

Abstract
Background
Calmodulin (CaM) mutations are associated with cardiac arrhythmia susceptibility including the congenital long QT syndrome (LQTS).
Objective
To determine the clinical, genetic and functional features of two novel CaM mutations in children with life-threatening ventricular arrhythmias.
Methods
The clinical and genetic features of two congenital arrhythmia cases associated with two novel calmodulin gene mutations were ascertained. Biochemical and functional investigations were done on the two mutations.
Results
A novel, de novo CALM2 mutation (D132H) was discovered by candidate gene screening in a male infant with prenatal bradycardia born to healthy parents. Postnatal course was complicated by profound bradycardia, prolonged QTc (651 msec), 2:1 atrioventricular block and cardiogenic shock. He was resuscitated and was treated with a cardiac device. A second novel, de novo mutation in CALM1 (D132V) was discovered by clinical exome sequencing in a three year-old boy who suffered witnessed cardiac arrest secondary to ventricular fibrillation. ECG recording after successful resuscitation revealed a prolonged QTc of 574 msec. The Ca2+ affinity of CaM-D132H and CaM-D132V revealed extremely weak binding to the C-domain with significant structural perturbations noted for D132H. Voltage-clamp recordings of human induced pluripotent stem cell (iPSC) derived cardiomyocytes transiently expressing wildtype or mutant CaM demonstrated that both mutations caused impaired Ca2+-dependent inactivation (CDI) of voltage-gated Ca2+ current. Neither mutant affected voltage-dependent inactivation.
Conclusion
Our findings implicate impaired CDI in human cardiomyocytes as the plausible mechanism for LQTS associated with two novel CaM mutations. The data further expand the spectrum of genotype and phenotype associated with calmodulinopathy.
Keywords: arrhythmia, calmodulin, long-QT syndrome, calcium channel
INTRODUCTION
Congenital arrhythmia syndromes are treatable causes of life threatening heart rhythm disturbances that occur during childhood and early adulthood.1 These include the congenital long QT syndrome (LQTS),2 catecholaminergic polymorphic ventricular tachycardia (CPVT),3 Brugada syndrome4 and others. Sudden cardiac death may be the first manifestation of the disease. Efforts to identify at-risk persons and to screen for known genetic mutations of arrhythmia syndromes are increasingly successful, but a genetic cause cannot always be found. When a mutation is identified, genotype-phenotype correlation can guide treatment and promote better understanding of the pathophysiological mechanism for arrhythmia susceptibility.5
Recently, mutations in genes encoding the ubiquitous Ca2+ sensing protein calmodulin (CaM), have been discovered as a novel genetic basis for congenital arrhythmia susceptibility.6–10 At least 12 mostly de novo mutations have been discovered in CALM1, CALM2, and CALM3 associated with LQTS, CPVT or idiopathic ventricular fibrillation. In some cases, mutations have been associated with features of more than one arrhythmia syndrome (e.g., LQTS + CPVT).9 In the reported cases of LQTS-associated CaM mutations, infants or young children presented with life-threatening cardiac arrhythmias and a markedly prolonged QT interval. Other notable features include prenatal bradycardia, second-degree heart block, T-wave alternans and cardiac arrest. Calmodulin mutations associated with LQTS reduce Ca2+ binding affinity and impair modulation of cellular targets, particularly the L-type voltage-gated Ca2+ channel (LTCC).11, 12 Specifically, CaM mutants associated with LQTS weaken Ca2+-dependent inactivation (CDI) and this effect has been demonstrated to prolong the plateau phase of the cardiac action potential and is predicted to promote ventricular arrhythmias.
In this paper, we report two novel de novo mutations discovered in two different CaM-encoding genes. Both novel CaM mutations are missense and alter the same aspartate residue at position 132. Biochemical analyses demonstrated substantial impairments in Ca2+ affinity for both mutations, and functional studies performed on human induced pluripotent stem cell (iPSC) derived cardiomyocytes demonstrated impaired CDI of the LTCC. These findings further expand the spectrum of genotype-phenotype relationships among the calmodulinopathies and demonstrate functional consequences of two novel CaM mutations in human cardiomyocytes.
