

Abstract
Osteoporosis, which is characterized by resorption of bone exceeding formation, remains a significant human health concern, and the impact of this condition will only increase with the “greying” of the worldwide population. This review focuses on current and emerging approaches for delivering therapeutic agents to restore bone remodeling homeostasis. Well-known antiresorptive and anabolic agents such as estrogen, estrogen analogs, bisphosphonates, calcitonin, and parathyroid hormone, along with newer modulators and antibodies, are primarily administered orally, intravenously, or subcutaneously. Although these treatments can be effective, continuing problems include patient non-compliance and adverse systemic or remote-site effects. Controlled drug delivery via polymeric, targeted, and active release systems extends drug half-life by shielding against premature degradation and improves bioavailability, while also providing prolonged, sustained, or intermittent release at therapeutic doses to more effectively treat osteoporosis and associated fracture risk.
Keywords: osteoporosis, antiresorptive, anabolic, drug delivery, controlled release, bone-targeting
1. Introduction
Osteoporosis, which means “porous bone”, is a widespread skeletal condition characterized by reduced bone strength, low bone mass, altered macro-geometry, and micro-architectural deterioration of bone tissue. This condition was initially considered a histological diagnosis, however reductions in both the quantity and quality of bone are recognized as adversely affecting mechanical properties [1, 2]. Reduced strength can ultimately lead to increased risk of fracture.
In 2014, the National Osteoporosis Foundation reported that nearly 43 million U.S. citizens had decreased bone density, and at least 9.9 million met diagnostic criteria for osteoporosis [2]. The World Health Organization reported that more than 75 million people in North America, Europe, and Japan have osteoporosis [3, 4]. The worldwide incidence of osteoporosis reached nearly 9 million fractures annually, resulting in an osteoporotic fracture every 3 seconds [5].
In addition to being among the most common bone diseases, osteoporosis significantly increases healthcare costs. Half of osteoporosis patients receive drug treatments, averaging $500 each, or $2 billion annually nationwide [6]. The American Association of Clinical Endocrinologists noted that as the population ages and osteoporosis likely becomes more common, costs are projected to reach nearly $25 billion by 2025. Lifetime risk percentages of hip, spine, and wrist fractures in women are 23, 29 and 21%, respectively, and 11, 14 and 5% in men [7, 8]. Every year in the U.S., 3.5 million hospital bed days are attributed to osteoporotic fractures and over 60,000 nursing home admissions are related to hip fractures. Trends are similar in Europe, where the estimated cost of osteoporotic fractures was €36 billion in 2000 and is expected to double to €77 billion by 2050 [9, 10].
2. Pathogenesis of osteoporosis
Bone mass is low at birth and increases for the next two to three decades as the activity of bone-forming osteoblasts surpasses that of the bone-resorbing osteoclasts, ultimately leading to peak bone density during young adulthood. Through this period, osteoclast and osteoblast activities are equivalent for some time, allowing sufficient bone density to be maintained [11, 12]. Afterward, the activity of osteoclasts begins to surpass that of osteoblasts, and bone density declines, which may lead to osteoporosis. Studies of bone microarchitecture have shown that trabecular bone loss begins in the third decade of life, before gonadal sex steroid deficiency develops, whereas cortical loss typically begins in the sixth decade, around menopause in women and at a similar age in men [13].
Figure 1 illustrates, in a simplified fashion, the complex interactions between bone cells. Coupled activity of osteoclasts and osteoblasts is responsible for maintenance of bone mass during remodeling. Once osteoclasts have completed resorption, osteoblast precursors are recruited to and adhere to the site in response to signals emitted by osteoclasts. In response to mechanical loading, osteocytes further regulate the differentiation and function of osteoclasts and osteoblasts through the release of signaling molecules. Numerous systemic molecules, such as steroid hormones, parathyroid hormone, vitamin D and its metabolites, and calcitonin, as well as locally secreted proteins, such as macrophage colony-stimulating factor (mCSF), osteoprotegerin (OPG), sclerostin, receptor activator of nuclear factor κ-B ligand (RANKL), and various growth factors, exhibit control over remodeling behaviors [12, 14, 15]. Dysregulation of these modulators, however, can lead to alteration of the balance between osteoclast and osteoblast activity, as is mainly the case for primary osteoporosis arising in postmenopausal women and elderly men [16]. Much has been learned about the pathogenesis of osteoporosis, and a detailed description is beyond the scope of this review (see for example [13,15]). How endogenous signals and exogenous factors such as diet and exercise influence bone physiology is a major focus of ongoing research.
Figure 1.
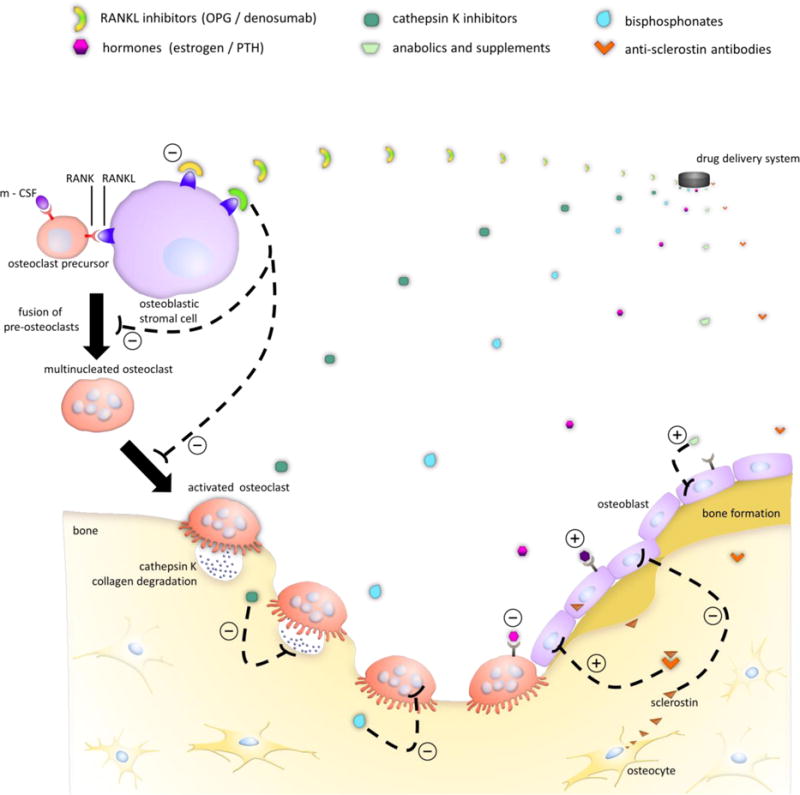
Controlled release of different antiresorptive and anabolic drugs and their mechanisms of action on specific cell types in bone for treatment of osteoporosis. In the presence of m-CSF and RANKL, osteoclast precursors differentiate and fuse to eventually form activated osteoclasts. Antiresorptives such as estrogen, bisphosphonates, OPG, and denosumab inhibit osteoclast development and activity, while anabolic therapies, which include PTH, primarily affect osteoblast activity. Cathepsin K and anti-sclerostin antibodies represent emerging therapies for osteoporosis treatment.
3. Osteoporosis Treatments
As described in Section 2, the onset of osteoporosis can be linked with poorly regulated levels of reproductive hormones, calcitonin, growth factors, thyroid hormones, vitamin D, and/or various cytokines influential in bone remodeling. The coupling imbalance between the resorptive and formative processes described in Section 2 is sought to be alleviated through therapy with antiresorptive agents or anabolic drugs (Figure 1). The following sections briefly review antiresorptive, anabolic, and emerging drug treatments (summarized in Table 1).
Table 1.
