesis. Breast Cancer (Auckl) 2011;5:163–74. doi: 10.4137/BCBCR.S7481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Roderick HL, Cook SJ. Ca2+ signalling checkpoints in cancer: remodelling Ca2+ for cancer cell proliferation and survival. Nat Rev Cancer. 2008;8:361–75. doi: 10.1038/nrc2374. [DOI] [PubMed] [Google Scholar]
- 44.Alonso MT, Manjarres IM, Garcia-Sancho J. Privileged coupling between Ca(2+) entry through plasma membrane store-operated Ca(2+) channels and the endoplasmic reticulum Ca(2+) pump. Mol Cell Endocrinol. 2012;353:37–44. doi: 10.1016/j.mce.2011.08.021. [DOI] [PubMed] [Google Scholar]
- 45.Arbabian A, Brouland JP, Gelebart P, Kovacs T, Bobe R, Enouf J, et al. Endoplasmic reticulum calcium pumps and cancer. Biofactors. 2011;37:139–49. doi: 10.1002/biof.142. [DOI] [PubMed] [Google Scholar]
- 46.Bublitz M, Musgaard M, Poulsen H, Thogersen L, Olesen C, Schiott B, et al. Ion pathways in the sarcoplasmic reticulum Ca2+-ATPase. J Biol Chem. 2013;288:10759–65. doi: 10.1074/jbc.R112.436550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Leprivier G, Remke M, Rotblat B, Dubuc A, Mateo AR, Kool M, et al. The eEF2 kinase confers resistance to nutrient deprivation by blocking translation elongation. Cell. 2013;153:1064–79. doi: 10.1016/j.cell.2013.04.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Proud CG. Signalling to translation: how signal transduction pathways control the protein synthetic machinery. Biochem J. 2007;403:217–34. doi: 10.1042/BJ20070024. [DOI] [PubMed] [Google Scholar]