

Abstract
Angiogenesis plays a critical role in wound healing after spinal cord injury. Therefore, understanding the events that regulate angiogenesis has considerable relevance from a therapeutic standpoint. We evaluated the contribution of matrix metalloproteinase (MMP)-2 to angiogenesis and vascular stability in spinal cord injured MMP-2 knockout and wildtype (WT) littermates. While MMP-2 deficiency resulted in reduced endothelial cell division within the lesioned epicenter, there were no genotypic differences in vascularity (vascular density, vascular area, and endothelial cell number) over the first two weeks post-injury. However, by 21 days post-injury MMP-2 deficiency resulted in a sharp decline in vascularity, indicative of vascular regression. Complementary in vitro studies of brain capillary endothelial cells confirmed MMP-2 dependent proliferation and tube formation. As deficiency in MMP-2 led to prolonged MMP-9 expression in the injured spinal cord, we examined both short-term and long-term exposure to MMP-9 in vitro. While MMP-9 supported endothelial tube formation and proliferation, prolonged exposure resulted in loss of tubes, findings consistent with vascular regression. Vascular instability is frequently associated with pericyte dissociation and precedes vascular regression. Quantification of PDGFrβ+pericyte coverage of mature vessels within the glial scar (the reactive gliosis zone), a known source of MMP-9, revealed reduced coverage in MMP-2 deficient animals. These findings suggest that acting in the absence of MMP-2, MMP-9 transiently supports angiogenesis during the early phase of wound healing while its prolonged expression leads to vascular instability and regression. These findings should be considered while developing therapeutic interventions that block MMPs.
Keywords: Matrix metalloproteinases, contusion injury, PDGFrβ positive pericytes, angiogenesis, vascularity, vascular regression, proliferation
Introduction
Angiogenesis is a complex multistep process involving endothelial cell activation, sprouting, regression, and maturation (Wietecha et al., 2013). The matrix metalloproteinases (MMPs) MMP-2 and -9 are integral to these processes (Verslegers et al., 2013). They participate in the degradation of the extracellular matrix (ECM) that in turn facilitates detachment of pericytes, and the subsequent directed migration of endothelial cells along a pro-angiogenic gradient (Carmeliet and Jain, 2011). MMPs also regulate endothelial cell proliferation, differentiation, and unmasking of cryptic sites to release pro/anti-angiogenic factors (Roy et al., 2006) and vascular stabilization (Davis and Saunders, 2006; Zhu et al., 2000). Though their contributions to angiogenesis have been studied in detail in other organ systems (Kessenbrock et al., 2010), there is little known about the participation of these MMPs in angiogenesis during wound healing in the injured spinal cord. We have previously shown that MMP-2 is expressed during the angiogenic phase of wound healing after spinal cord injury (SCI) and deletion of MMP-2 results in poorer functional outcomes (Hsu et al., 2006).
SCI causes direct physical damage to the vascular network resulting in progressive hemorrhagic necrosis beginning in the central gray matter and expanding into the pericentral white matter (Simard et al., 2007). Angiogenesis, presumably resulting from the sprouting of surviving vessels, begins 3-4 days after injury within the central-most part of the damaged cord, and peaks at 7 days, where it reaches control levels or higher (Benton et al., 2008; Casella et al., 2002; Loy et al., 2002; Whetstone et al., 2003). These vessels, surrounded by macrophages, predominantly bear an immature phenotype (Benton et al., 2008; Casella et al., 2002; Loy et al., 2002; Whetstone et al., 2003); they are characterized by abnormal leakiness, the absence of the glucose-1 transporter protein, and paucity of perivascular investments including astrocytes and pericytes (Goritz et al., 2011; Whetstone et al., 2003). In the injured murine spinal cord, these immature vessels are localized to heterodomains, a term used to describe the unique environment imposed by focal collections of macrophages that are entrapped by glial scarring (Whetstone et al., 2003). The presence of immature vascular phenotypes in heterodomains contrasts that of the environment within the surrounding glial scar where blood vessels express the glucose-1 transporter, and are associated with astrocytic endfeet that are in close proximity to the endothelial basal lamina (Whetstone et al., 2003).
Inherent to angiogenesis is vascular stabilization (von Tell et al., 2006). Pericytes, which share a common basement membrane with endothelial cells, play a key role in conferring vascular stability (von Tell et al., 2006) through the deposition of matrix and/or by the release, presentation and activation of signals that promote endothelial cell differentiation and quiescence (Armulik et al., 2005). Disruption of pericyte-endothelial cell relationships leads to vascular destabilization, which is characterized by increased permeability, as well as loss of vascular integrity and regression (Simonavicius et al., 2012). While MMP-2 and -9 are implicated in angiogenesis (Verslegers et al., 2013) including vascular stabilization (Davis and Saunders, 2006; Zhu et al., 2000), their relative contributions to this event in the injured spinal cord are unknown.
Here we consider the roles of MMP-2 and -9 in modulating angiogenesis in the injured spinal cord during wound healing when gelatinase activity is notably prominent within angiogenic regions of the contused spinal cord (Goussev et al., 2003; Noble et al., 2002). Our data suggest that vascular stability and vessel maturity are dependent on MMP-2 expression, and that an imbalance in the expression of MMP-2 and MMP-9 results in long-term vascular regression and instability.
Materials and Methods
Breeding
To minimize genetic variances, homozygous MMP-2 knockout (KO) mice and their wild-type (WT) littermates were generated by breeding heterozygous males and females on a C57BL/6 background as previously described (Itoh et al., 1997). The genotypes of animals were confirmed by PCR using specific oligonucleotide primers on genomic DNA (Hsu et al., 2006).
Randomization and Blinding
Congenic littermates were genotyped after weaning and housed till adulthood (3 months of age). Since only female mice that had homozygous expression were used in this study, we randomly allocated animals to the different time points and the order of injury. The surgeon was blinded to the genotype. All histological assessments were performed by an observer blinded to injury and genotype of these mice.
Animal Model
All procedures were approved by the Institutional Animal Care and Use Committee at the University of California, San Francisco. All surgeries were performed blinded to the genotype and randomized across genotypes and post-surgery survival time-points. Adult female WT and KO mice (n = 56) were anesthetized with 2.5% Avertin (0.02 ml/g body weight, intraperitoneally (i.p.), Sigma, St. Louis, MO) and maintained at 37°C throughout surgery and during recovery by use of a warming pad. A contusive injury was produced as described previously (Hsu et al., 2006). Briefly, using aseptic techniques, the spinous process and the lamina of the 8th thoracic vertebra (T8) were removed and a circular region of the dura mater (~ 2.4 mm in diameter) was exposed. After stabilization of the vertebral column, a 3-g weight was dropped from a height of 5 cm onto the exposed dura mater. Following injury, the skin was closed with wound clips. Post-operative care included subcutaneous administration of trimethoprim/sulfonamide (Tribrissen), at 30mg/kg, twice a day for 10 days post-injury and manual expression of the bladder twice/day till the end of the study.
Systemic BrdU Injections
Animals (n= 5/genotype/time point) were administered bromodeoxyuridine [(BrdU, Sigma, St. Louis) (100 mg/kg, i.p.)] 24 hours prior to euthanasia at 0, 3, 7, 14, or 21 days post-injury.
Histological Analyses
Tissue Preparation
Mice were perfused with 4% paraformaldehyde (PFA) and the spinal cord was excised and post-fixed for 4 hours at 4°C, followed by incubation in 20% sucrose for 3-5 days at 4°C. A segment, 1.5 cm in length and centered over the site of injury, was cryoprotected in tissue embedding medium (Triangle Biomedical Sciences, Durham, NC). Spinal cords were sectioned longitudinally at 30-μm thickness on a cryostat, then thaw mounted on Superfrost microscope slides (Fisher, Pittsburgh, PA) and stored at −80°C.
Immunohistochemistry
Every fifth longitudinal section per animal was used for analysis. To minimize variability, replicates from injured animals and one uninjured animal were processed together. Sections were stained with Armenian Hamster anti-platelet endothelial cell adhesion molecule 1 (CD31) Ab (Hamster anti mouse, Millipore, Bellerica, MA; 1:500), anti-bromodeoxyuridine (BrdU Ab, polyclonal sheep, Abcam, Cambridge, MA; 1:1000), Rabbit anti-platelet derived growth factor receptor-beta (PDGFrβ, Rabbit monoclonal anti human, Abcam; 1:100), and Rabbit anti-glial fibrillary acidic protein (GFAP, Rabbit polyclonal anti cow, Dako, Carpinteria, CA; 1:500). Secondary antibodies used were, Cy-3 conjugated Goat x Armenian Hamster IgG (Jackson Immunoresearch, West Grove, PA; 1:200), FITC conjugated Donkey x Sheep IgG (Jackson Immunoresearch; 1:500), Cy-3 conjugated Goat x Rabbit IgG (Jackson Immunoresearch; 1:200), AlexaFluor 488 conjugated Goat x Armenian Hamster IgG (Jackson Immunoresearch; 1:200), and Alexa Fluor-647 conjugated Donkey x Rabbit IgG (Jackson Immunoresearch; 1:200). Nuclei were visualized with DAPI (Invitrogen, Carlsbad, CA; at 300 nM concentration).
Image Acquisition
Images were captured at the epicenter, defined as the region of maximal damage. The epicenter was subdivided into 3 zones; a central “core” composed primarily of macrophages, endothelial cells, and PDGFrβ+ pericyte scarring (Figures 1 and 2), the surrounding glial scar (Zone 1) and the region immediately adjacent to the glial scar (Zone 2), that includes reactive astrocytes (Figure 1). An internal control consisted of two images, taken at approximately 8 mm rostral and caudal to the epicenter, where the spinal cord showed no overt damage.
Figure 1. The epicenter of the contused spinal cord at 3 weeks post-injury is characterized by distinct regions, defined by glial and pericyte scarring.

A, This montage illustrates the typical appearance of the core and Zones 1 and 2, demarcated by white lines. The core typically consists of scattered CD31+ vessels and PBGFrβ+ pericytes, enclosed within a prominent pericyte scar. This pericyte scar is surrounded by Zone 1, consisting of dense GFAP+ glial scarring. Zone 2 represents the region immediately peripheral to the astrocyte scar and consists of both grey and white matter structures. Arrows indicate orientation of the cord as dorsal (d) and caudal (c).
