

Introduction
Macular dystrophies are arbitrarily classified on the basis of their tissue of origin into those originating from nerve fibre layer, photoreceptors and retinal pigment epithelium (RPE), RPE alone, Bruch's membrane and choroid. Vitelliform dystrophy of the fovea is a separate entity among the inherited macular dystrophies. It was first reported by Adam [1] and the first pedigree was described by Best. It results in typical vitelliform lesions that are bilateral though asymmetrical and cause only moderate diminution of vision for a long time.
Case Report
A 42 year old woman presented with a six month old history of gradual, painless, progressive diminution of vision in both eyes. She gave no history of any trauma, redness, pain, watering or discharge from either eye. She was not a known diabetic, hypertensive and never suffered any chronic disorder. There was no similar history in the immediate family. On examination she was found to be orthotropic with a Snellen's vision of 6/24 that improved to 6/12 in both eyes. Both eyes were externally quiet with clear corneas, clear anterior chambers, briskly reacting pupils with normal iris morphology and transparent lenses. Fundus examination of the right eye (RE) revealed a clear media with a normal well-defined vertically oval optic disc. The macula showed a large (4-5 disc diameter), well-circumscribed, single, yellow, sub-retinal lesion of a variable density, over which normal retinal blood-vessels traversed (Fig.1). There was no evidence of past or current inflammation and the remaining retina appeared normal.
Fig. 1.
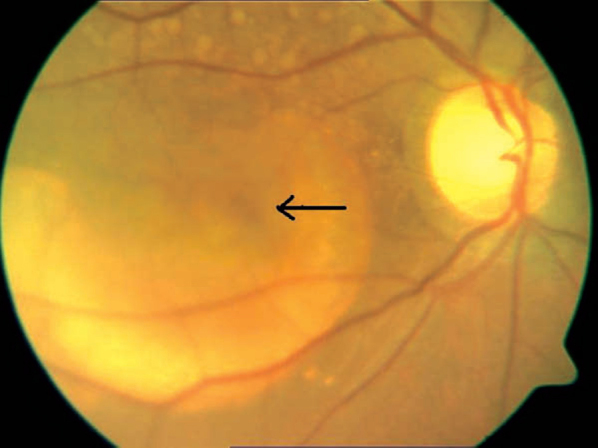
A large yellow macular lesion with a pseudohypopyon
Left eye (LE) also had a normal anterior segment with clear media. Fundus showed a normal optic disc with a large (2 DD) well-defined, elevated, yellow-orange central macular lesion that was surrounded by 14-15 small sized (1/3 DD) yellowish lesions (Fig. 2). The overlying retina was normal, as was the peripheral retina. The intra-ocular pressures were normal with applanation tonometry. The patient was investigated to rule out any specific cause of sub-retinal inflammation and all systemic investigations were normal. In view of the yellow vitelliform lesions in both eyes Best's vitelliform dystrophy was suspected and the patient was investigated with fundus fluorescein angiography (FFA), optical coherence tomography (OCT) and an electro-oculogram (EOG).
Fig. 2.
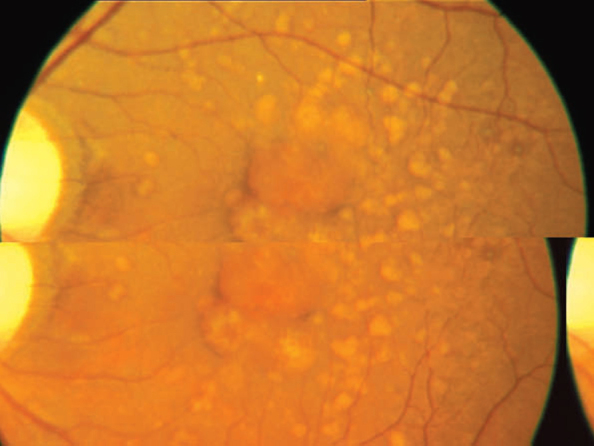
Central yellow orange lesion with multiple surrounding lesions
FFA of RE (Fig. 3) showed a normal optic disc, a large sub-retinal area of mottled hyperfluorescence that was well-defined and had a distinct upper level. There were numerous deposits seen around the macula. The overlying and peripheral retina appeared normal. FFA of LE (Fig. 4) showed a normal optic disc, numerous round sub-retinal hyperflourescent deposits that were encroaching the foveal avascular zone (FAZ). FAZ was still largely normal as was the central and peripheral retina.
Fig. 3.
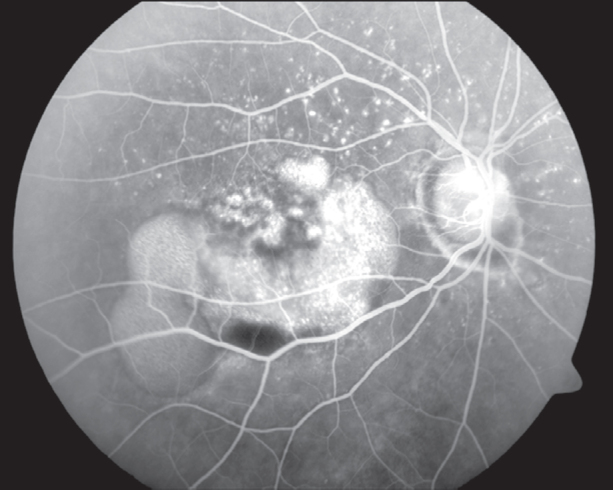
FFA showing mottled hyperflourescence with normal overlying retinal vasculature
Fig. 4.