Examples of Current and Emerging Agents Investigated for Osteoporosis*
| Classification | Compound | Mechanism | Type | Status |
|---|---|---|---|---|
|
Current Treatments
| ||||
| Hormones | Estrogen Calcitonin |
Activate estrogen or calcitonin receptors mediating cytokine responses and adenosine monophosphate (cAMP) kinase signaling, respectively, involved in osteoblast/clast function | Antiresorptive Antiresorptive |
Marketed |
| PTH (teriparatide) | Activate PTH-receptors in mediating kinase signaling involved in osteoblast/clast function | Anabolic | ||
|
| ||||
| SERMs | Raloxifene Bazedoxifene |
Activate or inhibit estrogen-specific receptor mediation of cytokine responses | Antiresorptive | Marketed |
|
| ||||
| Bisphosphonates | Alendronate Ibandronate Residronate Zoledronate |
Inhibitor of farnesyl pyrophosphate synthase, involved in osteoclast function | Antiresorptive | Marketed |
|
| ||||
| AMARTs | Denosumab | Inhibitor of RANKL, ligand involved with osteoclast formation and activity | Antiresorptive | Marketed |
|
| ||||
| Dietary Supplements | Calcium Vitamin D Calcitriol |
Intracellular receptor-mediated signaling | Aid in anabolic process | Marketed |
|
| ||||
| Other | Strontium ranelate | Inhibitor of RANKL, upregulates OPG, activates calcium receptors | Anabolic and antiresorptive | Marketed (in Europe) |
| Sodium fluoride | Activate Wnt signaling | Anabolic | ||
|
Emerging Treatments | ||||
| Cathepsin K inhibitor | Odanacatib | Inhibitor of cathepsin K, which degrades type I collagen in bone | Antiresorptive | Phase III, ended early, good results |
|
| ||||
| SERMs | Lasofoxifene Ospemifene Arzoxifene |
Activate or inhibit estrogen-receptor mediated cytokine responses | Antiresorptive | Phase III |
|
| ||||
| AMARTs | Romosozumab Blosozumab BPS804 |
Inhibitor of sclerostin, a Wnt inhibitor | Anabolic | Phase III Phase III Phase II |
|
| ||||
| PTHrP analogs | Abaloparatide | Mediate endochondral bone formation through paracrine signaling | Anabolic | Phase III |
|
| ||||
| β-arrestin analogs | PTH-barr | Activate transmembrane proteins in PTH signaling | Anabolic | Preclinical |
|
| ||||
| (Src) tyrosine kinase inhibitors | Saracatinib (AZD0530) AZD0424 |
Inhibit Src, expressed by osteoclasts, required for ruffled border development | Antiresorptive | Phase I Phase I |
|
| ||||
| Dickkopf-1 receptor antagonists | RN564 RH2-18 PF-04840082 BHQ880 |
Inhibit function of dickkopf-1, a Wnt inhibitor | Anabolic | Phase I Preclinical Preclinical Phase I |
|
| ||||
| Activin A receptor antagonists | Sotatercept (ACE-011) | Inhibit binding of activin A to Act RIIA receptor involved in osteoblast/clast formation | Anabolic and antiresorptive | Phase I |
|
| ||||
| Calcium-sensing receptor antagonists (calcylitics) | Ronacaleret (SB-751689) MK-5442 |
Alter calcium/PTH homeostasis by stimulating PTH secretion despite calcium serum levels | Anabolic | Phase II, discontinued phase II |
|
| ||||
| Statins | Simvastatin Lovastatin Rosuvastatin Fluvastatin |
3-hydroxy-3-methyl-glutaryl-coenzyme A (HMG CoA) reductase inhibitors | Anabolic | Preclinical Preclinical Phase III Preclinical |
3.1 Antiresorptive agents
Estrogen compounds were first approved in the U.S. for menopause in 1941, and noted for their effects on bone mass, but use of estrogen-based hormone replacement to prevent bone loss was not reported until 1982 [17]. Estrogen binds to α and β receptors on bone cells to reduce osteoclast production and activity, promote osteoblast production and activity, regulate calcium homeostasis, and suppress cytokine responses supporting resorption [18]. Consequently, estrogen treatment decreased the onset of osteoporotic fractures by approximately 50%, decreased bone turnover, and increased BMD in the lumbar spine by 5.1% in a one year study of early menopausal women [19, 20]. Because receptors on bone marrow and immune cells are also affected by estrogen, however, continued use can lead to increased risk of endometrial and breast cancer, stroke, thromboembolism, and coronary disease [21]. While it is still considered effective, estrogen has been withdrawn for treatment of osteoporosis in the U.S., but has instead been approved as a preventative measure [21].
Selective estrogen receptor modulators (SERMs) were developed as alternatives to estrogen. In these compounds, the steroidal moiety of estrogen has been removed to leave otherwise similar structures, allowing them to activate α and β estrogen receptors agonistically or antagonistically [18]. The U.S. Food and Drug Administration (FDA)-approved SERM raloxifene decreases the breast cancer risk posed by estrogen while significantly increasing BMD, especially in the vertebrae. However, thrombovascular risk still remains for SERMs [22].
Calcitonin, an FDA-approved peptide hormone, reduces loss of cancellous bone but is relatively ineffective in decreasing cortical bone loss [23]. Daily administration of low doses resulted in a 1% increase of spinal BMD, while high doses were ineffective after months of treatment [24]. By binding to calcitonin receptors in bone and the brain, osteoclastic activity is diminished and analgesic effects are mediated. Salmon calcitonin is currently marketed and has been shown to be significantly more potent than human calcitonin [24, 25]. Gastrointestinal symptoms have been observed, but these effects can be minimized with different modes of administration.
Bisphosphonates, first synthesized in the 1800s and introduced into the clinical setting in 1968, have become the first-line of FDA-approved drugs prescribed treatment for osteoporosis [26]. Bisphosphonates are derived from pyrophosphate, a potent inhibitor of bone mineralization. The phosphate moiety of bisphosphonates has a high affinity for hydroxyapatite crystals within bone, allowing binding to areas of mineralization. First generation bisphosphonates, which do not contain an amino group, cause osteoclast apoptosis following metabolization into toxic analogs of adenosine triphosphate (ATP) [27]. More recently developed nitrogen-containing compounds induce osteoclast apoptosis by inhibiting farnesyl pyrophosphate synthase in the mevalonate pathway that is vital for functioning osteoclasts [28]. This disrupted process leads to reduced resorption and increased bone mineral density. Due to decreased remodeling, however, bone microfractures and damage may accumulate, thereby compromising bone quality with continued use. Common side effects for bisphosphonates include osteonecrosis of the mandible and atypical low-energy subtrochanteric and femoral shaft fractures, along with gastrointestinal issues from poor phosphate absorption [29]. The nitrogen-containing compounds of alendronate, risedronate, ibandronate, and zoledronic acid are currently used for osteoporosis treatment in the U.S. due to their increased potency compared to clodronate and others lacking an amino group [13]. Overall, these commonly prescribed drugs have decreased vertebral fractures by 50%, other bone fractures by 20–25%, and hip fractures by about 50% in osteoporotic patients [30].
Denosumab is a human monoclonal antibody, also known as the first FDA-approved antibody-mediated anti-resorptive therapy (AMART) for osteoporosis [31]. The antibody inhibits RANKL, a potent mediator of osteoclast development and activity [32]. RANK has a much greater specificity for binding to the RANKL cell surface molecule than its natural competitor, OPG, which is secreted by bone marrow and a number of other cell types [33]. Denosumab promoted a higher BMD in the lumbar spine and hip and reduction in bone turnover markers than did alendronate, risendronate, ibandronate, zolendronate, raloxifene, and calcitonin throughout an exploratory three year study [34]. Another study, however, reported that denosumab and zolendronate produced similar BMD results in the lumbar spine of women [35].
Recombinant forms of OPG have also been investigated for osteoporosis treatment. Min et al. reported that administration of the protein to OPG-knockout mice with an onset of osteoporosis effectively increased the trabecular bone density in the proximal tibial metaphysis [36]. This effect was correlated with elevated serum levels of alkaline phosphatase (ALP) compared to OPG-treated wild-type controls that showed decreased ALP. A well-tolerated single dose of OPG led to immediate and subsequent decreases in resorption markers urinary N-telopeptide (NTX) and deoxypyridinoline (DPD) levels, respectively, in postmenopausal women [37]
Strontium ranelate, which is currently approved for use in Europe, is a bioactive agent derived from strontium. The compound exhibits both antiresorptive and osteogenic effects by inhibiting RANKL activity and upregulating osteoprotegerin while activating calcium-sensing receptors [38]. In phase III trials, strontium ranelate decreased vertebral, non-vertebral, and hip fracture risk by 41, 16, and 36%, respectively, in three year studies. Adverse effects associated with this therapy, such as nausea, diarrhea, and dermatitis, were reported to diminish shortly after initiating treatment. Thromboembolism risk is also associated with strontium ranelate in postmenopausal women [39].
3.2 Anabolic agents
The only approved anabolic drug currently used for treating severe stages of osteoporosis is recombinant human parathyroid hormone (rhPTH) 1–34, known as teriparatide [40]. PTH is known to exhibit resorptive properties in combination with vitamin D, which is important for regulating serum calcium and phosphate levels in the body. When intermittently administered, PTH(1–34) demonstrates bone stimulating properties [33, 41]. The complete hormone sequence, rhPTH(1–84), demonstrates increased anabolic effects in bone marrow cells in vitro [42]. Under normal to slightly elevated serum calcium levels, activation of PTH-receptors results in phospholipase C-stimulated production of inositol triphosphate and diacylglycerol, with subsequent intracellular calcium mobilization combined with protein kinase C activation [33]. These pathways activated by PTH subsequently affect lipoprotein receptor-related protein-5 or 6 (LRP5/6) mediated canonical wingless (Wnt) signaling, which promotes osteoblast development by downregulating sclerostin and RANKL expression. Sclerostin is known to be an antagonist of Wnt signaling and bone morphogenetic protein-induced osteogenesis and an upregulator of RANKL activation in osteoclasts [15, 43].