B, An internal control, located approximately 6700-8000 μm from core, represents baseline expression and distribution of blood vessels, pericytes, and astrocytes.
C-E, Examples of Zones 1 and 2, noting distance from the core, with white lines delineating the boundary between Zone 1 and the core.
GM: gray matter, WM: white matter
Figure 2. The epicenter core of the contused spinal cord at 3 weeks post-injury is primarily comprised of macrophages and thick pericyte scarring.
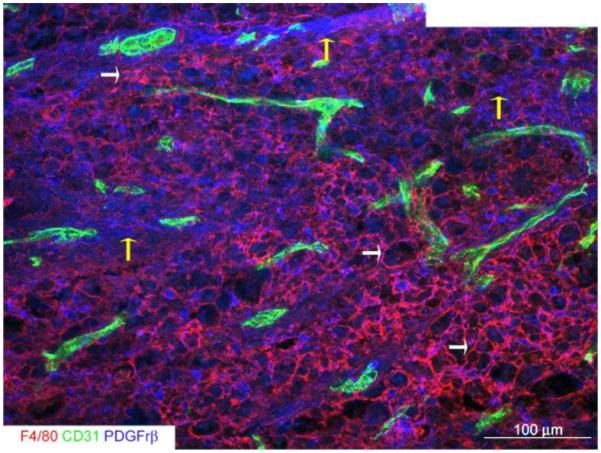
This montage illustrates the core of the epicenter where there is a characteristic dense accumulation of F4/80+ macrophages (white arrows), CD31+ vascular structures and pronounced pericyte scarring (yellow arrows).
All measurements of vascularity as defined by vascular density, area, and number of endothelial cells, were restricted to the core (Figure 2). Three non-overlapping images per section for a total of 8 sections per animal were captured with a 20x objective using a Zeiss LSM 510 confocal microscope (Zeiss, Thornwood, NY) using the following lasers: 405 nm for DAPI/Hoechst (10%); 488 nm for FITC/Alexa 488 (Argon tube current ~5.5A 5%); 543 nm for CY3 (80%). The Z-stage interval was set at 2 μm intervals. For the uninjured spinal cord, the same numbers of images were captured per section from the same segmental level of the spinal cord. Images were acquired using identical exposures for all groups and imported into the MetaMorph® Microscopy Automation and Image Analysis software (Molecular Devices, Downingtown, PA).
Non-overlapping images (635μm × 635μm) within Zones 1 and 2 (Figure 1) were captured with a Nikon C2 confocal microscope, equipped with 4 lasers (Nikon Inc., Melville, NY) for evaluation of pericyte investment of blood vessels and endothelial apoptosis. The Z-stage interval was set to a 1μm interval setting. The core was excluded from these analyses due to the dense expression PDGFrβ+ pericytes that obscured profiles of vessels.
Quantitative Analysis of Endothelial Cell Proliferation
All image analyses were performed blinded with respect to sample origin, and a total of 21 - 24 images per animal were evaluated. CD31, BrdU, and DAPI+ nuclei were identified using the Count Nuclei Module and the color combine function in MetaMorph® software. Set threshold parameters were used to identify BrdU+ and DAPI+ nuclei in CD31+ endothelial cells. Minimum and maximum nuclei width was set up by identifying positive nuclei in 6-8 images/animal. These set values were then used for all image analyses to obtain segmented images that identified only BrdU and DAPI+ nuclei. These three images (segmented-BrdU, segmented-DAPI and unmodified-CD31) were then overlaid using the color combine function. The total number of CD31+ cells that co-localized with BrdU and DAPI+ nuclei were counted using Metamorph®’s Count Nuclei Module. Proliferating endothelial cells are expressed as per μm2 CD31+ vascular area. Vascularity measurements for total numbers of endothelial cells (CD31+ cells with DAPI+ nucleus) were also counted in these images using a similar method of analysis.
Quantitative Analysis of Vascular Density
Intensity measurements were performed using MetaMorph® software. Vessel density, determined from CD31+ vessels, was based upon pixel intensity (Baluk et al., 2004) that was estimated using a set threshold (Benton et al., 2009; Benton et al., 2008; Fassbender et al., 2011; Lutton et al., 2012; Myers et al., 2011; Myers et al., 2012). Six to eight images per animal at 7 and 14 day time points (time points when we observed maximum CD31 staining) were used to set threshold parameters. Minimum pixel intensity was set such that most of the vessels were detected with minimum background detection while maximum pixel intensity was set to 255. An average of the minimum pixel intensity (determined to be 119 in this study) was then used as the minimum pixel intensity value for analyses of all images. Total intensity of each image was estimated by setting the minimum pixel intensity to 0 and maximum intensity to 255. Percentage vessel density was defined as (threshold pixel intensity/total pixel intensity)*100.
Quantitative Analysis of Vascular Area
Vascular area was defined as CD31+ area (Mahoney et al., 2009; Myers et al., 2012) and analyzed using the Angiogenesis Tube Formation application within MetaMorph®. Values for three parameters (minimum width, maximum width, and intensity above local background) were established for each time point after injury. The same settings were used for all images and data were expressed as the percentage (%) area covered by vessels [(Vascular area/total image area)*100].
Quantitative Analysis of Pericyte Coverage
Pericyte coverage of vessels was analyzed at 14 and 21 days post-injury. Images from a Nikon C2 confocal microscope were imported into Metamorph® and analyzed using the Manually Count Objects Module, where total numbers of vessels with and without pericyte coverage were determined. Percentage of vessels with pericyte coverage was calculated and compared within each zone between WT and KO.
Gelatin Zymography
Spinal cord lysates from 5mm segments, prepared from the epicenter (from sham and 21 days post-injury) were prepared as described previously (Hsu et al., 2006) and subjected to gelatin zymography. Equal amounts of protein (30 μg) were loaded on a 10% zymogram gel. For in vitro assays, conditioned media from cultured RBCEC-4 cells or activated RBCEC-4 cells were collected and subjected to gelatin zymography. Culture supernatants were concentrated using a 50 K Microcon filter (Millipore, Bellerica, MA), and equal amounts of protein (5 μg) were loaded on a 10% zymogram gel. After electrophoresis, the gel was incubated with renaturing buffer (Bio-Rad Laboratories, Hercules, CA) at room temperature for 30 min to restore the gelatinolytic activity of the proteins, and then incubated with developing buffer (Bio-Rad Laboratories) at 37°C for 48 h. The gel was then stained with Coomassie Blue and destained until clear bands became evident.
In Vitro Endothelial Cell Assays
Cell Lines and Culture Conditions
Rat brain capillary endothelial cells (RBCEC-4) (Blasig et al., 2001) were propagated in endothelial cell growth medium (Medium 131, Cascade Biologics, Portland, Oregon; 10% heat inactivated, fetal bovine serum, Hyclone, Logan, UT; 1X microvascular growth supplement (MVGS), Cascade Biologics; and 1X penicillin/streptomycin, UCSF Cell Culture Facility) on tissue culture dishes that were coated with 0.2 mg/ml of rat tail Collagen, Type I (Sigma, St. Louis, MO). The MMP-2 specific inhibitor, cis-9-octadecenoyl-N-hydroxylamide (OA-Hy, Calbiochem, San Diego, CA; Ki=1.7 μM) was dissolved in dimethylsulfoxide (DMSO) and used at a final concentration of 25 μM. MMP-9 inhibitor I (Calbiochem, C50 = 5 nM), MMP-1 (IC50 = 1.05 μM) and MMP-13 (IC50 = 113 nM), was dissolved in DMSO and used at a final concentration of 1 μM. DMSO served as the vehicle control. Endothelial cells were activated with 10 ng/ml tumor necrosis factor-alpha (TNF-α) and in some experiments (tube formation or proliferation) were exposed to purified recombinant human MMP-9 (Abcam, Cambridge, MA) at 10 pg/ml, 50 ng/ml or 100 ng/ml for 2h or 24h.
BrdU Assay
Endothelial cell proliferation was determined using a BrdU ELISA kit (Chemicon, Bellerica, MA) following a protocol recommended by the vendor. Cells were plated at the same conditions as described above in 96-well plates. MMP-9 was induced in cells by addition of 10 ng/ml TNF-α. Cultures were exposed to MMP-9 at 50 ng/ml in the absence of serum or growth factors, or MMP-2 or -9 specific inhibitors (25 μM OA-Hy or 1 μM MMP-9 inhibitor I in DMSO overnight). The following day BrdU was added and cells incubated at 37°C/5% CO2 overnight. Fixative/Denaturing Solution (Chemicon kit) was added to each well, followed by incubation with anti-BrdU antibody and Goat anti-Mouse IgG HRP Conjugate. The color was developed by addition of Substrate Solution and Stop Solution to each well. Absorbance was measured at dual wavelengths of 450-540 nm with a Thermomax plate reader (Molecular Devices, Sunnyvale, CA). Each assay was repeated twice with 4 wells/condition and absorbance normalized to medium or medium plus TNF-α / MMP-9.
Endothelial Cell Migration Assay
Migration was measured using Transwell culture chambers (polycarbonate filter with 8 μm pore size, 6.5 mm insert diameter (Costar, Corning, NY). Transwell chambers were coated with 0.2 mg/ml Collagen Type I and placed in a 24-well plate containing 600 μl of endothelial cell growth medium (Medium 131, Cascade Biologics, 10% heat inactivated, fetal bovine serum, Hyclone, 1X microvascular growth supplement (MVGS), Cascade Biologics; and 1X penicillin/streptomycin, UCSF Cell Culture Facility). 1×105 cells resuspended in serum free medium, medium containing DMSO or medium containing OA-Hy were placed in the upper compartment of the chamber and incubated at 37°C for 18-20 h to allow cell migration (3 Transwells per condition). F-10/alpha MEM medium without FBS was used as a negative control. Cells were fixed on the membrane with 4% PFA and stained with DAPI. All images were captured on a fluorescence microscope (Optiphot EF-D3; Nikon, Tokyo, Japan) equipped with a SPOT camera (SPOT™ Imaging solutions, Sterling Heights, MI) using the same exposure setting. The number of endothelial cells on the lower surface of the filter was determined by counting five randomly selected fields per membrane (100x magnification) using MetaMorph®’s Count Nuclei Module. Each experiment was performed three times using triplicate wells and migration was expressed as relative cells migrated as compared to medium alone.