METHODS
Mutation discovery
Genomic DNA was extracted from the peripheral blood lymphocytes of study participants and both sets of parents. Informed consent was obtained from the parents described in Case 1 by a protocol approved by the Ethics board of University Hospital Lausanne (CHUV), Lausanne, Switzerland. Approval for retrospective review of Case 2 was obtained from the Institutional Review Board of Lurie Children’s Hospital of Chicago. In Case 1, all coding exons and their boundaries of the KCNQ1, SCN5A, KCNH2, CALM1 and CALM2 genes of the study participant were screened by polymerase chain reaction (PCR) and direct nucleotide sequencing. Primer sequences and PCR conditions are available on request (contact Z. A. Bhuiyan). Mutational analysis was performed by bi-directional sequencing on an ABI 3500 sequencer (Thermo Fisher Scientific, Waltham, MA). Annotation of variants in CALM1 and CALM2 were based on accession numbers NM_006888.4 and NM_001743.4 (NCBI), respectively.
Mutagenesis
For measurement of Ca2+ affinity, the CaM-D132H mutation was engineered using site-directed mutagenesis as previous reported.7 For electrophysiological experiments, CaM mutations were generated using QuikChange in the mammalian expression plasmid pIRES2-EGFP, which enabled co-expression of CaM and EGFP.
Measurement of Ca2+ Affinity
Proteins were expressed in E. coli BL21 (DE3) grown at 37°C in LB broth. Expression was induced at an O.D. of 0.8 (600nm) using 1 mM IPTG for 4 h. Cells were harvested by centrifugation and purified as previously described.7 15N-enriched protein was produced in the same manner with the exception that cells were grown in minimal media supplemented with 0.5 g/l of 15NH4Cl and induced with 1 mM IPTG overnight at room temperature. Sample purity and molecular weight were confirmed by SDS gel electrophoresis and positive electrospray mass spectroscopy. Calcium ion binding was characterized using a fluorescence-based approach.13 NMR HSQC spectra were collected using a BRUKER Avance III 600 MHz spectrometer equipped with a cryoprobe. Sample conditions were 50 mM HEPES, 100 mM KCl, 50 μM EGTA, pH 7.4 and data were collected at 25°C. CaCl2 was titrated into the sample up to a 78:1 (Ca2+ to CaM) molar ratio. CaM backbone resonance assignments were determined from previously assigned conditions by tracking chemical shift changes over the course of temperature and pH titrations.
Electrophysiology on human cardiomyocytes
Human iPSC-derived cardiomyocytes were obtained from Cellular Dynamics International (Madison, WI) and cultured according to the supplier’s protocol. Cells were plated on 15 mm glass coverslips coated with 0.1% gelatin solution and incubated at 37°C in 5% CO2. Recordings were done between days 7–14 in culture. Transfection of cardiomyocytes was performed with TransIT-LT1 (Mirus Bio, Madison, WI) according to the manufacturer’s protocol. Cells were transfected with wildtype or mutant CaM in the pIRES2-EGFP vector 40–48 hours before electrophysiological recording experiments. Transfection efficiency was estimated at 50%.
Glass electrodes were pulled using a Model P-1000 micropipette puller (Sutter Instruments, Novato, CA) and flame-polished for a final resistance of 1–2 MΩ. Currents carried by Ca2+ or Ba2+ were recorded at room temperature in the whole-cell configuration, filtered at 5 kHz and leak subtracted using a P/4 method. Bath solution contained (in mM) 150 Tris, 10 glucose, 1 MgCl2, and either 10 CaCl2 or 10 BaCl2 and adjusted to pH 7.4 with methanesulfonic acid. Osmolarity of the bath solution was 290mOsm/L. The composition of the pipette solution was (in mM) 135 CsCl, 10 EGTA, 1 MgCl2, 4 Mg-ATP, and 10 HEPES and adjusted to pH 7.3 with CsOH. Osmolarity of the pipette solution was 280mOsm/L.
Data were acquired with an Axopatch 200B amplifier and Clampex 10.3 software (Molecular Devices, Sunnyvale, CA). Green fluorescent cells were identified immediately prior to patch-clamp experiments. Native (endogenous) Ca2+ currents were measured with 100 msec depolarizing voltage steps to between −80 and +60 mV from a holding potential of −80mV. Current carried by Ca2+ was recorded first, followed by recording of Ba2+ current. Non-inactivating current was defined as the amount of current remaining at the end of a 100 msec voltage step. Current evoked by pulses between −10 mV and 50 mV were fit with a single exponential function to determine inactivation time constants. Parameters describing the voltage dependence of activation were obtained by fitting conductance-voltage plots with a Boltzmann function. To evaluate voltage dependent inactivation, Ba2+ current was measured during 1000 msec depolarizing voltage steps between −80 and +60mV from a holding potential of −80mV.