Although calcium and vitamin D supplements administered independently are an insufficient means of treating osteoporosis, nutritional deficiencies of these agents can lead to hyperparathyroidism, hypocalcemia, and osteoporosis. Consequently, they have been administered in combination with stand-alone estrogen, PTH, and bisphosphonate therapies. Studies have also shown mild effects on increasing BMD and reducing fracture risk [21]. Calcium also supplements the use of sodium fluoride, shown to stimulate osteoblast proliferation via Wnt/β-catenin signaling and to increase vertebral BMD in women with osteoporosis by 8% for every consecutive year of use. However, decreased cortical BMD, increased atypical fractures, and gastrointestinal issues have prevented approval of sodium fluoride in the U.S. [44, 45]. Calcitriol, a metabolite of vitamin D, increases calcium absorption and reduces fracture risk in postmenopausal women compared to calcium alone, while also temporarily increasing bone mass in some studies [46, 47]. Administration of insulin like growth factor I (IGF-I) as an anabolic therapy to elderly women was associated with increased femoral and vertebral BMD in the Framingham Osteoporosis Study [48]. However, localized pain, carpal tunnel syndrome, venous thrombosis, cholestatic liver disease, and fractures, among other serious adverse effects, have been associated with growth hormone treatments [49].
3.3 Modified and Emerging Drug Therapies
To combat the disadvantages or side effects associated with existing treatments, modified therapies and new drugs are emerging. In addition, these approaches target newly discovered pathways involved with osteoclast formation, increase drug affinity, or improve bone targeting.
SERMs, such as bazedoxifene, have been combined with estrogen and estrogen analogs to minimize the adverse cardiovascular effects posed by the compounds individually while increasing BMD compared to placebo and raloxifene [50]. Combinations of hormone therapy with alendronate, risedronate, and calcitonin have shown additive effects in increasing BMD [21]. Sequential administration of alendronate, then PTH, followed again by alendronate to osteopenic rats led to the most trabecular bone growth and strength along with the best microarchitecture [51].
Among other PTH and parathyroid hormone-related protein (PTHrP) analogs investigated in preclinical and clinical studies [52], the targeting efficiency of PTH(1–33) was improved while removing the hypercalcemic effect by conjugation with a collagen-binding domain derived from bacterial collagenase with an affinity to bone and skin. A single dose administered to ovariectomized rats led to a maximum increase of 14% in vertebral BMD compared to a temporary 5% increase with daily PTH administration [53]. Various drugs have also been chemically modified or conjugated with the phosphate-carbon-phosphate (P-C-P) moiety that characterizes bisphosphonates to increase affinity for the bone surface. Example compounds include bisphosphonate-conjugated estradiol, prostaglandin E2, and estrogen analogs, of which a single dose of prostaglandin E2-bisphosphonate in ovariectomized rats inhibited 77% of BMD loss in preclinical trials [54]. More targeted approaches will be discussed more extensively in Section 4.
Currently, bioactive agents acting on new targets are in different stages of preclinical and clinical development. Odanacatib is among the cathepsin K inhibitors being investigated for antiresorptive purposes [55, 56]. Cathepsin-K is an enzyme secreted by osteoblasts that degrades type I collagen in bone. Promising new antibodies, such as romosozumab, blosozumab, and BPS804, act to directly inhibit sclerostin, a protein produced and secreted by osteocytes in bone [57]. Phase II trials showed 11 and 17% increases in vertebral BMD following treatment with maximum doses of romosumab and blosozumab, respectively, for 12 months [57]. Active agents still in early development include β-arrestin analogs, proto-oncogene tyrosine kinase inhibitors, dickkopf-1, activin A, and calcium-sensing receptor antagonists [55, 58–66]. Well-known drugs, such as statins, are also being considered as anabolic therapies for osteoporosis. While rosuvastatin did not reduce osteoporotic risk in phase III trials, simvastatin showed promising early results by enhancing bone mechanical properties and microarchitecture via osteoblast proliferation and differentiation in preclinical trials [67]. Lovastatin and fluvastatin have also been investigated in preclinical trials [68].
4. Drug Delivery Approaches for Osteoporosis
Ensuring the continuous delivery of therapeutic agents to osteoporotic bone is a major concern for physicians and researchers around the world, as any drug, regardless of potency, cannot exert positive change to bone quality if received off-target, metabolized, or skipped. Many of the currently available treatments for osteoporosis are prescribed in daily or weekly applications of oral tablets or injectable solutions for systemic delivery, however variability in patient compliance and absorptive efficiency may significantly reduce the bioavailability of agent to below clinically effective values, negatively impacting treatment efficacy [69, 70]. Patient compliance in particular is a significant obstacle, with noncompliance of treatment occurring in up to 65% of patients prescribed oral bisphosphonates, and rates of discontinuation of treatment reaching above 75% for daily regiments within the first year [69–71]. Even for quarterly or biannual administration, patient compliance does not exceed 70%, although compliance and persistence past the second year are significantly improved compared to more frequent dosing schedules [70]. To circumvent such issues, more efficient methods of therapeutic delivery are required to minimize patient intervention and to ensure the proper dosing and scheduling. Encapsulation of poorly bioavailable molecules into drug carriers, chemical modification of systemically introduced factors for tissue targeting, and controlled release of drug from slowly degrading “depot” systems or actively triggered devices are all methods to improve delivery efficiency beyond typical oral treatment schedules.
4.1 Current Approaches
Current clinical approaches to drug delivery are relatively straightforward in design, with oral and injectable bisphosphonate systems dominating the market (summarized in Table 2) [72]. Oral bisphosphonates are currently prescribed in tablet form for intestinal absorption and systemic application via regular dosing ranging from weekly (alendronate, risedronate) to monthly (risedronate, ibandronate) intervals [71]. Other bisphosphonates, such as zoledronate, that display lower oral viability are administered intravenously via prolonged infusion by healthcare providers, as is also the case for quarterly ibandronate [73]. When administered intravenously, doses are up to an order of magnitude lower than oral treatments with significantly less frequent scheduling because bisphosphonates have a relatively poor oral bioavailability of less than two percent. Once present in circulation, their high affinity for bone allows prolonged drug presence in the patient [74, 75]. Similarly, the SERMs raloxifene and bazedoxifene and the metallic salt strontium ranelate are currently administered in oral form [73]. The SERMs are prescribed in daily tablet form, while strontium ranelate is administered daily as a sachet of powder dissolved in drink [73]. Oral bioavailability of SERMs is similar to bisphosphonates, ranging from 2–6% of loaded drug [73]. Oral bioavailability of strontium ranelate is significantly higher in comparison, at 27% [76]. Over the counter dietary supplements for ameliorating osteoporosis, such as calcium, calcium phosphate, and vitamin D, are similarly available in oral tablet and capsule forms [73].
Table 2.