Endothelial Tube Formation Assay
Matrigel tube formation assays were performed in 96-well plates, coated with 50 μl Matrigel/well (BD Biosciences, Bedford, MA), and incubated at 37°C/5%CO2 incubator for 1 hour. Cells were plated at a density of 1.5 × 104 cells per well in endothelial cell basal medium containing 0.1% BSA, in the presence and absence of OA-Hy (25 μM). Endothelial cells were activated with 10 ng/ml TNF-α or incubated with MMP-9 at 10 pg/ml, 50 ng/ml or 100 ng/ml for 2h or 24h. MMP-9 inhibitor I (1 μM) was used with activated endothelial cells. Images were digitally captured on an inverted microscope (Olympus, Center Valley, PA) equipped with a SPOT camera (SPOT™ Imaging solutions) and analyzed using Angiogenesis Tube Formation Module (MetaMorph®). Total tube length, number of segments and number of branch points were determined. Each assay was repeated two to three times with 4-5 wells/condition and values normalized to medium or medium plus TNF-α/ MMP-9.
Statistical Analyses
Statistical analyses were performed using GraphPad Prism (Version 6.0, GraphPad Software, San Diego, CA). Unless otherwise stated, data showed a normal distribution based upon the Shapiro-Wilk test. All in vivo quantifications were analyzed by two-way analysis of variance (ANOVA) and where appropriate Sidak’s multiple comparisons test was used for within group and between group comparisons. For in vitro assays, 2 group comparisons were analyzed using unpaired two-tailed t-tests, whereas analyses of 3 or more groups were by one-way ANOVA and where appropriate, the Tukey’s post-hoc test. All data are represented as means + standard error of the mean with significance defined as p ≤ 0.05.
Results
Appearance of the injured epicenter
The epicenter of both MMP-2 KO and WT animals is comprised of a central “core”, composed of endothelial cells and macrophages that reside within a rich network of processes created by PDGFrβ+ pericyte scarring (Figures 1 and 2), the surrounding glial scar (Zone 1) and the region immediately adjacent to the glial scar (Zone 2) that include some reactive astrocytes (Figure 1).
Endothelial proliferation is dependent on MMP-2
We have previously reported a differential pattern of MMP-9 and MMP-2 expression following SCI in wild type mice. While both are expressed during angiogenesis, MMP-9 expression is most prominent within the acute period (up to 3 days post-injury) (Goussev et al., 2003; Noble et al., 2002), whereas MMP-2 exhibits pronounced expression from 3 up to at least 28 days post-injury (Goussev et al., 2003; Hsu et al., 2006; Noble et al., 2002). The prolonged expression of MMP-2 coincides with an increase in vascular density (Whetstone et al., 2003). To dissect out the roles of individual MMPs in supporting angiogenesis, we examined this wound healing process in the spinal cord injured MMP-2 KO. To determine the dependency of angiogenesis on MMP-2, we first evaluated endothelial proliferation within the lesion core (Figures 1-2), which is devoid of astrocytes and contains non-neural cells that are derived from intrinsic perivascular fibroblasts and pericytes, proliferating endothelial progenitors, and infiltrating fibrocytes (a bone marrow–derived precursor present in the systemic circulation) and inflammatory cells (Burda and Sofroniew, 2014; Faulkner et al., 2004; Goritz et al., 2011; Kawano et al., 2012; Silver and Miller, 2004; Silver et al., 2015; Soderblom et al., 2013; Wanner et al., 2013). The number of BrdU+CD31+DAPI+ endothelial cells, normalized to vascular area, was compared between genotypes (Figure 3A). Proliferation in the WT group increased over time and on average the values for the WT group were greater than that of the KO group [(two-way ANOVA, interaction, p=0.5934, F (4, 39) = 0.7049; effect of genotype, p=0.0083, F (4, 39) = 3.871; effect of time p=0.0096, F (1, 39) = 7.736)]. Main effects analysis revealed a significant effect of both time and genotype. Figures 3B-I are representative images from WT and KO mice showing the presence of BrdU+CD31+ cells in the WT but not in KO mice. Whereas, endothelial cell proliferation was observed in the WT group at 14 and 21 days post-injury, similar differences were not apparent in the KO group, suggesting that MMP-2 is critical to this process.
Figure 3. Endothelial cell proliferation is reduced in the spinal cord injured MMP-2 KO.

A, Endothelial proliferation is significantly lower in MMP-2 KO as compared to the WT group [(two-way ANOVA, effect of genotype, p=0.0083, Values are means + SEM, (n = 5 animals/genotype/time-point)].
B-E, A representative image from the core of the epicenter of a WT at 7 days post-injury. Note the presence of both proliferating endothelial cells (BrdU+, Dapi+, and CD31+ vessels) (white arrows, E) and non-endothelial cells (yellow arrow). Enclosed box in E is shown at higher magnification in e, illustrating the co-localization of BrdU+ Dapi+ CD31+ in a vascular segment.
F-I, A representative image from the core of the epicenter of a KO at 7 days post-injury. While there are prominent CD31+ vessels, none appear to co-localize with BrdU and there is evidence for BrdU+ CD31-cells (yellow arrow, I). Enclosed box in I is shown at higher magnification in i.
MMP-2 deficiency results in vascular regression
Given reduced endothelial cell proliferation in the KO group, we next determined if indices of vascularity (vascular density, vascular area, and number of endothelial cells) were likewise altered within the core over a period of 3 weeks post-injury (Figure 4). As there were no genotypic differences in vascular density in uninjured spinal cords, all measurements were normalized to their respective uninjured values.
Figure 4. Loss of MMP-2 results in long-term vascular regression and increased expression of MMP-9.

Vascularity was analyzed by densitometric (A) and morphometric (B, C) analyses within the core of the epicenter. Change in vascularity, expressed relative to uninjured mice, was analyzed over time across genotypes (two-way ANOVA followed Sidak’s multiple comparisons test).
A, While vascular density is increased in the MMP-2 KO at 7 (**p<0.01) and 14 (*p<0.05) days post injury relative to the WT group, there is a loss of vascular density by 21 days (**p<0.01).
B-C, There is a significant loss of vascular area (B, *p<0.05), and endothelial cells (C, ****p<0.0001) at 21 days post-injury in the MMP-2 KO mice. Values are mean + SEM, (n=5 animals/time point/genotype).
D, Gelatin zymography confirms greater expression of MMP-9 proforms (105kDa) in the injured (inj) MMP-2 KO at 21 days post-injury as compared to injured WT mice and sham (sh) controls.
Note that only the proenzyme form of MMP-9 protein is detected by zymography, an in vivo finding consistent with other studies (Hsu et al., 2008; Hsu et al., 2006; Wang et al., 2000). This is not surprising as the ability to detect active MMP-9 may be compromised by its rapid degradation after binding to the cell surface (Yu and Stamenkovic, 1999).
Purified human MMP-2 and MMP-9 served as MMP standards (MMP STD).
We performed a two way ANOVA that compares two variables, time and genotype. Since we observed a significant interaction between the variables, post-hoc tests for within group changes (time dependent effect) were performed to analyze the effect of time. In WT and MMP-2 KO group individually we asked the question as to how each of the vascular measures evolved over time. There was a significant genotype by time interaction in vascular density [(two-way ANOVA, interaction, p<0.0001, F (3, 67) = 9.284)] (Figure 4A). Within group comparisons revealed no differences over time in the WT group (Sidak’s multiple comparison test, p>0.05). In contrast, there was a transient increase in vascular density by 7 days in the KO group (p≤0.01), followed by a decrease at 21 days relative to all other time points (p≤0.05, 3 hours; p<0.0001, 7 and 14 days). Comparison of vascular density between genotypes at each of the time-points (Sidak’s multiple comparison test) demonstrated a robust increase in vascular density at 7 (p≤0.01) and 14 (p≤0.05) days post-injury followed by sharp decline by 21 days in the KO group (p≤0.01).
Analysis of vascular area across time-points showed a genotype by time point interaction [(two-way ANOVA, interaction, p=0.0009, F (3, 67) = 6.228)]. Vascular area increased over time relative to 3 days post-injury in WT mice (7 days, p<0.0001; 14 days, p≤0.01; 21 days, p≤0.05; Sidak’s multiple comparison test, Figure 4B). In the KO group, vascular area was transiently elevated (7 and 14 days, p<0.0001; 21 days, p>0.05) relative to 3 days with a marked decline in vascular area by 21 days relative to 7 and 14 days post-injury (p<0.0001). Whereas between genotype comparisons revealed no differences between WT and KO mice at 3, 7, and 14 days (p>0.05, Sidak’s multiple comparison test), there was a reduction in vascular area at 21 days post-injury in the KO relative to the WT group (p<0.05).
Next, the number of endothelial cells was compared across time-points and between genotypes (Figure 4C). There was a significant genotype by time interaction [(two-way ANOVA, interaction, p<0.0001, F (3, 67) = 12.76)]. In WT mice, there was an increase in the number of endothelial cells at all time points relative to 3 days post-injury (p<0.0001, Sidak’s multiple comparison test) (Figure 4C). In contrast, this increase was transient in the KO mice (7, 14, and 21 days vs. 3 days, p<0.0001), as evidenced by a reduction in endothelial cell number at 21 days post-injury as compared to early time periods post-injury (7 days, p<0.05; 14 days, p<0.0001). Comparisons between genotypes at each of the time-points (Sidak’s multiple comparison test) showed a significant reduction in endothelial cell number at 21 days post-injury in the KO relative to the WT group (p<0.0001) with no group differences at earlier time points (p>0.05).
Together these in vivo findings demonstrate that while MMP-2 is a determinant of endothelial cell division, formation of new vessels at early time-points proceeds in the absence of this protease. However, longer-term deficiency in MMP-2 culminates in vascular regression, as evidenced by a reduction in vascular density, area, and endothelial cell numbers.