Statistical analysis was done using OriginPro 9.1 (OriginLab, Northhampton, MA) using a one way ANOVA test with the Bonferroni correction for multiple samples.
RESULTS
Case presentations
Case 1
A male infant was born at term by uncomplicated vaginal delivery. He had been diagnosed with a moderate inlet ventricular septal defect and fetal bradycardia at 28 weeks gestation. His initial condition was good, but the ECG on postnatal day 1 revealed sinus bradycardia (83 bpm) and a very prolonged QTc (651 msec, Fig. 1). On postnatal day 2, he developed 2:1 atrioventricular (AV) block, profound bradycardia (50 bpm) complicated by cardiogenic shock with renal and hepatic failure. A temporary transvenous right ventricular pacing electrode was placed and adrenergic support was initiated with subsequent improvement of his systemic circulation. Beta-adrenergic receptor blockade with propranolol was introduced as soon as hemodynamic stability was achieved. Maternal anti-SSA and anti-SSB antibodies were not present. Family history was negative for syncope or sudden death, and both parents had normal ECG recordings with QTc intervals 391 msec (mother) and 406 msec (father).
Fig. 1. Electrocardiogram from Case 1.

Standard 12-lead ECG recorded at presentation. The rhythm was sinus at 83 bpm with a prolonged QT interval (QTc: 651 ms). Evidence for T-wave alternans was present in lead II.
On the fourth day of life, a permanent epicardial dual-chamber (DDD) pacemaker was implanted and his heart was paced at 110 bpm. Echocardiography showed closure of the perimembranous ventricular septal defect, left ventricular non-compaction and an ejection fraction of 58%. The infant progressively recovered from cardiogenic shock, renal and hepatic failure and was discharged on postnatal day 30 with a treatment regimen consisting of propranolol and mexiletine. A 12-lead ECG at this time demonstrated a prolonged QTc interval (Supplemental Fig. S1). At six and 12 months of age, no further arrhythmic events had occurred, but QTc prolongation persisted (Supplemental Fig. S2). However, because of the persistence of a prolonged QTc despite medication, the epicardial pacemaker was upgraded to an epicardial defibrillator.
Case 2
A 3 year-old boy suffered sudden, witnessed collapse while walking up a short flight of steps in a park and received bystander CPR. The initial cardiac rhythm recorded by paramedics was ventricular fibrillation (Fig. 2A). He received a single transcutaneous shock (1.3 Joules/kg), which converted him to asystole followed by intra-osseous epinephrine (0.01 mg/kg) after which he regained sinus rhythm with adequate perfusion following a brief episode of polymorphic ventricular tachycardia (Fig. 2A). The subject remained obtunded with an initial Glasgow Coma Scale score of 3. His first ECG in sinus rhythm revealed a QTc interval of 574 msec (Fig. 2B) with T-wave abnormalities that were considered multifactorial (e.g., recent cardiac arrest, acute neuronal injury). Broad spectrum viral PCR testing of nasal swabs revealed influenza A virus subtype H3 and metapneumovirus. He was treated prophylactically with pooled intravenous immunoglobulins for possible myocarditis. The subject was diagnosed with LQTS due to persistent QTc prolongation despite neurologic improvement, and no other features of myocarditis (negative inflammatory markers, normal echocardiography and magnetic resonance imaging, cardiac catheterization with unremarkable biopsy and coronary angiography).
Fig. 2. Electrocardiographic features of Case 2.

A, Tracings obtained from the defibrillator used to deliver a transcutaneous shock in the field. The rhythm in the top panel is ventricular fibrillation. The rhythm in the bottom panel is polymorphic ventricular tachycardia with cycle length variation between 240 and 340 msec. Sweep speed on the tracings is 25 mm/sec. The tracings are presented in chronological order. B, Standard 12-lead ECG 60 minutes after cardiac arrest showing QTc prolongation (574 ms).
His mother and father had normal QTc intervals of 438 msec and 384 msec, respectively. The father reported an episode of “arrhythmia with fever during childhood”, but no records were available and he was asymptomatic through age 38 years. A maternal first cousin had hypoplastic left heart syndrome. The family history was otherwise negative for cardiac or neurologic events.
