

Abstract
Norovirus full-genome sequencing is challenging due to sequence heterogeneity among genomes. Previous methods have relied on PCR amplification, which is problematic due to primer design, and transcriptome sequencing (RNA-Seq), which nonspecifically sequences all RNA, including host and bacterial RNA, in stool specimens. Target enrichment uses a panel of custom-designed 120-mer RNA baits that are complementary to all publicly available norovirus sequences, with multiple baits targeting each position of the genome, which overcomes the challenge of primer design. Norovirus genomes are enriched from stool RNA extracts to minimize the sequencing of nontarget RNA. SureSelect target enrichment and Illumina sequencing were used to sequence full genomes from 507 norovirus-positive stool samples with reverse transcription–real-time PCR cycle threshold (CT) values of 10 to 43. Sequencing on an Illumina MiSeq system in batches of 48 generated, on average, 81% on-target reads per sample and 100% genome coverage with >12,000-fold read depth. Samples included genotypes GI.1, GI.2, GI.3, GI.6, GI.7, GII.1, GII.2, GII.3, GII.4, GII.5, GII.6, GII.7, GII.13, GII.14, and GII.17. When outliers were accounted for, we generated >80% genome coverage for all positive samples, regardless of CT values. A total of 164 samples were tested in parallel with conventional PCR genotyping of the capsid shell domain; 164/164 samples were successfully sequenced, compared to 158/164 samples that were amplified by PCR. Four of the samples that failed capsid PCR analysis had low titers, which suggests that target enrichment is more sensitive than gel-based PCR. Two samples failed PCR due to primer mismatches; target enrichment uses multiple baits targeting each position, thus accommodating sequence heterogeneity among norovirus genomes.
INTRODUCTION
Norovirus is a leading cause of outbreaks of acute gastroenteritis (1, 2), with an estimated prevalence of 20% in cases of acute gastroenteritis in developed countries (3) and a large financial burden, associated with ward and hospital closures, in health care settings (4). In countries in which rotavirus vaccine has been introduced, norovirus is now the leading cause of medically attended gastroenteritis in children (5, 6).
Norovirus has a 7.5-kb single-stranded RNA genome organized into 3 open reading frames (ORFs), i.e., ORF1, ORF2, and ORF3. ORF1 encodes a nonstructural polyprotein that is cleaved posttranslationally and includes the RNA-dependent RNA polymerase. ORF2 encodes the major structural capsid protein, which is divided into shell (S) and protruding (P) domains. The P domain has two subdomains, P1 and P2. P2 is the most exposed antigenic site and contains immunogenic epitopes; consequently, it has the greatest sequence variation. ORF3 codes for a minor capsid protein.
Comparison of viral genetic sequences allows linking of previously unrecognized transmission events or exclusion of cases from an outbreak. Traditionally, norovirus genotyping has involved PCR amplification and capillary sequencing of partial regions of the polymerase and capsid sequences, followed by additional sequencing of the P2 region for outbreak investigations. This is a labor-intensive process that requires several rounds of PCR and sequencing, each requiring genogroup- or genotype-specific primers, and ultimately yields only partial genome sequences. Moreover, while the P2 domain can identify linked outbreak events with 64 to 73% specificity (assuming bootstrap support values of >70 or <70, respectively), the full capsid sequence can identify linked outbreak events with 100% specificity (7) and thus is more informative.
Whole-genome sequencing simplifies investigation of norovirus molecular epidemiology by generating all regions of interest in one step, thus allowing identification of the genotype, variant type, and full capsid sequence and negating the need for sequential PCR and sequencing reactions. However, unlike culture of bacteria (which can be isolated in pure culture), culture of norovirus is difficult (8). Moreover, as norovirus replicates within the host cell, viral nucleic acid extracts are contaminated by host DNA and, if obtained from clinical specimens, by DNA and RNA from enteric bacteria.
To date, norovirus sequencing from clinical material has been achieved by two methods, namely, sequencing of overlapping PCR fragments (9–12) and direct sequencing of total RNA (13–16). The former generates a pure viral template, which improves the quality of sequencing but requires multiple PCR amplifications; the latter necessitates great depth of sequencing to generate the target norovirus genome. Here we describe the application of a third method, SureSelect target enrichment (Agilent), which has been used successfully to generate full pathogen genomes for difficult-to-culture bacteria and DNA and RNA viruses directly from clinical samples (17–19). Norovirus genomes are enriched directly from stool RNA extracts by using a panel of custom-designed 120-mer RNA baits that are complementary to all publicly available norovirus sequences, with multiple baits targeting each position of the genome. This approach overcomes the problems of primer design in PCR and of nontarget sequencing in transcriptome sequencing (RNA-Seq).
MATERIALS AND METHODS
Samples.
A total of 507 norovirus-positive stool samples from 382 patients in four health care centers in the United Kingdom were processed for whole-genome sequencing. Samples included genotypes GI.1, GI.2, GI.3, GI.6, GI.7, GII.1, GII.2, GII.3, GII.4, GII.5, GII.6, GII.7, GII.13, GII.14, and GII.17, as detailed in Table 1. The presence of norovirus was verified in all samples using a multiplex, norovirus GI- and GII-specific, one-step, real-time reverse transcription-PCR (RT-qPCR) assay; the primer and probe sequences and cycling conditions have been described previously (24). For 78/507 samples provided by one of the centers, the presence of norovirus RNA was not verified in the reextracted residual specimens; for those samples, the RT-qPCR cycle threshold (CT) values correspond to the original extracts used as part of the diagnostic service. RT-qPCR CT values are used in this study as semiquantitative indicators of viral titers.
TABLE 1.