Upregulation of MMP-9 in the injured spinal cord
We have previously reported a compensatory increase in MMP-9 at 1 and 2 weeks post-injury in the spinal cord injured MMP-2 KO (Hsu et al., 2006), a period of time corresponding to angiogenesis (Whetstone et al., 2003). Here we show that MMP-9 is likewise elevated at 3 weeks post-injury (Figure 4D) and as such may not only support angiogenesis in the absence of MMP-2, but may contribute to vascular regression. Here we dissect out the MMP-directed pathways that modulate angiogenesis after spinal cord injury. In the in vitro assays we are mimicking the wound-healing phase of spinal cord injury, where MMP-2 expression dominates. To determine if MMP-2 is involved in endothelial cell proliferation and angiogenesis as assayed by tube formation, we perform in vitro assays using endothelial cells that constitutively express MMP-2.
MMP-2 and MMP-9 exhibit both redundant as well as unique roles in angiogenesis
We have previously shown that MMP-9 protein immunolocalizes with blood vessels (Noble et al., 2002) and prominent gelatinase activity, which consists of both MMP-2 and MMP-9, is observed in the blood vessels at the injury epicenter up to 21 days post-injury (Goussev et al., 2003). Thus, using an in vitro system where we can isolate the different gelatinase activities we are able to address the role of each of the gelatinases in modulating angiogenesis. To determine the dependency of angiogenesis on MMP-2 and MMP-9, cell proliferation, migration, and tube formation were evaluated in an immortalized rat brain capillary endothelial cell line (RBCEC-4) (Blasig et al., 2001) that constitutively expresses MMP-2 in the absence of MMP-9 (Figure 5A). Both cell proliferation, as assessed by BrdU incorporation, and cell migration were reduced by 92% and 40%, respectively, in the presence of the selective MMP-2 inhibitor, OA-Hy [(Cell proliferation: Unpaired two-tailed t-test for proliferation, p<0.001, t=70.31, df=6; Figure 5B and Cell migration: One-way ANOVA p<0.0001, F (2, 126) = 21.35, Bonferroni’s multiple comparisons test, p<0.001 OA-Hy vs. vehicle or medium, Figure 5C-D)].
Figure 5. MMP-2 induces endothelial cell proliferation and migration and facilitates tube formation in vitro.

A, While RBCEC-4 cells endogenously express MMP-2 at high levels by gelatin zymography, MMP-9 is not detected (RBCEC4, lanes 1 and 2). In contrast, bone marrow derived macrophages (BMDM, lanes 3 and 4) express high levels of MMP-9 but no detectable levels of MMP-2.
B, RBCEC-4 cell proliferation, based upon BrdU ELISAs, is reduced in the presence of an MMP-2 inhibitor (OA-Hy), whereas an MMP-9 inhibitor (Inh.I) has no detectable effect. (unpaired two-tailed t-test, ***p<0.001).
C, Representative images of migrating RBCEC-4 cells on a transwell membrane in the presence of the MMP-2 inhibitor, OA-Hy, or vehicle (DMSO).
D, Migrating RBCEC4 cells, expressed relative to medium alone, are reduced in the presence of the MMP-2 inhibitor, OA-Hy, relative to vehicle (DMSO), [(one-way ANOVA followed by Bonferroni’s multiple comparisons test, ###p<0.001 vs. medium, ***p<0.001 vs. vehicle)]. Bars represent mean + SEM averaged over 5 fields/membrane, 3 transwells/condition, and 3 independent experiments.
E, Representative images of RBCEC-4 cells plated on Matrigel and treated with the MMP-2 inhibitor, OA-Hy, or vehicle (DMSO).
F-H, Tube formation is attenuated in the presence of the MMP-2 inhibitor OA-Hy relative to the vehicle, DMSO (one-way ANOVA followed by Bonferroni’s multiple comparisons test), as assessed by F, measures of tube length (###p<0.001 vs. medium, **p<0.01 vs. vehicle), G, number of segments (##p<0.01 vs. medium, **p<0.01 vs. vehicle) and H, number of branch points (###p<0.001 vs. medium, ***p<0.001 vs. vehicle). Values were normalized to medium alone. Bars represent mean + SEM averaged over 4 wells/condition and 3 independent experiments.
Next, we examined the contribution of MMP-2 to tube formation. RBCEC-4 cells were plated on BD Matrigel in the presence of vehicle (DMSO) or OA-Hy. Blockade of MMP-2 resulted in an 18-22% decrease in tube length (Figure 5E-F), [(One-way ANOVA p=0.0002, F (2, 20) = 12.94, Bonferroni’s multiple comparisons test, p<0.01 OA-Hy vs. vehicle; p<0.001 OA-Hy vs. medium)]. Inhibition of MMP-2 also affected the number of segments and branch points. There was a 22% loss in number of segments (Figure 5G), [(One-way ANOVA, p=0.0011, F (2, 20) = 9.800, Bonferroni’s multiple comparisons test, OA-Hy vs. medium or vehicle, p<0.01)], and 24% reduction in the number of branch points (Figure 5H), [(One-way ANOVA, p=0.0002, F (2, 20) = 13.74, Bonferroni’s multiple comparisons test, OA-Hy vs. medium or vehicle, p<0.0001)]. Together, these in vitro data demonstrate that angiogenesis, as defined by proliferation, migration and tube formation, is modulated by MMP-2.
To evaluate the effect of MMP-9, RBCEC-4 cells were activated with TNF-α to induce expression of this protease (Figure 6A). Endothelial proliferation was unchanged in activated endothelial cells treated with an MMP-9 inhibitor I at a concentration of either 1 μM [(Figure 6B, Mann Whitney test, p=0.7428)] or10 μM of this inhibitor (data not shown). In contrast, exposure to the MMP-2 selective inhibitor, OA-Hy, reduced proliferation of activated endothelial cells [(Figure 6B, Unpaired two-tailed t-test, p<0.0001; t=77.51, df=14)]. These data confirm that MMP-9 is not required for cell proliferation, whereas, MMP-2 is able to sustain endothelial cell proliferation in the absence of MMP-9. These data are consistent with in vivo findings of reduced endothelial cell proliferation in MMP-2 KO mice that overexpress MMP-9.
Figure 6. MMP-9, in the absence of MMP-2, does not modulate endothelial cell proliferation but does support endothelial tube formation.

A, By gelatin zymography, MMP-9 is upregulated in RBCEC-4 cells in the presence of TNF-α. Molecular weight markers (M) and purified MMP-2 and MMP-9 served as controls (C), and medium was prepared from RBCEC-4 cells, treated with (+) and without (-) TNF-α.
B, Whereas inhibition of MMP-9 (Inh. I) has no effect on proliferation of activated (TNF-α treated) RBCEC-4 cells, OA-Hy inhibits cell proliferation in both the presence and absence of Inh I. (Unpaired two-tailed t-test, ***p<0.001, ****p<0.0001). Absorbance was normalized to medium + TNF-α wells.
C-E, Inhibition of MMP-2 with OA-Hy in activated (MMP-9-expressing) RBCEC-4 cells has no effect on tube formation (p>0.05), whereas inhibition of both MMP-2 and MMP-9 resulted in a decrease (one-way ANOVA followed by Tukey’s multiple comparisons test) in C, tube length (**p<0.01 vs. medium, #p<0.05 vs. OA-Hy), D, number of segments (*p<0.05 vs. medium, #p<0.05 vs. OA-Hy) and E, number of branch points (*p<0.05 vs. medium, #p<0.05 vs. OA-Hy). All measures were normalized to medium + TNF-α wells. Bars represent mean + SEM averaged over 4 wells/condition and 3 independent experiments.
To determine the contribution of MMP-2 and MMP-9 to the formation of vascular tubes, activated RBCEC-4 cells (expressing both MMP-9 and MMP-2) were plated on BD Matrigel in the presence of vehicle, OA-Hy, or both inhibitors (OA-Hy and MMP-9 inhibitor I). The MMP-2 inhibitor OA-Hy had no effect on tube length of activated endothelial cells (Figure 6C), [(One-way ANOVA, p=0.0025, F (2, 25) = 7.684, Tukey’s multiple comparisons test, p>0.05 vs. vehicle)], the number of vascular segments, (Figure 6D), [(One-way ANOVA, p=0.0131, F (2, 25) = 5.184, Tukey’s multiple comparisons test, p>0.05 vs. vehicle)], or the relative number of branch points, (Figure 6E), [(One-way ANOVA, p=0.0093, F (2, 25) = 5.676, Tukey’s multiple comparisons test, p>0.05 vs. vehicle)]. Inhibition of both MMPs resulted in reduction in tube formation for all the outcomes measured, tube length (Figure 6C) [(Tukey’s multiple comparisons test, p<0.01 vs. vehicle)], the number of vascular segments, (Figure 6D) [(Tukey’s multiple comparisons test, p<0.05 vs. vehicle)], and the relative number of branch points, (Figure 6E) [(Tukey’s multiple comparisons test, p<0.05 vs. vehicle)]. Figures 6C to 6E middle bar represents tube measures in the absence of MMP-2 but in the presence of MMP-9. There is no difference between vehicle treated group and OA-Hy treated wells, suggesting that tube formation proceeds normally in the absence of MMP-2, but in the presence of MMP-9. These data are consistent with our in vivo observations, where we reported similar vascularity between WT and MMP-2 KO early after injury, suggesting that MMP-9 may be compensating for this early angiogenic response. Thus, while MMP-9, in the absence of MMP-2, supports tube formation, inhibition of both proteases (Figure 6C-6E, third bar) results in a reduction in tube length, number of segments and branch points. These data suggest that both proteases may be involved in tube formation.