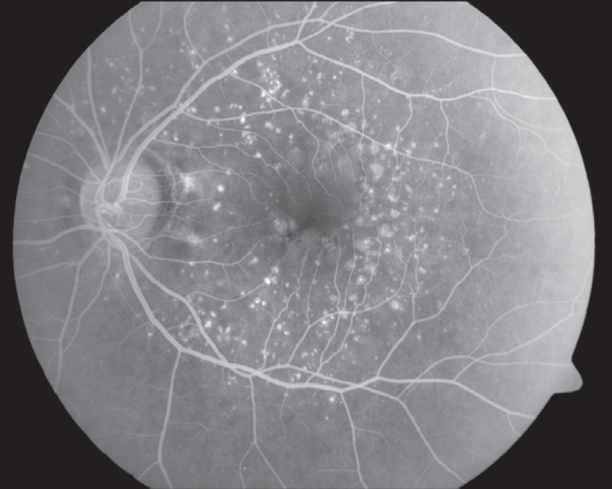
Normal appearance of the fovea, foveal avascular zone is distorted
OCT of RE (Fig. 5) showed two large “mounds” reflecting the RPE detachment with normal retinal morphology. At one point we could detect a breach in the continuity of RPE and a cystic change above it. There was no evidence of a choroidal neo-vascular membrane (CNVM). OCT of LE showed an entirely different picture that showed two “mounds” that were distinctly smaller than those in RE and were much better circumscribed. There was no apparent breach in the RPE (Fig.6).
Fig. 5.
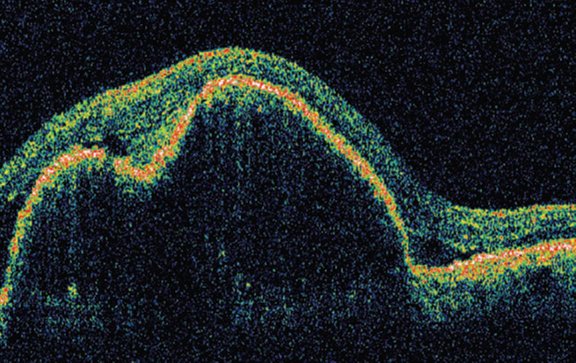
Large retinal pigment epithelium detachment with a breach
Fig. 6.
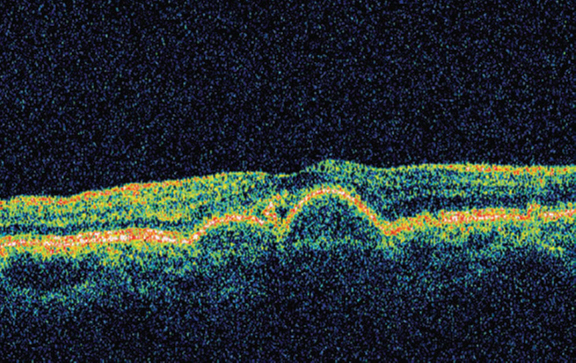
Multiple retinal pigment epithelium detachments
EOG showed light peak values of 217 uV and dark trough value of 143 uV thus giving an Arden ratio of 1.51 which was significantly below the normal of 2. Electro-retinogram (ERG) showed value of 210 uV for ‘a’ wave and a value of 410 uV for ‘b’ wave under mesopic conditions. ERG values were well within normal limits.
Thus on basis of bilateral multiple well-circumscribed yellow-orange macular sub-retinal lesions, that showed RPE detachments on OCT and revealed EOG-ERG dissociation on electro-physiology, a diagnosis of Best's vitelliform dystrophy was made. RE probably had a more advanced disease and had reached Stage 3 or pseudohypopyon (Fig. 1 – arrow) stage while LE had a typical multi-focal vitelliform Stage 2 lesion.
Discussion
Best disease, also termed vitelliform macular dystrophy, is an autosomal dominant disorder, which classically presents in childhood with the striking appearance of a yellow or orange yolk-like lesion in the macula. A hallmark of the disease is a markedly abnormal EOG in all stages of progression and in phenotypically normal carriers.
Usual age of onset is from 3-15 years, with an average of six years. The condition is often detected late in life because visual acuity remains good for many years. The atrophic stage usually occurs after 40 years. There is no known gender predilection. Many individuals initially are asymptomatic, with fundus lesions noted on examination. Visual symptoms include decreased acuity and metamorphopsia. These symptoms may worsen if the disease progresses to the atrophic stage.
This disorder has variable clinical expression. Some carriers have a normal examination and remain asymptomatic. Findings are usually bilateral and can be asymmetric.
Fundus appearance progresses through several stages, though all individuals may not progress beyond the early stages. Other individuals can skip from the earliest stages to an atrophic-appearing macula. Unilateral findings and multifocal lesions have been described [2].