Metrics of norovirus whole-genome sequencing for all samples (total) and for each genotype
| Genotypea | No. of samples (%)b |
% OTRs (median [range]) | Read depth (median [range]) (fold) | Genome coverage (median [range]) (%) | CT (median [range]) | |||
|---|---|---|---|---|---|---|---|---|
| Sequenced | Pass | Suboptimal | Fail | |||||
| GI.1 | 2 | 2 (100) | 0 (0) | 0 (0) | 63.05 (43.85–82.25) | 11,194 (7,239–15,149) | 100 (100–100) | 31 (30–32) |
| GI.2 | 4 | 4 (100) | 0 (0) | 0 (0) | 77.60 (2.17–94.70) | 11,464 (379–21,843) | 100 (99–100) | 29 (24–33) |
| GI.3 | 15 | 15 (100) | 0 (0) | 0 (0) | 74.13 (1.08–93.25) | 13,157 (246–27,569) | 100 (90–100) | 27 (17–35) |
| GI.6 | 1 | 1 (100) | 0 (0) | 0 (0) | 86.56 (NA) | 8,642 (NA) | 100 (NA) | 29 (NA) |
| GI.7 | 1 | 1 (100) | 0 (0) | 0 (0) | 83.88 (NA) | 18,414 (NA) | 100 (NA) | 21 (NA) |
| GI.ut | 2 | 1 (50) | 0 (0) | 1 (50) | 40.34 (9.50–71.18) | 7,000 (42–13,957) | 91 (83–100) | 29 (23–35) |
| GII.1 | 3 | 3 (100) | 0 (0) | 0 (0) | 95.61 (20.06–97.04) | 11,990 (4,365–16,506) | 100 (99–100) | 15 (14–31) |
| GII.13 | 1 | 1 (100) | 0 (0) | 0 (0) | 77.44 (NA) | 10,043 (NA) | 100 (NA) | 21 (NA) |
| GII.14 | 6 | 6 (100) | 0 (0) | 0 (0) | 53.31 (4.20–81.60) | 10,238 (1,081–15,215) | 100 (100–100) | 27 (21–32) |
| GII.17 | 2 | 2 (100) | 0 (0) | 0 (0) | 63.30 (40.27–86.33) | 13,204 (8,598–17,811) | 100 (100–100) | 24 (21–27) |
| GII.2 | 24 | 21 (88) | 0 (0) | 3 (12.5) | 57.60 (0.60–99.47) | 4,717 (7–23,889) | 100 (64–100) | 24 (18–32) |
| GII.3 | 105 | 91 (87) | 3 (2.9) | 11 (10.5) | 85.00 (0.02–99.36) | 16,034 (7–38,843) | 100 (3–100) | 21 (10–38) |
| GII.4 | 281 | 250 (89) | 12 (4.3) | 19 (6.8) | 83.75 (0.02–99.63) | 12,465 (1–46,996) | 100 (5–100) | 22 (12–43) |
| GII.5 | 6 | 5 (83) | 0 (0) | 1 (16.7) | 70.21 (0.04–97.13) | 16,468 (1–29,488) | 100 (49–100) | 19 (16–23) |
| GII.6 | 40 | 38 (95) | 0 (0) | 2 (5) | 70.32 (0.45–98.23) | 9,356 (3–31,643) | 100 (22–100) | 21 (13–33) |
| GII.7 | 10 | 9 (90) | 0 (0) | 1 (10) | 53.14 (2.72–83.88) | 12,779 (2,106–26,914) | 100 (96–100) | 25 (22–30) |
| GII.ut | 4 | 3 (75) | 1 (25) | 0 (0) | 49.02 (0.59–92.61) | 11,356 (98–23,588) | 100 (94–100) | 25 (19–35) |
| NegEx | 2 | 0 (0) | 0 (0) | 2 (100) | 26.30 (16.18–36.42) | 42 (3–81) | 11 (9–12) | Not detected |
| Total | 509 | 453 (89) | 16 (3) | 40 (8) | 81.22 (0.02–99.63) | 12,227 (1–46,996) | 100 (3–100) | 22 (10–43) |
| Total excluding runs 30 and 31 | 413 | 381 (92) | 16 (4) | 16 (4) | 84.45 (0.02–99.63) | 14,341 (1–46,996) | 100 (13–100) | 22 (10–40) |
GI.ut, genogroup I untypeable; GII.ut, genogroup II untypeable; NegEx, negative control.
Pass, >90% genome coverage and >100-fold read depth; suboptimal, >90% genome coverage or >100-fold read depth; fail, <90% genome coverage and <100-fold read depth; NA, range not applicable for the single sample; CT, real-time PCR cycle threshold.
All specimens were residual diagnostic specimens obtained from patients with confirmed norovirus infections. Specimens were submitted to the University College London (UCL) Infection DNA Bank for use in this study. All samples were supplied to the study in an anonymized form; the use of the specimens for research was approved by the National Research Ethics Service (NRES) Committee London-Fulham (Research Ethics Committee [REC] reference no. 12/LO/1089). All stool samples were stored at −80°C between diagnostic testing and RNA extraction for full-genome sequencing.
A total of 164 stool samples were genotyped using capsid PCR and Sanger sequencing in parallel with SureSelect target enrichment whole-genome sequencing. PCR primer sequences and cycling conditions for genotyping have been described previously (submitted for publication). Briefly, GI- or GII-specific primers were used to amplify a 597- or 468-nucleotide region of the norovirus capsid shell domain, respectively; amplicons were capillary sequenced in the forward and reverse directions. Generated sequences were submitted to the norovirus genotyping tool to identify the capsid genotype (20).
RNA extraction.
RNA was purified from 200 μl of a clarified 10% (wt/vol) stool suspension using the Qiagen EZ1 virus minikit or the QIAsymphony DSP virus/pathogen kit, with a 90-μl elution volume. All purified RNA was stored at −80°C prior to cDNA synthesis.
cDNA synthesis.