To study the long-term consequences of MMP-9 on tube formation, RBCEC4 cells were activated with TNF-α, which induced MMP-9. Prolonged expression of MMP-9 resulted in a substantial decrease in vessel length [(Figure 7A, unpaired two-tailed t-test p<0.0001, t=5.624, df=15)], number of segments [(Figure 7B, unpaired two-tailed t-test p<0.0001, t=6.018, df=15)], and number of branch points [(Figure 7C, unpaired two-tailed t-test p<0.0001, t=5.50,7 df=15)] as compared to non-activated cells. As TNF-α may also induce expression of other proteins beyond MMP-9 that may be involved in angiogenesis, similar outcomes were evaluated in cultures exposed to recombinant MMP-9. MMP-9 did not alter cell proliferation [(Figure 7D, One-way ANOVA, p=0.8867, F (2, 12) = 0.1215)]. While short-term exposure (2h) had no effect on tube formation [(One-way ANOVA, p=0.3614, F (3, 11) = 1.181)], as demonstrated by similar tube lengths between treated and untreated cells, prolonged exposure to MMP-9 (24h) led to decreased tube length [(One-way ANOVA, p=0.0003, F (3, 11) = 15.07, Tukey’s multiple comparisons test, 10pg/ml vs. 0pg/ml, p≤0.001; 50ng/ml or 100ng/ml vs. 0pg/ml, p≤0.01; Figure 7E)].
Figure 7. Prolonged exposure to TNF-α or exogenously added MMP-9 leads to a decrease in tube formation but does not alter endothelial cell proliferation.
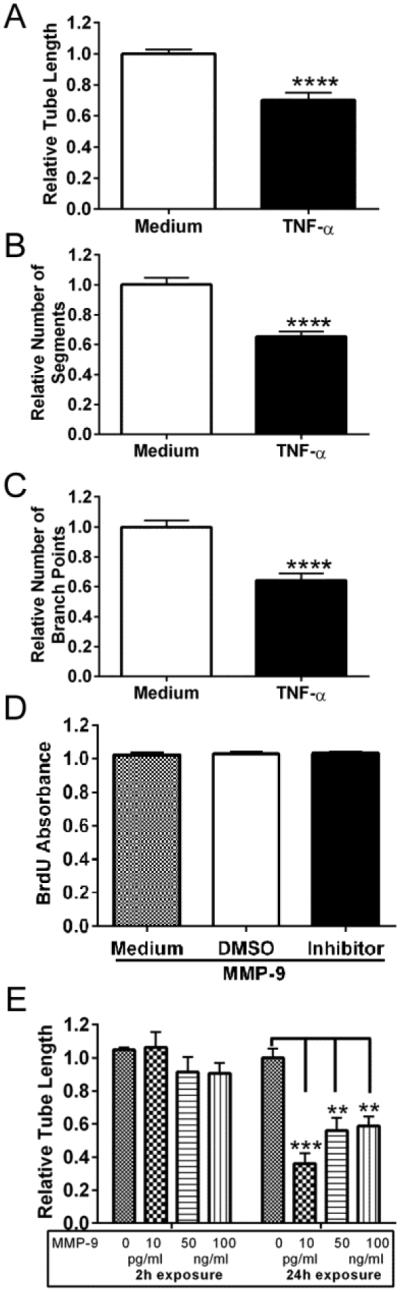
A-C, There is a reduction in tube length, number of segments and number of branch points in response to prolonged MMP-9 expression (unpaired two-tailed t-test, ****p<0.0001). All measures were normalized to medium alone. Bars represent mean + SEM averaged over 4 wells/condition and 3 independent experiments.
D, Proliferation of RBCEC-4 cells, exposed to exogenously added MMP-9 for 24 hours, was analyzed by BrdU ELISA. Long-term exposure to MMP-9 did not alter cell proliferation (one-way ANOVA, p>0.05). Absorbance was normalized to medium. Bars represent mean + SEM averaged over 8 replicates.
E, Formation of tubes by RBCEC-4 cells was analyzed by a Matrigel assay after either short-term (2h) or prolonged (24h) exposure to MMP-9. Prolonged exposure to MMP-9 leads to a decrease in tube length (one-way ANOVA followed by Tukey’s multiple comparisons test, ***p<0.001; **p<0.01). Measures of tube length were normalized to medium alone. Bars represent mean + SEM, averaged over 8 replicates.
Together, in vivo and in vitro findings demonstrate a temporal specificity of MMP-9 - directed angiogenesis. While early exposure to this protease supports this wound healing event, prolonged exposure results in loss of vascular structures. Such findings may at least in part explain vascular regression that is seen in the epicenter core of the MMP-2 KO by 21 days post-injury.
MMP-2 deficiency is associated with a loss of pericytes surrounding vessels in the glial scar
Given the emergence of regression within vessels in the core that lack barrier properties (Whetstone et al., 2003), we next examined the consequences of MMP-2 deficiency on vascular stability within Zone 1, a region immediately adjacent to the lesion core that is comprised of a mature, newly proliferative astrocyte scar (Wanner et al., 2013) and expresses abundant MMP-9 (Hsu et al., 2008; Hsu et al., 2006) and the adjacent Zone 2 that contains hypertrophic astrocytes (Figure 1).
Stabilization of endothelial cells is dependent upon pericytes (Bergers et al., 2003; Kessenbrock et al., 2010; von Tell et al., 2006) and destabilization is a hallmark of vascular regression (Hammes et al., 2004; Pfister et al., 2010). Thus, we evaluated PDGFrβ pericyte/vessel contact (Figures 8A-F) and quantified the percentage of vessels with pericyte coverage across time-points and between genotypes (Figure 8G-I). Representative images from zones 1, 2 and internal control at 21 days post-injury from WT and KO mice are shown in figures 8A-F. The morphology of PDGFrβ positive cells at zones 1 and 2 differed from pericytes in control region, often showing a ramified morphology and bridging between different vessels (Figure 8c). In zone 1 from WT mice pericytes with a normal phenotype are in direct contact with vessels (Figure 8a). In contrast in KO mice, pericyte staining is only punctate and no pericytes are attached to the vessels (Figure 8b). In zone 2, attached pericytes were observed in both WT and KO mice and had normal as well as ramified phenotypes. In a region away from the damaged area (internal control), all vessels displayed pericyte attachment (Figure 8E-F). Pericyte coverage of endothelial cells was similar between genotypes and across time-points in Zone 2 (Figure 8H) [(Two-way ANOVA, Interaction, p=0.0523, F (1, 15) = 4.441; effect of time, p=0.7880, F (1, 15) = 0.07495; effect of genotype, p=0.9364, F (1, 15) = 0.006577)] and the internal control (Figure 8I) [Two-way ANOVA, Interaction, p=0.0305, F (1, 15) = 5.705, ns post hoc analysis]. In contrast, there was a significant effect of genotype in Zone 1 (Figure 8G) [(Two-way ANOVA, Interaction, p=0.1065, F (1, 13) = 3.008; effect of time, p=0.1071, F (1, 13) = 2.997; effect of genotype, p=0.0128, F (1, 13) = 8.324)]. Post-hoc analyses (Sidak’s multiple comparisons test) revealed a loss of pericyte coverage over time in the KO group (p<0.05) and a greater loss of pericyte coverage at 21 days post-injury in the KO as compared to WT group (p<0.01). These findings, demonstrating a progressive loss of pericyte coverage of blood vessels between 14 and 21 days post injury in the KO group, suggests the emergence of vascular instability. As MMP-9 is prominently expressed within astrocytes in Zone 1 in the MMP-2 KO mice (Hsu et al., 2008; Hsu et al., 2006), this protease may be a candidate mediator of pericyte detachment.
Figure 8. Blood vessels associated with pericytes within the glial scar of the MMP-2 KO are reduced in numbers by 21 days post-injury.

A-F, Immunolocalization of CD31+ endothelial cells and PBGFrβ+ pericytes within Zone 1, Zone 2, and in an internal control in representative sections, prepared from spinal cord injured WT and KO mice. Enclosed boxes are shown at higher magnification in a, c, and e (WT) and b, d, and f (KO) showing CD31+ vessels and PBGFrβ+ pericytes. Filled arrows (c, e and f) show pericytes in close proximity to blood vessels. Open arrows (c, d) delineate ramified pericytes. G, Blood vessels in the MMP-2 KO mice show reduced pericyte coverage of vessels in Zone 1 as compared to WT mice at 21 days post-injury (**p<0.01) and over time (*p<0.05, 21 days post-injury as compared to 14 days post-injury) [(two-way ANOVA followed by Sidak’s multiple comparisons test)]. H-I, No differences in pericyte coverage was observed in both genotypes over time (14 and 21 days post-injury) or between WT and MMP-2 KO mice at each of the time points in zone 2 or the internal control. Bars represent mean + SEM, (n=5 mice/genotype/time-point).
Discussion
While MMP-9 and MMP-2 have established roles in disruption of the barrier, degradation of myelin and glial scar formation in the injured spinal cord, no studies to date have considered their contributions to angiogenesis and vascular stability. This study addresses the temporal contributions of MMP-2 and -9 to angiogenesis in the injured spinal cord, as reestablishment of vascular network is paramount to a fully functional spinal cord. In vitro studies reveal redundancy of these proteases in supporting endothelial tube formation and this may account for the ability of early angiogenesis to proceed in the injured cord in the absence of MMP-2. However, in vitro and in vivo studies demonstrate that prolonged expression of MMP-9 in the absence of MMP-2 results in vascular instability and regression. Collectively, these findings suggest that angiogenesis in the injured spinal cord is dependent upon the coordinated activity of both MMPs in order to achieve a stable vascular network.
Angiogenesis and SCI
We addressed the contribution(s) of MMP-2 and MMP-9 to angiogenesis during wound healing in the injured spinal cord, focusing on the lesion core and the surrounding astrocytic scar. SCI results in loss of blood vessels at the injury epicenter. Beginning several days after SCI, there is proliferation of endothelial cells (Casella et al., 2002) and the subsequent emergence of fibrotic scar forming cells (Goritz et al., 2011) and scar-forming astrocytes (Burda and Sofroniew, 2014). The final phase of tissue remodeling begins at the end of the first week within the lesioned segment and is characterized by the formation of a well-organized, compact astrocytic scar that serves as a protective barrier, segregating inflammatory cells and blood vessels from segments immediately rostral and caudal to the injury (Bush et al., 1999; Faulkner et al., 2004; Wanner et al., 2013). Both MMP-2 and MMP-9 are elevated during this period of wound healing, where they are expressed in astrocytes, and in the case of MMP-9, in both endothelial cells and infiltrating macrophages (Goussev et al., 2003; Hsu et al., 2008; Hsu et al., 2006; Zhang et al., 2011). Thus, these proteases may be critical to the formation of a stable vascular network.