An epicardial implantable cardioverter-defibrillator (ICD) was placed 27 days after the event and he was treated with atenolol. Through 13 months of clinical follow-up, he had no cardiac events captured by his ICD and QTc values ranged between 464 ms and 482 ms (Supplemental Fig. S1). A summary of clinical features and other findings for both cases is presented in Table 1.
Table 1.
Clinical features associated with novel CaM mutations.
| Subject | Sex | Age at Diagnosis | Clinical Presentation | QTc (msec) | Treatment | Mutation | Additional Findings |
|---|---|---|---|---|---|---|---|
| Case 1 | M | 1 day | 2:1 AV block, cardiogenic shock | 661 | Propranolol Mexiletine ICD | CALM2-D132H | Left ventricular non compaction |
| Case 2 | M | 3 years | Cardiac arrest | 674 | Atenolol ICD | CALM1-D132V | Additional mutations of unknown significance: GJA5-A96S, DSP-P2777H |
Discovery of novel calmodulin mutations
Targeted mutation screening performed on Case 1 and his parents revealed a heterozygous de novo missense CALM2 mutation (c.394G>C; p.Asp132His). No other mutations were found in KCNQ1, SCN5A, KCNH2 and CALM1. In Case 2, clinical exome sequencing with trio analysis of maternal and paternal samples demonstrated a heterozygous de novo missense mutation in CALM1 (c.395 A>T; p.Asp132Val). Additional mutations were found in both Case 2 and his mother but were of unknown clinical significance (GJA5 p.Ala96Ser, DSP p.Pro2777His). No mutations were observed in CALM2, SCN5A, KCNQ1 and KCNH2.
Aspartic acid is completely conserved at position 132 in calmodulin across species. This residue is critical for Ca2+ ion binding in the fourth EF-hand motif of the C-domain (CaM-C). Neither variant was found in the 1000 genomes,14 NHLBI-Exome Sequencing Project15 or Exome Aggregation Consortium (http://exac.broadinstitute.org/) databases.
Biochemical consequences of mutant calmodulins
The Ca2+ affinity of recombinant wildtype (WT) and mutant CaM was measured by a standard fluorescence approach.13 We observed substantially lower Ca2+ affinity in CaM-C for both D132V (146 ± 61 μM) and D132H (177 ± 48 μM) relative to WT (2.3 ±0.2 μM) CaM (Fig. 3A). To further understand the structural implications, additional Ca2+ titrations of the D132H mutant were performed and monitored by 2D 15N-1H NMR. In the apo-CaM state (Ca2+ free), the majority of backbone cross peaks were visible with only a few resonances missing or shifted compared to the WT CaM spectra (Fig. 3B). Addition of Ca2+ caused many cross peaks in the spectrum to disappear or shift to a location different from that observed for WT CaM (Fig. 3C). Notably, significant perturbations were observed for resonances throughout the C-terminal domain and the linker region between the domains. These effects were present at a stoichiometric 4:1 ratio of Ca2+ to CaM as well as in the presence of a large excess of Ca2+ (78:1; Fig. 3C, D). These results are consistent with a significant disruption of Ca2+ binding by CaM-C and of the corresponding shift from a population of largely closed to largely open conformations in this domain, which is known to facilitate the engagement of target proteins. Differences with respect to WT CaM N-terminal domain (CaM-N) were also observed in the NMR titration up to a 4:1 molar ratio. Hence, Ca2+ binding is also impaired in the CaM-N. However, upon addition of a sufficient excess of Ca2+, all CaM-N domain cross peaks eventually matched those of WT CaM (Fig. 3C, D), indicating that the effects were substantially smaller than those observed in CaM-C.
Fig. 3. Impaired Ca2+ binding by mutant calmodulin.

A, Overlay of intrinsic fluorescence reporting on CaM-C domain for WT (black), D132H (red) and D132V (green) mutant CaM during the course of Ca2+ titration. B, C, and D, Overlay of 15N-1H HSQC NMR spectra for WT (black) and D132H mutant (red) in the absence of Ca2+ (B), at a 4:1 molar ratio of Ca2+ to CaM (C), and in the presence of excess Ca2+ (78:1 molar ratio of Ca2+ to CaM) (D).