RNA extracts were concentrated to 11 μl using a vacuum centrifuge at 65°C prior to first-strand cDNA synthesis. First-strand cDNA was synthesized using random primers and SuperScript III (Life Technologies), according to the manufacturer's instructions. Briefly, 1 μl of 10 mM (each) deoxynucleoside triphosphate (dNTP) mixture and 1 μl of 3 μg/ml random primers were incubated with 11 μl of RNA for 5 min at 65°C to anneal the primers to the RNA template, followed by incubation on ice for 1 min. RNA-primer templates were mixed with 4 μl of 5× first-strand buffer, 1 μl of 0.1 M dithiothreitol (DTT), 1 μl of RNaseOUT, and 1 μl of SuperScript III at 25°C for 5 min, followed by cDNA synthesis at 50°C for 1 h and enzyme inactivation at 70°C for 15 min. Second-strand cDNA was synthesized using the mRNA Second Strand Synthesis module (NEBNext), according to the manufacturer's instructions. Briefly, 20 μl of first-strand cDNA was incubated with 48 μl of water, 8 μl of 10× second-strand buffer, and 4 μl of second-strand enzyme mixture at 16°C for 2.5 h. Double-stranded cDNA was purified and concentrated with Genomic DNA Clean and Concentrator (Zymo Research), according to the manufacturer's instructions, with a 30-μl elution volume, and was quantified with a Qubit dsDNA high-sensitivity kit (Invitrogen).
SureSelect target enrichment. (i) RNA bait design.
Overlapping 120-mer RNA baits complementary to and spanning the length of 622 norovirus partial or complete genomes from GenBank were designed using an in-house PERL script. Briefly, a 120-nucleotide sliding window was scanned along each reference genome at intervals of 10 nucleotides. If a 120-mer was sufficiently different from other 120-mer sequences in the bait set (as assessed by BLAT [21]), then it was retained in the bait set; otherwise, that 120-mer was discarded. Therefore, the bait set spans the diversity in all of the included reference genomes. The bait set is available upon request. The reference genomes included samples from polymerase genotypes GI.P1, GI.P2, GI.P3, GI.P4, GI.P6, GI.P8, GI.Pb, GI.Pc, GI.Pd, GI.Pf, GII.P1, GII.P2, GII.P3, GII.P4, GII.P5, GII.P6, GII.P7, GII.P8, GII.P11, GII.P12, GII.P15, GII.P16, GII.P17, GII.P18, GII.P21, GII.P22, GII.Pc, GII.Pe, GII.Pg, GII.Pp, GIII, GIV, GV, and GVI and capsid genotypes GI.1, GI.2, GI.3, GI.4, GI.5, GI.6, GI.8, GII.2, GII.3, GII.4, GII.5, GII.6, GII.7, GII.8, GII.10, GII.11, GII.12, GII.13, GII.14, GII.15, GII.16, GII.17, GII.18, GII.21, GII.22, GIII, GIV, GV, and GVI. The GII.4 reference genomes included samples from all major GII.4 strains, including CHDC1970s, Bristol 1993, Camberwell 1994, US95/96, Farmington Hills 2002, Lanzhou 2002, Asia 2003, Hunter 2004, Yerseke 2006a, Den Haag 2006b, Osaka 2007, Apeldoorn 2007, New Orleans 2009, and Sydney 2012. The custom-designed norovirus bait library was uploaded to Agilent SureDesign and synthesized by Agilent Biotechnologies.
(ii) Library preparation, hybridization, and enrichment.
Norovirus cDNA samples were quantified, and carrier G147 human genomic DNA male (Promega) was added if necessary to obtain a total of 200 ng. All DNA samples were mechanically sheared for 150 s, using a Covaris E210 focused ultrasonicator (duty cycle, 5%; peak incident power, 175 W; cycles per burst, 200), to yield a fragment size of approximately 270 bp. End repair, nontemplated addition of the 3′-A adapter, ligation, hybridization, enrichment PCR, and all postreaction cleanup steps were performed according to the SureSelectXT Illumina paired-end sequencing library protocol. All recommended quality control steps were performed between steps.
Negative controls.
All RNA extraction batches included a negative extract control, consisting of sterile Qiagen buffer ASL extracted with the Qiagen EZ1 virus minikit alongside stool samples. All negative extracts were tested by norovirus-specific real-time RT-qPCR to verify the absence of contaminating RNA. To determine the level of contaminating norovirus RNA in the sequencing pipeline, two negative extracts were processed for sequencing.
Illumina sequencing.
Samples were multiplexed with 48 samples per run. Paired-end sequencing was performed on an Illumina MiSeq sequencing platform, with the 500-cycle v2 reagent kit. Base calling and sample demultiplexing were performed as standard for the MiSeq platform, producing paired FASTQ files for each sample.
Sequence assembly.
All assemblies were performed in CLC Genomics Workbench v8, as summarized in Fig. 1. All reads were quality trimmed, and adapter sequences were removed. Trimmed reads were mapped to a curated reference list consisting of all norovirus complete-genome and complete-gene sequences in GenBank as of 14 July 2015 (n = 688). All paired-end reads mapping to the reference list (filtered reads) were taken forward to de novo assembly using Workbench default parameters and a minimum contig length of 200 nucleotides. Contigs generated from the de novo assembly were aligned to a single GenBank reference sequence of the relevant genotype, to check the orientation of the contig and, when multiple contig sequences were generated, the position of each contig relevant to the reference. Multiple contig sequences were joined on the basis of overlapping nucleotide sequences or with a manually inserted gap. All trimmed reads (prefiltering) were mapped to the full-length contig sequence generated from the de novo assembly, to generate a final consensus sequence. Areas of low coverage (<10-fold) were assigned the ambiguity symbol N.
FIG 1.

Schematic of the norovirus full-genome assembly pipeline.
Simulated mixed infection.
To assess whether a reliable consensus sequence could be generated from a mixed infection, the reads generated from two single infections (one GII.3 and one GII.4) were merged into a single assembly pipeline. The consensus sequences generated from the single infections (original) and the mixed infection (simulated) were aligned to identify the number of differences between the two consensus sequences.
Statistical analysis.