Our data suggest that MMP-2 acts as a regulator of angiogenesis and not as a pro-angiogenic molecule or switch and is also important for vascular stabilization. MMP-2 is one of the ligands that bind to αvβ3 receptor to activate angiogenesis (Silletti et al., 2001) and leads to activation of P13K/AKT pathway to induce expression of VEGF, a known pro-angiogenic factor (Chetty et al., 2009). Consistent with our study, activation of αvβ3 integrin has been shown to rescue angiogenesis after SCI. It is also noteworthy that this activation also resulted in improved white matter sparing and locomotor function (Han et al., 2010), outcomes that highlight the importance of angiogenesis to both structural and functional recovery.
Our in vivo and in vitro studies revealed the dependency of endothelial cell proliferation on MMP-2. However, we also found that indices of vascularity (vascular density, vascular area, and number of endothelial cells) proceeded in the absence of MMP-2, for up to two weeks post-injury. The process of angiogenesis need not be dependent on endothelial cell proliferation. Sprouting angiogenesis, which involves endothelial cell division and migration, is only one of many mechanisms that promote angiogenesis (Folkman, 2003). Intussusceptive angiogenesis (Mentzer and Konerding, 2014), vasculogenic mimicry (Soda et al., 2013), vessel cooption (Alameddine et al., 2014), and recruitment of endothelial progenitor cells (Hillen and Griffioen, 2007) are not dependent upon endothelial cell proliferation. Regarding the latter, bone marrow derived endothelial progenitor cells have been shown to participate in SCI-induced neovascularization (Kamei et al., 2012). These cells are recruited to the injured cord as early at 3 days post-injury and markedly increase by 7 days post-injury (Kamei et al., 2012). As we find a significant increase in vascular density at 7 days post injury, it is conceivable that this increase is at least in part attributed to the recruitment of endothelial progenitor cells. Furthermore, MMP-9 may be critical to this recruitment. There is precedent for this protease in the mobilization of circulating endothelial progenitor cells from the bone marrow after myelosuppression (Heissig et al., 2002). And, in a model of cortical cerebral ischemia, systemic administration of endothelial progenitor cells, results in an MMP-9 dependent enhanced vascular density in the peri-infarcted area (Morancho et al., 2015).
MMPs and vascular regression
We found that MMP-2 deficiency resulted in vascular regression within the injured core. The extended expression of MMP-9 in the injured spinal cord may serve to signal this vascular regression. This hypothesis is supported by in vitro studies showing that prolonged exposure of endothelial cell cultures to MMP-9, resulted in reduced endothelial tube formation. Macrophages, major constituents of the core, express MMP-9 (Hsu et al., 2006) and thus have opportunity to interact with the local vasculature and influence vascular integrity.
MMP-directed regression is exemplified in diverse biological processes including involution of the corpus luteum (Duncan et al., 1998), developmental remodeling of the vasculature (Baluk et al., 2004; Benjamin, 2000) and during the process of lactation in the mammary gland (Ambili and Sudhakaran, 1998). In each of these events regression is necessary to prune newly formed leaky vessels to yield stable, well-perfused vessels that confer homeostasis (Wietecha et al., 2013). However, excessive MMP-9 activity directs pathologic vascular regression through mechanisms that are primarily related to degradation of the extracellular matrix. Such a relationship is seen in a 3-dimensional angiogenesis model where MMP-9 triggered collagen proteolysis resulted in capillary regression and endothelial cell loss (Davis et al., 2001). Similarly in a model of angiogenesis using the aorta, excessive and prolonged exposure to MMP-9 leads to the reabsorption of the neovasculature (Zhu et al., 2000). Recent studies reported an increase in MMP-9 activity in the brain after chronic mild hypoxia that corresponds to post-hypoxic vascular pruning resulting from the degradation of both laminin and claudin-5 (Boroujerdi et al., 2015).
Vascular regression may occur by a variety of MMP-9 directed proteolytic activities, including the generation of soluble and extracellular matrix-derived anti-angiogenic mediators. Anti-angiogenic factors such as endostatin, (Heljasvaara et al., 2005), arresten, canstatin, and tumstatin (Mundel and Kalluri, 2007) inhibit endothelial cell proliferation, migration, and tube formation, and induce endothelial apoptosis (Panka and Mier, 2003). These anti-angiogenic peptides may competitively bind to the free integrin receptors on endothelial cells, initiating anti-angiogenic signaling pathways (Nyberg et al., 2005). In addition, MMPs promote the instability of extracellular contacts with endothelial cell integrins (Wietecha et al., 2013), and widespread proteolysis of various components of the extracellular matrix scaffold, resulting in loss of support to the vascular network, and the resultant vascular regression (Davis and Saunders, 2006).
MMPs and vascular instability
Vascular instability precedes vascular regression (Saint-Geniez and D'Amore, 2004) and is characterized in part by the loss of pericyte/endothelial contacts (Geevarghese and Herman, 2014). Pericytes play a critical role in the formation, maturation, and maintenance of the blood-brain barrier (Daneman et al., 2010; Winkler et al., 2012) and in promoting vascular stability (Geevarghese and Herman, 2014). The latter occurs through multiple bidirectional signaling pathways between endothelial cells and pericytes (Armulik et al., 2011; von Tell et al., 2006). Recruitment of PDGFRβ-positive pericytes to vessels is regulated by PDGF (produced by the migrating tip cells), an important vascular stabilizing factor, that induces upregulation of MMP-2 (Risinger et al., 2010). In addition, several other ligand–receptor systems, including, TGFβ, Ang 1 and 2, Sprouty2, and endosialin have been implicated in regulating vascular maturation and stability through signaling between endothelial cells and pericytes (von Tell et al., 2006).
Here we examined the pericyte investments of blood vessels within the astrocytic scar. In previous studies of wild type mice we noted that these vessels exhibit features consistent with a mature phenotype including astrocytic investments and expression of the glucose-1 transporter (Whetstone et al., 2003). In the current study, we also found that these vessels display pericyte investments. These findings are in sharp contrast to the MMP-2 KO where there is reduced endothelial-pericyte association by 21 days post injury. While pericyte detachment is common to diverse disease models including glioblastoma (Du et al., 2008), diabetic retinopathy (Hammes et al., 2004; Pfister et al., 2010), and Alzheimer’s disease (Halliday et al., 2015), this is the first study to show that deficiency in MMP-2 in the injured spinal cord, likewise culminates in delayed pericyte detachment in the glial scar.
Excess expression of MMP-9 in astrocytes comprising the astrocytic scar (Hsu et al., 2008) is a likely mechanism underlying reduced pericyte coverage. In transgenic mice expressing APOE4, a model of Alzheimer’s disease, increased MMP-9 activity leads to breakdown of tight junction and basement membrane proteins, resulting in diminished pericyte coverage (Bell et al., 2012; Halliday et al., 2015). In addition, NG2, a proteoglycan, expressed on the surface of pericytes, is a substrate for MMP-9. Increased expression of MMP-9 in pericytes leads to cleavage of NG2 and pericyte detachment from endothelial cells (Schultz et al., 2014).
Conclusion
Together our in vivo and in vitro studies support redundancy of MMP-9 and -2 in supporting angiogenesis. However, deficiency in MMP-2 results in altered wound healing characterized by vascular regression and instability. While the mechanisms underlying these changes remain unclear, we speculate that prolonged exposure to MMP-9, known to be expressed in macrophages and the astrocytic scar (Hsu et al., 2008; Hsu et al., 2006; Zhang et al., 2011), promotes vascular regression of immature vessels in the macrophage-rich core and destabilization of mature vessels in the glial scar. Vascular regression and instability may result in loss of functional recovery and reduced white matter sparing observed in MMP-2 KO mice (Hsu et al., 2006).
Highlights.
There are both redundant and diverse roles for MMP-2 and MMP-9 in angiogenesis.
Genetic deletion of MMP-2 results in long-term vascular instability and regression.
Prolonged expression of MMP-9 may generate this aberrant wound healing.
Our study warrants a cautionary note for drugs intended to inhibit these MMPs.
Acknowledgements
The authors thank Christine Cun for performing all the surgeries and Larry Ackerman for capturing the confocal images. Thanks to Christopher Czisch for assistance with Nikon C2 confocal images.
Funding: This work was supported by a grant from the National Institutes of Health, NINDS, R01 NS39278 (LJN).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosure/Conflict of Interest:
The authors declare no competing financial interests.