Effect of novel CaM mutations on human cardiac calcium channels
Prior studies have demonstrated that overexpression of mutant protein in adult guinea pig ventricular cardiomyocytes and fetal mouse ventricular cardiomyocytes significantly impairs CDI of the cardiac LTCC.11, 12 We investigated this phenomenon using human iPSC-derived cardiomyocytes transiently expressing WT or mutant CaM. For these experiments, we elected to employ a lipid-based transfection reagent to avoid extreme overexpression that might occur with adenovirus as was used for a previous study.11 Using this experimental approach, we were able to investigate the effects of recombinant CaM on the behavior of native (endogenous) Ca2+ currents exhibited by human cardiac myocytes. Figure 4A depicts average Ca2+ currents evoked by depolarizing voltage steps from −80mV to +60mV. The rapid inactivation evident in cells expressing WT CaM is markedly blunted in cells transfected with D132H or D132V consistent with impaired CDI. In Fig. 4B, the average normalized Ca2+ current at +20mV is superimposed onto the Ba2+ current recorded at the same potential. Expression of D132H or D132V CaM had no effect on peak Ca2+ current density (Fig. S3A, Table S1). However, Fig. 5 shows that the ratio of the non-inactivating current to peak current (IEnd/IPeak) is significantly greater in cells expressing mutant CaM, which is consistent with impaired CDI. Current-voltage relationships and peak amplitudes for Ba2+ currents were not different among cells expressing WT or mutant CaM (Fig. S3B, Table S2). Comparison of the inactivation time constants for Ca2+ current reveals significantly larger values in cells expressing mutant CaM as compared to WT, indicative of slower inactivation (p<0.05, Fig. 6A). Inactivation time constants for Ba2+ currents measured in cells expressing WT or mutant CaM were similar at all potentials indicating no effect on voltage dependence of inactivation (Fig. 6B). Averaged conductance-voltage relationships were also not different among the groups (Fig. S4).
Fig. 4. Calcium current recordings from human iPSC-cardiomyocytes expressing WT or mutant CaM.

A, Average Ca2+ currents from cells expressing WT or mutant (D132H, D132V) CaM. Current density is reported as pA/pF. Current was elicited by 100 msec depolarizations from −80 to 60 mV every 10 sec (holding potential was −80 mV). B, Comparison between Ca2+ (red trace) and Ba2+ (black trace) currents recorded at 20 mV. Currents were normalized to peak to compare kinetics. Number of replicates: WT (Ca2+ n = 25, Ba2+ n = 20), D132H (Ca2+ n =9, Ba2+ n = 8), D132V (Ca2+ n = 7, Ba2+ n = 7).
Fig. 5. Impaired Ca2+-dependent inactivation of human cardiac calcium channels.
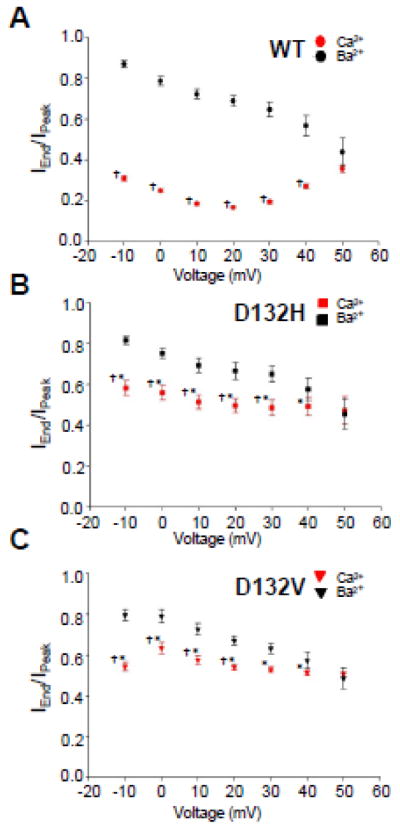
Ratios of non-inactivating current to peak current (IEnd/IPeak) for cells expressing mutant CaM are consistent with impaired CDI. A, IEnd/IPeak ratios determined for cells expressing WT CaM (Ca2+ current, n = 25; Ba2+ current, n = 20). B, IEnd/IPeak ratios determined for cells expressing D132H (Ca2+ current, n = 9; Ba2+ current, n = 8). C, IEnd/IPeak ratios determined for cells expressing D132V (Ca2+ current, n = 7; Ba2+ current n = 7). Significant differences (p < 0.05) between IEnd/IPeak ratios for Ca2+ and Ba2+ currents determined for the same CaM allele are indicated by single daggers (†). Significant differences (p < 0.05) between IEnd/IPeak ratios for Ca2+ currents between cells expressing WT vs mutant CaM are indicated by asterisks (*).