All statistical analysis was performed with SPSS v23, using two-tailed tests at the 5% significance level. The differences in the percentage of on-target reads (OTRs), read depth, and percent genome coverage among norovirus genotypes and in PCR CT values among pass, suboptimal, and failed samples were tested by Kruskal-Wallis analysis of variance (ANOVA), with pairwise multiple comparisons of significant results and P values adjusted for multiple comparisons. The relationships between PCR CT values and percentage of OTRs, read depth, and percent genome coverage were assessed by Spearman's correlations.
A simple linear regression model (independent variable, PCR CT value; dependent variable, logit-transformed percent genome coverage) was fitted to generate prediction intervals for percent genome coverage from the PCR CT values. The percent genome coverage was transformed using the formula tr_genome = [% genome coverage × (n − 1) + 0.5]/n, where n is the total number of sequences, to ensure that there were no proportions of 0 or 1, and then were transformed again using the logit function logit-transformed % genome coverage = log[tr_genome/(1 − tr_genome)], where log is the natural logarithm with base e. Outliers (highlighted in Fig. SA3 in the supplemental material) were excluded from regression analysis.
RESULTS
Overall sequencing outcomes.
Since the aim was to generate full genome sequences, we defined the cutoff value for sequencing success as >90% coverage of the full norovirus genome with >100-fold mean read depth, to ensure robust consensus sequences. Samples that met only one of these criteria were categorized as suboptimal, and those that did not meet either criteria were considered a fail.
Of 507 samples across all sampled genotypes, 453 (89%) passed, i.e., had >90% genome coverage and >100-fold read depth (Table 1 and Fig. 2; also see Fig. SA1 in the supplemental material). In total, 93% of samples had genome coverage of >90% at any depth. A median of 81.22% of the total sequencing reads generated for each sample mapped to the norovirus genome, referred to as the percentage of OTRs. On average, 100% of the full genome was covered, with a median read depth of 12,227-fold (Table 1). There were no significant differences in the percentage of OTRs (P = 0.127), mean read depth (P = 0.398), or percent genome coverage (P = 0.203) among norovirus genotypes (Fig. 3a to c).
FIG 2.
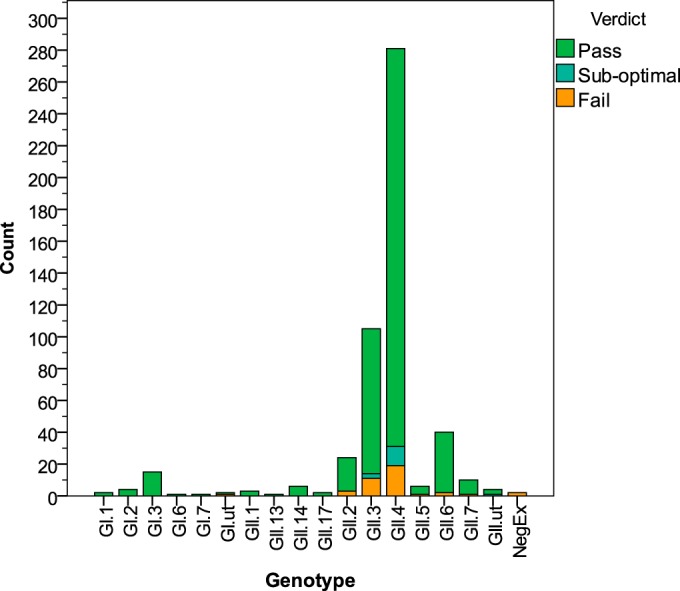
Numbers of samples sequenced according to norovirus genotype, classified by sequencing outcome. Pass, >90% genome coverage and >100-fold read depth; suboptimal, >90% genome coverage or >100-fold read depth; fail, <90% genome coverage and <100-fold read depth. Genotype refers to capsid genotype only.
FIG 3.

Norovirus full-genome sequencing outcome metrics according to norovirus genotype (a to c) and RT-qPCR CT values (d to f). OTR, on-target reads; CT, cycle threshold; NegEx, negative control. Red lines, median values.
Significant correlations were found between the percentage of OTRs and read depth (R = 0.757; P < 0.001) (see Fig. SA2 in the supplemental material) and between PCR CT values and (i) percentage of OTRs (R = −0.536; P < 0.001), (ii) read depth (R = −0.468; P < 0.001), and (iii) percent genome coverage (R = −0.223; P < 0.001) (Fig. 3d to f). It follows that there were significant differences in PCR CT values between samples that passed and those that were suboptimal (P < 0.001) or failed (P < 0.001), with median CT values of 22, 32, and 32, respectively (Fig. 4). There is an inverse relationship between CT values and viral loads (22); therefore, samples with lower CT values (higher viral titers) demonstrated greater percentage of OTRs, read depth, and genome coverage.
FIG 4.
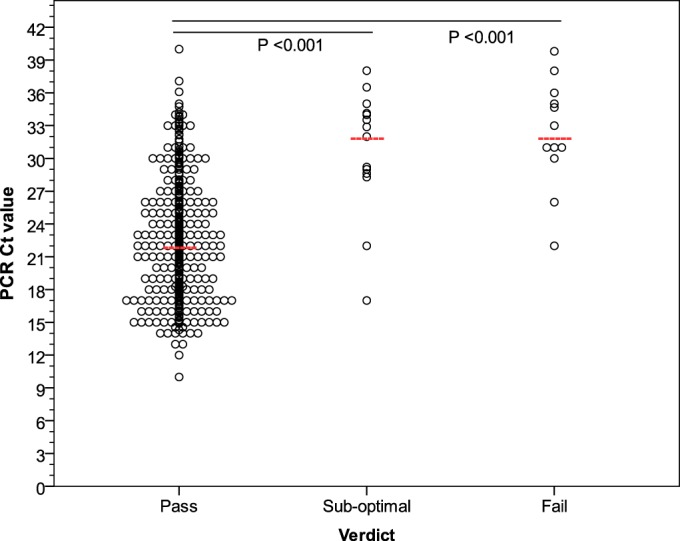
RT-qPCR CT values for all samples, excluding runs 30 and 31 (n = 413), sequenced with SureSelect target enrichment. Pass, >90% genome coverage and >100-fold read depth; suboptimal, >90% genome coverage or >100-fold read depth; fail, <90% genome coverage and <100-fold read depth. Red lines, median values.