References
- Alameddine RS, Hamieh L, Shamseddine A. From sprouting angiogenesis to erythrocytes generation by cancer stem cells: evolving concepts in tumor microcirculation. BioMed research international. 2014;2014:986768. doi: 10.1155/2014/986768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ambili M, Sudhakaran PR. 60K gelatinase in involuting rat mammary gland is produced as a 90K proenzyme. Biochemistry and molecular biology international. 1998;45:389–399. doi: 10.1080/15216549800202772. [DOI] [PubMed] [Google Scholar]
- Armulik A, Abramsson A, Betsholtz C. Endothelial/pericyte interactions. Circulation research. 2005;97:512–523. doi: 10.1161/01.RES.0000182903.16652.d7. [DOI] [PubMed] [Google Scholar]
- Armulik A, Genove G, Betsholtz C. Pericytes: developmental, physiological, and pathological perspectives, problems, and promises. Developmental cell. 2011;21:193–215. doi: 10.1016/j.devcel.2011.07.001. [DOI] [PubMed] [Google Scholar]
- Baluk P, Raymond WW, Ator E, Coussens LM, McDonald DM, Caughey GH. Matrix metalloproteinase-2 and -9 expression increases in Mycoplasma-infected airways but is not required for microvascular remodeling. Am J Physiol Lung Cell Mol Physiol. 2004;287:L307–317. doi: 10.1152/ajplung.00404.2003. [DOI] [PubMed] [Google Scholar]
- Bell RD, Winkler EA, Singh I, Sagare AP, Deane R, Wu Z, Holtzman DM, Betsholtz C, Armulik A, Sallstrom J, et al. Apolipoprotein E controls cerebrovascular integrity via cyclophilin A. Nature. 2012;485:512–516. doi: 10.1038/nature11087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamin LE. The controls of microvascular survival. Cancer metastasis reviews. 2000;19:75–81. doi: 10.1023/a:1026552415576. [DOI] [PubMed] [Google Scholar]
- Benton RL, Maddie MA, Gruenthal MJ, Hagg T, Whittemore SR. Neutralizing endogenous VEGF following traumatic spinal cord injury modulates microvascular plasticity but not tissue sparing or functional recovery. Current neurovascular research. 2009;6:124–131. doi: 10.2174/156720209788185678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benton RL, Maddie MA, Minnillo DR, Hagg T, Whittemore SR. Griffonia simplicifolia isolectin B4 identifies a specific subpopulation of angiogenic blood vessels following contusive spinal cord injury in the adult mouse. J Comp Neurol. 2008;507:1031–1052. doi: 10.1002/cne.21570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergers G, Song S, Meyer-Morse N, Bergsland E, Hanahan D. Benefits of targeting both pericytes and endothelial cells in the tumor vasculature with kinase inhibitors. The Journal of clinical investigation. 2003;111:1287–1295. doi: 10.1172/JCI17929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blasig IE, Giese H, Schroeter ML, Sporbert A, Utepbergenov DI, Buchwalow IB, Neubert K, Schonfelder G, Freyer D, Schimke I, et al. *NO and oxyradical metabolism in new cell lines of rat brain capillary endothelial cells forming the blood-brain barrier. Microvascular research. 2001;62:114–127. doi: 10.1006/mvre.2001.2318. [DOI] [PubMed] [Google Scholar]
- Boroujerdi A, Welser-Alves JV, Milner R. Matrix metalloproteinase-9 mediates post-hypoxic vascular pruning of cerebral blood vessels by degrading laminin and claudin-5. Angiogenesis. 2015;18:255–264. doi: 10.1007/s10456-015-9464-7. [DOI] [PubMed] [Google Scholar]
- Burda JE, Sofroniew MV. Reactive gliosis and the multicellular response to CNS damage and disease. Neuron. 2014;81:229–248. doi: 10.1016/j.neuron.2013.12.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bush TG, Puvanachandra N, Horner CH, Polito A, Ostenfeld T, Svendsen CN, Mucke L, Johnson MH, Sofroniew MV. Leukocyte infiltration, neuronal degeneration, and neurite outgrowth after ablation of scar-forming, reactive astrocytes in adult transgenic mice. Neuron. 1999;23:297–308. doi: 10.1016/s0896-6273(00)80781-3. [DOI] [PubMed] [Google Scholar]
- Carmeliet P, Jain RK. Molecular mechanisms and clinical applications of angiogenesis. Nature. 2011;473:298–307. doi: 10.1038/nature10144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casella GT, Marcillo A, Bunge MB, Wood PM. New vascular tissue rapidly replaces neural parenchyma and vessels destroyed by a contusion injury to the rat spinal cord. Experimental neurology. 2002;173:63–76. doi: 10.1006/exnr.2001.7827. [DOI] [PubMed] [Google Scholar]
- Chetty C, Lakka SS, Bhoopathi P, Rao JS. MMP-2 alters VEGF expression via alphaVbeta3 integrin-mediated PI3K/AKT signaling in A549 lung cancer cells. Int J Cancer. 2009;127:1081–1095. doi: 10.1002/ijc.25134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daneman R, Zhou L, Kebede AA, Barres BA. Pericytes are required for blood-brain barrier integrity during embryogenesis. Nature. 2010;468:562–566. doi: 10.1038/nature09513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis GE, Pintar Allen KA, Salazar R, Maxwell SA. Matrix metalloproteinase-1 and -9 activation by plasmin regulates a novel endothelial cell-mediated mechanism of collagen gel contraction and capillary tube regression in three-dimensional collagen matrices. Journal of cell science. 2001;114:917–930. doi: 10.1242/jcs.114.5.917. [DOI] [PubMed] [Google Scholar]
- Davis GE, Saunders WB. Molecular balance of capillary tube formation versus regression in wound repair: role of matrix metalloproteinases and their inhibitors. J Investig Dermatol Symp Proc. 2006;11:44–56. doi: 10.1038/sj.jidsymp.5650008. [DOI] [PubMed] [Google Scholar]
- Du R, Petritsch C, Lu K, Liu P, Haller A, Ganss R, Song H, Vandenberg S, Bergers G. Matrix metalloproteinase-2 regulates vascular patterning and growth affecting tumor cell survival and invasion in GBM. Neuro-oncology. 2008;10:254–264. doi: 10.1215/15228517-2008-001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duncan WC, McNeilly AS, Illingworth PJ. The effect of luteal "rescue" on the expression and localization of matrix metalloproteinases and their tissue inhibitors in the human corpus luteum. The Journal of clinical endocrinology and metabolism. 1998;83:2470–2478. doi: 10.1210/jcem.83.7.4950. [DOI] [PubMed] [Google Scholar]
- Fassbender JM, Myers SA, Whittemore SR. Activating Notch signaling post-SCI modulates angiogenesis in penumbral vascular beds but does not improve hindlimb locomotor recovery. Experimental neurology. 2011;227:302–313. doi: 10.1016/j.expneurol.2010.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faulkner JR, Herrmann JE, Woo MJ, Tansey KE, Doan NB, Sofroniew MV. Reactive astrocytes protect tissue and preserve function after spinal cord injury. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2004;24:2143–2155. doi: 10.1523/JNEUROSCI.3547-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Folkman J. Fundamental concepts of the angiogenic process. Current molecular medicine. 2003;3:643–651. doi: 10.2174/1566524033479465. [DOI] [PubMed] [Google Scholar]
- Geevarghese A, Herman IM. Pericyte-endothelial crosstalk: implications and opportunities for advanced cellular therapies. Translational research : the journal of laboratory and clinical medicine. 2014;163:296–306. doi: 10.1016/j.trsl.2014.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goritz C, Dias DO, Tomilin N, Barbacid M, Shupliakov O, Frisen J. A pericyte origin of spinal cord scar tissue. Science. 2011;333:238–242. doi: 10.1126/science.1203165. [DOI] [PubMed] [Google Scholar]
- Goussev S, Hsu JY, Lin Y, Tjoa T, Maida N, Werb Z, Noble-Haeusslein LJ. Differential temporal expression of matrix metalloproteinases after spinal cord injury: relationship to revascularization and wound healing. J Neurosurg. 2003;99:188–197. doi: 10.3171/spi.2003.99.2.0188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halliday MR, Rege SV, Ma Q, Zhao Z, Miller CA, Winkler EA, Zlokovic BV. Accelerated pericyte degeneration and blood-brain barrier breakdown in apolipoprotein E4 carriers with Alzheimer's disease. Journal of cerebral blood flow and metabolism : official journal of the International Society of Cerebral Blood Flow and Metabolism. 2015 doi: 10.1038/jcbfm.2015.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammes HP, Lin J, Wagner P, Feng Y, Vom Hagen F, Krzizok T, Renner O, Breier G, Brownlee M, Deutsch U. Angiopoietin-2 causes pericyte dropout in the normal retina: evidence for involvement in diabetic retinopathy. Diabetes. 2004;53:1104–1110. doi: 10.2337/diabetes.53.4.1104. [DOI] [PubMed] [Google Scholar]
- Han S, Arnold SA, Sithu SD, Mahoney ET, Geralds JT, Tran P, Benton RL, Maddie MA, D'Souza SE, Whittemore SR, et al. Rescuing vasculature with intravenous angiopoietin-1 and alpha v beta 3 integrin peptide is protective after spinal cord injury. Brain. 2010;133:1026–1042. doi: 10.1093/brain/awq034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heissig B, Hattori K, Dias S, Friedrich M, Ferris B, Hackett NR, Crystal RG, Besmer P, Lyden D, Moore MA, et al. Recruitment of stem and progenitor cells from the bone marrow niche requires MMP-9 mediated release of kit-ligand. Cell. 2002;109:625–637. doi: 10.1016/s0092-8674(02)00754-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heljasvaara R, Nyberg P, Luostarinen J, Parikka M, Heikkila P, Rehn M, Sorsa T, Salo T, Pihlajaniemi T. Generation of biologically active endostatin fragments from human collagen XVIII by distinct matrix metalloproteases. Experimental cell research. 2005;307:292–304. doi: 10.1016/j.yexcr.2005.03.021. [DOI] [PubMed] [Google Scholar]
- Hillen F, Griffioen AW. Tumour vascularization: sprouting angiogenesis and beyond. Cancer metastasis reviews. 2007;26:489–502. doi: 10.1007/s10555-007-9094-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu JY, Bourguignon LY, Adams CM, Peyrollier K, Zhang H, Fandel T, Cun CL, Werb Z, Noble-Haeusslein LJ. Matrix metalloproteinase-9 facilitates glial scar formation in the injured spinal cord. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2008;28:13467–13477. doi: 10.1523/JNEUROSCI.2287-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu JY, McKeon R, Goussev S, Werb Z, Lee JU, Trivedi A, Noble-Haeusslein LJ. Matrix metalloproteinase-2 facilitates wound healing events that promote functional recovery after spinal cord injury. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2006;26:9841–9850. doi: 10.1523/JNEUROSCI.1993-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itoh T, Ikeda T, Gomi H, Nakao S, Suzuki T, Itohara S. Unaltered secretion of beta-amyloid precursor protein in gelatinase A (matrix metalloproteinase 2)-deficient mice. The Journal of biological chemistry. 1997;272:22389–22392. doi: 10.1074/jbc.272.36.22389. [DOI] [PubMed] [Google Scholar]