Stage 1 (previtelliform stage) consists of normal macula or subtle RPE pigment changes, EOG is abnormal and visual acuity (VA) is 20/20.
Stage 2 (vitelliform stage) consists of well-circumscribed, 0.5-5 mm round, elevated, yellow or orange lesion bearing an egg-yolk appearance; usually centered on the fovea; may be multifocal; rest of the fundus has a normal appearance. VA is 20/20 to 20/50.
Stage 3 (pseudohypopyon stage) consists of yellow material breaks through the RPE which accumulate in the subretinal space in a cyst with a fluid level. The yellow material shifts with extended changes in position (60-90 min). This stage most often is found in the teenage years, but it has been described in individuals aged 8-38 years. VA is 20/20 to 20/50.
Stage 4 (vitelloruptive stage) consists of scrambled egg appearance due to break up of the uniform vitelliform lesion. Pigment clumping and early atrophic changes may be noted. Visual acuity may deteriorate moderately. VA is 20/20 to 20/100.
Stage 5 (atrophic stage) consists of disappearance of the yellow material over time and an area of RPE atrophy remains. This appearance is difficult to distinguish from other causes of macular degeneration. Visual acuity can deteriorate more markedly at this stage. VA may reduce to less than 20/200.
Stage 6 (CNVM/cicatricial stage) occurs after the atrophic stage, where choroidal neovascularisation may develop and leading to a whitish subretinal fibrous scar.
Best disease is autosomal dominant disorder with a variable penetrance [2]. Genetic linkage has mapped the disease to the long arm of chromosome 11 (11q12-q13) [3]. The abnormality is in the RPE, as noted on histopathology and electrophysiology testing. Lesions are restricted to the eye without any systemic associations. Petrukhin et al [5], identified the retina-specific gene and designated it as the VMD2. The protein encoded by this gene was proposed to be called bestrophin.
Weingeist et al [4], reported an abnormal accumulation of lipofuscin granules within the RPE. The RPE have degenerative changes in some cases and there may be secondary loss of photoreceptor cells. Marmorstein et al [6], demonstrated that bestrophin localised in the plasma membrane of the RPE cells may be responsible for EOG changes.
This disease has mixed prognosis. Some carriers never express their phenotype, while others never progress beyond the earliest stages and maintain better than 20/40 vision in both eyes. In general, most affected will maintain reading vision in at least one eye throughout life. In one study, 88% of patients retained 20/40 or better visual acuity and only 4% had 20/200 or worse visual acuity in the better eye.
The vitelliform stage has to be differentiated from adult foveo-macular dystrophy (AOFVD), central serous retinopathy with a fibrinous exudate, pigment epithelial detachment, coalescence of basal laminar drusen and solar retinopathy. AOFVD is a pattern dystrophy affecting mid to advanced aged adults which is associated with deposition of yellowish material in a region 0.25 to 1 disk diameter in size under the macula. This material appears to be lipofuscin by histopathologic examination, is auto-fluorescent and is not associated with profound decrease in the EOG. In the atrophic stage the various differential diagnoses are age-related macular degeneration (AMD), stargardt disease and chronic central serous chorio-retinopathy (CSC).
There is no known cure for the disease. The patient has to be observed for development of CNVM that may have to be treated using photodynamic therapy or intra-vitreal anti-vascular endothelial growth factors.
Conflicts of Interest
None identified
References
- 1.Deutman AF, Hoyng CB. Macular Dystrophies. In: Schachat AP, Hengst TC, editors. Medical Retina. 3rd ed. Mosby; London: 2001. p. 1225. [Google Scholar]
- 2.Deutman AF, Hoyng CB. Macular Dystrophies. In: Schachat AP, Hengst TC, editors. Medical Retina. 3rd ed. Mosby; London: 2001. p. 1226. [Google Scholar]
- 3.Stone EM, Nichols BE, Sterb LM, Kimura AE, Sheffield VC. Genetic linkage of vitelliform macular degeneration to chromosome 11q13. Nat Genet. 1992;1:246–250. doi: 10.1038/ng0792-246. [DOI] [PubMed] [Google Scholar]
- 4.Weingeist TA, Kobrin JL, Watzke RC. Histopathology of Best's macular dystrophy. Arch Ophthalmol. 1982;100:1108. doi: 10.1001/archopht.1982.01030040086016. [DOI] [PubMed] [Google Scholar]
- 5.Petrukhin K, Koisti MJ, Bakall B, Li W. Identification of the gene responsible for Best's macular dystrophy. Nature Genet. 1998;19:241–247. doi: 10.1038/915. [DOI] [PubMed] [Google Scholar]
- 6.Marmorstein LY, Rayborn M, Wang X, Hollyfiekd JJ, Petrukhin K. Bestrophin the product of Best vitelliform dystrophy gene (VMD2) , localises to the basolateral plasma membrane of the retinal pigment epithelium. Proc Nac Acad Sci. 2000;97:12758–12763. doi: 10.1073/pnas.220402097. [DOI] [PMC free article] [PubMed] [Google Scholar]