Fig. 6. Inactivation time constants for Ca2+ and Ba2+ currents.

Inactivation time constants were fit by a single exponential function from peak current to the end of a 100 msec (Ca2+) or 1000 msec (Ba2+) step to the indicated voltage. A, Cells expressing mutant CaM have a larger time constant than cells expressing WT CaM, consistent with a slower inactivation rate (p<0.05 at all voltages). Differences between D132H and D132V expressing cells were not significant (p > 0.1) at any voltage. B, Inactivation time constants determined for Ba2+ currents were not significantly different among cells expressing WT, D132H and D132V (p > 0.1).
DISCUSSION
Congenital LQTS is a monogenic arrhythmia susceptibility syndrome with a high level of genetic and allelic heterogeneity.2, 5 Sixteen genes have been associated with LQTS and more than 1000 mutations have been identified. CALM1, CALM2, and CALM3 encoding identical calmodulin polypeptides are the most recent genes to be implicated in the disorder and there is an emerging genotype-phenotype correlation.16 Clinical presentation of children with LQTS-associated CaM mutations typically includes syncope or cardiac arrest during exertion with a seemingly disproportionate representation of de novo mutations in neonates and infants afflicted with life-threatening arrhythmias as compared to other genetic subsets of LQTS. The spectrum of clinical presentations of CaM mutations has probably not been fully elucidated and it remains valuable to ascertain additional cases.
In this paper, we described two cases of long QT syndrome associated with two novel CaM mutations (CALM1-D132V, CALM2-D132H) affecting a highly conserved aspartic acid residue in the same position in the two encoded proteins. We further elucidated the functional and biochemical consequences of the mutations including the demonstration of impaired LTCC function evoked by expressing CaM mutants in human iPSC-derived cardiomyocytes. These findings emphasize the value of considering CALM1-3 genes in the setting of early onset and severe LQTS, and also reinforce the contribution of impaired calcium channel gating as the primary pathogenic mechanism in this form of calmodulinopathy.
Clinical Features of Calmodulinopathic LQTS
The main clinical features of the two cases we presented resemble those of previously reported CaM mutation-associated LQTS.7, 9, 10 In particular, presentation in early life with severe, life-threatening ventricular arrhythmias, cardiac arrest and a markedly prolonged QTc interval are common to most of these cases. In a few of the reported cases and our Case 1, fetal bradycardia was observed. Generally, CaM mutations are associated with a structurally normal heart, but there have been two cases with CALM2 mutations at position 132 (Case 1 reported herein, Case 3 reported by Makita, et al.9) in which left ventricular non-compaction (LVNC) was observed. Case 2, which had a CALM1 mutation at this same amino acid position, did not exhibit LVNC. Whether this structural abnormality is directly or indirectly related to CaM mutation is unknown.
Features of persistent neurological deficits and delayed neurodevelopment were emphasized in the first report of CaM mutations associated with LQTS.7 In Case 1, there were no neurologic sequelae. In Case 2, there was transient profound neurologic injury and subtle long-term neurodevelopmental delay (primarily motor delays). The current hypothesis to explain the neurological phentype associated with CaM mutation is that these were sequelae of multiple cardiac arrests. In situations where a single episode of cardiac arrest was promptly treated or when cardiac arrest did not occur, neurological features have not been apparent or more transient as in Case 2. Further, brain calcium channels may not be as strongly modulated by calmodulins as they are in heart owing to the expression of a family of Ca2+ binding proteins that can antagonize CaM-dependent CDI in central neurons.17, 18
Molecular Pathogenesis of Calmodulinopathic LQTS
Previous studies have demonstrated that overexpression of CaM mutants associated with LQTS in HEK cells, adult guinea pig ventricular myocytes (aGPVM) and mouse fetal ventricular myocytes (FVM) impairs CDI of the cardiac LTCC.11, 12 Attenuated CDI of the LTCC leads to increased Ca2+ influx during channel activation and this prolongs phases 2 and 3 of the cardiac action potential. Although these studies were crucial for elucidating the functional consequences of CaM mutants, the translation of these findings to human cardiomyocytes has not been addressed until now. We utilized human iPSC-derived cardiomyocytes as a platform with which to determine the effect of CaM mutants D132H and D132V on function of the LTCC. This human cardiomyocyte system offers practical advantages for the study of LTCC, which are difficult to express in non-cardiac cells (e.g., HEK-293 cells) because of their multi-subunit architecture. However, cardiomyocytes derived from iPSCs have an immature cellular phenotype including a depolarized diastolic membrane potential that hinders reliable measurements of action potential morphology and duration.19, 20 Fortunately, these limitations did not impede our ability to record robust endogenous Ca2+ currents and to determine the functional impact of CaM mutants.