Examples of Current and Emerging Delivery Methods**
|
Current Treatments
| ||||
|---|---|---|---|---|
| Drug Type | Examples | Delivery Method | Frequency of Dosage | Typical Dose |
| Bisphosphonates | Alendronate | Oral tablet | Daily Weekly |
10 mg 70 mg |
| Ibandronate | Oral tablet Intravenous infusion |
Monthly Quarterly |
150 mg 3 mg |
|
| Risedronate | Oral tablet | Daily Weekly Monthly |
5 mg 35 mg 150 mg |
|
| Zoledronate | Intravenous infusion | Annual | 5 mg | |
|
| ||||
| Hormones | Estrogen | Oral tablet Transdermal film |
Daily Weekly |
0.5 mg 25–100 μg |
| Calcitonin | Subcutaneous or intramuscular injection | Daily Alternating daily |
50–200 IU 100–400 IU |
|
| Intranasal spray | Daily | 200 IU | ||
| PTH (teriparatide) | Subcutaneous thigh/abdominal injection | Daily | 20 μg | |
|
| ||||
| SERMs | Raxilofene | Oral tablet | Daily | 60 mg |
| Bazedoxifene | Oral tablet | Daily | 0.45–20 mg | |
|
| ||||
| AMARTs | Denosumab | Subcutaneous injection | Biannual | 60 mg |
|
| ||||
| Dietary Supplements | Calcium | Oral tablet | Daily | 1000–1300 mg |
| Vitamin D | Oral tablet | Daily | 200–800 IU | |
| Calcitriol | Oral tablet | Daily | 0.25 μg | |
|
| ||||
| Other | Strontium ranelate | Oral aqueous solution | Daily | 2 g |
|
Emerging Treatments
| ||||
|---|---|---|---|---|
| Drug | Delivery Method | Examples | Status | |
| Alendronate | Depot | Intraosseous injection of PLGA nanoparticles | Preclinical (in vitro) | |
| Depot | Intramuscular injection of PLGA microparticles | Preclinical (rat) | ||
| Ceramic carrier | Adsorption onto intravenously injected hydroxyapatite nanoparticle | Preclinical (rabbit, rat) | ||
|
| ||||
| Zoledronate | Polymeric carrier | Oral via lysine-deoxycholic acid conjugation | Preclinical (rat) | |
|
| ||||
| PTH (teriparatide) | Actively triggered | Wireless release from implanted metallic microchip wells | Clinical (Phase I) | |
| Wireless release from implanted lipid membranes | Preclinical pig) | |||
|
| ||||
| Estrogens | Depot | Incorporation in implantable calcium deficient hydroxyapatite scaffolds | Preclinical (in vitro) | |
| Polymeric carrier | Intravenous injection of loaded PEG nanoparticles | Preclinical (rat) | ||
|
| ||||
| Simvastatin | Depot | Hydrolytic release from conjugated polymer | Preclinical (in vitro) | |
| Depot | Intraosseously injected PLGA microspheres | Preclinical (minipig) | ||
| Polymeric carrier | Injectable drug-PEG micelles | Preclinical (in vitro) | ||
| Targeted polymeric carrier | Injectable PLGA-tetracycline functionalized nanoparticles | Preclinical (mouse) | ||
|
| ||||
| miRNA (miR-26a) | Depot, polymeric carrier | PLGA microsphere release from PLLA scaffolds | Preclinical (mouse) | |
|
| ||||
| 6-bromoindirubin-3′-oxime (GSK3β inhibitor) | Targeted polymeric carrier | Intravenous injectable aspartic acid-conjugated micelles | Preclinical (mouse) | |
The two most commonly used biologically-derived osteoporosis treatments denosumab and teriparatide are available only in subcutaneous injectable dosage forms [77]. Denosumab is administered biannually in 60 mg doses. In contrast, teriparatide is prescribed as daily subcutaneous injections of 20 μg. As is typical of injectable treatments, bioavailability of denosumab and teriparatide is in excess of 90%.
Of currently approved osteoporosis therapies, only estradiol and calcitonin are not regularly delivered via oral or injectable methods. Estradiol is available in a topically applied film for transdermal delivery, while calcitonin is available in a nasal spray for inhalation. Both treatments are able to exploit the relatively high diffusive capacity relative to typical therapeutics that each hormone possesses to transport across the epidermis and mucous membranes, respectively [78, 79]. Estrogen’s status as a steroid hormone imparts high cellular permeability, allowing transdermal bioavailability to be approximately 20 times that of oral bioavailability [78]. Similarly, calcitonin appears to be actively endocytized by nasal epithelial cells, producing unusually high absorptive capacity relative to its size [79].
4.2 Emerging Approaches
A variety of approaches are being investigated to address the drawbacks of existing delivery methods as well as to enhance therapeutic efficacy. Table 2 summarizes select emerging approaches that are further described in the following subsections.
4.2.1 Drug Carriers
Use of drug carriers to improve the pharmacokinetics of therapeutic agents is a widespread and common strategy that includes a broad range of techniques from simple adsorption of bioactive molecules onto particle surfaces to covalent incorporation of drug molecules into polymer networks [80, 81]. Effective use of drug carriers can mitigate undesirable side effects of the prescribed drug while simultaneously improving its circulatory lifetime and release profile [81–83]. Common to almost all drug carrier methods is the generation of micro-and nano-particles, within or upon which drug can be loaded [81, 82]. These particles can be of polymeric, ceramic, or biological origin, allowing for a broad range of drug and delivery options [81, 83]. Examples of delivery vehicles being developed include poly(lactic-co-glycolic acid) nano- and microspheres containing bisphosphonates, which are typically prepared via emulsion-solvent evaporation and are amenable to aerosolized orotracheal and intravenous delivery [81].
Polymeric carriers are widely used for their relative ease of manufacture and tailorable physical qualities [81]. Synthetic polymers allow for a high degree of control of chain length and structure, including monomer distribution within copolymers and chemical modification of sidechains. Control of side chain functionalization can greatly contribute to carrier viability and targeting ability via addition of cell-adhesive moieties, such as Arg-Gly-Asp (RGD) peptides, or lineage-specific antibodies [82]. Furthermore, polymeric carriers can be designed to covalently bind and incorporate their drug load as side groups or as part of their chemical backbone, as shown by Asafo-Adjei et al. for a polymerized form of simvastatin (“polysimvastatin”) in a poly(ethylene glycol-block-simvastatin) copolymer [80]. Such systems minimize burst release typically associated with diffusion from particles encapsulating drug by enforcing a degradation-dependent limitation upon drug release. In contrast to traditional drug carriers, such drug conjugated systems provide significantly steadier, longer term release profiles.
Ceramic drug carriers, especially hydroxyapatite nanoparticles, are attractive drug carriers for their ability to provide osteoconductive, space-filling function to the application site in conjunction with controlled release capability [83]. Ceramic particles have benefited greatly from advances in nanoscale production and functionalization, allowing for strict control of particle size, shape, porosity, and chemical features. Particle outer and inner surfaces can be separately functionalized to optimize drug carrying capacity without sacrificing osteoconductive or biocompatible outer surfaces. Ceramic particles are capable of carrying a wide variety of agents for release, including not only directly bioactive agents such as hormones, but also other drug loaded microparticles that lack the stability or functionalization for proper targeting [84, 85]. Titania nanotubes have been demonstrated to protect and release proteins, such as BMP-2, as well as drug-loaded polymeric coatings and nanoparticles, such as vancomycin in poly(ethyleneimine)-human serum albumin, for burst and sustained release profiles, respectively [82]. Thin coats of unloaded polymer act as caps for the titania nanotubes, providing a temporarily seal that prevents premature release or hydrolytic degradation of loaded compounds prior to desired schedule [82].
Biologically derived polysaccharide, gelatin, and lipid-based carriers possess unrivaled biocompatibility and minimal cytotoxicity compared to synthetic systems [81, 83, 86]. Polysaccharide and liposome carriers are currently under investigation for use in inhalable systems due to their high biocompatibility and ease of uptake [81, 87]. Micellar systems are relatively simple to manufacture and can substantially improve the effective solubility of hydrophobic drugs in aqueous environments [84]. Simvastatin has been successfully delivered via injected calcium phosphate-conjugated simvastatin-deoxycholic acid micelles in mouse models, and successfully released from poly(ethylene glycol-caprolactone) (PEG-PCL) micelles loaded in titania nanotube arrays in vitro [84, 85]. Beyond their high biocompatibility relative to other systems, biologically derived systems benefit from broad resource bases and relatively low cost to manufacture [81, 86].
4.2.2 Tissue Targeting
Drug modification to improve tissue specificity is another common strategy to increase drug bioavailability. Drug targeting allows for selective local activity of therapeutic agents that minimizes nonspecific interactions and systemic toxicity. As described in Section 3.1, bisphosphonates possess an innate affinity for bone, and are prime examples of bone-targeted drugs, displaying binding affinity for bone calcium so strong that it actually interferes with ability of the bisphosphonate to interact with the desired protein targets [88]. Exploitation of the calcium affinity of phosphate groups allows enhancement of bone targeting via covalent substitution of carboxylic acids on bioactive molecule [88]. The addition of monophosphate functionality has been demonstrated to significantly improve hydroxyapatite binding affinity of benzoindole, salicylic acid, and quinolone compounds by Jahnke et al., who also demonstrate that the binding affinity improves with addition of flexible bridging chains between the phosphate group and core molecule, with direct attachment of phosphate groups to aromatic rings failing to confer bone specificity [88]. Similarly, conjugation to tetracycline or minimized derivatives enhances bone conjugation via chelation of calcium ions on hydroxyapatite surfaces [89, 90]. Biologically derived tetracycline acts as a phosphate mimic, substituting two such groups when interacting with three surface ions [90]. Negatively charged and phosphorylated peptides such as aspartic acid and phosphorylated serine exhibit similar calcium binding affinity in a more biomimetic modality, along with improved degradability and lowered cytotoxicity [72, 89–91]. Hydrophobic therapeutics such as GSK3β inhibitors have significantly improved solubility and affinity for bone fracture sites following covalent conjugation to aspartic acid peptides in a rat model [74]. Control of peptide targeting sequence length and composition can provide further specificity of surface binding; six repeat sequences of aspartic acid have a higher affinity for bone formation surfaces compared to eight repeat sequences, which have a greater affinity for resorptive surfaces often associated with fracture sites [74, 91]. Appropriate conjugation of these targeting moieties to anabolic or antiresorptive molecules may greatly improve therapeutic efficacy and local distribution, minimizing the required concentration and frequency of drug doses [72, 90, 91]. Lastly, mono- and polysaccharides display similarly high binding affinities for calcium and hydroxyapatite, providing non-peptide based readily degradable targeting systems [90].