Predicted genome coverage.
The estimated linear regression model is y = 7.432 − 0.059x, where the dependent variable y is the logit-transformed genome coverage proportion and the independent variable x is the PCR CT value (n = 477, R2 = 0.058; P < 0.001). Prediction intervals generated using the linear regression model predicted that stool samples with norovirus RT-qPCR CT values of <40 would generate 92 to 100% of the full genome sequence, with 95% certainty (Fig. 5).
FIG 5.
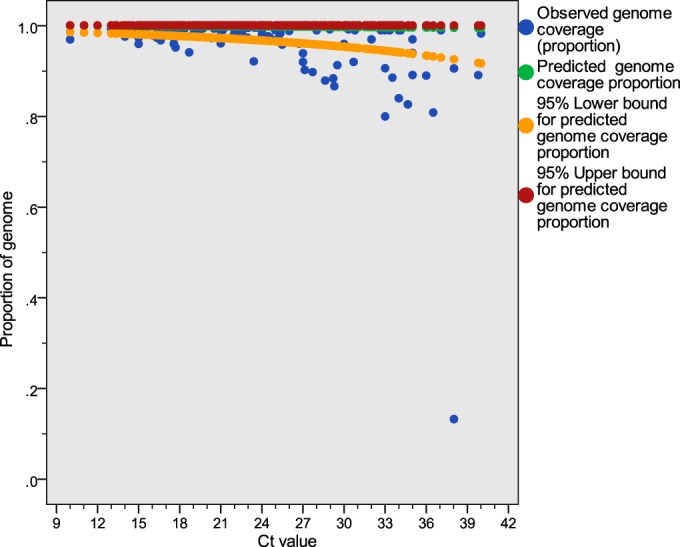
Observed and predicted genome coverage values with 95% prediction intervals, excluding outliers identified in Fig. SA3 in the supplemental material. The fitted linear regression model is y = 7.432 − 0.059x, where the dependent variable y is the logit-transformed genome coverage proportion and the independent variable x is the PCR CT value (n = 477).
Failed samples.
The outliers in Fig. 3f were dominated by samples from two sequencing runs (runs 30 and 31) (see Fig. SA3 in the supplemental material), which were known to have had processing problems during cDNA preparation. Six of the 16 samples with CT values of <30 and genome coverage of <80% had sufficient residual specimens for repeat testing; all of those samples passed in repeat testing.
Three samples (highlighted in Fig. SA3 and detailed in Table SA1 in the supplemental material) generated unexpectedly low values for percent genome coverage (range, 49 to 73%), given their RT-qPCR CT values (range, 22 to 29), but were not part of sequencing run 30 or 31. Sequences from all three samples were fragmented throughout ORF1, with ORF3 and ORF2 absent downstream of the P1 and P2 protruding domains (see Fig. SA4 in the supplemental material). In all three cases, the percentage of OTRs (0.01, 2.53, and 6.76%) and average read depth (1-, 120-, and 137-fold) were low for ORF1, despite apparently good CT values. Coverage of ORF1 and the 5′ end of ORF2 was sufficient to confirm two samples as GII.4 and one as GII.5 using the norovirus genotyping tool; we have shown good sequencing outcomes for both genotypes in other samples (Table 1). It is not possible to exclude the possibility of a novel recombinant strain, with recombination at the P1-P2 junction in ORF2 and subsequent failure due to missing complementary baits in the enrichment; if this were the case, however, we would expect to see good coverage of the enriched region (in this case ORF1), which we do not. Moreover, all three samples had been reextracted at referring centers, and the CT values supplied were obtained from PCRs carried out with the original diagnostic extracts. This, combined with the low coverage of ORF1, suggests that extraction failure at the local hospitals may explain the unexpected sequencing failures. It has not been possible to test either possibility, since none of the original samples remains.
Low-titer samples.
Seven samples generated full genome sequences despite low viral titers (PCR CT values of ≥36). To determine whether those samples had misleadingly high CT values due to a mismatch in the RT-qPCR primer target region, the seven genome sequences were aligned with the RT-qPCR primer and probe sequences used to generate the CT values. There were no mismatches in the primer or probe sites (see Fig. SA5 in the supplemental material), which suggests that the samples were genuinely low-titer samples and confirms the sensitivity of the method for low-titer samples.
Comparison with capsid genotyping.
A total of 96% (158/164 samples) and 100% (164/164 samples) of the samples processed in parallel were successfully genotyped by PCR with Sanger sequencing and by our method, respectively (see Table SA2 in the supplemental material). For the 158 samples typed by both methods, there was 100% agreement in the respective genotypes. Of the 6 samples that failed capsid typing by PCR, 4 were GII.4, 1 was GII.7, and 1 was GI.3 (see Table SA3 in the supplemental material). Two of the failed samples, with CT values of 20 and 27, had mismatches at the genotyping primer sites (see Fig. SA6 in the supplemental material), which accounted for the genotyping failures in those instances. The remaining four of the six samples that failed genotyping had CT values of >30 (range, 31 to 37), which suggests that the genotyping PCR is less sensitive than sequencing by target enrichment.
Contamination.
Two negative-extract samples, consisting of buffer ASL that was treated in the same way as, and alongside, the stool samples, were negative for norovirus RNA by RT-qPCR. However, target enrichment and sequencing generated 16 to 36% OTRs, with 3- to 81-fold read depth. The genome coverage values for the samples were only 9 and 12%, with reads fragmented across the genome (see Fig. SA7 and SA8 in the supplemental material). The mapped regions did not correspond to PCR amplicon sites.
Mixed infections.