A potential limitation of our in vitro study relates to the challenge of replicating the in vivo ratio of mutant to WT CaM. Three separate genes encode CaM, and the proportion of mutant CaM depends on the expression of five other WT alleles. In a transient expression paradigm, it is not possible to finely control the amount of mutant protein expressed in each cell. To more accurately model the in vivo situation, reprogramming patient-derived fibroblasts to iPSCs as the starting point for cardiomyocyte differentiation might more accurately represent the disease state in both WT:mutant allelic ratio and genetic background.21 Our study was also limited to investigation of plasma membrane Ca2+ currents. Additional experiments to elucidate effects of these novel CaM mutations on sarcoplasmic reticulum Ca2+ release could provide complementary findings.
As stated previously, the reason that CDI is attenuated by CaM mutations associated with LQTS is an ineffective response to Ca2+ signals. The D132H mutation results in a substantial reduction of Ca2+ binding by CaM-C. At pH 7.4 there is very little charge on the histidine ring and therefore the neutralization of one charge in the highly negative electrostatic field in and around the Ca2+ binding site is not likely to greatly alter the overall attraction of the Ca2+ ion to the site. Rather, defective Ca2+ binding to CaM mutated at the D132 site is most likely due to the lack of any side chain oxygen atom to directly chelate the Ca2+ ion. In addition, such a mutation is expected to perturb the extensive network of hydrogen bonds formed in the binding site when a Ca2+ ion is bound. Importantly, because the effects on Ca2+ affinity extend to CaM-N, the ability of CaM to regulate cellular targets may be further compromised.
Summary
Our work expands the number of CaM mutations known to cause congenital arrhythmia susceptibility to 14. Calmodulin mutations associated with LQTS impair Ca2+ binding affinity, which attenuates CDI of the LTCC. In human cardiomyocytes, mutant CaM perturbs Ca2+ channel function and causes greater Ca2+ flux over time. These effects are predicted to cause prolongation of action potential duration and a propensity for cardiac arrhythmia.
Supplementary Material
Acknowledgments
FUNDING SOURCES
This work was supported by NIH grant HL083374 (A.L.G.), grant 29283 from Fondation Suisse de Cardiologie (Z.A.B.), endowment funding from Vanderbilt University (W.J.C.), NIH postdoctoral fellowship F32-HL117612 (C.N.J.) and a Howard Hughes Medical Institute Medical Research Fellowship (D.C.P.).
Footnotes
DISCLOSURES
The authors have no financial disclosures related to this work
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.George AL., Jr Molecular and genetic basis of sudden cardiac death. J Clin Invest. 2013;123:75–83. doi: 10.1172/JCI62928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schwartz PJ, Crotti L, Insolia R. Long-QT syndrome: from genetics to management. Circ Arrhythm Electrophysiol. 2012;5:868–877. doi: 10.1161/CIRCEP.111.962019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Venetucci L, Denegri M, Napolitano C, Priori SG. Inherited calcium channelopathies in the pathophysiology of arrhythmias. Nat Rev Cardiol. 2012;9:561–575. doi: 10.1038/nrcardio.2012.93. [DOI] [PubMed] [Google Scholar]
- 4.Mizusawa Y, Wilde AA. Brugada syndrome. Circ Arrhythm Electrophysiol. 2012;5:606–616. doi: 10.1161/CIRCEP.111.964577. [DOI] [PubMed] [Google Scholar]
- 5.Schwartz PJ, Ackerman MJ, George AL, Jr, Wilde AA. Impact of genetics on the clinical management of channelopathies. J Am Coll Cardiol. 2013;62:169–180. doi: 10.1016/j.jacc.2013.04.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nyegaard M, Overgaard MT, Sondergaard MT, et al. Mutations in calmodulin cause ventricular tachycardia and sudden cardiac death. Am J Hum Genet. 2012;91:703–712. doi: 10.1016/j.ajhg.2012.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Crotti L, Johnson CN, Graf E, et al. Calmodulin mutations associated with recurrent cardiac arrest in infants. Circulation. 2013;127:1009–1017. doi: 10.1161/CIRCULATIONAHA.112.001216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Marsman RF, Barc J, Beekman L, Alders M, Dooijes D, van den WA, Ratbi I, Sefiani A, Bhuiyan ZA, Wilde AA, Bezzina CR. A mutation in CALM1 encoding calmodulin in familial idiopathic ventricular fibrillation in childhood and adolescence. J Am Coll Cardiol. 2014;63:259–266. doi: 10.1016/j.jacc.2013.07.091. [DOI] [PubMed] [Google Scholar]