For active molecules that cannot be functionalized with phosphate groups due to loss of function or lack of appropriate functionalization sites, addition of bone targeting agents to drug carrying materials may be a more desirable option. PEGylation of nanoparticle carriers with copolymer poloxamer systems, notably poloxamer-407, can confer general resistance to adsorption via strong hydrophilic character of polyethylene glycol segments while simultaneously providing selective uptake by bone marrow sinusoidal endothelial cells, as shown by Porter et al. using surface-labeled poloxamer coated polystyrene microspheres as a proof of concept [72, 92–94]. This selective uptake by sinusoidal cells results in verifiable accumulation of nanoparticles in the sternum, long bones, and spinal column [92, 93]. Once sequestered, nanoparticles deliver loaded drugs in a local fashion, minimizing off target effects for less selective drug systems [93].
4.2.3 Depot Systems
Injectable and implantable depot systems are ideal examples of drug delivery systems that minimize the need for patient compliance; depot systems offer controlled long term release of drug from one or more strategically placed delivery sites via active or passive systems, circumventing issues relating to oral viability or patient noncompliance [95]. Depot systems are typically designed for local release of drug rather than systemic distribution, and as such are well suited for use with medications that may otherwise distribute broadly. While injectable systems are more commonly thought of in reference of depot systems, it is also important to consider the role of surgically placed depots as well; drug-loaded polymeric and ceramic scaffolds are an ideal approach to drug delivery to fracture sites and bone surfaces following surgical intervention. Drug loading into supportive structures for continuous release with or without drug carriers provides local chemical treatment as the graft is resorbed, allowing for ensured delivery of therapeutics to the desired site, as demonstrated by Orellana et al. using simvastatin and a model protein [96]. Further, certain ceramic materials may act as depots for bioactive molecules without additional loading or modification; silicon, magnesium, and strontium based ceramics release metal ions that promotes osteogenic function and inhibit resorptive behavior [97–99]. Use of such materials in concert with traditional drug therapies may substantially improve bone quality following trauma without need for additional patient intervention.
Actively triggered release systems are promising emerging options for drug delivery, providing possibilities for highly localized, high concentration drug delivery in a controlled manner. Lee et al. developed a radiofrequency-sensitive implantable device that sequesters drug into lipid membrane reservoirs, where the drug is held with minimal release until activated by lysing of the lipid membrane via frequency resonance heating of a metallic coil [95]. Use of multiple independent reservoirs with differently tuned coils allows for controllable sequential delivery of high concentration therapeutics in a noninvasive manner. Additionally, the system displays long term stability and is biodegradable once triggered, eliminating the need for surgical retrieval.
Similarly, Ferra et al. reported clinical testing of a microchip-based drug delivery device that uses metallic membranes to seal drug within multiple distinct reservoirs for controllable release via thermal electroablation of the metallic seal [100]. This design forgoes the use of lipid membranes in favor of impermeable metallic seals to ensure the stability of the reservoir seals against environmental factors. Similar to the system of Lee et al. [95], this microchip design utilizes wireless systems to trigger release of PTH(1–34) in a controllable daily fashion for up to 20 days [100]. An on board battery and control electronics allows the device to autonomously trigger drug release at preprogrammed intervals, thereby assuring compliance with reduced risk of over- or under-dosing [100]. PTH release and distribution from the device was comparable to typical subcutaneous injection while being more consistent between doses [100]. Elaboration upon this or similar electronically controlled devices may enable greater density of loaded doses, incorporation of sensing systems, and possible self-regulation of the release schedule. These systems hold great promise as pulsatile and autonomous delivery systems.
5. Conclusion
Drug therapies for treatment of osteoporosis are directed at restoring bone balance by inhibiting osteoclasts or stimulating osteoblasts. Approaches, such as oral administration of hormones or bisphosphonates, intravenous infusion of bisphosphonates, and injection of teriparatide, are subject to disadvantages of patient noncompliance or poor bioavailability. Transdermal delivery of steroidal hormones and inhalation of peptide hormones only partially avoid these problems. As new modulators of bone cell differentiation and activity are identified, such as antibody-mediated therapies or receptor-specific agonists and antagonists, advanced methods are increasingly useful for delivering these bioactive agents. Bone-targeting, encapsulation in or conjugation to degradable polymers, and both passive and actively triggered depot systems can deliver the drug where it is needed, protect it from damage, and maintain therapeutic doses while minimizing problems with patient compliance.
Acknowledgments
Funding from the National Institutes of Health (AR060964 and EB017902) is gratefully acknowledged.
Footnotes
Conflict of Interest
T.A. Asafo-Adjei has a patent, Polymeric Prodrug, pending. D.A. Puleo reports equity interest from Regenera Materials, LLC, outside the submitted work. In addition, Dr. Puleo has a patent, Polymeric Prodrug, pending.
A.J. Chen, and A. Najarzadeh declare that they have no conflict of interest.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
References
Papers of particular interest, published recently, have been highlighted as:
• Of importance
•• Of major importance
- 1.Assessment of fracture risk and its application to screening for postmenopausal osteoporosis. Report of a WHO Study Group. World Health Organ Tech Rep Ser. 1994;843:1–129. [PubMed] [Google Scholar]
- 2.National Osteoporosis Foundation. and American Academy of Orthopaedic Surgeons. Physician’s guide to prevention and treatment of osteoporosis. Washington, D.C.: National Osteoporosis Foundation; 1998. p. 30. [Google Scholar]
- 3.Wright NC, et al. The recent prevalence of osteoporosis and low bone mass in the United States based on bone mineral density at the femoral neck or lumbar spine. J Bone Miner Res. 2014;29(11):2520–6. doi: 10.1002/jbmr.2269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Johnell O, Kanis JA. An estimate of the worldwide prevalence and disability associated with osteoporotic fractures. Osteoporos Int. 2006;17(12):1726–33. doi: 10.1007/s00198-006-0172-4. [DOI] [PubMed] [Google Scholar]
- 5.Blume SW, Curtis JR. Medical costs of osteoporosis in the elderly Medicare population. Osteoporos Int. 2011;22(6):1835–44. doi: 10.1007/s00198-010-1419-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Burge R, et al. Incidence and economic burden of osteoporosis-related fractures in the United States, 2005-2025. J Bone Miner Res. 2007;22(3):465–75. doi: 10.1359/jbmr.061113. [DOI] [PubMed] [Google Scholar]
- 7.Osteoporosis prevention, diagnosis, and therapy. NIH Consens Statement. 2000;17(1):1–45. [PubMed] [Google Scholar]
- 8.Borgstrom F, et al. The societal burden of osteoporosis in Sweden. Bone. 2007;40(6):1602–9. doi: 10.1016/j.bone.2007.02.027. [DOI] [PubMed] [Google Scholar]
- 9.Kanis JA, Johnell O. Requirements for DXA for the management of osteoporosis in Europe. Osteoporos Int. 2005;16(3):229–38. doi: 10.1007/s00198-004-1811-2. [DOI] [PubMed] [Google Scholar]
- 10.Pietschmann P, et al. Osteoporosis: an age-related and gender-specific disease–a mini-review. Gerontology. 2009;55(1):3–12. doi: 10.1159/000166209. [DOI] [PubMed] [Google Scholar]