- Kamei N, Kwon SM, Kawamoto A, Ii M, Ishikawa M, Ochi M, Asahara T. Contribution of bone marrow-derived endothelial progenitor cells to neovascularization and astrogliosis following spinal cord injury. Journal of neuroscience research. 2012;90:2281–2292. doi: 10.1002/jnr.23113. [DOI] [PubMed] [Google Scholar]
- Kawano H, Kimura-Kuroda J, Komuta Y, Yoshioka N, Li HP, Kawamura K, Li Y, Raisman G. Role of the lesion scar in the response to damage and repair of the central nervous system. Cell and tissue research. 2012;349:169–180. doi: 10.1007/s00441-012-1336-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kessenbrock K, Plaks V, Werb Z. Matrix metalloproteinases: regulators of the tumor microenvironment. Cell. 2010;141:52–67. doi: 10.1016/j.cell.2010.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loy DN, Crawford CH, Darnall JB, Burke DA, Onifer SM, Whittemore SR. Temporal progression of angiogenesis and basal lamina deposition after contusive spinal cord injury in the adult rat. J Comp Neurol. 2002;445:308–324. doi: 10.1002/cne.10168. [DOI] [PubMed] [Google Scholar]
- Lutton C, Young YW, Williams R, Meedeniya AC, Mackay-Sim A, Goss B. Combined VEGF and PDGF treatment reduces secondary degeneration after spinal cord injury. Journal of neurotrauma. 2012;29:957–970. doi: 10.1089/neu.2010.1423. [DOI] [PubMed] [Google Scholar]
- Mahoney ET, Benton RL, Maddie MA, Whittemore SR, Hagg T. ADAM8 is selectively up-regulated in endothelial cells and is associated with angiogenesis after spinal cord injury in adult mice. J Comp Neurol. 2009;512:243–255. doi: 10.1002/cne.21902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mentzer SJ, Konerding MA. Intussusceptive angiogenesis: expansion and remodeling of microvascular networks. Angiogenesis. 2014;17:499–509. doi: 10.1007/s10456-014-9428-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morancho A, Ma F, Barcelo V, Giralt D, Montaner J, Rosell A. Impaired vascular remodeling after endothelial progenitor cell transplantation in MMP9-deficient mice suffering cortical cerebral ischemia. Journal of cerebral blood flow and metabolism : official journal of the International Society of Cerebral Blood Flow and Metabolism. 2015;35:1547–1551. doi: 10.1038/jcbfm.2015.180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mundel TM, Kalluri R. Type IV collagen-derived angiogenesis inhibitors. Microvascular research. 2007;74:85–89. doi: 10.1016/j.mvr.2007.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myers SA, DeVries WH, Andres KR, Gruenthal MJ, Benton RL, Hoying JB, Hagg T, Whittemore SR. CD47 knockout mice exhibit improved recovery from spinal cord injury. Neurobiology of disease. 2011;42:21–34. doi: 10.1016/j.nbd.2010.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myers SA, DeVries WH, Gruenthal MJ, Andres KR, Hagg T, Whittemore SR. Sildenafil improves epicenter vascular perfusion but not hindlimb functional recovery after contusive spinal cord injury in mice. Journal of neurotrauma. 2012;29:528–538. doi: 10.1089/neu.2011.2036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noble LJ, Donovan F, Igarashi T, Goussev S, Werb Z. Matrix metalloproteinases limit functional recovery after spinal cord injury by modulation of early vascular events. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2002;22:7526–7535. doi: 10.1523/JNEUROSCI.22-17-07526.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nyberg P, Xie L, Kalluri R. Endogenous inhibitors of angiogenesis. Cancer research. 2005;65:3967–3979. doi: 10.1158/0008-5472.CAN-04-2427. [DOI] [PubMed] [Google Scholar]
- Panka DJ, Mier JW. Canstatin inhibits Akt activation and induces Fas-dependent apoptosis in endothelial cells. The Journal of biological chemistry. 2003;278:37632–37636. doi: 10.1074/jbc.M307339200. [DOI] [PubMed] [Google Scholar]
- Pfister F, Wang Y, Schreiter K, vom Hagen F, Altvater K, Hoffmann S, Deutsch U, Hammes HP, Feng Y. Retinal overexpression of angiopoietin-2 mimics diabetic retinopathy and enhances vascular damages in hyperglycemia. Acta diabetologica. 2010;47:59–64. doi: 10.1007/s00592-009-0099-2. [DOI] [PubMed] [Google Scholar]
- Risinger GM, Jr., Updike DL, Bullen EC, Tomasek JJ, Howard EW. TGF-beta suppresses the upregulation of MMP-2 by vascular smooth muscle cells in response to PDGF-BB. American journal of physiology Cell physiology. 2010;298:C191–201. doi: 10.1152/ajpcell.00417.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy R, Zhang B, Moses MA. Making the cut: protease-mediated regulation of angiogenesis. Experimental cell research. 2006;312:608–622. doi: 10.1016/j.yexcr.2005.11.022. [DOI] [PubMed] [Google Scholar]
- Saint-Geniez M, D'Amore PA. Development and pathology of the hyaloid, choroidal and retinal vasculature. The International journal of developmental biology. 2004;48:1045–1058. doi: 10.1387/ijdb.041895ms. [DOI] [PubMed] [Google Scholar]
- Schultz N, Nielsen HM, Minthon L, Wennstrom M. Involvement of matrix metalloproteinase-9 in amyloid-beta 1-42.induced shedding of the pericyte proteoglycan NG2. Journal of neuropathology and experimental neurology. 2014;73:684–692. doi: 10.1097/NEN.0000000000000084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silletti S, Kessler T, Goldberg J, Boger DL, Cheresh DA. Disruption of matrix metalloproteinase 2 binding to integrin alpha vbeta 3 by an organic molecule inhibits angiogenesis and tumor growth in vivo. Proc Natl Acad Sci U S A. 2001;98:119–124. doi: 10.1073/pnas.011343298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silver J, Miller JH. Regeneration beyond the glial scar. Nature reviews Neuroscience. 2004;5:146–156. doi: 10.1038/nrn1326. [DOI] [PubMed] [Google Scholar]
- Silver J, Schwab ME, Popovich PG. Central nervous system regenerative failure: role of oligodendrocytes, astrocytes, and microglia. Cold Spring Harbor perspectives in biology. 2015;7:a020602. doi: 10.1101/cshperspect.a020602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simard JM, Tsymbalyuk O, Ivanov A, Ivanova S, Bhatta S, Geng Z, Woo SK, Gerzanich V. Endothelial sulfonylurea receptor 1-regulated NC Ca-ATP channels mediate progressive hemorrhagic necrosis following spinal cord injury. The Journal of clinical investigation. 2007;117:2105–2113. doi: 10.1172/JCI32041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simonavicius N, Ashenden M, van Weverwijk A, Lax S, Huso DL, Buckley CD, Huijbers IJ, Yarwood H, Isacke CM. Pericytes promote selective vessel regression to regulate vascular patterning. Blood. 2012;120:1516–1527. doi: 10.1182/blood-2011-01-332338. [DOI] [PubMed] [Google Scholar]
- Soda Y, Myskiw C, Rommel A, Verma IM. Mechanisms of neovascularization and resistance to anti-angiogenic therapies in glioblastoma multiforme. Journal of molecular medicine. 2013;91:439–448. doi: 10.1007/s00109-013-1019-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soderblom C, Luo X, Blumenthal E, Bray E, Lyapichev K, Ramos J, Krishnan V, Lai-Hsu C, Park KK, Tsoulfas P, et al. Perivascular fibroblasts form the fibrotic scar after contusive spinal cord injury. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2013;33:13882–13887. doi: 10.1523/JNEUROSCI.2524-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verslegers M, Lemmens K, Van Hove I, Moons L. Matrix metalloproteinase-2 and -9 as promising benefactors in development, plasticity and repair of the nervous system. Progress in neurobiology. 2013 doi: 10.1016/j.pneurobio.2013.03.004. [DOI] [PubMed] [Google Scholar]
- von Tell D, Armulik A, Betsholtz C. Pericytes and vascular stability. Experimental cell research. 2006;312:623–629. doi: 10.1016/j.yexcr.2005.10.019. [DOI] [PubMed] [Google Scholar]
- Wang X, Jung J, Asahi M, Chwang W, Russo L, Moskowitz MA, Dixon CE, Fini ME, Lo EH. Effects of matrix metalloproteinase-9 gene knock-out on morphological and motor outcomes after traumatic brain injury. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2000;20:7037–7042. doi: 10.1523/JNEUROSCI.20-18-07037.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wanner IB, Anderson MA, Song B, Levine J, Fernandez A, Gray-Thompson Z, Ao Y, Sofroniew MV. Glial scar borders are formed by newly proliferated, elongated astrocytes that interact to corral inflammatory and fibrotic cells via STAT3-dependent mechanisms after spinal cord injury. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2013;33:12870–12886. doi: 10.1523/JNEUROSCI.2121-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whetstone WD, Hsu JY, Eisenberg M, Werb Z, Noble-Haeusslein LJ. Blood-spinal cord barrier after spinal cord injury: relation to revascularization and wound healing. Journal of neuroscience research. 2003;74:227–239. doi: 10.1002/jnr.10759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wietecha MS, Cerny WL, DiPietro LA. Mechanisms of vessel regression: toward an understanding of the resolution of angiogenesis. Current topics in microbiology and immunology. 2013;367:3–32. doi: 10.1007/82_2012_287. [DOI] [PubMed] [Google Scholar]
- Winkler EA, Sengillo JD, Bell RD, Wang J, Zlokovic BV. Blood-spinal cord barrier pericyte reductions contribute to increased capillary permeability. Journal of cerebral blood flow and metabolism : official journal of the International Society of Cerebral Blood Flow and Metabolism. 2012;32:1841–1852. doi: 10.1038/jcbfm.2012.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Q, Stamenkovic I. Localization of matrix metalloproteinase 9 to the cell surface provides a mechanism for CD44-mediated tumor invasion. Genes & development. 1999;13:35–48. doi: 10.1101/gad.13.1.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Trivedi A, Lee JU, Lohela M, Lee SM, Fandel TM, Werb Z, Noble-Haeusslein LJ. Matrix metalloproteinase-9 and stromal cell-derived factor-1 act synergistically to support migration of blood-borne monocytes into the injured spinal cord. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2011;31:15894–15903. doi: 10.1523/JNEUROSCI.3943-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu WH, Guo X, Villaschi S, Francesco Nicosia R. Regulation of vascular growth and regression by matrix metalloproteinases in the rat aorta model of angiogenesis. Lab Invest. 2000;80:545–555. doi: 10.1038/labinvest.3780060. [DOI] [PubMed] [Google Scholar]