Three samples (3/507 samples) were identified as having sequences from more than one genotype during the assembly pipeline (see Table SA4 in the supplemental material). For two of the samples, the mixed infections were evident during the step of mapping to the reference list in the de novo pipeline (Fig. 1), in which reads were mapped to reference sequences corresponding to multiple norovirus genotypes (see Table SA4 in the supplemental material). For the third sample, the mixed infection was evident during the step of aligning contigs to a single reference of the appropriate genotype, in which a full-length contig was mapped to the reference sequence at ORF1 but not at ORF2 and ORF3. Comparison of the consensus sequences generated from a single infection and from a simulated mixed infection showed 178 to 332 single-nucleotide polymorphisms (SNPs) and 95.53 to 97.61% sequence identity between the consensus sequences from the single- and mixed-infection data sets (see Table SA5 in the supplemental material).
Turnaround times and costs.
The turnaround time associated with full-genome sequencing by SureSelect target enrichment was 6 days, 3 days longer than with genotyping (RNA-dependent RNA polymerase and capsid regions) by PCR and Sanger sequencing, with an extra associated cost of £54 when reagents are purchased in bulk (Table 2).
TABLE 2.
Turnaround times and costs associated with norovirus genotyping by PCR and Sanger sequencing versus SureSelect target enrichment full-genome sequencing
| Genotyping method | Hands-on time (h) | Total turnaround time (days) | Reagent costs per sample (£) |
|---|---|---|---|
| PCR and Sanger sequencinga | 7 | 3 | 32 |
| Full-genome sequencing with SureSelect target enrichment | 11.5 | 6 | 86–93b |
PCR amplification of three sites of interest for norovirus genotyping, i.e., RNA-dependent RNA polymerase, capsid shell domain, and capsid P2 domain, including one round of nested PCR, assuming that the RNA-dependent RNA polymerase and capsid shell domain targets are amplified and sequenced simultaneously.
Costs based on batches of 96 or 48 samples and sequencing with an Illumina MiSeq system.
DISCUSSION
Target enrichment is a highly effective method for sequencing full norovirus genomes across genotypes, with high read depth values (averaging >12,000-fold) and complete or almost complete genomes in 89% of samples. We report median genome coverage of 100% across all sequenced samples and, when outliers were accounted for, >80% genome coverage regardless of the viral titer.
Despite good molecular practice, however, low-level contamination did occur. Since negative extracts were RT-qPCR negative but target enrichment yielded reads that mapped to the norovirus genome, we suspect the source of contamination to be the automated equipment used for target enrichment and sequencing library preparation. In the context of norovirus-positive specimens, the level of contamination was low; reads were fragmented and mapped to only 9 to 12% of the genome with <100-fold read depth, significantly below the observed median percent genome coverage and read depth values seen for norovirus-positive samples (100% and >12,000-fold, respectively) and below the 95% prediction intervals for percent genome coverage (92 to 100% for samples with CT values of <40). These findings support our acceptance criteria for downstream analysis, i.e., >100-fold read depth and >90% genome coverage. When a complete genome sequence is not critical for downstream analysis, >60% genome coverage would be acceptable if the read depth was >100-fold, based on the 95% prediction intervals. Due to the potential for low-level contamination, however, specimens for which norovirus RNA is not detectable by real-time PCR should not be sequenced.
Previous reports described norovirus whole-genome sequencing with overlapping PCR amplicons or RNA-Seq, the findings of which are summarized in Table SA6 in the supplemental material. PCR-based methods yield high read depth values; however, due to sequence heterogeneity among genotypes, primers generally need to be genotype specific (9). Although broad-range primers were reported by Cotten et al. (10), the approach retained a limited success rate; full genome sequences were amplified from comparable proportions of samples of GII.13 (83% versus 100% in this study), GII.6 (88% versus 95%), and GII.4 (92% versus 89% or 93% irrespective of read depth). PCR fared worse, however, recovering fewer full genomes from GI (20% versus 100% in this study), GII.2 (40% versus 88%), GII.3 (77% versus 87% or 90% irrespective of read depth), and GII.7 (0% versus 90%). Norovirus whole-genome sequencing from a single 7.5-kb amplicon was also described and was used to generate 25 full-genome sequences (23); the authors did not report the success rate using this approach, however, and it is generally very difficult to amplify fragments of such a size. Here we report complete or nearly complete genome sequences in 93% of processed samples. In target enrichment, baits are designed using all publically available norovirus sequences, across all GI and GII genotypes; unlike PCR, which uses a single primer at each target site, multiple baits are designed to cover each position in the genome, thus accounting for sequence variations among norovirus genomes. This allows unbiased sequencing across known genotypes in a single reaction. A disadvantage of the method is that it may fail to generate sequences for a newly emerging genotype when the existing baits are a poor match.
Whole-transcriptome sequencing (RNA-Seq) involves sequencing of the total RNA or mRNA content of a stool specimen. The advantage of RNA-Seq is that there is no requirement for PCR primers; therefore, it is completely unbiased. Although all whole genomes determined by RNA-Seq that have been reported to date are predominantly GII.4, it is theoretically possible to sequence all genotypes with equal success, as evidenced by the work of Bavelaar et al., who successfully sequenced five non-GII.4 genomes (16). The data generated by RNA-Seq are sufficient to generate almost-complete norovirus genome sequences; 40 to 100% of reported samples achieved >90% genome coverage (13–16) (summarized in Table SA6 in the supplemental material). However, the median percentage of OTRs across all reported samples was only 2 to 3% with a MiSeq or HiSeq system (13, 15) or 28% with an Ion Torrent PGM system (16), compared to 81% OTRs with SureSelect target enrichment. The large proportion of nontarget data obtained using RNA-Seq makes the technique uneconomical and, critically, results in low read depth values, i.e., on average, only 9- to 259-fold using a MiSeq or HiSeq system (13–15) or 1,309-fold using an Ion Torrent PGM system (16). In contrast, the median read depth using target enrichment is >12,000-fold, which allows large sample batches to be sequenced in a single MiSeq run and allows downstream analysis of minority variants.