- 11.Drake MT, Clarke BL, Lewiecki EM. The Pathophysiology and Treatment of Osteoporosis. Clin Ther. 2015;37(8):1837–50. doi: 10.1016/j.clinthera.2015.06.006. [DOI] [PubMed] [Google Scholar]
- 12.Lupsa BC, Insogna K. Bone Health and Osteoporosis. Endocrinol Metab Clin North Am. 2015;44(3):517–30. doi: 10.1016/j.ecl.2015.05.002. [DOI] [PubMed] [Google Scholar]
- 13••.Canalis E. Wnt signalling in osteoporosis: mechanisms and novel therapeutic approaches. Nat Rev Endocrinol. 2013;9(10):575–83. doi: 10.1038/nrendo.2013.154. This paper provides an in-depth overview of bone remodeling at the cellular level, explaining the actions of osteoclast, osteoblast, and osteocyte bone-cell types in response to each other throughout the remodeling process. Various Wnt signaling-associated bone diseases including osteoporosis, and the roles of secreted signaling proteins influencing the pathway, such as sclerostin and macrophage colony stimulating factor, are discussed. New drug therapies developed targeting the pathway to promote anabolic processes, are covered in the review. [DOI] [PubMed] [Google Scholar]
- 14••.Rivadeneira F, Makitie O. Osteoporosis and Bone Mass Disorders: From Gene Pathways to Treatments. Trends Endocrinol Metab. 2016;27(5):262–81. doi: 10.1016/j.tem.2016.03.006. This paper explains in depth the genes encoding factors in signaling pathways crucial for mesenchymal cell differentiation, skeletal development, bone remodeling and metabolism. It focuses in several of the remaining discovered genes to expose their role in bone biology. The intuition provided by genetic studies helps in identifying of biomarkers predictive of disease, redefining disease, response to treatment, and discovery of novel drug targets for skeletal disorders. [DOI] [PubMed] [Google Scholar]
- 15.Stefanick ML. Estrogens and progestins: background and history, trends in use, and guidelines and regimens approved by the US Food and Drug Administration. Am J Med. 2005;118(Suppl 12B):64–73. doi: 10.1016/j.amjmed.2005.09.059. [DOI] [PubMed] [Google Scholar]
- 16.Gennari L, Merlotti D, Nuti R. Selective estrogen receptor modulator (SERM) for the treatment of osteoporosis in postmenopausal women: focus on lasofoxifene. Clin Interv Aging. 2010;5:19–29. doi: 10.2147/cia.s6083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ettinger B, Genant HK, Cann CE. Long-term estrogen replacement therapy prevents bone loss and fractures. Ann Intern Med. 1985;102(3):319–24. doi: 10.7326/0003-4819-102-3-319. [DOI] [PubMed] [Google Scholar]
- 18.Lufkin EG, et al. Treatment of postmenopausal osteoporosis with transdermal estrogen. Ann Intern Med. 1992;117(1):1–9. doi: 10.7326/0003-4819-117-1-1. [DOI] [PubMed] [Google Scholar]
- 19.Mauck KF, Clarke BL. Diagnosis, screening, prevention, and treatment of osteoporosis. Mayo Clin Proc. 2006;81(5):662–72. doi: 10.4065/81.5.662. [DOI] [PubMed] [Google Scholar]
- 20.Maximov PY, Lee TM, Jordan VC. The discovery and development of selective estrogen receptor modulators (SERMs) for clinical practice. Curr Clin Pharmacol. 2013;8(2):135–55. doi: 10.2174/1574884711308020006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Overgaard K, et al. Effect of salcatonin given intranasally on early postmenopausal bone loss. BMJ. 1989;299(6697):477–9. doi: 10.1136/bmj.299.6697.477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Overgaard K, Riis BJ. Nasal salmon calcitonin in osteoporosis. Calcif Tissue Int. 1994;55(2):79–81. doi: 10.1007/BF00297178. [DOI] [PubMed] [Google Scholar]
- 23.Carstens JH, Jr, Feinblatt JD. Future horizons for calcitonin: a U.S. perspective. Calcif Tissue Int. 1991;49(Suppl 2):S2–6. doi: 10.1007/BF02561368. [DOI] [PubMed] [Google Scholar]
- 24.Francis MD, Valent DJ. Historical perspectives on the clinical development of bisphosphonates in the treatment of bone diseases. J Musculoskelet Neuronal Interact. 2007;7(1):2–8. [PubMed] [Google Scholar]
- 25.Marini F, Brandi ML. Pharmacogenetics of osteoporosis. Best Pract Res Clin Endocrinol Metab. 2014;28(6):783–93. doi: 10.1016/j.beem.2014.07.004. [DOI] [PubMed] [Google Scholar]
- 26.Luckman SP, et al. Nitrogen-containing bisphosphonates inhibit the mevalonate pathway and prevent post-translational prenylation of GTP-binding proteins, including Ras. J Bone Miner Res. 1998;13(4):581–9. doi: 10.1359/jbmr.1998.13.4.581. [DOI] [PubMed] [Google Scholar]
- 27.Kennel KA, Drake MT. Adverse effects of bisphosphonates: implications for osteoporosis management. Mayo Clin Proc. 2009;84(7):632–7. doi: 10.1016/S0025-6196(11)60752-0. quiz 638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Murad MH, et al. Clinical review. Comparative effectiveness of drug treatments to prevent fragility fractures: a systematic review and network meta-analysis. J Clin Endocrinol Metab. 2012;97(6):1871–80. doi: 10.1210/jc.2011-3060. [DOI] [PubMed] [Google Scholar]
- 29.Farrier AJ, et al. New anti-resorptives and antibody mediated anti-resorptive therapy. Bone Joint J. 2016;98-B(2):160–5. doi: 10.1302/0301-620X.98B2.36161. [DOI] [PubMed] [Google Scholar]
- 30.Lacey DL, et al. Osteoprotegerin ligand is a cytokine that regulates osteoclast differentiation and activation. cell. 1998;93(2):165–176. doi: 10.1016/s0092-8674(00)81569-x. [DOI] [PubMed] [Google Scholar]
- 31.Hammer GD, McPhee SJ. Pathophysiology of Disease: An Introduction to Clinical Medicine. McGraw Hill Professional; 2010. [Google Scholar]
- 32••.Mandema JW, et al. Time course of bone mineral density changes with denosumab compared with other drugs in postmenopausal osteoporosis: a dose-response-based meta-analysis. J Clin Endocrinol Metab. 2014;99(10):3746–55. doi: 10.1210/jc.2013-3795. This paper compares lumbar spine and total hip BMD changes in postmenopausal women from current treatment dose regimens of bisphosphonates, SERMs, PTH, calcitonin and denosumab. Nonlinear least-squares random-effects meta-regression regression analysis was conducted on a culmination of data in reported randomized controlled clinical trials representative of over 113,000 study participants. [DOI] [PubMed] [Google Scholar]
- 33.Anastasilakis AD, et al. Denosumab versus zoledronic acid in patients previously treated with zoledronic acid. Osteoporos Int. 2015;26(10):2521–7. doi: 10.1007/s00198-015-3174-2. [DOI] [PubMed] [Google Scholar]
- 34.Min H, et al. Osteoprotegerin reverses osteoporosis by inhibiting endosteal osteoclasts and prevents vascular calcification by blocking a process resembling osteoclastogenesis. J Exp Med. 2000;192(4):463–74. doi: 10.1084/jem.192.4.463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bekker PJ, et al. The effect of a single dose of osteoprotegerin in postmenopausal women. J Bone Miner Res. 2001;16(2):348–60. doi: 10.1359/jbmr.2001.16.2.348. [DOI] [PubMed] [Google Scholar]
- 36.Brennan TC, et al. Osteoblasts play key roles in the mechanisms of action of strontium ranelate. Br J Pharmacol. 2009;157(7):1291–300. doi: 10.1111/j.1476-5381.2009.00305.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cianferotti L, D’Asta F, Brandi ML. A review on strontium ranelate long-term antifracture efficacy in the treatment of postmenopausal osteoporosis. Ther Adv Musculoskelet Dis. 2013;5(3):127–39. doi: 10.1177/1759720X13483187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bazaldua OV, Bruder J. Teriparatide (Forteo) for osteoporosis. Am Fam Physician. 2004;69(8):1983–4. [PubMed] [Google Scholar]
- 39.Ishizuya T, et al. Parathyroid hormone exerts disparate effects on osteoblast differentiation depending on exposure time in rat osteoblastic cells. J Clin Invest. 1997;99(12):2961–70. doi: 10.1172/JCI119491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang H, et al. Recombinant human parathyroid hormone related protein 1-34 and 1-84 and their roles in osteoporosis treatment. PLoS One. 2014;9(2):e88237. doi: 10.1371/journal.pone.0088237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.van Bezooijen RL, et al. Wnt but not BMP signaling is involved in the inhibitory action of sclerostin on BMP-stimulated bone formation. Journal of Bone and Mineral Research. 2007;22(1):19–28. doi: 10.1359/jbmr.061002. [DOI] [PubMed] [Google Scholar]