- 9.Makita N, Yagihara N, Crotti L, et al. Novel calmodulin mutations associated with congenital arrhythmia susceptibility. Circ Cardiovasc Genet. 2014;7:466–474. doi: 10.1161/CIRCGENETICS.113.000459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Reed GJ, Boczek NJ, Etheridge S, Ackerman MJ. CALM3 mutation associated with long QT syndrome. Heart Rhythm. 2014;12:419–422. doi: 10.1016/j.hrthm.2014.10.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Limpitikul WB, Dick IE, Joshi-Mukherjee R, Overgaard MT, George AL, Jr, Yue DT. Calmodulin mutations associated with long QT syndrome prevent inactivation of cardiac L-type Ca currents and promote proarrhythmic behavior in ventricular myocytes. J Mol Cell Cardiol. 2014;74:115–124. doi: 10.1016/j.yjmcc.2014.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yin G, Hassan F, Haroun AR, Murphy LL, Crotti L, Schwartz PJ, George AL, Satin J. Arrhythmogenic calmodulin mutations disrupt intracellular cardiomyocyte Ca2+ regulation by distinct mechanisms. J Am Heart Assoc. 2014;3:e000996. doi: 10.1161/JAHA.114.000996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.VanScyoc WS, Sorensen BR, Rusinova E, Laws WR, Ross JB, Shea MA. Calcium binding to calmodulin mutants monitored by domain-specific intrinsic phenylalanine and tyrosine fluorescence. Biophys J. 2002;83:2767–2780. doi: 10.1016/S0006-3495(02)75286-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Abecasis GR, Auton A, Brooks LD, DePristo MA, Durbin RM, Handsaker RE, Kang HM, Marth GT, McVean GA. An integrated map of genetic variation from 1,092 human genomes. Nature. 2012;491:56–65. doi: 10.1038/nature11632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fu W, O’Connor TD, Jun G, et al. Analysis of 6,515 exomes reveals the recent origin of most human protein-coding variants. Nature. 2013;493:216–220. doi: 10.1038/nature11690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.George AL., Jr Calmodulinopathy: a genetic trilogy. Heart Rhythm. 2015;12:423–424. doi: 10.1016/j.hrthm.2014.11.017. [DOI] [PubMed] [Google Scholar]
- 17.Oz S, Tsemakhovich V, Christel CJ, Lee A, Dascal N. CaBP1 regulates voltage-dependent inactivation and activation of CaV1. 2 (L-type) calcium channels. J Biol Chem. 2011;286:13945–13953. doi: 10.1074/jbc.M110.198424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Oz S, Benmocha A, Sasson Y, Sachyani D, Almagor L, Lee A, Hirsch JA, Dascal N. Competitive and non-competitive regulation of calcium-dependent inactivation in CaV1. 2 L-type Ca2+ channels by calmodulin and Ca2+-binding protein 1. J Biol Chem. 2013;288:12680–12691. doi: 10.1074/jbc.M113.460949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Knollmann BC. Induced pluripotent stem cell-derived cardiomyocytes: boutique science or valuable arrhythmia model? Circ Res. 2013;112:969–976. doi: 10.1161/CIRCRESAHA.112.300567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sinnecker D, Goedel A, Laugwitz KL, Moretti A. Induced pluripotent stem cell-derived cardiomyocytes: a versatile tool for arrhythmia research. Circ Res. 2013;112:961–968. doi: 10.1161/CIRCRESAHA.112.268623. [DOI] [PubMed] [Google Scholar]
- 21.Rocchetti M, Sala L, Ronchi C, Altomare C, Mura M, Gullo F, Moretti A, Gnecchi M, Crotti L, Schwartz PJ, Zaza A. Genotype-phenotype correlation in induced pluripotent stem cell (iPSC) derived cardiomyocytes carrying calmodulin mutations. Biophys J. 2014;106:333A. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.