Our de novo assembly pipeline identified mixed-genotype infections in three samples. However, with as many as 332 SNPs among the consensus sequences generated from single infections and a simulated mixed infection, we suggest that a reliable consensus sequence cannot be generated using this assembly pipeline. This is due to mismapping of reads in relatively conserved regions, as evidenced by the majority of SNPs being found in ORF1 (163/178 SNPs and 284/332 SNPs in the GII.3 and GII.4 consensus sequences, respectively). Thus, while this pipeline can identify infections with a mixture of genotypes, an alternative approach is required for assembly and generation of the consensus sequence, possibly involving the use of minority variants and haplotype reconstruction.
We have shown target enrichment to be superior to PCR capsid amplification for genotyping; all samples (164/164 samples) that were processed in parallel successfully generated genome sequences by target enrichment, whereas 96% (158/164 samples) were successfully amplified by capsid typing PCR. Four of the six samples that failed capsid genotyping but were sequenced with target enrichment had low norovirus titers (based on PCR CT values), which suggests that target enrichment is more sensitive than conventional genotyping methods. The remaining two failed samples had primer mismatches that accounted for the amplification failures. Target enrichment overcomes the limitations of primer design by allowing multiple baits with different sequences to target each region of the genome, thus accounting for sequence heterogeneity in a way that PCR primers cannot.
Unlike classic genotyping, which requires sequential PCR and sequencing reactions that yield only fragments of the genome, full-genome sequencing can, in a single reaction, provide the RNA polymerase and capsid sequences, which are important for genotyping and also can identify recombinations and reveal minority variants in an intrahost viral population. The cost of whole-genome sequencing with target enrichment is around £50 more expensive than PCR genotyping of the capsid and polymerase genes. However, whole-genome sequencing using overlapping amplicons is comparable in cost to enrichment methods. The turnaround time for the target enrichment method is 6 days, compared to 3 days for capsid and polymerase genotyping. The hands-on time for semiautomated target enrichment is 4 h more than that for conventional genotyping and comparable to that for RNA-Seq. A current drawback is the need for batch processing of samples to achieve the costs savings. This would be feasible for a regional sequencing service or a named study but might be difficult for a diagnostic laboratory. Further developments to shorten hybridization and sequencing times and to enable random-access processing would address these drawbacks.
The advancement of sequencing techniques, from PCR with capillary sequencing to target enrichment with deep sequencing, facilitates the use of full norovirus genomes in clinical practice. In conjunction with growing expertise, lower costs, and faster turnaround times, full genomes can be sequenced for under £100 in less than 1 week; this makes full-genome sequencing a reality not just for academic settings but also for informing public health practice in real time.
Supplementary Material
ACKNOWLEDGMENTS
We thank the Great Ormond Street Hospital Virology Department, the Royal Free Hospital Virology Department, the Norfolk and Norwich University Hospital Microbiology Department, and the Public Health England Virus Reference Department for supplying specimens for sequencing.
We declare no conflicts of interest.
Funding Statement
This work was funded by the PATHSEEK FP7 EU grant. PATHSEEK is funded by the European Union's Seventh Programme for research, technological development, and demonstration under grant agreement no. 304875. J.R.B. is funded by a National Institute for Health Research (NIHR) doctoral fellowship (NIHR-HCS-D12-03-15). J.R.B. and D.S. are supported by the NIHR Biomedical Research Centre (BRC) at Great Ormond Street Hospital for Children NHS Foundation Trust and University College London (UCL). J.B. receives funding from the NIHR UCL/UCLH BRC. We acknowledge the infrastructure support from the UCL Pathogen Genomics Unit (PGU) the NIHR UCL/UCLH BRC and the UCL MRC CMMV. The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JCM.01052-16.
REFERENCES
- 1.de Wit MA, Koopmans MP, Kortbeek LM, Wannet WJ, Vinje J, van Leusden F, Bartelds AI, van Duynhoven YT. 2001. Sensor, a population-based cohort study on gastroenteritis in the Netherlands: incidence and etiology. Am J Epidemiol 154:666–674. doi: 10.1093/aje/154.7.666. [DOI] [PubMed] [Google Scholar]
- 2.Scallan E, Hoekstra RM, Angulo FJ, Tauxe RV, Widdowson MA, Roy SL, Jones JL, Griffin PM. 2011. Foodborne illness acquired in the United States: major pathogens. Emerg Infect Dis 17:7–15. doi: 10.3201/eid1701.P11101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ahmed SM, Hall AJ, Robinson AE, Verhoef L, Premkumar P, Parashar UD, Koopmans M, Lopman BA. 2014. Global prevalence of norovirus in cases of gastroenteritis: a systematic review and meta-analysis. Lancet Infect Dis 14:725–730. doi: 10.1016/S1473-3099(14)70767-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lopman BA, Reacher MH, Vipond IB, Hill D, Perry C, Halladay T, Brown DW, Edmunds WJ, Sarangi J. 2004. Epidemiology and cost of nosocomial gastroenteritis, Avon, England, 2002–2003. Emerg Infect Dis 10:1827–1834. doi: 10.3201/eid1010.030941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Koo HL, Neill FH, Estes MK, Munoz FM, Cameron A, Dupont HL, Atmar RL. 2013. Noroviruses: the most common pediatric viral enteric pathogen at a large university hospital after introduction of rotavirus vaccination. J Pediatr Infect Dis Soc 2:57–60. doi: 