- 42.Pan L, et al. Fluoride promotes osteoblastic differentiation through canonical Wnt/β-catenin signaling pathway. Toxicology letters. 2014;225(1):34–42. doi: 10.1016/j.toxlet.2013.11.029. [DOI] [PubMed] [Google Scholar]
- 43.Riggs BL, et al. Effect of fluoride treatment on the fracture rate in postmenopausal women with osteoporosis. N Engl J Med. 1990;322(12):802–9. doi: 10.1056/NEJM199003223221203. [DOI] [PubMed] [Google Scholar]
- 44.Tilyard MW, et al. Treatment of postmenopausal osteoporosis with calcitriol or calcium. N Engl J Med. 1992;326(6):357–62. doi: 10.1056/NEJM199202063260601. [DOI] [PubMed] [Google Scholar]
- 45.Civitelli R, et al. Bone turnover in postmenopausal osteoporosis. Effect of calcitonin treatment. J Clin Invest. 1988;82(4):1268–74. doi: 10.1172/JCI113725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Langlois JA, et al. Association Between Insulin-Like Growth Factor I and Bone Mineral Density in Older Women and Men: The Framingham Heart Study 1. The Journal of Clinical Endocrinology & Metabolism. 1998;83(12):4257–4262. doi: 10.1210/jcem.83.12.5308. [DOI] [PubMed] [Google Scholar]
- 47.Saaf M, et al. Growth hormone treatment of osteoporotic postmenopausal women - a one-year placebo-controlled study. Eur J Endocrinol. 1999;140(5):390–9. doi: 10.1530/eje.0.1400390. [DOI] [PubMed] [Google Scholar]
- 48.Lindsay R, et al. Efficacy of tissue-selective estrogen complex of bazedoxifene/conjugated estrogens for osteoporosis prevention in at-risk postmenopausal women. Fertil Steril. 2009;92(3):1045–52. doi: 10.1016/j.fertnstert.2009.02.093. [DOI] [PubMed] [Google Scholar]
- 49•.Amugongo SK, et al. Effects of sequential osteoporosis treatments on trabecular bone in adult rats with low bone mass. Osteoporos Int. 2014;25(6):1735–50. doi: 10.1007/s00198-014-2678-5. This paper investigates the combination of anti-resorptive (alendronate or raloxifene) and anabolic (PTH) treatment courses as a potentially more synergistic therapy option for osteoporosis treatment compared to single drug therapies. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Leder BZ, et al. Effects of abaloparatide, a human parathyroid hormone-related peptide analog, on bone mineral density in postmenopausal women with osteoporosis. J Clin Endocrinol Metab. 2015;100(2):697–706. doi: 10.1210/jc.2014-3718. [DOI] [PubMed] [Google Scholar]
- 51•.Ponnapakkam T, et al. Treating osteoporosis by targeting parathyroid hormone to bone. Drug Discov Today. 2014;19(3):204–8. doi: 10.1016/j.drudis.2013.07.015. This paper describes a more targeted therapy of PTH by conjugating to a collagen-binding domain with high bone affinity to enhance the effectiveness of PTH treatment. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Fujisaki J, et al. Osteotropic drug delivery system (ODDS) based on bisphosphonic prodrug. V. Biological disposition and targeting characteristics of osteotropic estradiol. Biol Pharm Bull. 1997;20(11):1183–7. doi: 10.1248/bpb.20.1183. [DOI] [PubMed] [Google Scholar]
- 53•.Makras P, Delaroudis S, Anastasilakis AD. Novel therapies for osteoporosis. Metabolism. 2015;64(10):1199–214. doi: 10.1016/j.metabol.2015.07.011. This paper provides a thorough and updated overview of the effects of specific novel PTH and β-arrestin analogs and Src tyrosine kinase, dickkopf-1, activin A, calcium-sensing receptor antagonists on bone mineral density in clinical trials, along with the effects of established treatments. It also delves into the consideration of existing agents utilized for other therapeutic applications, being reinvented for osteoporosis therapy. [DOI] [PubMed] [Google Scholar]
- 54.Mukherjee K, Chattopadhyay N. Pharmacological Inhibition of Cathepsin K: a Promising Novel Approach for Postmenopausal Osteoporosis Therapy. Biochem Pharmacol. 2016 doi: 10.1016/j.bcp.2016.04.010. [DOI] [PubMed] [Google Scholar]
- 55.Appelman-Dijkstra NM, Papapoulos SE. Sclerostin Inhibition in the Management of Osteoporosis. Calcif Tissue Int. 2016;98(4):370–80. doi: 10.1007/s00223-016-0126-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gesty-Palmer D, et al. A beta-arrestin-biased agonist of the parathyroid hormone receptor (PTH1R) promotes bone formation independent of G protein activation. Sci Transl Med. 2009;1(1):1ra1. doi: 10.1126/scitranslmed.3000071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Roskoski R., Jr Src protein-tyrosine kinase structure, mechanism, and small molecule inhibitors. Pharmacol Res. 2015;94:9–25. doi: 10.1016/j.phrs.2015.01.003. [DOI] [PubMed] [Google Scholar]
- 58.Hannon RA, et al. Effects of the Src kinase inhibitor saracatinib (AZD0530) on bone turnover in healthy men: a randomized, double-blind, placebo-controlled, multiple-ascending-dose phase I trial. J Bone Miner Res. 2010;25(3):463–71. doi: 10.1359/jbmr.090830. [DOI] [PubMed] [Google Scholar]
- 59.Glantschnig H, et al. A rate-limiting role for Dickkopf-1 in bone formation and the remediation of bone loss in mouse and primate models of postmenopausal osteoporosis by an experimental therapeutic antibody. J Pharmacol Exp Ther. 2011;338(2):568–78. doi: 10.1124/jpet.111.181404. [DOI] [PubMed] [Google Scholar]
- 60.Betts AM, et al. The application of target information and preclinical pharmacokinetic/pharmacodynamic modeling in predicting clinical doses of a Dickkopf-1 antibody for osteoporosis. J Pharmacol Exp Ther. 2010;333(1):2–13. doi: 10.1124/jpet.109.164129. [DOI] [PubMed] [Google Scholar]
- 61.Iyer SP, et al. A Phase IB multicentre dose-determination study of BHQ880 in combination with anti-myeloma therapy and zoledronic acid in patients with relapsed or refractory multiple myeloma and prior skeletal-related events. Br J Haematol. 2014;167(3):366–75. doi: 10.1111/bjh.13056. [DOI] [PubMed] [Google Scholar]
- 62.Ruckle J, et al. Single-Dose, Randomized, Double-Blind, Placebo-Controlled Study of ACE-011 (ActRIIA-IgG1) in Postmenopausal Women. Journal of Bone and Mineral Research. 2009;24(4):744–752. doi: 10.1359/jbmr.081208. [DOI] [PubMed] [Google Scholar]
- 63.Fitzpatrick LA, et al. Ronacaleret, a calcium-sensing receptor antagonist, increases trabecular but not cortical bone in postmenopausal women. J Bone Miner Res. 2012;27(2):255–62. doi: 10.1002/jbmr.554. [DOI] [PubMed] [Google Scholar]
- 64.Halse J, et al. A phase 2, randomized, placebo-controlled, dose-ranging study of the calcium-sensing receptor antagonist MK-5442 in the treatment of postmenopausal women with osteoporosis. J Clin Endocrinol Metab. 2014;99(11):E2207–15. doi: 10.1210/jc.2013-4009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Dai L, et al. The functional mechanism of simvastatin in experimental osteoporosis. J Bone Miner Metab. 2016;34(1):23–32. doi: 10.1007/s00774-014-0638-y. [DOI] [PubMed] [Google Scholar]
- 66.Pena JM, et al. Statin therapy and risk of fracture: results from the JUPITER randomized clinical trial. JAMA Intern Med. 2015;175(2):171–7. doi: 10.1001/jamainternmed.2014.6388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Cheng L, et al. Persistance and Compliance with Osteroporosis Therapies Among Women in a Commercially Insured Population in the United States. JOURNAL OF MANAGED CARE & SPECIALTY PHARMACY. 2015;21(9):824–U322. doi: 10.18553/jmcp.2015.21.9.824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Lakatos P, et al. A Retrospective Longitudinal Database Study of Persistence and Compliance with Treatment of Osteoporosis in Hungary. Calcif Tissue Int. 2016;98(3):215–25. doi: 10.1007/s00223-015-0082-6. [DOI] [PubMed] [Google Scholar]
- 69.Luhmann T, et al. Bone targeting for the treatment of osteoporosis. J Control Release. 2012;161(2):198–213. doi: 10.1016/j.jconrel.2011.10.001. [DOI] [PubMed] [Google Scholar]
- 70.Various. Medications for Osteoporosis. 2016 Apr; 2016 [cited 2016; Available from: http://www.drugs.com/condition/osteoporosis.html.
- 71•.Low SA, et al. Biodistribution of Fracture-Targeted GSK3beta Inhibitor-Loaded Micelles for Improved Fracture Healing. Biomacromolecules. 2015;16(10):3145–53. doi: 10.1021/acs.biomac.5b00777. This paper presents a method providing a fourfold increase in drug delivery to fracture sites over distribution in undamaged bone. Authors introduce a relatively facile, amino acid based method of generating micelles that carry and delivery anabolic GSKbeta inhibitors to fracture sites. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Reginster J. Strontium Ranelate in Osteoporosis. Current Pharmaceutical Design. 8(21):1907–1916. doi: 10.2174/1381612023393639. [DOI] [PubMed] [Google Scholar]
- 73.Ramachandran C, Fleisher D. Transdermal delivery of drugs for the treatment of bone diseases. Adv Drug Deliv Rev. 2000;42:197. doi: 10.1016/s0169-409x(00)00062-4. [DOI] [PubMed] [Google Scholar]
- 74.Ozsoy Y, Gungor S, Cevher E. Nasal delivery of high molecular weight drugs. Molecules. 2009;14(9):3754–79. doi: 10.3390/molecules14093754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Tan J, et al. A single CT-guided percutaneous intraosseous injection of thermosensitive simvastatin/poloxamer 407 hydrogel enhances vertebral bone formation in ovariectomized minipigs. Osteoporos Int. 2016;27(2):757–67. doi: 10.1007/s00198-015-3230-y. [DOI] [PubMed] [Google Scholar]
- 76••.Lee CH, et al. Biological lipid membranes for on-demand, wireless drug delivery from thin, bioresorbable electronic implants. NPG Asia Materials. 2015;7(11):e227. doi: 10.1038/am.2015.114. This paper presents a wholly biodegradable implantable triggered pulsatile release system for use in precision drug delivery without requiring surgical removal once depleted. Elaboration of biodegradable electronic systems may substantially improve post-surgical patient compliance and outcome. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Jia Z, et al. Simvastatin prodrug micelles target fracture and improve healing. J Control Release. 2015;200:23–34. doi: 10.1016/j.jconrel.2014.12.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Posadowska U, et al. Injectable nanoparticle-loaded hydrogel system for local delivery of sodium alendronate. Int J Pharm. 2015;485(1–2):31–40. doi: 10.1016/j.ijpharm.2015.03.003. [DOI] [PubMed] [Google Scholar]
- 79.Farra R, et al. First-in-human testing of a wirelessly controlled drug delivery microchip. Sci Transl Med. 2012;4(122):12. doi: 10.1126/scitranslmed.3003276. [DOI] [PubMed] [Google Scholar]
- 80.Bae J, Park JW. Preparation of an injectable depot system for long-term delivery of alendronate and evaluation of its anti-osteoporotic effect in an ovariectomized rat model. Int J Pharm. 2015;480(1–2):37–47. doi: 10.1016/j.ijpharm.2015.01.020. [DOI] [PubMed] [Google Scholar]
- 81.Orellana BR, Hilt JZ, Puleo DA. Drug release from calcium sulfate-based composites. J Biomed Mater Res Part B. 2015;103B:135–142. doi: 10.1002/jbm.b.33181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Jeon OC, et al. Oral delivery of zoledronic acid by non-covalent conjugation with lysine-deoxycholic acid: In vitro characterization and in vivo anti-osteoporotic efficacy in ovariectomized rats. Eur J Pharm Sci. 2016;82:1–10. doi: 10.1016/j.ejps.2015.11.004. [DOI] [PubMed] [Google Scholar]
- 83.Tripathi G, Raja N, Yun HS. Effect of direct loading of phytoestrogens into the calcium phosphate scaffold on osteoporotic bone tissue regeneration. J Mater Chem B. 2015;3(44):8694–8703. doi: 10.1039/c5tb01574j. [DOI] [PubMed] [Google Scholar]
- 84.Hu Y, et al. 17beta-Estradiol-Loaded PEGlyated Upconversion Nanoparticles as a Bone-Targeted Drug Nanocarrier. ACS Appl Mater Interfaces. 2015;7(29):15803–11. doi: 10.1021/acsami.5b02831. [DOI] [PubMed] [Google Scholar]
- 85.Shi S, et al. The application of nanomaterials in controlled drug delivery for bone regeneration. J Biomed Mater Res A. 2015;103(12):3978–92. doi: 10.1002/jbm.a.35522. [DOI] [PubMed] [Google Scholar]
- 86.Asafo-Adjei TA, Dziubla TD, Puleo DA. Synthesis and Characterization of a Poly(ethylene glycol)-Poly(simvastatin) Diblock Copolymer. RSC Adv. 2014;4(102):58287–58298. doi: 10.1039/C4RA10310F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Miladi K, et al. Drug carriers in osteoporosis: preparation, drug encapsulation and applications. Int J Pharm. 2013;445(1–2):181–95. doi: 10.1016/j.ijpharm.2013.01.031. [DOI] [PubMed] [Google Scholar]
- 88.Smith JR, Lamprou DA. Polymer coatings for biomedical applications: a review. Transactions of the IMF. 2014;92(1):9–19. [Google Scholar]
- 89.Ito T, et al. Preparation of calcium phosphate nanocapsules including simvastatin/deoxycholic acid assembly, and their therapeutic effect in osteoporosis model mice. J Pharm Pharmacol. 2013;65(4):494–502. doi: 10.1111/jphp.12008. [DOI] [PubMed] [Google Scholar]
- 90.Liu X, L Xiaoran, Li Shaobing, Zhou Xiaosong, Li Sha, Wang Qiangbin, Dai Jianwu, Lai Renfa, Xie Li, Zhong Mei, Zhang Ye, Zhou Lei. An In Vitro Study of a Titanium Surface Modified by Simvastatin-Loaded Titania Nanotubes-Micelles. Journal of Biomedical Nanotechnology. 2014;10(2):11. doi: 10.1166/jbn.2014.1810. [DOI] [PubMed] [Google Scholar]
- 91.Cunha L, Grenha A. Sulfated Seaweed Polysaccharides as Multifunctional Materials in Drug Delivery Applications. Mar Drugs. 2016;14(3) doi: 10.3390/md14030042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92•.Dolatabadi JEN, Hamishehkar H, Valizadeh H. Development of dry powder inhaler formulation loaded with alendronate solid lipid nanoparticles: solid-state characterization and aerosol dispersion performance. Drug Development and Industrial Pharmacy. 2015;41(9):1431–1437. doi: 10.3109/03639045.2014.956111. This paper describes the preparation and characterization of aerosol capable solid-lipid alendronate nanoparticles for orotracheal delivery. Although not tested in vivo, promising aerodynamic performance was observed. [DOI] [PubMed] [Google Scholar]
- 93.Jahnke W, et al. A General Strategy for Targeting Drugs to Bone. Angew Chem Int Ed Engl. 2015;54(48):14575–9. doi: 10.1002/anie.201507064. [DOI] [PubMed] [Google Scholar]
- 94.Low SA, Kopecek J. Targeting polymer therapeutics to bone. Adv Drug Deliv Rev. 2012;64(12):1189–204. doi: 10.1016/j.addr.2012.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Wang D, et al. Bone-targeting macromolecular therapeutics. Adv Drug Deliv Rev. 2005;57(7):1049–76. doi: 10.1016/j.addr.2004.12.011. [DOI] [PubMed] [Google Scholar]
- 96.Aoki K, et al. Peptide-based delivery to bone. Adv Drug Deliv Rev. 2012;64(12):1220–38. doi: 10.1016/j.addr.2012.05.017. [DOI] [PubMed] [Google Scholar]
- 97.Porter CJH, et al. The polyoxyethylene/polyoxypropylene block co-polymer Poloxamer-407 selectively redirects intravenously injected microspheres to sinusoidal endothelial cells of rabbit bone marrow. FEBS Letters. 1992;305(1):62–66. doi: 10.1016/0014-5793(92)80655-z. [DOI] [PubMed] [Google Scholar]
- 98.Lin K, et al. Enhanced osteoporotic bone regeneration by strontium-substituted calcium silicate bioactive ceramics. Biomaterials. 2013;34(38):10028–42. doi: 10.1016/j.biomaterials.2013.09.056. [DOI] [PubMed] [Google Scholar]
- 99.Qu H, Bhattacharyya S, Ducheyne P. Silicon oxide based materials for controlled release in orthopedic procedures. Adv Drug Deliv Rev. 2015;94:96–115. doi: 10.1016/j.addr.2015.05.015. [DOI] [PubMed] [Google Scholar]
- 100.Razavi M, et al. In vivo biocompatibility of Mg implants surface modified by nanostructured merwinite/PEO. J Mater Sci Mater Med. 2015;26(5):184. doi: 10.1007/s10856-015-5514-3. [DOI] [PubMed] [Google Scholar]