10.1093/jpids/pis070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Payne DC, Vinje J, Szilagyi PG, Edwards KM, Staat MA, Weinberg GA, Hall CB, Chappell J, Bernstein DI, Curns AT, Wikswo M, Shirley SH, Hall AJ, Lopman B, Parashar UD. 2013. Norovirus and medically attended gastroenteritis in U.S. children. N Engl J Med 368:1121–1130. doi: 10.1056/NEJMsa1206589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Verhoef L, Williams KP, Kroneman A, Sobral B, van Pelt W, Koopmans M. 2012. Selection of a phylogenetically informative region of the norovirus genome for outbreak linkage. Virus Genes 44:8–18. doi: 10.1007/s11262-011-0673-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jones MK, Watanabe M, Zhu S, Graves CL, Keyes LR, Grau KR, Gonzalez-Hernandez MB, Iovine NM, Wobus CE, Vinje J, Tibbetts SA, Wallet SM, Karst SM. 2014. Enteric bacteria promote human and mouse norovirus infection of B cells. Science 346:755–759. doi: 10.1126/science.1257147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kundu S, Lockwood J, Depledge DP, Chaudhry Y, Aston A, Rao K, Hartley JC, Goodfellow I, Breuer J. 2013. Next-generation whole genome sequencing identifies the direction of norovirus transmission in linked patients. Clin Infect Dis 57:407–414. doi: 10.1093/cid/cit287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cotten M, Petrova V, Phan MV, Rabaa MA, Watson SJ, Ong SH, Kellam P, Baker S. 2014. Deep sequencing of norovirus genomes defines evolutionary patterns in an urban tropical setting. J Virol 88:11056–11069. doi: 10.1128/JVI.01333-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Won YJ, Park JW, Han SH, Cho HG, Kang LH, Lee SG, Ryu SR, Paik SY. 2013. Full-genomic analysis of a human norovirus recombinant GII.12/13 novel strain isolated from South Korea. PLoS One 8:e85063. doi: 10.1371/journal.pone.0085063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chhabra P, Walimbe AM, Chitambar SD. 2010. Complete genome characterization of genogroup II norovirus strains from India: evidence of recombination in ORF2/3 overlap. Infect Genet Evol 10:1101–1109. doi: 10.1016/j.meegid.2010.07.007. [DOI] [PubMed] [Google Scholar]
- 13.Nakamura S, Yang CS, Sakon N, Ueda M, Tougan T, Yamashita A, Goto N, Takahashi K, Yasunaga T, Ikuta K, Mizutani T, Okamoto Y, Tagami M, Morita R, Maeda N, Kawai J, Hayashizaki Y, Nagai Y, Horii T, Iida T, Nakaya T. 2009. Direct metagenomic detection of viral pathogens in nasal and fecal specimens using an unbiased high-throughput sequencing approach. PLoS One 4:e4219. doi: 10.1371/journal.pone.0004219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wong TH, Dearlove BL, Hedge J, Giess AP, Piazza P, Trebes A, Paul J, Smit E, Smith EG, Sutton JK, Wilcox MH, Dingle KE, Peto TE, Crook DW, Wilson DJ, Wyllie DH. 2013. Whole genome sequencing and de novo assembly identifies Sydney-like variant noroviruses and recombinants during the winter 2012/2013 outbreak in England. Virol J 10:335. doi: 10.1186/1743-422X-10-335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Batty EM, Wong TH, Trebes A, Argoud K, Attar M, Buck D, Ip CL, Golubchik T, Cule M, Bowden R, Manganis C, Klenerman P, Barnes E, Walker AS, Wyllie DH, Wilson DJ, Dingle KE, Peto TE, Crook DW, Piazza P. 2013. A modified RNA-Seq approach for whole genome sequencing of RNA viruses from faecal and blood samples. PLoS One 8:e66129. doi: 10.1371/journal.pone.0066129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bavelaar HH, Rahamat-Langendoen J, Niesters HG, Zoll J, Melchers WJ. 2015. Whole genome sequencing of fecal samples as a tool for the diagnosis and genetic characterization of norovirus. J Clin Virol 72:122–125. doi: 10.1016/j.jcv.2015.10.003. [DOI] [PubMed] [Google Scholar]
- 17.Brown AC, Bryant JM, Einer-Jensen K, Holdstock J, Houniet DT, Chan JZ, Depledge DP, Nikolayevskyy V, Broda A, Stone MJ, Christiansen MT, Williams R, McAndrew MB, Tutill H, Brown J, Melzer M, Rosmarin C, McHugh TD, Shorten RJ, Drobniewski F, Speight G, Breuer J. 2015. Rapid whole-genome sequencing of Mycobacterium tuberculosis isolates directly from clinical samples. J Clin Microbiol 53:2230–2237. doi: 10.1128/JCM.00486-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Christiansen MT, Brown AC, Kundu S, Tutill HJ, Williams R, Brown JR, Holdstock J, Holland MJ, Stevenson S, Dave J, Tong CY, Einer-Jensen K, Depledge DP, Breuer J. 2014. Whole-genome enrichment and sequencing of Chlamydia trachomatis directly from clinical samples. BMC Infect Dis 14:591. doi: 10.1186/s12879-014-0591-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Depledge DP, Palser AL, Watson SJ, Lai IY, Gray ER, Grant P, Kanda RK, Leproust E, Kellam P, Breuer J. 2011. Specific capture and whole-genome sequencing of viruses from clinical samples. PLoS One 6:e27805. doi: 10.1371/journal.pone.0027805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kroneman A, Vennema H, Deforche K, Avoort HVD, Penaranda S, Oberste MS, Vinje J, Koopmans M. 2011. An automated genotyping tool for enteroviruses and noroviruses. J Clin Virol 51:121–125. doi: 10.1016/j.jcv.2011.03.006. [DOI] [PubMed] [Google Scholar]
- 21.Kent WJ. 2002. BLAT: the BLAST-like alignment tool. Genome Res 12:656–664. doi: 10.1101/gr.229202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Brown JR, Gilmour K, Breuer J. 2016. Norovirus infections occur in B-cell-deficient patients. Clin Infect Dis 62:1136–1138. doi: 10.1093/cid/ciw060. [DOI] [PubMed] [Google Scholar]
- 23.Eden JS, Tanaka MM, Boni MF, Rawlinson WD, White PA. 2013. Recombination within the pandemic norovirus GII.4 lineage. J Virol 87:6270–6282. doi: 10.1128/JVI.03464-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Brown JR, Shah D, Breuer J. Viral gastrointestinal infections and norovirus genotypes in a paediatric UK hospital, 2014–2015 J Clin Virol, in press. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.